

Abstract
Selective inhibition of Cyclin-dependent kinase 8 and cyclin C (CDK8/CycC) has been suggested as a promising strategy for reducing mitogenic signals in cancer cells with reduced toxic effects on normal cells. We developed a novel virtual screening protocol for drug development and applied it to discovering new CDK8/CycC type II ligands, which is likely to achieve long residence time and specificity. We first analyzed binding thermodynamics of 11 published pyrazolourea ligands using molecular dynamics simulations and a free energy calculation method, VM2, and extracted the key binding information to assist virtual screening. The urea moiety was found to be the critical structural contributor of the reference ligands. Starting with the urea moiety we conducted substructure-based searches with our newly developed Superposition and Single-Point Energy Evaluation method, followed by free energy calculations, and singled out three purchasable compounds for bio-assay testing. The ranking from the experimental result is completely consistent with the predicted rankings. A potent drug-like compound has a Kd value of 42.5 nM, which is comparable to the most potent reference ligands and provided a good starting point for further improvement. This study shows that our novel virtual screening protocol is an accurate and efficient tool for drug development.
Keywords: structure-based drug design, compound fragment, computer simulation, in silico screening, binding affinity, kinetics
A potent drug-like compound with CDK8/CycC suggested by our novel vitual screening tool which has a Kd value of 42.5 nM. Our work first analyzed binding thermodynamics of 11 published pyrazolourea ligands using molecular dynamics simulations and a free energy calculation method, VM2. Key urea moiety was selected for substructure-based searches with our newly developed Superposition and Single-Point Energy Evaluation method, followed by free energy calculations, and singled out purchasable compounds for bio-assay testing.
Introduction
Cyclin-dependent kinase 8 and cyclin C (CDK8/CycC) associate with the mediator complex and regulate gene transcription of nearly all RNA polymerase II-dependent genes.[1–5] A number of studies have shown that CDK8/CycC modulates the transcriptional output from distinct transcription factors involved in oncogenic control,[6] which include the Wnt/β-catenin pathway, Notch, p53, and TGF-β.[7,8] Compared with other CDKs, CDK8 is a more gene-specific expression regulator [9, 10] and is differently expressed in cancer.[2] In this view, selective inhibition of CDK8/CycC could be a promising strategy for reducing mitogenic signals in cancer cells with reduced toxic effects on normal cells.[11,12] The steroidal natural product cortistatin A is the first reported potent and selective ligand of CDK8/CycC.[13] Inhibition of CDK8/CycC with cortistatin A suppresses AML cell growth and has anticancer activity in animal models of AML.[14]
The existing ligands fall into two categories based on the major conformations of CDK8 to which they bind. Type I ligands bind to the DMG-in (Aspartate-Methionine-Glycine near the N-terminal region of the activation loop) conformation and occupy the ATP-binding site. The Senexin-type, CCT series, and COT series compounds, which possess 4-aminoquinazoline,[15] 3,4,5-trisubstituted pyridine [16] and 6-azabenzothiophene [17] scaffolds, respectively, belong to this category. Senex company identified Senexin B with an IC50 value of 24 nM. The R&D for new type I CDK8 ligands made a significant progress in the past a couple of years and many promising compounds were identified.[18, 19] In 2016 new potent and selective CDK8 ligands with benzylindazole scaffold previously reported as HSP90 ligands were described by Schiemann et al.[20] One of the most promising molecules showed an IC50 value against CDK8/CycC of 10 nM. More recently 4,5-dihydrothieno[3’,4’:3,4]benzo[1,2-d] isothiazole derivatives were found to have sub-nanomolar in-vitro potency (IC50: 0.46 nM) against CDK8/CycC and high selectivity.[21]
Schneider et al. published the first pyrazolourea series of type II CDK8 ligands in 2013.[22] Type II ligands bind to DMG-out conformation and occupy the ATP-binding site and the allosteric site (deep pocket). The deep pocket is adjacent to the ATP-binding site and is accessible in CDK8 by the rearrangement of the DMG motif from the active (DMG-in) to the inactive state (DMG-out). This pocket is inaccessible in the DMG-in conformation, where the Met174 side-chain is reoriented to make the site available to ATP.[23] Many well-known kinase ligands such as sorafenib and imatinib belong to the type II category. The ligands found by Schneider et al. anchored in the CDK8 deep pocket and extended with diverse functional groups toward the hinge region and the front pocket. These variations can cause the ligands to change from fast to slow binding kinetics, resulting in an improved residence time which is defined as the period for which a protein is occupied by a ligand.[24] As compared with type I ligands, binding of a type II compound to the DMG-out conformation often achieves longer residence time.[21,22,25,26] Residence time is considered to be a key success factor for drug discovery in addition to binding affinity.[27,28]
Inspired by the discovery of the pyrazolourea ligands we furthered the research to discover new type II CDK8 ligands. We investigated the thermodynamics of the binding between CDK8 and the 11 published type II ligands (Table 1) using a rigorous free energy calculation method with the VM2 package.[29] The knowledge gained from our molecular dynamics (MD) simulations and free energy calculations was then used with our recently developed substructure-based screening protocol, followed by free energy calculation to select tight binders. This paper is organized as below. We first present the analysis of binding free energies and the energy components of the 11 CDK8-ligand complexes with the help of both free energy calculations and MD simulations, from which we extract the key interactions as well as the mechanism in the binding between CDK8 and the ligands. Then we present our substructure-based screening and free energy calculation protocols, as illustrated in Figure 1 and detailed in the method section, for compound selection which is verified by experiments, and discussion of its application in the discovery of new type II CDK8 ligands. As a conclusion, we explain the implication of this novel method as a general tool to the drug discovery field.
Table 1.
Structures of the 11 reference ligands published by Schneider et al.,[22] and their corresponding available PDB IDs, experimental free energies (ΔGexp), and the length of MD simulations.
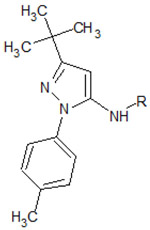 | ||||
|---|---|---|---|---|
| R= | PDB ID |
ΔGexp (Kcal/mol) |
MD (ns) |
|
| 1 | 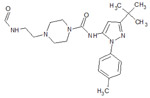 |
4F6W | −10.324 | 500 |
| 2 | 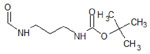 |
4F7L | −10.979 | 500 |
| 3 |  |
4F7J | −7.184 | |
| 4 | 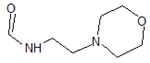 |
4F70 | −7.877 | |
| 5 |  |
4F6U | −8.447 | 500 |
| 6 | H | No bind | ||
| 7 | 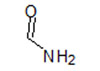 |
4F6S | −7.533 | |
| 8 |  |
−7.965 | ||
| 9 | 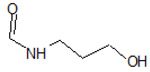 |
−7.482 | ||
| 10 |  |
−8.078 | 500 | |
| 11 | 4F7N | −9.739 | 500 | |
Figure 1.

