

Abstract
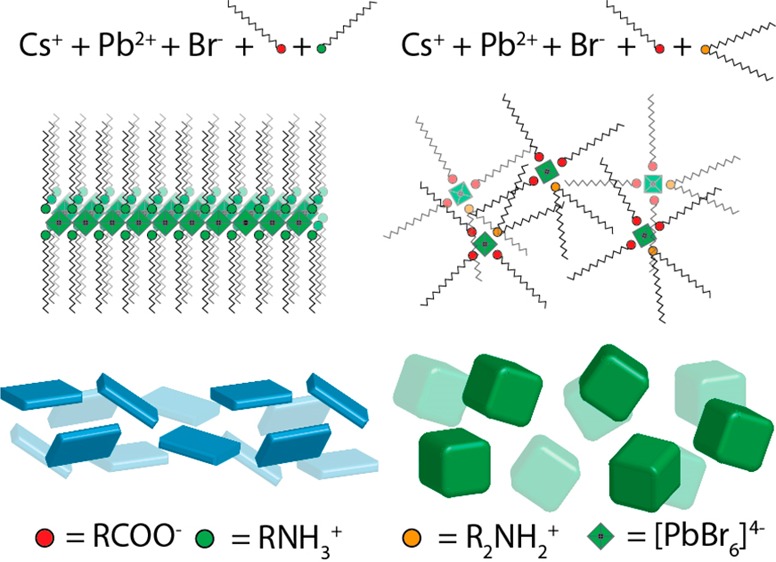
Fully inorganic cesium lead halide perovskite (CsPbX3) nanocrystals (NCs) have been extensively studied due to their excellent optical properties, especially their high photoluminescence quantum yield (PLQY) and the ease with which the PL can be tuned across the visible spectrum. So far, most strategies for synthesizing CsPbX3 NCs are highly sensitive to the processing conditions and ligand combinations. For example, in the synthesis of nanocubes of different sizes, it is not uncommon to have samples that contain various other shapes, such as nanoplatelets and nanosheets. Here, we report a new colloidal synthesis method for preparing shape-pure and nearly monodispersed CsPbBr3 nanocubes using secondary amines. Regardless of the length of the alkyl chains, the oleic acid concentration, and the reaction temperature, only cube-shaped NCs were obtained. The shape purity and narrow size distribution of the nanocubes are evident from their sharp excitonic features and their ease of self-assembly in superlattices, reaching lateral dimensions of up to 50 μm. We attribute this excellent shape and phase purity to the inability of secondary amines to find the right steric conditions at the surface of the NCs, which consequently limits the formation of low-dimensional structures. Furthermore, no contamination from other phases was observed, not even from Cs4PbBr6, presumably due to the poor ability of secondary aliphatic amines to coordinate to PbBr2 and, hence, to provide a reaction environment that is depleted in Pb.
Keywords: Secondary amines, nanocubes, inorganic metal halides, perovskites, colloidal synthesis, superlattices
The striking performance of lead halide perovskites (LHPs) in thin films in solar cells,1−3 as well as the works of both Schmidt et al.4 and Protesescu et al.,5 has inspired various groups to develop synthesis methods that produce high-quality LHP nanocrystals (NCs) (e.g., CsPbX3, MAPbX3, and FAPbX3; MA, methylammonium; FA, formamidinium; and X being Cl–, Br–, and I–).6−9 Among the various LHP NCs, CsPbX3 ones have been investigated the most owing to their high stability, narrow emission line widths and high photoluminescence quantum yield (PLQY, 65–95%).5,6,10,11 CsPbX3 nanocubes were initially prepared by injecting Cs-oleate into a hot solution (140–200 °C) of PbX2 (which serves as a source of both Pb2+ and X– ions) that had been dissolved in a mixture of oleic acid (OA) and oleylamine (OLAm). However, several groups have now shown that choosing an appropriate combination of primary amines and protic acids (in terms of chain length and molar ratio), adjusting the reaction temperature results in the synthesis of differently shaped LHPs [namely nanocubes, nanoplatelets (NPLs), nanorods (NRs), nanowires (NWs), and nanosheets (NSs)], or both.12−20 Unfortunately, this shape variability can also be detrimental, as it is not always easy to obtain homogeneous samples in terms of shape purity. Moreover, attempts to vary the size of the nanocubes (by reducing the reaction temperature12 or increasing the concentration of the acid)17,21 often lead to the contamination of the sample with NPLs. This is attributed to an increase in the concentration of oleylammonium species, which start competing with Cs+ ions, in the addition to the surface of the growing NCs.21,22 Shortening the hydrocarbon chain length of the primary amine also leads to the formation of NPLs, irrespective of the synthesis temperature.23 Finally, the use of excess amines promotes the formation of Cs-rich (Pb-depleted) phases, such as Cs4PbX6,24,25 which can also be an undesired product. Ideally, one would need a robust synthesis scheme that is not limited by all these reaction parameters so that cubes of various sizes can be prepared in a reproducible manner by sampling a broad set of reaction conditions, with no contamination from NCs with other shapes and crystal phases.
