

Abstract
Chimeric antigen receptors (CAR)-T cell therapy has recently made promising advances towards treatment of B-cell malignancies. This approach makes use of an antibody-derived single chain variable fragment (scFv)-based CAR to target the CD19 antigen. Currently scFvs are the most common strategy for creation of CARs, but tumor cells can also be targeted using non-antibody based approaches with designs focused on the interaction between natural receptors and their ligands. This emerging strategy has been used in unique ways to target multiple tumor types, including solid and haematological malignancies. In this review, we will highlight the performance of receptor-ligand combinations as designs for CARs to treat cancer, with a particular focus on haematologic malignancies.
Keywords: NKG2D, cytokines, cell therapy, immunotherapy, T cell
Introduction
The goal of T cell engineering is to generate large numbers of tumor-specific T cells that attack the tumor directly and promote a potent anti-tumor immune response in the patient. The earliest clinical trials of engineered T cells in cancer relied on the expression of cloned T cell receptors (TCRs) that were targeted against tumor antigen peptides presented by MHC molecules. However, one disadvantage of this strategy was the ability of tumor cells to downregulate MHC class I expression, therefore limiting the effectiveness of TCR-engineered T cells. The next step in development of T cell engineering was the creation of artificial receptors that were highly specific for tumor antigens in a MHC-independent manner. These receptors are known as chimeric antigen receptors (CAR), and have triggered an enormous interest from both academia and industry due to their potential to eliminate cancer, even in tumor-relapsed patients. Clinical trials using CAR T cells have been particularly successful in treating haematological malignancies, with the most impressive results seen against CD19+ malignancies [1–5].
CARs are frequently composed of an extracellular single-chain variable fragment (scFv) derived from an antigen-specific monoclonal antibody (mAb), followed by a spacer region, a transmembrane domain, and intracellular signaling domains with or without costimulatory domains. In the most common form, the specificity of the CAR is conferred by the conformation of a variable heavy and variable light chains that are fused together by a flexible linker. The idea of leveraging specificity and affinity of mAbs is an enormous advantage to the design of CARs, yet repurposing variable chains from a full mAb into a scFv may not necessarily translate into a functional CAR. Some of the reasons for this include instability and aggregation behaviors of scFvs that can result in loss of CAR specificity and thus potentially leading to off-target toxicity and poor persistence in vivo [6, 7], Furthermore, scFvs may function differently when used in membrane-bound proteins compared to soluble proteins, suggesting that immunogenicity, affinity, and specificity of scFvs should be carefully assessed and examined in the context of CAR T cell responses.
An alternative approach is the use of extracellular domains of natural receptors or ligands for the design of CARs. Although the pool of known candidates is considered small, this approach has been used by several groups to target multiple types of cancer, including haematological and solid malignancies [8–11], The range of natural receptors and ligands that can be converted into CARs will depend on the antigen expression pattern in healthy tissue, abundant availability of the antigen on tumor cells and the specificity of receptor-ligand interaction. Examples detailed in this review are described in Table 1.
Table 1.
Receptor and ligand -based CARs currently in development.
| CAR (based on receptor) | Target (ligand) | Costimulatory domains | Indication | Phase of development | |
|---|---|---|---|---|---|
| CARs based on natural receptors | NKp30 | B7116, BAG6, Gal3 | CD28.CD3z | Liquid and solid tumors | Preclinical |
| NKG2D | MICA, MICB, ULBP 1–6 | CD3z/DaplO assoc | Liquid and solid tumors | Clinical | |
| DNAM-1 | PVR and Nectin-2 | CD28.CD3z | Solid tumors | Preclinical | |
| CD27 | CD70 | 41BB.CD3z | Liquid and solid tumors | Clinical | |
| CD16 | Fc region of Abs | 41BB.CD3z | Liquid and solid tumors | Clinical | |
| CARs based on receptor-binding domain of ligands | GM-CSF | CD116 | CD28.CD3Z | Liquid tumors | Preclinical |
| Adnectin | EGFR | CD28.41BB.CD3z | Solid tumors | Preclinical | |
| IL-13 | IL-13Rα2 | 41BB.CD3z | Solid tumors | Clinical | |
| IL-11 | IL-11Rα | CD28.CD3z | Solid tumors | Preclinical | |
| FSH | FSHR | CD28.CD3Z | Solid tumors | Preclinical | |
| T1E | ErbB family | CD28.CD3z | Solid tumors | Clinical |
Here we will summarize the current knowledge on the use of natural receptors and ligands as the basis for CAR T cell therapy to treat malignancies. We will discuss key points associated with receptor-ligand CAR design, manufacturing, pre-clinical and clinical development of this approach to therapy.
Receptor-ligand CAR design and manufacturing
Chimeric antigen receptors are an efficient way to create a large number of T cells with the capability of specific tumor recognition. Significant progress has been made in the use of natural receptor-ligand interactions as the basis for the design of new chimeric receptors. Based on the structural design of these molecules, members of this class can be divided in two categories: natural receptor-based CARs or ligand-based CARs. Independent of the nature of the target antigen, i.e. receptor or ligand, these should ideally follow the same rules applied to scFv-based CARs, which requires selective surface expression of target antigens on tumor cells or an increased expression on tumor cells with limited expression on healthy tissues. One of the potential advantages in the use of natural receptor- or ligand-based CARs is their limited immunogenicity, in particular for CARs where the extent of the extracellular domain uses a full-length native protein. Some CAR constructs use scFv derived from murine antibodies, which can increase the risk of developing an immune response against the genetically modified cells, leading to reduced effectiveness [12], ScFvs have been documented for aggregation, poor expression, instability, and tendency to dimerize [13], Therefore, the requirement for optimal choices for hinge/spacer region and transmembrane domain, making the identification of functional CARs laborious [14, 15], Rules have yet to be defined for the most effective CAR designs based on scFvs. Alternatively, receptor-based CARs are built based on the full extracellular binding domain of these proteins, harnessing specificity and structural stability, in particularly when associate with their native transmembrane domain.