Workflow of the novel virtual screening protocol for drug development. The numbers in ovals are the compounds obtained in this study.
Results and Discussion
Analysis on binding thermodynamics of the reference complexes
1. Overall analysis on binding free energies and conformations
Computed and measured binding free energies for the reference CDK8 pyrazolourea ligands are compared and plotted in Figure 2, and the corresponding free energy components are presented in Table 2. The free energy convergence plots can be found in Figure S1. A significant correlation is observed, as indicated by the correlation coefficient of 0.71, which is comparable to those obtained for other protein-ligand systems via the VM2 method.[29,30] According to the free energy components in Table 2, it appears that the major driving force for the bindings of all the reference ligands is the van der Waals interactions. Pyrazolourea ligand 6 has the least van der Waal interaction with the protein. As a result, its calculated ΔG is −6.45 kcal, which is equivalent to 20 μM, consistent with the experimental result of no binding. The overall electrostatic interactions (Coulomb + desolvation) provide a negative contribution to the binding. The entropy penalties correctly reflect the flexibility of the ligands. For example, the variable substituents are linear and become longer on pyrazolourea ligands 6, 7, 8, 3, 9, 10, 11, and their entropy penalties become larger in the same order. The same phenomenon is also observed on pyrazolourea ligands 4 and 5. It is noteworthy that pyrazolourea ligands 1 and 2 have the strongest binding affinities with CDK8 among the 11 pyrazolourea ligands but pay above-average entropy penalties.
Figure 2.

The correlation between the experimental and calculated free Energies. Dark fitting line and R2 value are for the 11 reference ligands; red fitting line and R2 value are for 11 reference ligands and 3 tested compounds.
Table 2.
Detailed energy breakdowns of computed CDK8-pyrazolourea ligands binding free energies ΔGcalc, along with experimental binding free energies, ΔGexp. Change in mean energy associated with force-field bond-stretch, angle-bend and dihedral terms (ΔEValence); change in mean force-field Coulombic energy (ΔECoulomb); change in mean Poisson-Boltzmann solvation energy (ΔEPB); change in mean nonpolar surface energy (ΔENP); change in mean force-field Lennard-Jones energy (ΔEVDW); change in mean total energy ΔH, the sum of the prior five terms; and change in configurational entropy contribution to the free energy (−TΔS). Unit is kcal/mol. RMSD: root-mean-square deviation of most stable computed conformation of the complex for ligand alone/all mobile atoms (Angstroms).
| ligand | ΔEValence | ΔECoulomb | ΔEPB | ΔENP | ΔEVDW | ΔH | −TΔS | ΔGcalc | ΔGexp | RMSD |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1.031 | −19.845 | 60.764 | −8.548 | −93.837 | −60.435 | 39.487 | −20.948 | −10.324 | 1.457/1.753 |
| 2 | 1.498 | −15.235 | 39.754 | −6.577 | −68.517 | −49.077 | 30.610 | −18.467 | −10.979 | 1.810/0.728 |
| 3 | 2.338 | −20.051 | 36.290 | −4.754 | −52.065 | −38.242 | 28.983 | −9.259 | −7.184 | 1.727/0.600 |
| 4 | 2.217 | −16.014 | 41.236 | −5.760 | −63.637 | −41.958 | 26.605 | −15.353 | −7.877 | 1.637/1.318 |
| 5 | 2.065 | −14.742 | 38.291 | −5.760 | −62.614 | −42.760 | 28.792 | −13.968 | −8.447 | 1.851/2.492 |
| 6 | 2.512 | −6.755 | 25.610 | −3.652 | −45.295 | −27.580 | 21.130 | −6.450 | No bind | |
| 7 | 1.448 | −14.167 | 31.055 | −4.049 | −49.417 | −35.130 | 23.157 | −11.973 | −7.533 | 1.841/0.414 |
| 8 | 2.911 | −16.977 | 33.393 | −4.265 | −53.090 | −38.028 | 24.416 | −13.612 | −7.965 | |
| 9 | 2.409 | −21.251 | 37.597 | −4.978 | −54.164 | −40.387 | 31.244 | −9.143 | −7.482 | |
| 10 | 4.992 | −19.615 | 39.659 | −5.191 | −62.883 | −43.038 | 32.392 | −10.646 | −8.078 | |
| 11 | 1.566 | −29.122 | 43.449 | −5.654 | −58.240 | −48.001 | 33.962 | −14.039 | −9.739 | 1.675/0.318 |
Figure 3 provides a sense for the conformational variation among the 11 bound pyrazolourea ligands via an overlay of their most stable predicted conformations. The common scaffold which occupies the region called “deep pocket” takes a uniform pose, while, not surprisingly, the variable substituents which occupy the region called “front pocket” show a wider range of positions. RMSDs for all the mobile atoms (i.e., mobile protein atoms plus ligand atoms) are quite similar and all less than 2.0 Å, indicating a good matching between the predicted poses and the crystal structures. Ligand 1 has the lowest RMSD for all the mobile atoms presumably because we used the protein in 4F6W, which is the co-crystal structure of the protein and ligand 1, in the VM2 calculations for all the pyrazolourea ligands while the RMSD was calculated between a ligand together with the protein and its available crystal structure. The ligands with RMSDs for ligand alone less than 1.0 Å have nearly identical poses as their crystal structures. For the ligands with larger RMSDs for ligand alone, a portion of their structures deviate from the crystal structures. Ligand 5 has the largest RMSD for ligand alone because in the predicted pose its morpholine moiety moves away from A100 and thus loses the H bonding with this residue. Similarly the morpholine ring in pyrazolourea ligand 4 deviates from its crystal counterpart as well. For ligand 1, it is the piperazine ring that has a large deviation.
Figure 3.

The overlay of the most stable conformations of the 11 reference ligands in the binding pocket of CDK8. The protein is not shown for clarity.
Since our free energy calculation shows that the binding is mainly driven by van der Waals interaction and ligand sizes have strong correlation with van der Waals interactions, we plotted the relationship between ligand sizes (i.e., the number of non-hydrogen atoms) and the experimental free energies to check how well they correlate. Many scoring functions use ligand sizes to assess ligands.[31] The plot can be found in the supplement materials (Figure S2). The correlation coefficient is 0.48, far below the correlation coefficient of 0.71 between the experimental and calculated free Energies. Interestingly the data points on the plot have very similar distribution as those in Figure 2. This result suggests that the molecular size is not the only key determinant of binding, and even for the congeneric ligand series the driving force of which is van der Waals interaction other factors are still important to rank ligands accurately. It is thus of interest to examine the relationship between ΔH, which includes not only van der Waals but also all other energy terms except entropy, and the experimental free energies. The resulting correlation coefficient is improved to 0.61 and again the data points on the plot have very similar distribution as those on the two above-mentioned plots (refer to Figure S3). The new correlation coefficient is lower than the one for the full computed free energies (0.71) by 15%, echoing the results in the previous studies.[29,30] This result highlights the importance of the configurational entropy to the correlation between calculation and experiment.