Here, we report a general synthesis procedure for nearly monodispersed shape- and phase-pure CsPbBr3 nanocubes. The key to achieving shape and phase purity is by omitting oleylamine, a primary amine that promotes both the anisotropic growth and the formation of unwanted phases. The CsPbBr3 NCs reported in this work can be synthesized using an extremely simple synthesis scheme that employs a protic acid such as OA as the only surfactant that is present in the reaction environment, delivering phase-pure CsPbBr3 NCs that are capped only with oleate molecules. However, by using only OA, the tunability of the NC size is not optimal. Thus, we introduce a secondary aliphatic amine in addition to OA to modulate the growth of the NCs. By varying the chain length of the secondary amine and the reaction temperature, we could tune the edge length of the cubes from 5 to 25 nm (in the range of conditions that are explored here). The size distributions of the NCs were also narrow, and the PL emission line widths were as low as 68 meV. Remarkably, secondary amines in the form of protonated ammonium ions are not able to bind to the surface of CsPbBr3 NCs nearly as effectively as oleylamine (or as effectively as a primary amine in general); see Scheme 1. Hence, they do not compete effectively with oleate molecules for binding to the surface of the NCs, and this inhibits the growth of NPLs under any of the conditions tested: different temperatures, ligand concentrations, and lengths of the amines’ aliphatic chains. In our NCs, the surface coverage (6–8%) by secondary ammonium ions is much lower than that of oleate molecules (92–94%), as is supported by nuclear magnetic resonance (NMR) measurements and X-ray photoelectron spectroscopy (XPS). Density functional theory (DFT) calculations indicate that dialkylammonium molecules cannot bind to the {100} facets of CsPbBr3 without heavily distorting the lattice, suggesting that their binding to the surface of NCs is weak and secondary to that of oleates. The homogeneity in the size and shape of the CsPbBr3 NCs is clear from the ability of the nanocubes to spontaneously self-assemble into superlattices with lateral dimensions of up to 50 μm. These are the largest dimensions that have been reported to date for superlattices (SLs) of LHP NCs. Notably, this record SL size was achieved without any size-selective precipitation or washings steps beyond the NCs’ isolation after synthesis.
Scheme 1. Synthesis of CsPbBr3 NCs in the Presence of Protonated Primary and Secondary Amines.
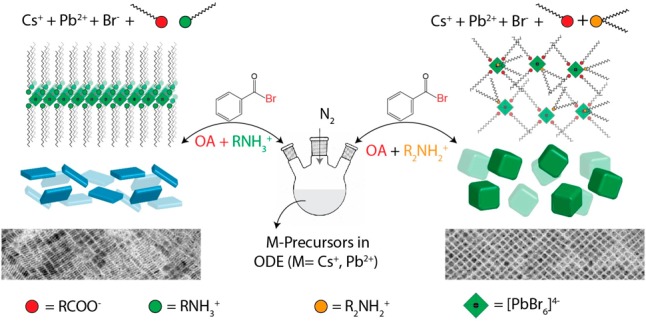
In this work, we used a modified version of our recently developed synthesis of CsPbX3 NCs, in which lead acetate trihydrate (PbAc2·3H2O) and a benzoyl halide are used separately as precursors for Pb and X (Cl, Br, and I), respectively,8 instead of the more common approach in which PbX2 is introduced as a combined source of Pb and X. In our revised scheme, benzoyl bromide is injected into a solution containing cesium and lead oleates in either the presence or the absence of secondary amines. After the synthesis, the NCs were separated and cleaned by adding a 1:6 volume ratio mixture containing toluene and ethyl acetate (or acetone) to the crude solution, which was then centrifuged and redispersed in toluene. A major finding is that no amine or ammonium ligands are actually needed for this type of synthesis. Pb and Cs salts are perfectly soluble in a mixture of ODE and OA alone because they form the corresponding metal oleates. Then, once the bromide precursor is injected, CsPbBr3 NCs are immediately formed. These NCs have cubic shapes, as can be seen by transmission electron microscopy (TEM) (Figure S1a). The absence of platelets under these synthesis conditions is most likely due to the fact that alkyl carboxylates are not known to be involved in the formation of layered halide perovskites. The NCs have an overall good optical quality, as is attested by UV–vis optical absorption and photoluminescence (PL) spectra (Figure S1c). Their X-ray diffraction (XRD) pattern conforms to the orthorhombic phase (Figure S1b). These NCs are passivated with either oleate or oleic acid because no other surfactants are used in the synthesis. This is in agreement with other studies that report the same type of NC surface passivation, although those NCs were grown in mixtures containing quaternary ammonium ions in addition to OA.26 In our “minimal” reaction scheme, the surface passivation by oleate species only occurs as a direct consequence of our decision to use only the minimum amount of chemicals that are required for the synthesis. Further attempts to tune the size of OA capped NCs (for example, by performing syntheses at different concentrations of OA; see Figure S2) were unsuccessful. It appears, therefore, that this oversimplified scheme, despite providing some basic understanding of the growth and surface chemistry of the NCs, does not provide size tunability.
To overcome these issues without using primary amines, we tested the effect of introducing secondary aliphatic amines to our synthesis under similar reaction conditions. In a first series of experiments, we systematically varied the hydrocarbon chain length of the secondary amine, from dihexylamine to dioctadecylamine, while all of the other reaction conditions were kept constant. (See Figure 1 for details and the caption of Figure 1 for acronyms of the various secondary amines used). We observed the formation of nearly monodisperse nanocubes in every case regardless of the type of dialkylamine that was used, as is shown in the TEM images in Figure 1a–e. By an increase of the secondary amine chain length, the size of the NCs decreased from 17 nm in the case of dihexylamine, to 7.5 nm in case of dioctadecylamine. The XRD patterns evidence the crystalline nature of the nanocubes, which closely match the orthorhombic perovskite phase (Figure 1f). The size uniformity in all of the samples is also evident from the optical absorption spectra (Figure 1g, gray-shadowed spectra), which show that the nanocubes have distinctive excitonic features, and from the PL spectra, which demonstrate that they have narrow emission lines with line widths in the range of 68–75 meV. The PL quantum yield (PLQY) of the various washed CsPbBr3 samples was in the range of 48–80%, and the average life times were approximately 5–10 ns (see Figure S3 and Table S1). Such PLQYs are comparable with those in previous reports by people who employ similar washing protocols using ethyl acetate or acetone.26,27
Figure 1.