In terms of target recognition, the use of some natural receptor-based CARs can potentially result in improved biological activity as they recognize multiple targets on the same tumor, or the same targets across multiple types of tumors. Targeting single antigens carries a risk for antigen loss leading to tumor immune escape [16, 17], whereas there is thought to be less risk of immune escape when multiple antigens are targeted. One example is the NKG2D CAR, which is known to bind eight different ligands, and has been shown to recognize and eliminate myeloma, lymphoma, and ovarian cancer tumors [10, 18–22].
While the design of CARs based on natural receptors or ligands offer many therapeutic advantages, a downside risk is the possibility of target antigen expression on healthy cells and the potential for on-target, off-tumor toxicity. In this context, toxicity may be accentuated by particular CAR designs, especially those with costimulatory domains associated with expansion in vivo. Despite the risk, the most impressive clinical results with CAR-T cells have been seen with second generation CD19 CAR-T cells, with either CD28 or41BB costimulatory signaling domains followed by the CD3 zeta signaling domain, for treatment of several haematologic malignancies, where anti-tumor activity was dependent on significant T-cell expansion in vivo, and in many cases associated with a life-threatening cytokine storm [5].
One unique challenge with manufacturing natural receptor-ligand based CARs is the inability to differentiate the CARs from the endogenous receptor or ligand, if the receptor is also expressed on normal effector cells. A few approaches can be adopted to allow precise quantification of transduction efficiency. One approach is to develop an antibody designed to recognize the chimeric junction, the area linking the receptor and costimulatory domain, which is located intracellularly. This can be a challenging strategy requiring a stringent screening method. An alternative approach is to determine the expression on transduced cells above the threshold expression of the endogenous protein under the manufacturing conditions. This can be achieved using mock-transduced cells for endogenous expression, so that any receptor expression above the endogenous expression is considered CAR-specific expression.
Natural receptor-based CARs
One method of generating CARs includes the fusion of native receptors, which have evolved over time to bind to their ligands on opposing cells, with the intracellular signaling domains required to induce T cell activation (Figure 1 A, B). One growing class of natural receptor CARs has been based on the use of NK cells and their receptors. NK cells have broad cytotoxic activity against tumor cells and no requirement for MHC restriction, therefore the use of NK receptors as CARs has the potential to yield robust anti-tumor activity. These receptors can also recognize various stress-induced or overexpressed ligands on several types to tumors, which results in a broad range of cancer indications [23, 24], This class has multiple members either in preclinical or clinical development stage. Examples of CARs designed based on these receptors include NKp30, DNAM-1, and NKG2D CARs [9–11,25], These CARs were designed in a way that the extracellular domain of their respective NK receptor remains intact, yet linked to a variety of cytoplasmic costimulatory and/or signaling domains. NKG2D CAR has been shown to recognize and kill lymphomas, myelomas, and many solid tumors, while significantly altering the tumor microenvironment and promoting the development of anti-tumor immunity [10, 18–20, 22, 26–28], One potential limiting aspect of this approach is the fact that NK receptor-based CARs may also recognize their ligands when expressed on non-tumor cells, leading to unexpected toxicity. Therefore, the off-tumor effects of these NK receptor based CAR T cells should be understood in order to use them safely [22].
Figure 1.
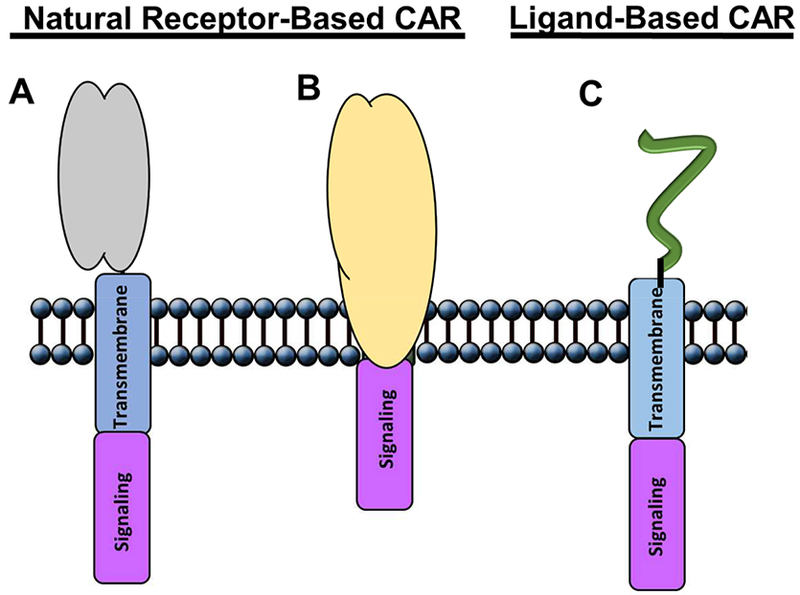
Schematic structure of receptor- and ligand-based chimeric antigen receptors. Receptor-based CARs: (A) native protein extracellular domain bound to either CD8 or CD28 transmembrane, followed by intracellular signaling domain; and (B) complete native receptor, including both extracellular and transmembrane domains, followed by intracellular signaling domain. (C) Ligand-based CAR: ligand molecule is bound to either CD8 or CD28 transmembrane, followed by intracellular signaling domain.