We calculated the MMPBSA energies for pyrazolourea ligands 1, 2, 5, 10 and 11 with the trajectories from the previous study,[32], and plotted the relationship between the MMPBSA energies and the experimental free energies as well (Figure S4). The correlation coefficient is poorly 0.4, much worse than the correlation coefficient of 0.72 when only the same five ligands are considered in the VM2 plot. The MMPBSA method is also an end-point method that uses a force-field and an implicit solvent model to estimate binding free energies. A key difference from the VM2 method is that MMPBSA calculations typically either neglect configurational entropy or approximate it as average vibrational entropy over essentially randomly selected, energy-minimized molecular dynamics snapshots. Furthermore, the MMPBSA method samples conformations by molecular dynamics, which is far less thorough than the aggressive conformational searches in the VM2 method. It is thus not surprising that the MMPBSA method had a much worse performance than the VM2 method.
2. Close-up analysis on selected reference complexes
Figure 4 presents the major interactions in the predicted conformation of CDK8 and pyrazolourea ligand 1. The residues E66 and D173 form strong H bonding with the urea linker on 1, and K52 has a salt bridge with E66. R356 has a cation-π interaction with the benzene ring on 1. All of these interactions can be found in the crystal structure 4F6W as well. Ligand 1 has strong van der Waals interaction with the protein. The free energy decomposition result (refer to Table S1) shows that residues E66, L70, A172, D173, M174 and R356 contribute the most to van der Waals energy. Ligand 1 pays the largest entropy penalty among the reference ligands, presumably due to its large yet relatively flexible structure. In addition, ligand 1 pays much larger desolvation penalty than other ligands but its Columbic energy is only on the average level, resulting in the poorest overall electrostatic energy. This indicates that many polar groups on ligand 1 don’t contribute to binding.
Figure 4.

Major interactions found in the predicted lowest energy conformation of CDK8 and ligand 1.
Pyrazolourea ligand 2 has similar binding mode as 1, except the cation-π interaction with R356. Instead, its t-butyl group interacts with R356 through van der Waals interaction. Schneider et al. thought this interaction plays an important role in the binding of ligand 2.[22] However, according to the free energy decomposition result residues E66, L70, I79, D173 and M174 contribute the largest van der Waals energy to the binding. The contribution from R356 is relatively small. Our calculation shows that the strong affinity of ligand 2 comes from the well-balanced energy and entropy terms. It has the second largest molecular size and favourable van der Waals energy; it pays relatively small entropy penalty; its desolvation penalty is much smaller than that of ligand 1 and on the same level as those of other strong binders such as pyrazolourea ligands 5, 10 and 11.
As discussed in the last subsection, pyrazolourea ligand 5 has the largest RMSD for ligand alone among all the reference ligands. In the most stable predicted conformation, the morpholine ring loses the H bonding interaction with A100 which is assumed to be important to the binding of ligand 5 to CDK8.[22] As a result, its calculated Coulombic interaction is relatively small, which causes the underestimation of its computed binding free energy. The MD simulation didn’t catch this H bonding interaction either. It is possible that the parameters of GAFF force field for morpholine need to be tweaked to pick up this H bonding. The major van der Waals contributors to ligand 5 are E66, L70, I79, F97 and D173. But the total van der Waals energy is much weaker than those of ligands 1 and 2.
The VM2 method accurately predicted the binding pose of pyrazolourea ligand 11. Its RMSD for ligand alone is the smallest among the reference ligands. Its hydroxyl group forms strong H bonding interactions with D98 and A100. Probably for this reason it achieves the strongest Coulombic energy and the most favorable overall electrostatic energy. Its van der Waals interaction is much smaller than those of other strong binders though, and it pays large entropy penalty due to its long and flexible carbon chain. Therefore Coulombic energy plays an important role in the binding of ligand 11 to CDK8. It has the perfect length of the carbon chain that helps the strong binding to occur when compared with ligands 3, 9 and 10.
Key information learned from the computational binding thermodynamics study
The VM2 method predicted the binding free energies with relatively high correlation with the experimental data for the pyrazolourea ligands. It also revealed information that is not available from experiments. The driving force for the binding of the reference ligands to CDK8/CycC is van der Waals interaction, and the overall electrostatic interaction has a negative contribution to the binding affinity. The analysis on the strong binders suggests that there is room for all of them to improve their binding affinities. Specifically, ligand 1 pays extremely large desolvation and entropy penalties. This can be improved by making the structure less flexible and position the polar groups at places that promote H bonding. Ligands 2 and 5 have poor overall electrostatic energy. Polar groups can be introduced to form H bonding with the protein to improve the electrostatic energy. Ligand 11 pays large entropy penalty. Its linear carbon chain can be modified to a more rigid structure to reduce the entropy penalty.
On the predicted lowest energy conformations of all the reference ligands, the urea moiety forms H bonding with residues E66 and D173, and accounts for the majority of H bonding between the protein and the ligands. The 500 ns MD simulations[32] show that these hydrogen bonds are highly stable. Their occurrence percentages are roughly 90~96% and 76~93% respectively for all the reference ligands. Therefore, we used the urea moiety to initiate our virtual screening research for new type II CDK8 ligands.
Substructure -based Virtual Screening
As the first step the urea moiety was used in the substructure search on ChemDiv database. This resulted in 187,000 compounds. These compounds were screened with the criteria detailed in experimental section including a moiety specific requirement: each nitrogen atom of the urea moiety must have one attached hydrogen atom. These hydrogen atoms are needed in H bonding with E66. After this initial screening step, the pool of candidates is reduced to 9,914 compounds.
The reduced pool was then processed with the Superposition and Single-Point Energy Evaluation method (refer to experimental section). It took 5 to 10 minutes to process one compound on one core with a Xeon E5–2640 v2 @ 2.00GHz CPU. With 20 cores we finished this energy evaluation step in two days. The binding energies of top 100 compounds are listed in Table S2, and the molecular structures of top 20 compounds are listed in Table S3. Top 20 compounds have highly diverse molecular structures. We also applied this evaluation method on ligand 1, and found it has a lower binding energy than all of the candidates. After superposition and energy minimization, all of them were found to have the similar binding mode as pyrazolourea ligand 1. Figure 5 presents the conformations of pyrazolourea ligand 1 and the compound ranked #1 (called CL1 hereafter) by the Superposition and Single-Point Energy Evaluation method. The predicted conformation of pyrazolourea ligand 1 can be aligned with the crystal structure 4F6W very well. For CL1, besides the key interactions with E66 and D173 it also forms a hydrogen bond with A100, a residue in the hinge region which is important to ligand binding and residence time.[22] The predicted conformations of CL2, CL3, CL4 and CL5 with CDK8 are demonstrated in Figure S5. CL2 and CL4 have the same binding mode as CL1, interacting with E66, D173 and A100 through hydrogen bonds. CL3 doesn’t have the interaction with A100, but its 1,3,4-thiadiazole forms H bonding with K52. CL5 doesn’t have contact with A100 either. Instead its indole ring forms another H bonding with D173.