(a–e) TEM images of CsPbBr3 NCs synthesized by varying the alkyl chain length of secondary amines. (a) dihexylamine (DHAm), (b) dioctylamine (DOAm), (c) didecylamine (DDAm), (d) didodecylamine (DDDAm), and (e) dioctadecylamine (DODAm). (f) Corresponding XRD patterns with reference (CsPbBr3, reference code. 96-451-0746). The presence of only the strong order peaks in the 12 nm sample (prepared with DOAm) is most likely the effect of preferential orientation for this sample. (g) Absorbance and PL spectra. The average sizes of the nanocubes measured from TEM images are written in panel f. The reaction conditions used in all these syntheses were as follows: the amount of each precursor used was 0.2 mmol of PbAc2·3H2O, 0.05 mmol of Cs2CO3, 4.75 mmol of OA, and 1.25 mmol of dialkylamine; the reaction temperature was 80 °C; 50 μL of benzoyl bromide was diluted in 0.5 mL of octadecene; the reaction time was 15 s.
We measured the reaction yield in terms of Pb present in the NCs compared to Pb added as a precursor [by inductively coupled plasma optical emission spectroscopy (ICP-OES) elemental analysis]. We have also calculated the total number of NCs (expressed as nanomoles of NCs) produced using a short (DHAm), intermediate (DOAm), and longer chain (DDDAm) amine in the synthesis. Results are reported in Table 1. The overall reaction yield decreases steadily (albeit not so strikingly) with increasing length of the alkyl chain of the amine, but the number of NCs increases considerably. It appears, therefore, that longer-chain amines promote a higher nucleation rate than shorter amines, but then the formed nuclei have to compete more for the remaining monomers; hence, their growth rate is reduced.
Table 1. NC Size, Reaction Yield, and Nanomoles of NCs Synthesized Using Three Different Secondary Amines.
| amine used | DHAm | DOAm | DDDAm |
| NC size | 17 | 13 | 8.2 |
| reaction yield | 52% | 49% | 43% |
| nanomoles of NCs synthesized | 3.5 | 7.1 | 25 |
Our reaction scheme is quite flexible in terms of the parameters that can be tuned to modify the size of the NCs, and it is quite robust because nanocubes are always formed. For example, using didodecylamine and varying the reaction temperature (from 50 to 140 °C) enables the synthesis of monodisperse nanocubes with sizes ranging from 6.2 to 19 nm (Figure 2a–d). XRD analysis indicates that all the samples have orthorhombic perovskite phase (Figure 2e). The degree of size monodispersity in the samples that are characterized by smaller sizes is evident from the multiple excitonic peaks; the energy separation between the peaks increases as the size decreases, which is in accordance with what has been observed in ensemble optical spectra of classic quantum dots. The size of the nanocubes could be further decreased to 5.1 nm by replacing didodecylamine with dioctadecylamine and working at 50 °C (Figure S4). Moreover, the size of the nanocubes could be further tuned from 9 to 25 nm by using didecylamine and by varying the reaction temperature from 80 to 140 °C (Figure S5).
Figure 2.
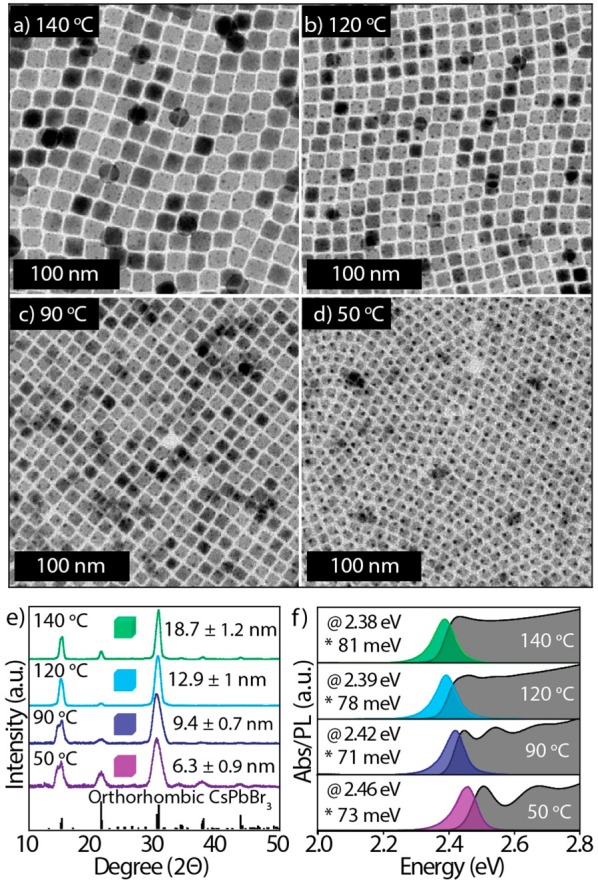
Data on CsPbBr3 NCs synthesized by using DDDAm at various temperatures, while the rest of the reaction conditions remained unchanged, as is illustrated in Figure 1. (a–d) TEM images of CsPbBr3 NCs, (e) XRD patterns of CsPbBr3 NCs that match the orthorhombic perovskite crystal structure (CsPbBr3, reference code. 96-451-0746), (f) absorption, PL spectra, PL max (@), and full width at half maxima (fwhm, *) of the corresponding samples. The average sizes of the nanocubes measured from TEM images are written in panel e. Notice that higher excitonic features are absent in the two samples with PL peaked at 2.39 and 2.38 eV, similar to the sample of Figure 1f prepared with DHAm (top spectrum). This is most likely due to a change in the electronic structure for samples of this size (the NCs entering a weak quantum confinement regime).