CD70 has been identified as the ligand for CD27, a co-stimulatory receptor involved in T cell proliferation and survival [29], A number of human tumors have been shown to express CD70, including solid and haematological malignancies [30], Recent studies suggest that the mechanism of expression of CD70 is closely associated with the pVHL/HIF pathway, an important mediator of hypoxia, a survival mechanism of tumor cells [31, 32], Due to a restricted expression pattern in healthy tissues and abundant expression in malignant cells, CD70 can be considered as a possible target for the development of cell based therapies. Preclinical studies performed with T cells genetically engineered with different CAR constructs designed to target CD70 showed improved in vitro activity using a CD27–41BB-zeta CAR. This construct consists of the CD27 molecule minus its intracellular domain, fused with the co-stimulatory of 41BB and the CD3 zeta signaling domains [33], Data showed curative effects and long-term survival of tumor-bearing NSG immunodeficient mice. A syngeneic mouse model was used to test efficacy and toxicity in vivo, where toxicity caused by cytokine release was observed at cell doses 100-fold higher than needed to show efficacy. These preclinical data were used in support of a Phase I clinical trial to test safety of CD27 CAR T cells against five indications of solid malignancies (NCT02830724).
Another class of receptor-based CARs makes use of the extracellular binding region of FcyRIII (CD16), which can be expressed in the surface of several immune cells, including monocytes, neutrophils, macrophages and NK cells [34, 35], This receptor mediates antibody-dependent cellular cytotoxicity (ADCC) through binding to Fc region of antibodies, therefore enhancing the effects of therapeutic antibodies regardless of the target tumor antigen. The ability of CD16 to bind IgG antibodies results in a potential universal CAR, where only one type of engineered T cells is needed to treat multiple types of cancer when given with the appropriate IgG antibodies that target that cancer. One potential challenge is for an undesirable activation of the CAR by antibody-antigen aggregates that regularly occur as part of a normal immunity against pathogens. These Fc- targeted CARs could potentially be triggered by antibodies that could aggregate at tissue sites throughout the body without association of a tumor antigen. In such cases, the unintended activation of immune system may result in severe off-target toxicities.
Ligand-based CARs
Another approach involves the use of receptor-binding domain of ligands in the design of CARs (Figure 1 C). This approach takes advantage of several classes of ligand molecules, including cytokines, immunoglobulin superfamily proteins and peptides [36–39], The use of cytokine receptors as targets for CAR-based T cells therapies has been a useful tool to treat specific types of tumors. Interleukin 11 (IL-11) demonstrates functional activity in vitro on many different systems, including bone, hepatic, neuronal, haematopoietic, lymphopoietic, and adipose, either alone or in combination with other growth factors [40, 41], IL-11 plays a major role in epithelial cancer biology through activation of STAT3, leading to improved survival and proliferation of neoplastic cells, if they express the ligand-specific IL-11Rα receptor subunit [42, 43], Overexpression of human IL-11Rα has been shown in multiple cancers of epithelial and mesenchymal origin, including colon, gastric, breast, prostate, and osteosarcoma (OS) [42, 44], Relapsed OS patients frequently have lung metastasis that are resistant to available chemotherapy treatments.
IL-11Rα was shown to be expressed on a variety of human OS cell lines and on 88% of OS pulmonary metastases from patients. Therefore, an IL-11Rα CAR has been created by connecting an IL-11 peptide as an extracellular domain to the T cell activation endodomains. These IL-11Rα CAR T cells have been shown to kill OS cells in vitro and accumulate in OS lung tumors when delivered intravenously in vivo. Treatment of mice with established OS lung metastases resulted in significant tumor cell apoptosis and tumor regression [39], The development of an IL-11 based CAR T cell therapy can be a desirable therapeutic option for these patients considering the direct correlation between peripheral lymphocyte numbers and OS patient outcomes, with high lymphocyte counts associated with better outcome [45].
Another example of a cytokine receptor used in the design of CAR is the adoptive T-cell therapy against glioblastoma with the use of cells engineered to target IL-13Rα2 [36, 46], IL-13Rα2 is expressed on more than 80% of high-grade gliomas, is associated with a reduced rate of survival, and has low expression in normal tissue. Physiologically, IL-13 binds to IL-13Rα2 with a much higher affinity compared to IL-13Rα1, the form more widely expressed in tissues, making IL-13Rα2 a more suitable target candidate for immunotherapy [47, 48], The initial design of the IL-13Rα2 CAR used IL-13 with an E13Y mutation fused to a hinge region from human γ4Fc, the CD4 transmembrane domain, and the signaling domain of CD3 zeta. CAR T cell-mediated specific lysis of multiple tumor cell types in vitro, including Daudi lymphoma, THP-1 (acute myeloid leukemia - AML), and U251 glioma cells, with natural or artificial expression of IL-13Rα2 [49], Control target cells that expressed either IL-13Rα1 or no IL-13 receptor were not killed, demonstrating the specificity of the CAR to the target IL-13Rα2. Efficacy in vivo was demonstrated using labeled-U87 glioma cells implanted into the forebrain of NOD-SCID mice. IL-13Rα2 CAR T cell-treated mice demonstrated a loss of tumor signal by three days, and the signal remained low for many weeks after treatment. In contrast, mice treated with control CAR T cells had increased tumor growth. [50], An IL-13Rα2-specific CAR has been created with two amino acid mutations in the IL-13 ligand [49]. The mutations were specifically designed to increase affinity to IL-13Rα2 while decreasing affinity to IL-13Rα1. A second generation IL-13Rα2-specific CAR was created with a 4–1BB costimulatory signaling domain. Data comparing the activity of first and second generations of IL-13Rα2-specific CAR T cells showed the second generation was superior in vivo [51].
Adnectins are a class of synthetic peptides derived from the tenth type III domain of human fibronectin (10Fn3) and have been shown to bind to epidermal growth factor receptor (EGFR), a transmembrane receptor that mediates proliferation and cell survival. Genetic modifications in the binding region of 10Fn3 has shown to improve the specificity without compromising the structure of the protein [52], Adnectin has been used as the binding portion of a CAR in combination of CD28, 41BB and CD3 zeta signaling molecules, with the binding and signaling units connected by CD8 hinge and transmembrane domains. In terms of efficacy, adnectin-based CAR T cells demonstrated comparable cell killing activity both in vitro and in vivo against EGFR-expressing cells, when compared to the standard scFv-based CAR T cells. Furthermore, adnectin-based CAR T cells showed lower binding affinity towards EGFR relative to the scFv-based version, leading to a superior safety profile for this therapy [38].