Figure 5.

Predicted binding modes of ligand 1 (top) and compound CL1 (bottom) by the Superposition and Single-Point Energy Evaluation method. The crystal structure of the reference ligand is colored green for comparison.
We picked 19 compounds from the top ones by the Superposition and Single-Point Energy Evaluation method, plus two selection rules: (1) candidates are able to form at least one hydrogen bond with the hinge region (residues 97 to 100); (2) candidates don’t have toxic substructures. We eye inspected the predicted conformations of the top compounds until the pool was filled up. Table 3 lists the molecular structures of the candidates. When compared with the top 20 compounds ranked by the Superposition and Single-Point Energy Evaluation method, the 19 candidates for VM2 evaluation are more similar to each other. For instance, CL1 and CL2 are different by only one atom; CL4, CL21, CL37, and CL79 share the same scaffold. Similarly, CL34 and CL119, CL36, CL46 and CL83, CL125 and CL128 belong to 3 different congeneric series. The VM2 free energy calculation took 3 to 7 days per compound on 4 CPU cores as above.
Table 3.
Molecular structures of the 19 candidate compounds picked for VM2 free energy evaluation. The rankings are from the Superposition and Single-Point Energy Evaluation method.
| Ranking | Chemical structure | Ranking | Chemical Structure |
|---|---|---|---|
| 1 |  |
68 | 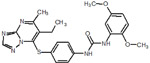 |
| 2 | 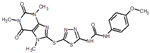 |
79 | |
| 4 | 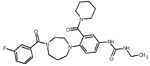 |
82 |  |
| 19 | 83 | 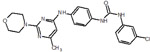 |
|
| 21 | 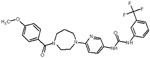 |
88 | 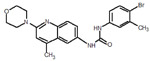 |
| 23 | 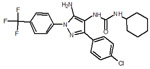 |
119 | 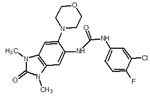 |
| 34 | 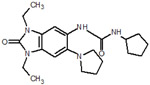 |
125 | 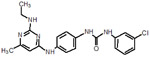 |
| 36 | 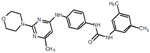 |
128 | 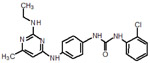 |
| 37 | 142 | ||
| 46 |
Table 4 presents the calculated binding free energies (ΔG) and their energy components of the 19 candidates. Only 8 candidates have negative ΔG and none of them have ΔG lower than ligand 1. Some candidates have a large valence energy term which includes all the bonded interactions. This indicates that these compounds have much internal stress at the protein binding site. This energy term can be considered as an indicator of how well a compound can fit the binding site. The candidates with positive ΔG pay large desolvation penalty and their overall electrostatic energies are consistently greater than 30.0 kcal/mol, about 10.0 kcal/mol higher than those of the candidates with negative ΔG. CL1 and CL2 were also ranked as the top 2 compounds by VM2. When compared with ligand 1, they have much higher (less favorable) ΔEVDW presumably due to their smaller molecular sizes, but they pay less entropy penalty and gain much lower (more favorable) ΔECoulomb. CL88 was ranked the 3rd by VM2. Its Coulombic energy is on the same level as that of ligand 1 and its van der Waals contribution is similar to that of CL1 and CL2. It has reduced desolvation penalty and entropy penalty that boost its calculated binding free energy.
Table 4.
The calculated free energy and energy components of the 19 candidate compounds by VM2. Please refer to Table 1 for the meaning of each energy term.
| compound | ΔEValence | ΔECoulomb | ΔEPB | ΔENP | ΔEVDW | ΔH | −TΔS | ΔGcalc | ΔGexp |
|---|---|---|---|---|---|---|---|---|---|
| CL1 | 2.575 | −37.351 | 59.884 | −6.063 | −66.181 | −47.136 | −28.567 | −18.569 | −10.116 |
| CL 2 | 5.835 | −30.900 | 51.406 | −5.825 | −69.650 | −49.134 | −31.329 | −17.805 | −9.528 |
| CL 88 | 5.437 | −20.290 | 46.567 | −5.697 | −68.374 | −42.357 | −25.080 | −17.277 | −6.784 |
| CL 21 | 12.007 | −93.269 | 109.888 | −7.106 | −71.414 | −49.894 | −34.696 | −15.198 | |
| CL 37 | 8.164 | −88.559 | 110.296 | −6.768 | −62.763 | −39.630 | −29.486 | −10.144 | |
| CL 79 | 15.828 | −90.286 | 106.577 | −6.888 | −62.442 | −37.211 | −28.617 | −8.594 | |
| CL 68 | 8.595 | −19.085 | 47.124 | −6.280 | −65.626 | −35.272 | −28.657 | −6.615 | |
| CL 125 | 5.678 | −17.151 | 38.969 | −5.712 | −55.414 | −33.630 | −32.058 | −1.572 | |
| CL 128 | 0.682 | −11.092 | 43.001 | −5.845 | −55.046 | −28.300 | −31.304 | 3.004 | |
| CL 83 | 10.269 | −9.602 | 42.038 | −6.105 | −63.284 | −26.684 | −31.977 | 5.293 | |
| CL 36 | 11.726 | −9.625 | 43.528 | −6.369 | −65.677 | −26.417 | −32.157 | 5.740 | |
| CL 46 | 11.314 | −14.672 | 50.305 | −6.328 | −64.128 | −23.509 | −31.125 | 7.616 | |
| CL 119 | 5.942 | −11.856 | 49.080 | −5.776 | −58.916 | −21.526 | −30.401 | 8.875 | |
| CL 142 | 6.350 | −8.376 | 42.859 | −5.845 | −53.171 | −18.183 | −29.681 | 11.498 | |
| CL 34 | 16.957 | −10.557 | 40.449 | −5.221 | −63.213 | −21.585 | −35.780 | 14.195 | |
| CL 4 | 13.136 | −10.238 | 45.821 | −6.210 | −58.802 | −16.293 | −30.728 | 14.435 | |
| CL 23 | 7.329 | −22.129 | 66.030 | −6.459 | −58.193 | −13.422 | −32.146 | 18.724 | |
| CL 19 | 14.920 | −16.982 | 50.464 | −6.551 | −57.835 | −15.984 | −37.405 | 21.421 | |
| CL 82 | 62.593 | −5.770 | 58.124 | −6.423 | −29.028 | 79.496 | −42.367 | 121.863 | |
| reference | 1.031 | −19.845 | 60.764 | −8.548 | −93.837 | −60.435 | −39.487 | −20.948 | −10.324 |
The lowest energy conformation of CL1 with CDK8 (Figure 6) predicted by VM2 is similar to the one predicted by the Superposition and Single-Point Energy Evaluation method. The key hydrogen bonds with E66, D173 and A100 are well kept. Furthermore, its 1,3,4-thiadiazole forms H bonding with K52. This may explain its highly favorable Coulombic contribution. CL1 features large aromatic scaffold, which make the whole molecular structure relatively rigid and reduce the entropy penalty. The extended conformation helps the bulky aromatic rings on both ends stay at the hydrophobic deep pocket and front pocket respectively. They are surrounded by nonpolar residues and make extensive van der Waals interactions with them. CL1 has a relatively small molecular weight (MW), 473 daltons. As a comparison, the MW of ligand 1 is 640 daltons. CL2 is different from CL1 by only one atom, and it has the exactly same binding mode as CL1. The predicted lowest energy conformation of CL88 can be found in Figure S6. The starting conformation of CL88 for the VM2 free energy calculation has H bonding with A100. But VM2 ended up with the morpholine ring rotating by 90° and losing the contact with A100. CL88 has a bulky quinoline ring in the middle of its backbone. This helps to reduce its entropy penalty, but also limits the arrangement of the whole molecular structure to accommodate H bonding between the morpholine ring and A100.