Another parameter that we tested is the concentration of OA. Even when the concentration of OA was increased 4-fold (from 3 mmol to 12.6 mmol), which strongly promotes the formation of NPLs in the case of primary amines,21 no NPLs were observed with didodecylamine (Figure S6). To compare the primary and secondary amines, several control syntheses were carried out by using primary and secondary amines separately under fully protonated conditions. In two series of experiments, we directly compared two pairs of amines: oleylamine versus dioctadecylamine and dodecylamine versus didodecylamine. OA was used as the carboxylic acid in all the syntheses, and all other reaction conditions were kept the same. We performed the reaction at a temperature of 100 °C, which is known to promote the synthesis of NSs and NPLs in the case of primary amines.23 Indeed, when primary amines were used, we observed the formation of CsPbBr3 NSs or NPLs, as was evidenced by TEM, optical absorbance, and PL spectroscopies (Figures 3a,b and S7a,c). XRD analysis closely match the orthorhombic perovskite phase (Figure S8). However, monodispersed nanocubes were formed in the case of secondary amines (Figures 3c-d and S7b,d). To further study the effect of the dialkylamine, and its inability to form 2D perovskites, we performed the synthesis of NCs in the absence of a Cs+ precursor. These conditions, when using primary amines, are known to promote the synthesis of a 2D hybrid layered phase with formula L2PbBr4 (L denotes a primary alkylammonium ligand).28−30 Indeed, we observed the formation of a white precipitate immediately after the injection of benzoyl bromide in case of dodecylamine (Figure S9a). TEM, XRD, UV–vis absorption, and PL analyses confirmed that the white precipitate corresponds to the 2D (dodecylammonium)2PbBr4 phase (Figure S9). Under the same reaction conditions, but working with didodecylamine, no precipitate was collected and the reaction mixture remained clear and colorless (Figure S9b).
Figure 3.
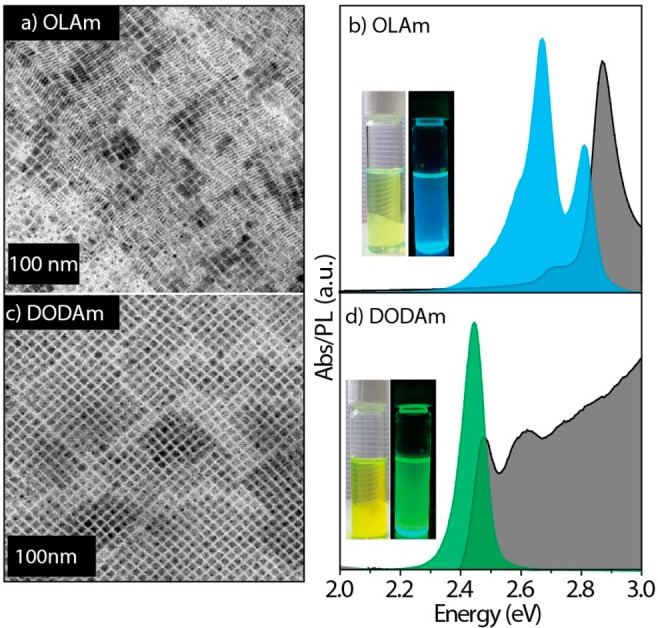
(a, c) TEM images and (b, d) absorption and PL spectra of CsPbBr3 NCs synthesized by using OLAm and DODAm. The synthesis was carried out under standard reaction conditions (described in Figure 1) except for the reaction temperature, which was 100 °C. The insets in panels b and d are the photographs of corresponding NC solutions under ambient and UV light.
We also ran syntheses under conditions that promote the formation of Pb-depleted Cs4PbBr6 phases in the case of primary aliphatic amines (that is, in the presence of a large excess of amine). Primary aliphatic amines can stabilize PbBr2, and therefore, they deprive the reaction environment of Pb2+ species and promote the formation of a Cs4PbBr6 phase over a CsPbBr3 one. However, with our secondary amines, only CsPbBr3 nanocubes were obtained (Figure S10). We ascribe this behavior to the inability of secondary amines to stabilize PbBr2. This was also proven in a control experiment in which CsPbBr3 NCs were mixed with a large excess of a secondary amine (didodecylamine). Notably, the NCs remained stable and UV–vis absorption spectra did not show any sign of a Cs4PbBr6 phase. The same experiment produced a Cs4PbBr6 phase when it was carried out with oleylamine (Figure S12 and Video 1), as has already been reported previously.31−33 Section 13 of the Supporting Information (and the associated Figure S13) discusses additional experiments with regard to the solubilization of PbBr2 by aliphatic amines.