A follicle-stimulating hormone (FSH) CAR is an example of ligand-based CAR design to target ovarian cancer cells known to express high amounts of FSH receptor [53], Human T cells transduced with FSH CAR were specifically cytotoxic against FSHR-expressing human ovarian cancer cell lines in vitro. Similarly, FSH CAR T cell treatment led to significant anti-tumor efficacy in a xenograft model of ovarian cancer. Evaluation of patient samples for FSHR expression showed no expression of the target outside ovarian tissue. Reinforcing the safety profile of this target, monitoring of tissue pathology in ovarian cancer murine models did not reveal harmful effects associated with the delivery of a fully murine FSHR-targeted T cells, supporting further development of this approach for ovarian cancer [54].
An approach was designed by Nakazawa etal. to target GM-CSF receptor (GMR) via its ligand GM-CSF to treat Juvenile Myelomonocytic Leukemia (JMML), a fatal, myelodysplastic/myeloproliferative neoplasm of early childhood [8]. The GMR-specific CAR contains the full-length sequence of human GM-CSF linked to the transmembrane and cytoplasmic domain of human CD28, and followed by the human CD3 zeta signaling domain. GMR-specific CAR T cells showed specific cytotoxicity and growth inhibition of JMML progenitor cells (CD34+) compared to normal haematopoietic progenitors, a feature that might be explained by difference in GMR conformation between JMML and normal cells [55, 56]. The ability to distinguish structural changes based on the level of receptor activation makes the GMR-specific CAR T cell therapy an attractive option for the treatment of this disease.
Natural Receptor- and ligand-based CARs tested in the clinic
Compared to anti-CD19 CARs and the clinical outcomes in the treatment of B-cell malignancies, relatively little is known about the clinical efficacy of natural receptor-based or ligand-based CARs. Amongst the CARs brought into the clinic (Table 1), the ligand-based CARs IL-13Rα and T1E are the ones with clinical information available [36, 46, 57–59].
Malignant glioma is the most common primary brain tumor in the United States with an annual incidence of two to three per 100,000 adults per year. Adoptive CAR T cell therapy represents a promising approach to treat patients with glioblastoma as these cells have potent cytotoxic capabilities and the potential of trafficking to distant sites in response to tumor secreted factors [60], This ability is particularly relevant given the glioblastoma’s invasive phenotype.
Initial clinical approaches evaluated intracranial administration of CD8 T cells that expressed a first generation IL-13Rα2-specific CAR. This approach showed transient anti-glioma responses with no “high-grade” therapy-related side effects [36], Based on these outcomes, a second generation modified IL13Rα2-specific CAR T cell was created to improve anti-tumor potency and T-cell persistency. The new approach involved the addition of 41BB costimulation, and the binding motif was combined with a mutated lgG4-Fc linker to reduce off-target Fc-receptor interactions [61], Clinical findings from a single patient demonstrated a dramatic anti-tumor response, combined with higher numbers of immune cells and greater inflammatory cytokines in the cerebrospinal fluid. To note, CAR T cell activity remained restricted to the brain, as no signs of cytokine accumulation or traces of CAR+ T cells were found outside the cerebrospinal fluid [46].
There are very few tumor-specific antigens, so target selection requires balancing potential for anti-tumor efficacy against the risk of unacceptable toxicity. CARs have been designed to recognize targets that are over-expressed on tumor tissues compared to healthy tissues. One class of CAR targets is the ErbB receptor family, which comprises of four members including epidermal growth factor receptor (EGFR or ErbB-1), ErbB-2 (HER2 or neu), ErbB-3 and ErbB-4 [62–65], To note, one of the mechanisms of tumor resistance is the common increased expression of the non-targeted members of the same family in order to compensate the elimination of the targeted member [66], Hence, the approach to target multiple members from the same family using a CAR that recognize multiple motifs provides substantial benefits, by increasing specificity and abrogating tumor resistance. To accomplish this, a second generation CAR named T1 E28z was created where the signaling endodomain contains CD28 followed by CD3 zeta [37], The target recognition domain was based on the chimeric polypeptide T1E. This peptide consists of the N-terminal portion of TGF-α fused to the C-terminal portion of mature EGF. T1E binds to ErbB1-based homo and heterodimers with high affinity, and T1E also binds ErbB2/3 heterodimers and all ErbB4 containing dimers [67].
T1 E28z CAR T cells have demonstrated anti-tumor efficacy in several preclinical models of solid malignancies, including breast, head and neck, and ovarian cancers [37, 68], Additional in vitro studies using T cells derived from patients with epithelial ovarian cancer showed cytotoxicity activity against autologous tumor spheres [68], Despite being highly expressed in tumor tissues, ErbB receptors are found in several healthy tissues, raising concerns about the toxic potential of targeting these with CAR T cells. Van Der Stegen et al. took advantage of the fact that T1E28Z CAR recognizes mouse ErbB receptors to understand the potential for on-target off-tumor effects of this therapeutic approach [69], The study revealed that administration of CAR T cells either i.v. or intratumorally did not result in clinical or histopathologic toxicity. However, when delivered i.p., anti-tumor efficacy was accompanied by dose-dependent side effects. Toxicity was mediated by target recognition of CAR T cells, which led to release of both human IL-2 and IFN-γ, and murine IL-6 cytokines. In cases of very high tumor burden, the treatment with T1E28z CAR T cells was lethal. These data demonstrate that CAR-induced cytokine release syndrome can be modeled in mice that express target antigen in an appropriate expression. It also highlights the importance for the proper choice of route of administration and cell dose of CAR T cells to balance the therapeutic benefits versus any potential toxicity.