Figure 6.

Predicted lowest energy conformation of CL1 with CDK8 by VM2.
Among the 19 compounds evaluated with VM2, only the top 3 compounds, i.e. CL1, CL2 and CL88, kept their starting poses, i.e., the key urea moiety had the expected contacts with CDK8, while the rest lost such contacts in their predicted lowest energy conformations. In this research we focused on finding actives with our virtual screening tool. Therefore, we only purchased the top 3 compounds ranked by VM2, and had bio-assay testing with them. The result (ΔGexp) can be found in Table 4. CL1 and CL2 show strong binding affinity with CDK8, the Kd values of which are 42.5 nM and 114 nM respectively. The Kd value of CL88 is 11.4 μM, which is barely lower than the no-binding cutoff (20 μM) in the testing. Both the Superposition and Single-Point Energy Evaluation method and the VM2 method ranked the 3 compounds correctly. CL1 achieved a binding affinity comparable to those of the two most potent reference ligands (Kd values of 10 nM and 30 nM for ligand 2 and ligand 1 respectively). It has a drug-like structure with dimethyl-xanthine, 1,3,4-thiadiazole and benzene moieties on its backbone. Figure 2 presents the correlation between the calculated and experimental free energies when the data points of the 3 tested compounds are plotted together with the reference ligands. The correlation coefficient drops from 0.71 to 0.47 after these data points are added. The poorer correlation is mainly caused by the overestimated binding free energy of CL88. If this data point is removed from the plot, the correlation coefficient becomes 0.72. Despite the overestimation, VM2 predicts that CL88 have no contact with the hinge region, which is probably correct and explains the weak affinity of this compound. The interaction with the hinge region is important to CDK8 ligand binding and is found in the binding mode of the two strong binders, CL1 and CL2.
With our substructure-based virtual screening protocol, we successfully discovered a new potent CDK8/CycC type II ligand, CL1, which is comparable to the published reference ligands. The effectiveness of this novel method is originated from the combination of two powerful computational methods and the knowledge learned from the thermodynamics analysis on reference ligands. The Superposition and Single-Point Energy Evaluation method was able to efficiently estimate the binding poses for candidate compounds with a co-crystal structure as reference. The binding energies obtained by this method are based upon force field energy equations and PB solvation model. Energy minimization is implemented on candidate compounds and binding site residues until convergence. Therefore, this method can offer high accuracy in energy prediction while maintaining relatively high speed. The VM2 free energy calculation method carries out thorough conformation sampling and considers both enthalpy and entropy contributions to free energies. This method is fast and accurate enough to be used as the last check point in virtual screening. And since it provides not only the total free energy and its components but also the energy contributions from the portions of the molecular systems, it is a useful tool for ligand optimization as well.
This novel substructure-based drug screening tool is also valuable in the context of fragment-based drug discovery. The structural moieties that make key interactions with a target protein in existing co-crystal structures can be employed separately in similarity or substructure database searches for fragments, which can then be merged or linked together to generate new ligands. Fragments generally have weak affinities with target proteins and pose significant challenges for screening through biophysical techniques.[33,34] The two energy evaluation methods in the virtual screening tool have the accuracy to solve the problem of fragment screening.
Conclusions
We developed a novel virtual screening method and applied it to the discovery of new CDK8/CycC type II ligands. The core of this method consists of two energy evaluation methods: Superposition and Single-Point Energy Evaluation, and VM2 free energy calculation. They together are able to efficiently and accurately screen candidate compounds. In this research work we first analyzed binding free energies and the energy components of 11 reference CDK8/CycC type II ligands with VM2, and extracted the information which was proved helpful for virtual screening.
The VM2 method accurately predicted the binding modes for the reference ligands, and the RMSDs were all less than 2.0 Å for the ligand atoms and atoms of binding site residues. The correlation coefficient is 0.71 between the calculated and measured free energies. The free energy and MD calculations successfully revealed the factors that play important roles in the ligand binding with CDK8 DMG-out conformation. The overall driving force of the binding is van der Waals interaction, but for some ligands Coulombic energy is also important to make the binding to occur. The urea moiety contributes the majority of H bonding between the reference ligands and CDK8 and acts as the anchorage to stabilize the ligands. The analysis on the strong binders also suggests that there is room for all of them to improve their binding affinities.
Starting with the urea moiety, we implemented the virtual screening method and singled out three compounds for bio-assay testing. The ranking from the experimental result is completely consistent with the predicted rankings by both Superposition and Single-Point Energy Evaluation method and VM2 free energy calculation method. We successfully discovered a new potent drug-like compound with a Kd value of 42.5 nM. Interestingly, top 2 compounds are different by only one atom but have a nearly 3-fold difference in binding affinity. This was accurately predicted by both energy evaluation methods. Therefore, our novel virtual screening method is accurate and efficient enough to be used in drug design projects. We believe this work has significant impact to the field of drug discovery.
Experimental Section
Reference CDK8-Ligand Complexes
Among the eleven ligands published by Schneider et al.,[22] shown as in Table 1, seven have co-crystal structures with the CDK8 DMG-out conformation. The PDB IDs are 4F6W, 4F7L, 4F7J, 4F70, 4F6U, 4F6S, and 4F7N for ligands 1, 2, 3, 4, 5, 7, and 11, respectively. The crystal structure 4F6W was employed in all of the energy computations in this research because ligand 1 in this crystal structure has the most extensive interactions with the protein and is one of the most potent ligands in this congeneric series. Ligand 1 was also used as the reference ligand in this study for the comparison purpose.