To better explain the way that primary and secondary amines interact with oleic acid, we resorted to nuclear magnetic resonance (NMR) spectroscopy. 1H NMR was used to investigate the protonation of dodecylamine and didodecylamine in the presence of OA. Details of this study are reported in Figures S14–17. According to the 1H NMR data, at an acid-to-amine (primary or secondary) ratio of 3:1 or higher, as is usually employed in our syntheses, the amine is fully protonated (Figures S14–16). However, while an environment that is rich in fully protonated amines is ideal for the synthesis of NCs with two-dimensional shapes or layered phases,21 it seems to have no influence on the shape control for secondary amines because only cubes are formed. We also performed nuclear Overhauser effect spectroscopy (NOESY) on washed CsPbBr3 nanocubes that had been synthesized using didodecylamine, dispersed in deuterated toluene to investigate the nature of the surface ligands. The NOESY spectrum (Figure S17a) indicates that both the acid and the secondary amine interact with the NC surface, suggesting that both ligands are bound species, at least to a certain extent. Heteronuclear single quantum coherence (1H–13C HSQC) was also performed to confirm that the signals of interest that were observed in the NOESY belong to the acid and amine ligands (Figure S17b). The 1H NMR spectrum of this sample (Figure S17c) shows that the signals of both ligands broaden, and hence, both species are in active dynamic processes with the NC surface [with longest correlation times (τc), compared to free ligands]. To estimate the ratio between the two ligands that are present on the surface of the NCs, namely OA and didodecylamine, we ran a quantitative 1H NMR on washed CsPbBr3 nanocube samples dissolved in deuterated DMSO. A quantitative analysis of the alkene protons of OA and the α-CH2 protons of the amine indicated that the majority of the ligands that are present in the sample belong to OA species (92–94%), while only 6–8% are amine species (Figure S17d). Similar values were obtained from a quantitative analysis (i.e., by X-ray photoelectron spectroscopy, XPS) of the signals due to N (which we chose as a “marker” for the presence of didodecylamine molecules) and C, which focuses on the spectral component due to the acidic COOH moiety of OA (Figure S18). It should be noted that the position of this C component is (288.1 ± 0.2) eV in our sample, corresponding to carbon atoms in COO– groups.34
No signal could be detected from carbons in COOH groups at approximately 289 eV, indicating that the signal to be considered for XPS quantitative analysis only originates from bound molecules in the form of oleates. We also investigated the surface ligand ratios of CsPbBr3 nanocubes that were prepared by using primary amine.8 Remarkably, XPS did not detect any COOH moiety signals of OA, but a strong N signal was observed, a clear sign that these NCs are mainly capped with ammonium species (Figure S18). This is in line with previously reported NMR studies on CsPbBr3 NCs synthesized using oleic acid and primary amines, indicating that only ammonium species are bound and no oleate is present on the surface.22,35 The great excess of oleate species in the NCs that were prepared in the presence of secondary amines indicates that we are dealing with cation-rich NCs. In particular, the as-prepared NCs most likely have a Pb-rich surface, and the Cs/Pb/Br ratio is close to 1:1.1:2.4, as is suggested by the elemental composition that was obtained via XPS. However, the bromine content could be underestimated here due to X-ray radiation induced Br desorption.
To further gain mechanistic insight into how secondary amines can be bound to the surface of CsPbBr3 NCs, and how this differs from the way in which primary amines bind to it, we carried out DFT calculations. We modeled a CsPbBr3 slab, employing 4 × 4 unit cells in the x and y periodic directions and 2 unit cells in the z finite direction, that was cut along the cubic (100) direction. This model is in line with our high-resolution TEM (HR-TEM) analysis of various CsPbBr3 nanocubes (Figure S19), ensuring CsBr termination on both sides. The passivation of primary and secondary amines was reproduced by replacing surface Cs+ ions with methylammonium and diethylammonium ions as primary and secondary ammonium ions, respectively. Due to the small size of the alkyl chain, these calculations mainly take into account the effect of the anchoring group on the surface, while they neglect most of the chain-to-chain interactions. Further details on the calculations are provided in the Experimental section. Taking a slab passivated with Cs+ ions as a point of reference, we could calculate the effect of the lattice strain induced by the ammonium ions that are bound to the surface by analyzing the pair radial distribution functions (rdf) for the Pb–Pb atomic distance. We quantified the changes in the lattice by performing ab initio molecular dynamic simulations for 2 ps and 2.5 fs time steps in an isothermal–isobaric ensemble at a constant pressure (1 atm) and temperature (300 K). We chose the Pb–Pb distance because this is associated with the lattice vector of the cubic perovskite unit cell. Although the lattice distorts to an orthorhombic conformation, it can still be considered a good reference parameter for comparing the passivation with ammonium ions to the unpassivated crystal structure (Figure 4a). As is illustrated in Figure 4b, the passivation with methylammonium provides rdf(Pb–Pb) with well-resolved and narrow peaks, while the diethylammonium passivated surface presents much broader and unresolved peaks (Figure 4c), which is a sign that the lattice is more deformed and in two points also breaks apart (Figure 4c, left panel). This deformation/breaking of the lattice is mainly attributed to the binding of the ion’s anchoring group to the surface. Indeed, one of the diethylammonium ion’s two hydrogen atoms is almost perpendicular to the surface, whereas the two ethyl groups (i.e., the ones that emulate the long alkyl chains in the experiments) and the remaining hydrogen atom rearrange themselves so as to minimize steric interactions with nearby ammonium ligands (Figure 4d,e). In our computational model, diethylammonium ions attempt to reduce the strain by heavily distorting the lattice. In a more realistic environment as in the experiments, these ligands, rather than stretching the lattice, would likely become detached from the surface. We also analyzed the rdf(N–N), i.e., the distance between the nitrogen atoms in the ammonium anchoring groups. On average, this rdf presents less resolved and broader peaks than the ones obtained from the rdf values of the inorganic core due to the greater mobility of the amine ligands at the surface. However, while the closest distance between the N atoms is 5.8 Å in the case of the methylammonium ions, it reaches significantly longer distances (up to 7.1 Å) for diethylammonium ions. This supports the idea that large steric repulsions are present between the ammonium groups. This theory was also confirmed by our analysis of the effect of reducing the surface concentration of secondary amines on the nanoplatelet surface. We indeed reduced the ligand concentration from 100%, where each cation site at the surface is filled with diethylammonium ions, up to 25%, where 3 out of 4 ligands have been detached from the surface as ammonium–Br ion pairs. We noticed that the lattice strain diminishes and the cubic shape is recovered when secondary amines are detached (Figure S20).
Figure 4.