These T1E28z CAR preclinical data supported the initiation of a Phase I clinical trial to investigate the safety of intratumoral injections of T1E28z CAR T cells in patients with locally advanced/recurrent head and neck squamous cell carcinoma (HNSCC) [58], This is an autologous cell therapy in which peripheral blood T-cells were genetically engineered using a retroviral vector to express two chimeric receptors: (1) T1E28z to engage multiple ErbB dimers that are commonly upregulated in HNSCC; (2) 4αβ, composed of the ectodomain of IL-4Rα linked with the endodomain of the shared IL-2/15 βc subunit, to convert the weak mitogenic stimulus provided by interleukin (IL)-4 into a strong and selective growth signal, allowing preferential expansion and enrichment of CAR-specific T cells ex vivo [70], These cells were administered in a single sitting to several sites around the viable tumor, and patients received a total dose of up to 10Λ9 cells, in a 3+3 dose escalation design distributed through 5 cohorts. To date, ten patients have been treated and no dose-limiting toxicities have been observed [71], Upon completion of the Phase I, the next expansion phase will potentially include mesothelioma and/or ovarian cancer as target indications for this therapeutic approach.
Safe clinical implementation of CAR T-cells requires a cautious and considered approach, given the fact that some of these target antigens might be naturally expressed at low levels on various healthy tissues. One approach to mitigate this concern is through the use of regional delivery to potentially mitigate the risks associated with on-target off-tumor toxicity [46, 57, 58], Despite severe toxic effects associated with high levels of proinflammatory cytokines, treatment of a recurrent glioblastoma patient with localized autologous CAR T cells targeting IL-13Rα2 resulted in a transient complete response, with dramatic improvements in the patient’s quality of life [46], In the case of ErBB targeted CAR T cells, disease control was achieved in 6 out of 10 head and neck squamous cell carcinoma (SNSCC) patients with no dose-limiting toxicity reported [58],
Another promising therapy for the treatment of haematological malignancies is using a NKG2D receptor based CAR. Many studies have been conducted using murine tumor models and primary human T cells that show the potential for this CAR therapy to be effective in treating cancer. Preclinical studies have shown efficacy in several syngeneic tumor models of solid and haematological malignancies [18, 72–74], In vivo experiments showed that NKG2D CART cell injection led to complete overall survival in murine lymphoma, multiple myeloma, and ovarian cancer models compared to injection of control wildtype NKG2D T cells. Data have demonstrated that NKG2D CAR T cell cytotoxicity and cytokine production are both important for complete anti-tumor efficacy, and that activation of host cytotoxicity and cytokines can also be activated as a part of the anti-tumor activity in vivo [20, 73, 75, 76], Furthermore, animals in long-term remission efficiently responded to rechallenge even in the absence of CAR T cells. This finding was dependent upon host T cells, indicating that NKG2D CAR T cells generated an adaptive tumor-specific immune response [26].
In terms of safety, two preclinical studies have been performed in order to understand the potential for toxic effects in humans. The first study combined evaluation of efficacy and toxicity of three distinct NKG2D-based CARs: (1) a fusion between the NKG2D receptor and the CD3 zeta chain (NKz); (2) Co-expression of NKz with DAP10 to increase cell surface expression (NKz10); and (3) a conventional second generation CAR, where the extracellular domain of the NKG2D receptor was fused to CD28, followed by CD3zeta (NK28z) [77], Delivery of NKG2D CAR T cells that had additional costimulation built into the receptor (e.g. CD28) caused significant morbidity and mortality, and the extent of toxicity varied between CAR designs and mouse strains tested. Additionally, pre-treatment with cyclophosphamide prior to delivery of the cells exacerbated the toxicity, a not unexpected finding considering that chemotherapy may induce upregulation of NKG2D ligands [23, 78], The second study focused on the evaluation of maximum tolerated dose and mechanism of toxicity associated with delivery of NKG2D CAR T cells [79], Experiments were performed with a NKz construct, which contained the full NKG2D receptor followed by the CD3 zeta signaling domain, and dose was escalated up to 2×107 cells per mouse as a single i.v. dose. The MTD was determined to be 107 cells per mouse, and repeated administration of this dose to healthy and tumor-bearing mice did not produce major toxicities. In contrast, i.v. injection of a higher dose of NKG2D CAR T cells resulted in acute inflammatory distress associated with the production of pro-inflammatory cytokines, a pattern consistence with cytokine storm. In order to understand the mechanism of toxicity and narrow down potential key players of this effect, several knockout or artificially depleted-mouse models were used either as recipients or donors of CAR T cells. NKG2D CAR T cells that were deficient in either perforin or GM-CSF alone did not result in acute toxicity when administered at high cell doses, however, the loss of IFN-γ, which is essential for anti-tumor activity, did not alter toxicity. These preclinical data were used to support the initiation of two Phase I clinical trials to evaluate primarily safety, pharmacokinetics, and potential efficacy of NKG2D CAR T cells in multiple cancer indications, including solid and haematological malignancies. The initial clinical study was designed to evaluate the safety of a single intravenous dose administration of up to 30 million CAR T cells (NCT02203825) in patients with Acute Myeloid Leukemia (AML) and Multiple Myeloma (MM), followed later by a multi-dose study of up to 3×10Λ9 CAR T cells in patient with AML, MM, and five solid tumor indications (NCT03018405).
Summary
The majority of the CARs used in the clinic contain a scFv for antigen recognition and specificity, with the anti-CD19 CAR leading the race with two approved products (Tisagenlecleucel and Axicabtagene Ciloleucel). Since many of the scFvs being used in preclinical and clinical development are derived from antibodies raised in mice, the issue with potentially immunogenicity is still a challenge. CARs derived from naturally occurring proteins may eliminate the concerns associated with immunogenicity, which represents a hurdle for this line of therapy. Some of these natural receptors or ligands are known to bind multiple targets, which can be an advantage to avoid the risk of immune escape associated with antigen-loss. On the other hand, the ability to bind multiple targets can increase the risks of on-target off-tumor toxicity, considering that most of the targets known to date have some expression on healthy tissues. As the various CAR T cell therapy approaches move through the clinic, understanding why some CAR designs or treatment strategies work better than others, will enable the field to customize designs for specific malignancies. Looking forward, CAR therapies promise to revolutionize treatment options for cancer patients.