Methodologies to Investigate the Binding Thermodynamics of Reference Complexes
1. Free energy Calculation
A rigorous statistical thermodynamics method, called VM2,[29] was used to calculate the binding free energies of CDK8 and its ligands in silico. VM2 belongs to a class of methods that focus on the most stable conformations of the molecules, so they are sometimes called predominant states methods. They compute the standard chemical potential of the protein-ligand complex and of the free ligand and protein, and take the difference to obtain the standard free energy of binding,
| (1) |
The standard chemical potential of each molecular species (i.e. complex, protein and ligand) is obtained by finding its M most stable conformations (j = 1, M), integrating the Boltzmann factor within each energy well j to obtain a local configurational integral Zj, and combining these local configuration integrals according to the following formula, where X = complex, protein, or ligand
| (2) |
Here C0 is the standard concentration, which, combined with the factor of 8π2, accounts for the positional and orientational mobility of the free molecule at standard concentration, and the second form of the summation is given in terms of the chemical potentials of the individual conformations. The probability of energy well j can be approximated on the basis of Zj, and thus the mean potential energy <U> or solvation energy <W> can be obtained. The configurational entropy at standard concentration can be computed as . The configurational entropy includes both a conformational part, which reflects the number of energy wells (conformations), and a vibrational part, which reflects the average width of the energy wells. 〈U+W〉 implicitly includes the change in solvent entropy via the implicit solvent model. As a consequence, the configurational entropy values reported here should not be directly compared with experimental entropy changes of binding for these systems.
The VM2 calculations are moderately fast, in part because they use implicit solvent models, which are widely accepted as computationally efficient alternatives to more detailed solvent models, and in part because for large systems such as protein-ligand complexes only a subset of atoms (~500–5000, depending on the nature of the active site) are treated as mobile. The free energies calculated with VM2 are regarded as relative free energies instead of absolute ones. They, together with the energetic components, are used to rank the candidate ligands and analyse their binding modes. The quality of the calculated free energies can be judged through the Pearson correlation coefficient (R2) between ΔGcalc and ΔGexp.
Crystal structure 4F6W was used as a reference structure for VM2 free energy calculations for all of the complexes of CDK8 and the ligands in this study. The starting conformations of the ligands were generated by superimposing them onto ligand 1 in 4F6W. Since 4F6W misses residues 178–195, which form the main portion of the activation loop, a homology model was generated by SWISS_MODEL [35–37] to add the missing residues. The pdb file for the homology model is provided in Supporting Information. The addition of the missing residues is very important to our calculations for two reasons: (1) Without them the activation loop is broken, so its movement in calculation cannot reflect the reality; (2) Since the missing residues are in the periphery of the binding site of CDK8 and some are very close to ligands, they have significant contribution to free energies. Amber99SB and GAFF Force Field (GAFF) [38–40] were applied to CDK8 and the ligands, respectively. The Amber 14 package [41–43] was employed to assign partial charges (-bcc) to the ligands, and the partial charges for the protein were from the standard force field parameters. The live set — the set of binding-site atoms treated as mobile — was defined as all atoms within 7 Å of any atom of the ligands. The real set — the set of protein atoms treated as rigid and thus supporting the live set — comprised all protein residues having any atom within 5 Å of any live-set atom. The pdb files for the live set and real set can be found in Supporting Information. All other protein residues (total 194 out of 359 residues, or 54%) were deleted, in order to reduce the size of the non-bonded pair list and thus speed up the calculations. The interactions between atoms that are beyond 12 Å apart are negligible; therefore those 194 residues have little contribution to free energies and thus can be deleted/ignored. In order to diminish any initial stress in the starting conformations that might artifactually drive the binding site away from its crystallographic starting conformation, the protein models were subjected to an initial relaxation. Van der Waals radii were used in both GB and PB solvation energy calculations. The VM2 method runs cycles of conformational search and configurational integration until convergence criteria is met.[29]
2. Unbiased Molecular Dynamics (MD) Simulation
We used the trajectories from the previous study [32] to calculate MM/PBSA energies with the Amber 14 package [41–43] and analyse hydrogen bonding. The details of MD simulations can be found in Supporting Information. We performed hydrogen bonding (H bonding) analysis on the MD trajectories. In this study a H-bond (D-H…A, where D and A stand for donor and acceptor respectively) is considered formed if the distance between H and A is smaller than 2.5 Å and the angle of D-H…A is greater than 150º. We used an in-house script to scan the trajectories for direct H bonding between ligands and CDK8. H bonding between ligands and the same residues are merged into one residue-ligand H bonding formation. The occurrence percentage of a hydrogen bond is calculated as the number of the frames where the hydrogen bond is found divided by the total frames (i.e., 25000).
Substructure-based Virtual Screening
We developed a novel substructure-based virtual screening method to assist the discovery of new drug candidates through efficient and extensive database searches. The workflow of this method is presented in Figure 1. We first computationally study the binding thermodynamics of the existing protein-ligand complexes involving the target protein based on their co-crystal structures. The key moieties on the ligands that make critical contributions to protein binding are identified. Those moieties and their similar structures are used in substructure database searches. In this study the database we used was ChemDiv, which has a collection of over 1,700,000 lead-like, drug-like small molecules in pharmaceutical industry.[44] The compounds obtained after the substructure database searches are then screened with the following criteria:
No more than 5 hydrogen bond donors;
No more than 10 hydrogen bond acceptors;
A molecular mass less than 600 and greater than 160 daltons;
An octanol-water partition coefficient logP less than 5 and greater than −0.4;
Specific requirements on the key moieties.
These criteria can be viewed as a modified version of Lipinski’s rule of five.[45,46] The compounds passing this initial screening are then evaluated and ranked by the Superposition and Single-Point Energy Evaluation method, which is detailed as below. The top 20 to 30 compounds from this step are further analysed with the VM2 free energy calculation method, as afore described. The top compounds that have similar or better binding free energies than a reference ligand based on the VM2 evaluation are purchased (or synthesized), and tested with bio-assay to verify the predicted binding affinities.
1. Superposition and Single-Point Energy Evaluation
Our novel superposition method is able to quickly generate the correct conformations for candidate compounds at the binding site of a target protein. It doesn’t apply conformational searches on candidate compounds. Instead, for the compounds from sub-structural searches, superposition is implemented by aligning the sub-structure of a candidate compound and a reference ligand at its bound state in a co-crystal structure. This method is based on the assumption that the sub-structure provides the key interactions between the target protein and the ligands. Therefore, as long as we anchor the key sub-structure at the correct position and pose, the whole ligand would probably have the correct bound conformation as well. The information about the key sub-structure is extracted from the analysis on the existing ligands of the target protein. Our superposition method also works on the compounds from similarity searches. In this case, our method first finds the common part, which can be backbones, moieties or functional groups, between a candidate compound and a reference ligand, and then superimposes them based on the common part. In the case that there are more than one key sub-structures or common parts, superposition is implemented with one at a time; if a sub-structure or common part is symmetric, the candidate compound is rotated to generate all possible superimposed conformations. In this study ligand 1 in the crystal structure 4F6W was used as the reference ligand.