(a–c) Planar and side views of the slabs optimized at 0 K. Cs+ and primary (methylammonium) and secondary (dietyhlammonium) amine passivated structures are given from top to bottom. Panels a and b show the lattice vectors in reciprocal space that are replicated periodically, while panel c shows the lattice vector in the finite direction. On the right of each structure is their corresponding pair radial distribution functions for the Pb–Pb and Cs–Cs (or N–N) atomic distances, calculated by ab initio MD simulations at the DFT or PBE level of theory with the NPT ensemble (T = 300 K, P = 1 atm). (d, e) Side and planar view of the binding of primary (top) and secondary amines (bottom). In the case of secondary amines, the anchoring groups are closer to each other than in primary amines, indicating a larger steric hindrance.
Colloidal NCs with a good size and shape uniformity tend to self-assemble into ordered superstructures upon solvent evaporation. The absence of contamination from other shapes combined with the narrow size distribution of cubic CsPbBr3 NCs synthesized using secondary aliphatic amines gives us access to high-quality samples that are suitable for self-assembly. Previously, Kovalenko’s group has reported 1–10 μm square-shaped superlattices (SLs) of size-selected CsPbBr3 and CsPbBr2Cl NCs obtained by slowly evaporating the solvent on substrates,7,36,37 Feldmann and co-workers have reported the formation of CsPbBr3 NCs SLs smaller than 1 μm in situ during the NC synthesis,38 and Vanmaekelbergh et al. have reported the preparation of ∼1 μm SLs by adding methyl acetate antisolvent to NCs dispersed in hexane.39
The CsPbBr3 NCs used in our self-assembly experiments were synthesized with didodecylamine under standard reaction conditions without any size-selective precipitation or washings steps beyond the NCs’ isolation after synthesis. The average nanocube’s edge length, which was estimated using TEM, was 8.3 ± 0.6 nm and the PL was centered at 2.42 eV (Figure S21). The self-assembly of NCs was accomplished by a evaporating the solvent on top of a tilted Si wafer inside the glovebox. Cubic or rectangular shaped SLs with lateral dimensions ranging from 12 to 40 μm (Figure 5a,b) were formed upon drying a ∼6 μM NC solution in toluene overnight. To the best of our knowledge, these are the largest reported NC SLs for lead halide perovskites. The formation of SLs was also possible under ambient conditions on the lab bench instead of in the glovebox (see the details in the Experimental Section). For that experiment, we used a ∼4 μM toluene solution with similarly sized nanocubes (edge length of 8.5 ± 0.4 nm; see Figure S22). In some parts of the Si wafer, polygonal SLs with long edges reaching up to 50 μm were observed (Figure S23). Confocal PL microscopy was used to obtain PL images (Figure 5a) and to acquire spectra of individual SLs (Figure 5c) under 488 nm excitation (see the Experimental section for details). The PL images (Figure 5b) revealed that the SLs show a spatially uniform PL on a micron length scale. They incorporate virtually all of the emissive material, and there are no noticeable amorphous NC aggregates outside, which further highlights the shape purity and narrow size distribution of the NCs that were synthesized using secondary aliphatic amines. The PL spectra taken from individual SLs are practically indistinguishable from each other within the spectral resolution of the microscope detector (∼3 nm), and they show a 2.5 meV red shift compared with the starting NC solution or to a solution of NCs obtained by dissolving a small area of the self-assembled sample (Figure 5c). The latter comparison indicates that the red shift is specific to the SLs; therefore, it is not due to any transformation that might have occurred in the NCs. The red-shifted PL in the CsPbBr3 NC SLs has been observed previously,7,36−39 but its exact origins are unclear and remain a subject of further investigation. High-resolution SEM images of the individual SLs indicate that they have relatively flat surfaces, and images that were acquired at higher magnifications evidence that the SLs consist of tightly packed nanocubes (Figure 5d,e).
Figure 5.

(a) Low-magnification confocal PL microscope image (under 488 nm excitation) of superlattices made from 8.3 ± 0.6 nm CsPbBr3 nanocubes (see the Experimental section). (b) A confocal PL image of several square-shaped superlattices. (c) PL spectra of CsPbBr3 nanocubes before and after self-assembly. PL spectra from the three individual superlattices that are labeled in panel b. The PL of the superlattices is broadened and red-shifted ∼2.5 meV compared to the PL of NCs in solution. (d) A scanning electron microscope (SEM) image of an individual superlattice and (e) a high-resolution SEM image in the inset showing closely packed individual nanocubes.
We have tested our approach also on CsPbCl3 and CsPbI3 NCs. Initial synthesis results show that secondary amines, together with oleic acid, can be used to prepare CsPbCl3 NCs (Figure S24) while in the case of CsPbI3, NCs quickly degraded over time (hours). It is likely that the surface chemistry of iodides is much different from that of chlorides and bromides, and these syntheses will require further investigation.
In conclusion, in this work, we have substituted primary amines with secondary amines in a benzoyl halide based hot-injection synthesis of CsPbBr3 nanocubes. This substitution eliminates the possibility of nanocrystals forming non-cubic shapes, resulting in samples with remarkably uniform CsPbBr3 shapes. The shape uniformity and size tunability of the nanocubes are not dependent on the temperature of the bromide precursor’s injection (50 to 140 °C range) or on the hydrocarbon chain length of secondary amines (from dihexyl to dioctadecyl). The resulting nanocubes have narrow PL line widths (full width at half maxima 68–81 meV) and high PLQYs (48–80%) in solution. The DFT calculations revealed that the dialkylammonium molecules do not participate much in binding to the surface of the nanocrystals. The great uniformity of the obtained samples was utilized to obtain nanocrystal superlattices with lateral dimensions reaching up to 50 μm, the largest reported so far in the lead halide perovskite family. This approach should be extendable to halide perovskite nanocrystals with compositions other than CsPbX3 (for example, MAPbX3 and FAPbX3). We believe that not only will secondary amines expand the list of molecules for the synthesis of lead halide perovskite nanocrystals, but also, they will pave the way to a better understanding of the photophysical properties of shape pure materials.