Practice Points.
Natural receptors have evolved to interact with one or more target proteins, which may be over-expressed on many types of tumors.
Because they are based on natural, endogenous proteins, the use of a natural receptor or ligand based CAR may not induce a host anti-CAR immune response.
Off-tumor but on-target responses can occur and potential toxicity needs to be monitored closely.
Research Agenda.
Natural receptors and their ligands are a promising opportunity for the development of CAR-based therapies for multiple tumor indications.
The majority of the CARs tested clinically are scFv-based. Despite providing exciting clinical results, the approach still faces some challenges, such as potential immunogenicity and CAR self-aggregation.
Selection of suitable receptor candidates should be done carefully due to potential target expression patterns on non-tumor tissues.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
C.L.S. has patents and financial interests in NK receptor-based CAR therapies. C.L.S. is a scientific founder for Celdara Medical, a consultant, and receives research support from Celdara Medical. J.M.M. is employed by Celdara Medical, which has a material financial interest in NK receptor-based CAR intellectual property assigned to the Trustees of Dartmouth College.
References
- [1].Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Science translational medicine. 2013;5:177ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Science translational medicine. 2014;6:224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chang ZL, Chen YY. CARs: Synthetic Immunoreceptors for Cancer Therapy and Beyond. Trends Mol Med. 2017;23:430–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nature medicine. 2015;21:581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nakazawa Y, Matsuda K, Kurata T, Sueki A, Tanaka M, Sakashita K, et al. Anti-proliferative effects of T cells expressing a ligand-based chimeric antigen receptor against CD116 on CD34(+) cells of juvenile myelomonocytic leukemia. J Hematol Oncol. 2016;9:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wu MR, Zhang T, Alcon A, Sentman CL. DNAM-l-based chimeric antigen receptors enhance T cell effector function and exhibit in vivo efficacy against melanoma. Cancer Immunol Immunother. 2015;64:409–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhang T, Lemoi BA, Sentman CL. Chimeric NK-receptor-bearing T cells mediate antitumor immunotherapy. Blood. 2005;106:1544–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang T, Wu MR, Sentman CL. An NKp30-based chimeric antigen receptor promotes T cell effector functions and antitumor efficacy in vivo. J Immunol. 2012;189:2290–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. 2016;3:16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bradbury A scFvsand beyond. Drug Discov Today. 2003;8:737–9. [DOI] [PubMed] [Google Scholar]
- [14].Wang W, Singh S, Zeng DL, King K, Nema S. Antibody structure, instability, and formulation. J Pharm Sci. 2007;96:1–26. [DOI] [PubMed] [Google Scholar]
- [15].Gacerez AT, Arellano B, Sentman CL. How Chimeric Antigen Receptor Design Affects Adoptive T Cell Therapy. J Cell Physiol. 2016;231:2590–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. The Journal of clinical investigation. 2016;126:3814–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ruella M, Maus MV. Catch me if you can: Leukemia Escape after CD19-Directed T Cell Immunotherapies. Comput Struct Biotechnol J. 2016;14:357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Barber A, Meehan KR, Sentman CL. Treatment of multiple myeloma with adoptively transferred chimeric NKG2D receptor-expressing T cells. Gene therapy. 2011;18:509–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Barber A, Rynda A, Sentman CL. Chimeric NKG2D expressing T cells eliminate immunosuppression and activate immunity within the ovarian tumor microenvironment. J Immunol. 2009;183:6939–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Barber A, Sentman CL. Chimeric NKG2D T cells require both T cell- and host-derived cytokine secretion and perforin expression to increase tumor antigen presentation and systemic immunity. J Immunol. 2009;183:2365–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Barber A, Zhang T, Megli CJ, Wu J, Meehan KR, Sentman CL. Chimeric NKG2D receptor-expressing T cells as an immunotherapy for multiple myeloma. Experimental hematology. 2008;36:1318–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sentman CL, Meehan KR. NKG2D CARs as cell therapy for cancer. Cancer J. 2014;20:156–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Spear P, Wu MR, Sentman ML, Sentman CL. NKG2D ligands as therapeutic targets. Cancer immunity. 2013;13:8. [PMC free article] [PubMed] [Google Scholar]
- [24].Moretta L, Moretta A. Unravelling natural killer cell function: triggering and inhibitory human NK receptors. EMBO J. 2004;23:255–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Demoulin B, Cook WJ, Murad J, Graber DJ, Sentman ML, Lonez C, et al. Exploiting natural killer group 2D receptors for CAR T-cell therapy. Future Oncol. 2017;13:1593–605. [DOI] [PubMed] [Google Scholar]
- [26].Barber A, Zhang T, Sentman CL. Immunotherapy with chimeric NKG2D receptors leads to long-term tumor-free survival and development of host antitumor immunity in murine ovarian cancer. J Immunol. 2008;180:72–8. [DOI] [PubMed] [Google Scholar]
- [27].Fernandez L, Metais JY, Escudero A, Vela M, Valentin J, Vallcorba I, et al. Memory T Cells Expressing an NKG2D-CAR Efficiently Target Osteosarcoma Cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23:5824–35. [DOI] [PubMed] [Google Scholar]