After a candidate compound is placed into the binding site of a target protein with possibly multiple conformations, these conformations of the candidate compound together with the target protein are then energy minimized using the Amber 14 package until convergence. A live set is defined similarly as in the VM2 method. Only the compound atoms and the protein atoms in the live set are relaxed in energy minimization. The energy of the complex Ecomplex is calculated with the equation
| (3) |
where Eel and Evdw are electrostatic and van der Waals energy with parameters of Amber99SB for the protein and GAFF for the ligands; Gpb is the solvation energy computed by solving the Poisson Boltzmann (PB) equation;[47,48] Gnp is the nonpolar energy estimated from solvent accessible surface area.
The protein and the candidate compound are then separated from the complex, but their conformations are kept fixed. Their energies, Eprotein and Eligand, are calculated by equation (3) as well. The binding energy for the compound is then calculated as
| (4) |
Theoretically this Superposition and Single-Point Energy Evaluation method is faster and more accurate than the conventional docking and scoring method because of the following reasons. Firstly, the binding conformations of the candidate molecules are obtained by direct superposition to the available crystal structure of a reference molecule. It thus saves time of generating millions of random conformations as the general practice in docking and still has more accurate binding mode with the protein. Secondly, the binding energy includes the solvation energy, and is calculated with force-field based energy equations and the PBSA model, thus making more physical sense than scoring functions. Therefore, it is quite accurate and efficient for screening a large number of candidates. However, this method doesn’t include entropy contribution and only accounts for the energy contribution from a single conformation. The top compounds obtained from this method need to be further analysed and ranked using the more accurate VM2 free energy calculation method.
Compounds and Bio-assay
In this study the compounds selected for bio-assay testing were purchased from ChemDiv Inc. Bio-assay testing was conducted by Proteros biostructures GmbH.[49] The Proteros Reporter Displacement Assay was used to determine Kd. Briefly speaking, the Proteros reporter displacement assay is based on reporter probes that are designed to bind to the site of interest of the target protein. The proximity between reporter and protein results in the emission of an optical signal. Compounds that bind to the same site as the reporter probe displace the probe, causing signal diminution. Reporter displacement is measured over time after addition of compounds at various concentrations. In order to ensure that the rate of probe displacement reflects compound binding and not probe dissociation, probes are designed to have fast dissociation rates. Thus, compound binding and not probe dissociation is the rate limiting step of probe displacement. For Kd determination, percent probe displacement values are calculated for the last time point, at which the system has reached equilibrium. For each compound concentration, percent probe displacement values are calculated and plotted against the compound concentration.
Supplementary Material
Acknowledgements
This study was supported by the US National Institutes of Health (GM-109045). The VM2 software package was provided by VeraChem LLC and is available free for academic use at www.verachem.com.
References:
- (1).Galbraith MD, Donner AJ, Espinosa JM, CDK8: a positive regulator of transcription. Transcription 2010, 1, 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Tsutsui T, Fukasawa R, Tanaka A, Hirose Y, Ohkuma Y, Identification of target genes for the CDK subunits of the Mediator complex. Genes. Cells 2011, 16, 1208–1218. [DOI] [PubMed] [Google Scholar]
- (3).Allen BL, Taatjes DJ, The Mediator complex: a central integrator of transcription. Nat. Rev. Mol. Cell Biol 2015, 16, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Rickert P, Seghezzi W, Shanahan F, Cho H, Lees E, Cyclin C/CDK8 is a novel CTD kinase associated with RNA polymerase II. Oncogene 1996, 12, 2631–2640. [PubMed] [Google Scholar]
- (5).Xu W, Ji JY, Dysregulation of CDK8 and Cyclin C in tumorigenesis. J. Genet. Genomics 2011, 38(10), 439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Conaway RC, Sato S, Tomomori-Sato C, Yao T, Conaway JW, The Mammalian Mediator Complex and Its Role in Transcriptional Regulation. Trends Biochem. Sci 2005, 30, 250–255. [DOI] [PubMed] [Google Scholar]
- (7).Nemet J, Jelicic B, Rubelj I, Sopta M, The Two Faces of Cdk8, a Positive/Negative Regulator of Transcription. Biochimie 2014, 97, 22–27. [DOI] [PubMed] [Google Scholar]
- (8).Li N, Fassl A, Chick J, Inuzuka H, Li X, Mansour MR, Liu L, Wang H, King B, Shaik S, et al. Cyclin C Is a Haploinsufficient Tumour Suppressor. Nat. Cell Biol 2014, 16, 1080–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Knuesel MT, Meyer KD, Bernecky C, et al. The human CDK8 subcomplex is a molecular switch that controls mediator coactivator function. Genes. Dev 2009, 23(4), 439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Rzymski T, Mikula M, Wiklik K, et al. CDK8 kinase–An emerging target in targeted cancer therapy. Biochim. Biophys. Acta 2015, 1854(10 Pt B), 1617–1629. [DOI] [PubMed] [Google Scholar]
- (11).Giovanni CD, Novellino E, Chilin A, Lavecchia A, Marzaro G, Investigational drugs targeting cyclin-dependent kinases for the treatment of cancer: an update on recent findings (2013–2016), Expert Opin. Investig. Drugs, 2016, 25(10), 1215–1230. [DOI] [PubMed] [Google Scholar]
- (12).Philip S, Kumarasiri M, Teo T, Yu M, Wang S, Cyclin-Dependent Kinase 8: A New Hope in Targeted Cancer Therapy? Miniperspective. J. Med. Chem, 2017, 61(12), 5073–5092. [DOI] [PubMed] [Google Scholar]
- (13).Cee VJ, Chen DY, Lee MR, Nicolaou KC, Cortistatin A is a high-affinity ligand of protein kinases ROCK, CDK8, and CDK11. Angew. Chem., Int. Ed 2009, 48, 8952–8957. [DOI] [PubMed] [Google Scholar]