Experimental Section
Chemicals
Lead acetate trihydrate [(PbAc2·3H2O), 99.99%], cesium carbonate (Cs2CO3, reagent Plus, 99%), benzoyl bromide (C6H5COBr, 97%), acetone (99.5%), ethyl acetate (98.8%), toluene (anhydrous, 99.5%), octadecene (ODE, technical grade, 90%), toluene-d8 (99 atom. %D) oleylamine (OLAm, 98%) dihexylamine (DHAm, 97%), dioctylamine (DOAm, 97%), didecylamine (DDAm, 98%), dioctdecylamine (DODAm, 99%), and oleic acid (OA, 90%) were purchased from Sigma-Aldrich. Didodecylamine (DDDAm, 97%), was purchased from TCI. Oleic acid and oleylamine were dried at 120 °C for an hour and stored in a glovebox. All other chemicals were used without any further purification.
Synthesis of CsPbBr3 Nanocubes
A total of 76 mg of lead(II) acetate trihydrate, 16 mg of cesium carbonate, and 10 mL of octadecene were combined in a 25 mL 3-neck flask. The reaction mixture was degassed for 5 min at room temperature then for 1 h at 115 °C. The ligand mixture containing 1.5 mL of pre-dried OA and 1.25 mmol of dialkylamine dissolved in 1 mL of anhydrous toluene was rapidly injected under nitrogen. After the complete dissolution of the metal precursors, the temperature of the reaction mixture was decreased to 80 °C. A total of 50 μL of a benzoyl bromide precursor diluted in 500 μL of dried ODE was then injected into the mixture. The mixture was cooled down after 15 s by using a water bath. Subsequently, 20 mL of an ethyl acetae and toluene mixture (with a ratio of 6:1) was added into the crude solution to destabilize the colloids, and the NCs were collected by centrifugation at 9000 rpm for 10 min. Finally, the supernatant was discarded, and the precipitate was redispersed in toluene.
Elemental Analysis
Elemental analysis on solutions of NCs was carried out via inductively coupled plasma optical emission spectroscopy (ICP-OES) on a Thermo Fisher iCap 6000. NC solutions were digested in Aqua Regia.
X-ray Diffraction Characterization
The XRD analysis was performed on a PANanalytical Empyrean X-ray diffractometer, equipped with a 1.8 kW CuKα ceramic X-ray detector operating at 45 kV and 40 mA. Samples for the measurements were prepared by drop-casting a concentrated solution of NCs on a zero-diffraction silicon substrate. All of the diffraction patterns reported here were collected at room temperature under ambient conditions using parallel beam geometry and symmetric reflection mode. Postacquisition XRD data analysis was carried out using the HighScore 4.1 software from PANalytical.
Transmission Electron Microscopy Characterization
Bright-field TEM images of the NC samples were acquired by a JEOL-1100 transmission electron microscope operating at an acceleration voltage of 100 kV. Samples were prepared by drop-casting diluted solutions of NCs onto carbon film-coated 200 mesh copper grids for low-resolution TEM. For HR-TEM, we instead used ultrathin carbon and holey carbon coated 400 mesh copper grids. HR-TEM analysis was carried out on a JEOL JEM-2200FS microscope equipped with a Schottky emitter operating at 200 kV and a CEOS spherical aberration corrector for the objective lens.
UV–Vis Absorption and PL Measurements
The UV–visible absorption spectra were recorded using a Varian Cary 300 UV–vis absorption spectrophotometer. The PL spectra were measured on a Varian Cary Eclipse spectrophotometer using an excitation wavelength (λex) of 350 nm for all the samples. Samples were prepared by diluting NC solutions in toluene in quartz cuvettes with a path length of 1 cm.
PL Quantum Yields and Time-Resolved PL Measurements
Absolute photoluminescence quantum yields of NC samples were measured using an Edinburgh FLS900 fluorescence spectrometer equipped with a xenon lamp, a monochromator for steady-state PL excitation, and a time-correlated single photon counting unit coupled with a pulsed laser diode (λex = 405 nm and pulse width of 50 ps) for time-resolved PL. The PLQY was measured using a calibrated integrating sphere (λex = 400 nm for all CsPbBr3 nanocube samples). All solutions were diluted to an optical density of 0.1 at the corresponding excitation wavelength to minimize self-absorption.
X-ray Photoelectron Spectroscopy Characterization
Measurements were performed on a Kratos Axis Ultra DLD spectrometer using a monochromatic Al Kα source (15 kV, 20 mA). The photoelectrons were detected at a takeoff angle of φ = 0° with respect to the surface normal. The pressure in the analysis chamber was kept below 7 × 10–9 Torr for data acquisition. The data was converted to VAMAS format and processed using CasaXPS software, version 2.3.17. The binding-energy (BE) scale was internally referenced to the C1’s peak (BE for C–C = 284.8 eV).