- [28].Song DG, Ye Q, Santoro S, Fang C, Best A, Powell DJ Jr. Chimeric NKG2D CAR-expressing T cell-mediated attack of human ovarian cancer is enhanced by histone deacetylase inhibition. Human gene therapy. 2013;24:295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wajant H Therapeutic targeting of CD70 and CD27. Expert Opin Ther Targets. 2016;20:959–73. [DOI] [PubMed] [Google Scholar]
- [30].Shaffer DR, Savoldo B, Yi Z, Chow KK, Kakarla S, Spencer DM, et al. T cells redirected against CD70 for the immunotherapy of CD70-positive malignancies. Blood. 2011;117:4304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ruf M, Mittmann C, Nowicka AM, Hartmann A, Hermanns T, Poyet C, et al. pVHL/HIF-regulated CD70 expression is associated with infiltration of CD27+ lymphocytes and increased serum levels of soluble CD27 in clear cell renal cell carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21:889–98. [DOI] [PubMed] [Google Scholar]
- [32].Vaupel P. The role of hypoxia-induced factors in tumor progression. Oncologist. 2004;9 Suppl 5:10–7. [DOI] [PubMed] [Google Scholar]
- [33].Wang QJ, Yu Z, Hanada KI, Patel K, Kleiner D, Restifo NP, et al. Preclinical Evaluation of Chimeric Antigen Receptors Targeting CD70-Expressing Cancers. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23:2267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Clemenceau B, Congy-Jolivet N, Gallot G, Vivien R, Gaschet J, Thibault G, et al. Antibody-dependent cellular cytotoxicity (ADCC) is mediated by genetically modified antigen-specific human T lymphocytes. Blood. 2006;107:4669–77. [DOI] [PubMed] [Google Scholar]
- [35].D’Aloia MM, Caratelli S, Palumbo C, Battella S, Arriga R, Lauro D, et al. T lymphocytes engineered to express a CD16-chimeric antigen receptor redirect T-cell immune responses against immunoglobulin G-opsonized target cells. Cytotherapy. 2016;18:278–90. [DOI] [PubMed] [Google Scholar]
- [36].Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21:4062–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Davies DM, Foster J, Van Der Stegen SJ, Parente-Pereira AC, Chiapero-Stanke L, Delinassios GJ, et al. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol Med. 2012;18:565–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Han X, Cinay GE, Zhao Y, Guo Y, Zhang X, Wang P. Adnectin-Based Design of Chimeric Antigen Receptor for T Cell Engineering. Molecular therapy : the journal of the American Society of Gene Therapy. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Huang G, Yu L, Cooper LJ, Hollomon M, Huls H, Kleinerman ES. Genetically modified T cells targeting interleukin-11 receptor alpha-chain kill human osteosarcoma cells and induce the regression of established osteosarcoma lung metastases. Cancer Res. 2012;72:271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Du X, Williams DA. Interleukin-11: review of molecular, cell biology, and clinical use. Blood. 1997;89:3897–908. [PubMed] [Google Scholar]
- [41].Yang YC. Interleukin 11: an overview. Stem Cells. 1993;11:474–86. [DOI] [PubMed] [Google Scholar]
- [42].Johnstone CN, Chand A, Putoczki TL, Ernst M. Emerging roles for IL-11 signaling in cancer development and progression: Focus on breast cancer. Cytokine Growth Factor Rev. 2015;26:489–98. [DOI] [PubMed] [Google Scholar]
- [43].Ernst M, Putoczki TL. Molecular pathways: IL11 as a tumor-promoting cytokine-translational implications for cancers. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20:5579–88. [DOI] [PubMed] [Google Scholar]
- [44].Lewis VO, Ozawa MG, Deavers MT, Wang G, Shintani T, Arap W, et al. The interleukin-11 receptor alpha as a candidate ligand-directed target in osteosarcoma: consistent data from cell lines, orthotopic models, and human tumor samples. Cancer Res. 2009;69:1995–9. [DOI] [PubMed] [Google Scholar]
- [45].Moore C, Eslin D, Levy A, Roberson J, Giusti V, Sutphin R. Prognostic significance of early lymphocyte recovery in pediatric osteosarcoma. Pediatr Blood Cancer. 2010;55:1096–102. [DOI] [PubMed] [Google Scholar]
- [46].Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med. 2016;375:2561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Thaci B, Brown CE, Binello E, Werbaneth K, Sampath P, Sengupta S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro Oncol. 2014;16:1304–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Debinski W, Gibo DM, Hulet SW, Connor JR, Gillespie GY. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 1999;5:985–90. [PubMed] [Google Scholar]
- [49].Kong S, Sengupta S, Tyler B, Bais AJ, Ma Q, Doucette S, et al. Suppression of human glioma xenografts with second-generation IL13R-specific chimeric antigen receptor-modified T cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:5949–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64:9160–6. [DOI] [PubMed] [Google Scholar]
- [51].Brown CE, Aguilar B, Starr R, Yang X, Chang WC, Weng L, et al. Optimization of IL13Ralpha2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Molecular therapy : the journal of the American Society of Gene Therapy. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Emanuel SL, Engle LJ, Chao G, Zhu RR, Cao C, Lin Z, et al. A fibronectin scaffold approach to bispecific inhibitors of epidermal growth factor receptor and insulin-like growth factor-I receptor. MAbs. 2011;3:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Urbanska K, Stashwick C, Poussin M, Powell DJ Jr. Follicle-Stimulating Hormone Receptor as a Target in the Redirected T-cell Therapy for Cancer. Cancer Immunol Res. 2015;3:1130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Perales-Puchalt A, Svoronos N, Rutkowski MR, Allegrezza MJ, Tesone AJ, Payne KK, et al. Follicle-Stimulating Hormone Receptor Is Expressed by Most Ovarian Cancer Subtypes and Is a Safe and Effective Immunotherapeutic Target. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23:441–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hansen G, Hercus TR, McClure BJ, Stomski FC, Dottore M, Powell J, et al. The structure of the GM-CSF receptor complex reveals a distinct mode of cytokine receptor activation. Cell. 2008;134:496–507. [DOI] [PubMed] [Google Scholar]