- (14).Pelish HE, Liau BB, Nitulescu II, Tangpeerachaikul A, Poss ZC, Da Silva DH, et al. Mediator Kinase Inhibition Further Activates Super-Enhancer Associated Genes in AML. Nature, 2015, 526(7572), 273–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Porter DC, Farmaki E, Altilia S, Schools GP, West DK, Chen M, et al. Cyclin-dependent kinase 8 mediates chemotherapy-induced tumor-promoting paracrine activities. Proc. Natl. Acad. Sci. USA 2012, 109, 13799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Mallinger A, Schiemann K, Rink C, et al. Discovery of Potent, Selective, and Orally Bioavailable Small-Molecule Modulators of the Mediator Complex-Associated Kinases CDK8 and CDK19. J. Med. Chem 2016, 59(3), 1078–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Koehler MF, Bergeron P, Blackwood EM, Bowman K, Clark KR, Firestein R, Kiefer JR, Maskos K, McCleland ML, Orren L, Salphati L, Schmidt S, Schneider EV, Wu J, Beresini MH, Develoment of a Potent, Specific CDK8 Kinase Inhibitor Which Phenocopies CDK8/19 Knockout Cells. ACS Med. Chem. Lett 2016, 7(3), 223–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Czodrowski P, Mallinger A, Wienke D, Esdar C, Pöschke O, Busch M, Rohdich F, Eccles SA, Ortiz-Ruiz MJ, Schneider R, Raynaud FI, Structure-based optimization of potent, selective, and orally bioavailable CDK8 inhibitors discovered by high-throughput screening. J. Med. Chem, 2016, 59(20), 9337–9349. [DOI] [PubMed] [Google Scholar]
- (19).Wang T, Yang Z, Zhang Y, Yan W, Wang F, He L, Zhou Y, Chen L, Discovery of novel CDK8 inhibitors using multiple crystal structures in docking-based virtual screening. Eur. J. Med. Chem 2017, 129, 275–286. [DOI] [PubMed] [Google Scholar]
- (20).Schiemann K, Mallinger A, Wienke D, Esdar C, Poeschke O, Busch M, Rohdich F, Eccles SA, Schneider R, Raynaud FI, Czodrowski P, Discovery of potent and selective CDK8 inhibitors from an HSP90 pharmacophore. Bioorganic Med. Chem. Lett 2016, 26(5), 1443–1451. [DOI] [PubMed] [Google Scholar]
- (21).Ono K, Banno H, Okaniwa M, Hirayama T, Iwamura N, Hikichi Y, Murai S, Hasegawa M, Hasegawa Y, Yonemori K, Hata A, Aoyama K, Cary DR, Design and synthesis of selective CDK8/19 dual inhibitors: Discovery of 4,5-dihydrothieno[3’,4’:3,4] benzo[1,2-d] isothiazole derivatives. Bioorg. Med. Chem 2017, 25(8), 2336–2350. [DOI] [PubMed] [Google Scholar]
- (22).Schneider EV, Bottcher J, Huber R, Maskos K, Neumann L, Structure–kinetic relationship study of CDK8/CycC specific compounds. Proc. Natl Acad. Sci. USA 2013, 110, 8081–8086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kumarasiri M, Teo T, Yu M, Philip S, Basnet SK, Albrecht H, Sykes MJ, Wang P, Wang S, In Search of Novel CDK8 Inhibitors by Virtual Screening. J. Chem. Inf. Model 2017, 57(3), 413–416. [DOI] [PubMed] [Google Scholar]
- (24).Copeland RA, Pompliano DL, Meek TD, Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discov 2006, 5(9), 730–739. [DOI] [PubMed] [Google Scholar]
- (25).Kroe RR, et al. Thermal denaturation: A method to rank slow binding, high affinity P38alpha MAP kinase inhibitors. J. Med. Chem 2003, 46(22), 4669–4675. [DOI] [PubMed] [Google Scholar]
- (26).Fujimoto J, Hirayama T, Hirata Y, Hikichi Y, Murai S, Hasegawa M, Hasegawa Y, Yonemori K, Hata A, Aoyama K, Cary DR, Studies of CDK 8/19 inhibitors: Discovery of novel and selective CDK8/19 dual inhibitors and elimination of their CYP3A4 time-dependent inhibition potential. Bioorganic Med. Chem, 2017, 25(12), 3018–3033. [DOI] [PubMed] [Google Scholar]
- (27).Copeland RA, Pompliano DL, Meek TD, Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discov 2006, 5(9), 730–739. [DOI] [PubMed] [Google Scholar]
- (28).Tummino PJ, Copeland RA, Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry 2008, 47(20), 5481–5492. [DOI] [PubMed] [Google Scholar]
- (29).Chen W, Gilson MK, Webb SP, Potter MJ, Modeling Protein-Ligand Binding by Mining Minima. J. Chem. Theory Comput 2010, 6(11), 3540–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Huang YMM, Chen W, Potter MP, Chang C-EA, Insights from Free-Energy Calculations: Protein Conformational Equilibrium, Driving Forces, and Ligand-Binding Modes., Biophysical J 2012, 103, 342–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Pan Y, Huang N, Cho S, MacKerell AD Jr., Consideration of Molecular Weight during Compound Selection in Virtual Target-Based Database Screening. J. Chem. Inf. Comput. Sci 2003, 43, 267–272. [DOI] [PubMed] [Google Scholar]
- (32).Cholko T, Chen W, Tang Z, Chang C-EA, A molecular dynamics investigation of CDK8/CycC and ligand binding: conformational flexibility and implication in drug discovery. J. of Comput. Aided. Mol. Des 2018, 32, 671–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Johnson CN, Erlanson DA, Murray CW, Rees DC, Fragment-to-Lead Medicinal Chemistry Publications in 2015: Miniperspective. J. Med. Chem, 2016, 60(1), 89–99. [DOI] [PubMed] [Google Scholar]
- (34).Johnson CN, Erlanson DA, Jahnke W, Mortenson PN, Rees DC, Fragment-to-Lead Medicinal Chemistry Publications in 2016: Miniperspective. J. Med. Chem, 2018, 61(5), 1774–1784. [DOI] [PubMed] [Google Scholar]
- (35).Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, Schwede T, SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Research 2014, 42(W1), W252–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T, Protein structure homology modelling using SWISS-MODEL Workspace. Nature Protocols, 2009, 4(1), 1–13. [DOI] [PubMed] [Google Scholar]
- (37).Arnold K, Bordoli L, Kopp J, Schwede T, The SWISS-MODEL Workspace: A web-based environment for protein structure homology modelling. Bioinformatics. 2006, 22, 195–201. [DOI] [PubMed] [Google Scholar]
- (38).Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, et al. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins: Struct, Funct, Bioinf 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Wang JM, Wolf RM, Caldwell JW, Kollman PA, Case DA, Development and testing of a general amber force field. J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- (40).Ozpinar GA, Peukert W, Clark T, An improved generalized AMBER force field (GAFF) for urea. J. Mol. Model 2010, 16, 1427–1440. [DOI] [PubMed] [Google Scholar]
- (41).Amber 14 by Case DA, Babin V, Berryman JT, et al. University of California, San Francisco, 2014. [Google Scholar]
- (42).Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, et al. The Amber biomolecular simulation programs. J. Comput. Chem 2005, 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Goetz AW, Williamson MJ, Xu D, Poole D, Le Grand S, et al. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput 2012, 8, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44). http://www.chemdiv.com/
- (45).Lipinski CA, Lombardo F, Dominy BW, Feeney PJ, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev 2001, 46(1–3), 3–26. [DOI] [PubMed] [Google Scholar]
- (46).Lipinski CA, Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today: Technologies. 2004, 1(4), 337–341. [DOI] [PubMed] [Google Scholar]
- (47).Luo R, David L, Gilson MK, Accelerated Poisson-Boltzmann calculations for static and dynamic systems. J. Comput. Chem 2002, 23, 1244–1253. [DOI] [PubMed] [Google Scholar]
- (48).Cai Q, Hsieh M, Wang J, Luo R, Performance of Nonlinear Finite-Difference Poisson-Boltzmann Solvers. J. Chem. Theory Comput 2010, 6, 203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49). http://www.proteros.com/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.