- [56].Hercus TR, Thomas D, Guthridge MA, Ekert PG, King-Scott J, Parker MW, et al. The granulocyte-macrophage colony-stimulating factor receptor: linking its structure to cell signaling and its role in disease. Blood. 2009;114:1289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Papa S, van Schalkwyk M, Maher J. Clinical Evaluation of ErbB-Targeted CAR T-Cells, Following Intracavity Delivery in Patients with ErbB-Expressing Solid Tumors. Methods Mol Biol. 2015;1317:365–82. [DOI] [PubMed] [Google Scholar]
- [58].van Schalkwyk MC, Papa SE, Jeannon JP, Guerrero Urbano T, Spicer JF, Maher J. Design of a phase I clinical trial to evaluate intratumoral delivery of ErbB-targeted chimeric antigen receptor T-cells in locally advanced or recurrent head and neck cancer. Hum Gene Ther Clin Dev. 2013;24:134–42. [DOI] [PubMed] [Google Scholar]
- [59].Klampatsa A, Achkova DY, Davies DM, Parente-Pereira AC, Woodman N, Rosekilly J, et al. Intracavitary ‘T4 immunotherapy’ of malignant mesothelioma using pan-ErbB re-targeted CAR T-cells. Cancer Lett. 2017;393:52–9. [DOI] [PubMed] [Google Scholar]
- [60].Salsman VSC KK; Shaffer DR; Kadikoy H; Li XN; Gerken C; Perlaky L; Metelitsa LS; Gao X; Bhattacharjee M; et al. Crosstalk between medulloblastoma cells and endothelium triggers a strong chemotactic signal recruiting T lymphocytes to the tumor microenvironment. PloS One. 2011;6:e20267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Jonnalagadda M, Mardiros A, Urak R, Wang X, Hoffman LJ, Bernanke A, et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Molecular therapy : the journal of the American Society of Gene Therapy. 2015;23:757–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:474–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Maliar A, Servais C, Waks T, Chmielewski M, Lavy R, Altevogt P, et al. Redirected T cells that target pancreatic adenocarcinoma antigens eliminate tumors and metastases in mice. Gastroenterology. 2012;143:1375–84 e1–5. [DOI] [PubMed] [Google Scholar]
- [64].Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32:1059–70. [DOI] [PubMed] [Google Scholar]
- [65].Zhou X, Li J, Wang Z, Chen Z, Qiu J, Zhang Y, et al. Cellular immunotherapy for carcinoma using genetically modified EGFR-specific T lymphocytes. Neoplasia. 2013;15:544–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008;27:3944–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wingens M, Walma T, van Ingen H, Stortelers C, van Leeuwen JE, van Zoelen EJ, et al. Structural analysis of an epidermal growth factor/transforming growth factor-alpha chimera with unique ErbB binding specificity. J Biol Chem. 2003;278:39114–23. [DOI] [PubMed] [Google Scholar]
- [68].Parente-Pereira AC, Whilding LM, Brewig N, van der Stegen SJ, Davies DM, Wilkie S, et al. Synergistic Chemoimmunotherapy of Epithelial Ovarian Cancer Using ErbB-Retargeted T Cells Combined with Carboplatin. J Immunol. 2013;191:2437–45. [DOI] [PubMed] [Google Scholar]
- [69].van der Stegen SJ, Davies DM, Wilkie S, Foster J, Sosabowski JK, Burnet J, et al. Preclinical in vivo modeling of cytokine release syndrome induced by ErbB-retargeted human T cells: identifying a window of therapeutic opportunity? J Immunol. 2013;191:4589–98. [DOI] [PubMed] [Google Scholar]
- [70].Wilkie S, Burbridge SE, Chiapero-Stanke L, Pereira AC, Cleary S, van der Stegen SJ, et al. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem. 2010;285:25538–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Papa S, Adami AA, Metoudi M, et al. Online Proceedings of the Annual Meeting of the American Association for Cancer Research www.abstractsonline.com/pp8/#!/4292/presentation/12333 2017. [Google Scholar]
- [72].Barber A, Zhang T, DeMars LR, Conejo-Garcia J, Roby KF, Sentman CL. Chimeric NKG2D receptorbearing T cells as immunotherapy for ovarian cancer. Cancer Res. 2007;67:5003–8. [DOI] [PubMed] [Google Scholar]
- [73].Zhang T, Barber A, Sentman CL. Chimeric NKG2D modified T cells inhibit systemic T-cell lymphoma growth in a manner involving multiple cytokines and cytotoxic pathways. Cancer Res. 2007;67:11029–36. [DOI] [PubMed] [Google Scholar]
- [74].Zhang T, Sentman CL. Mouse tumor vasculature expresses NKG2D ligands and can be targeted by chimeric NKG2D-modified T cells. J Immunol. 2013;190:2455–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Spear P, Barber A, Rynda-Apple A, Sentman CL. Chimeric antigen receptor T cells shape myeloid cell function within the tumor microenvironment through IFN-gamma and GM-CSF. J Immunol. 2012;188:6389–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Spear P, Barber A, Sentman CL. Collaboration of chimeric antigen receptor (CAR)-expressing T cells and host T cells for optimal elimination of established ovarian tumors. Oncoimmunology. 2013;2:e23564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].VanSeggelen H, Hammill JA, Dvorkin-Gheva A, Tantalo DG, Kwiecien JM, Denisova GF, et al. T Cells Engineered With Chimeric Antigen Receptors Targeting NKG2D Ligands Display Lethal Toxicity in Mice. Molecular therapy : the journal of the American Society of Gene Therapy. 2015;23:1600–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Soriani A, Zingoni A, Cerboni C, lannitto ML, Ricciardi MR, Di Gialleonardo V, et al. ATM-ATR dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK cell susceptibility and is associated with a senescent phenotype. Blood. 2008. [DOI] [PubMed] [Google Scholar]
- [79].Sentman ML, Murad JM, Cook WJ, Wu MR, Reder J, Baumeister SH, et al. Mechanisms of Acute Toxicity in NKG2D Chimeric Antigen Receptor T Cell-Treated Mice. J Immunol. 2016;197:4674–85. [DOI] [PMC free article] [PubMed] [Google Scholar]