Computational Modeling
The simulations were performed in vacuum at the DFT level of theory using the CP2K quantum chemistry code40 and employing the PBE exchange-correlation functional41 and a double ζ basis set plus polarization functions.42 Scalar relativistic effects have been accounted for by using effective core potential functions in the basis set. Spin–orbit coupling effects were not included in the calculations. For ground-state relaxations, the default force convergence threshold of 4.5 × 10–4 Ha/bohr was used. Molecular dynamic (MD) simulations included an equilibration stage of 1000 fs using a thermostat and barostat annealing time constant of 150 fs, followed by a production run of 500 fs with a longer annealing time constant (500 fs). At first, we performed structural optimizations at 0 K, allowing the supercell to relax in every direction. For these preliminary calculations, we considered platelets with a maximum surface ligand coverage, i.e., each surface Cs ion is replaced by ammonium, following the same approach we reported in previous works.27,43 We then computed the elongation of the supercell along the diagonal direction against the cell with only Cs ions at the surface, and we noticed that the lattice expanded by about 32% for the secondary amines but only 1% for primary amines. We then performed an ab initio MD simulation and computed the Pb–Pb, Cs–Cs, and N–N pair distance distributions for the primary and secondary amine passivated cases.
Nuclear Magnetic Resonance
All NMR spectra were acquired on a Bruker Avance III 400 MHz spectrometer, equipped with a Broad Band Inverse probe (BBI) operating at 300 K. After the 1H 90° pulse was optimized, using an automatic pulse calculation routine,44 a quantitative 1H NMR (q-NMR) experiment was acquired. We used 16 transient, 64 000 data points, 30 s of inter-pulse delays, no steady-state scans, a spectral width of 20.55 ppm (offset at 6.175 ppm), and a fixed receiver gain of 64. 1H NMR spectra for the titration curves were obtained using identical acquisition parameters except for the receiver gain (1). A 2D 1H–1H NOESY experiment (noesygpphpp, Bruker library)45 was performed using a mixing time of 300 ms, 32 transients, 2048 data points, and 256 increments. A 2D 1H–13C HSQC (multiplicity-edited Heteronuclear Single Quantum Coherence, hsqcedetgpsp.3, Bruker library)46 spectrum was acquired using 12 transients, 2048 data points, 400 increments, and an automatic spectral width and transmitter frequency offset optimization for 1H. A line broadening of 0.3 Hz was applied to free induction decay before the Fourier transform. The NMR chemical shifts refer to the peak of residual nondeuterated toluene at 7.09 ppm (1H) and 129.2 ppm (13C).
Self-Assembly of CsPbBr3 Nanocubes
The self-assembly was accomplished by slowly evaporating the solvent, and the experiment was set up in a glass Petri dish (with a 60 mm diameter). A thin piece of glass (∼0.5 mm) was put underneath one side of a Si wafer (50.8 mm diameter), creating an inclination of the wafer of ∼0.5–1° with respect to the bottom of the Petri dish. A toluene solution of the nanocrystal sample (between 100 and 150 μL of ∼4 μM 8.5 ± 0.9 nm CsPbBr3 cubes) was carefully deposited onto the Si wafer, forming a large tilted “drop”. The Petri dish was covered with the glass lid, the entire setup was loosely wrapped in foil to protect it from ambient light and air currents, and the solution was left to evaporate overnight on the lab bench.
Scanning Electron Microscopy Characterization
A JEOL JSM-7500FA microscope (Jeol, Tokyo, Japan) in high-vacuum mode, with an acceleration voltage of 5 kV and backscattered electrons was used. No coating was required because the samples were already conductive.
Confocal Photoluminescence Microscopy of Superlattices
The photoluminescence imaging of CsPbBr3 NC superlattices grown on Si wafer was performed using a Nikon confocal microscope system (A1-plus-s, Nikon Instruments, Yokohama, Japan) equipped with a A1-DUS Spectral Detector Unit that allows for a parallel acquisition of 32 channel spectral images at a maximum wavelength resolution of 2.5 nm. Depending on the level of detail desired, we used a 10× air (Nikon CFI Plan Apo Lambda 10× 0.45 NA) and a 60× oil (Nikon CFI Plan Apo Lambda 60X Oil 1.4 NA) objective lens. The selected excitation wavelength was 488 nm. The spectrally resolved images were recorded with an emission detection bandwidth of 2.5 nm over a wavelength in the range of 498–578 nm. The resulting stacks (32 spectral slices per stack) were processed using an open-source Fiji distribution of ImageJ ver. 1.52e.47 The spectral profiles of individual superlattices were obtained by drawing polygonal regions of interest (ROIs) around selected superlattices and performing a “Plot Z-stack Profile” operation in Fiji. This yields a plot of the brightness of ROI (i.e., mean gray value, which is proportional to the number of emitted photons) as a function of the slice number (namely, the emission wavelength bin with the width of the spectral resolution). The overall spectrum-colored images were obtained using the “Temporal Color Code” hyperstack function in Fiji with the LUT setting “Spectrum.”
Acknowledgments
We acknowledge funding from the European Union under grant agreement no. 614897 (ERC Grant TRANS-NANO). The work of D.B. was supported by the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement no. 794560. I.I. acknowledges The Netherlands Organization of Scientific Research (NWO) for financial support through the Innovational Research Incentive (Vidi) Scheme (grant no. 723.013.002). We thank Zhiya Dang for help with transmission electron microscopy analysis, Simone Lauciello for assistance with the HRSEM of superlattices, and Guilherme Almeida for many inspiring discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nanolett.8b03598.
A detailed description of the synthesis and the results of the control syntheses that were conducted under various conditions as well as PL lifetime decay curves, PLQY data, additional XRD patterns, TEM images, XPS spectra, NMR spectra (1H, 1H-13 HSQC, 1H–1H NOESY), optical spectra, the self-assembly procedure, and optical microscopy images of NC superlattices (PDF)
A video displaying two experiments in which a colloidal suspension of CsPbBr3 NCs in toluene is mixed with a toluene solution containing a large excess of either a primary amine or a secondary amine (AVI)
The authors declare no competing financial interest.
Supplementary Material
References
- Park N. G.; Gratzel M.; Miyasaka T.; Zhu K.; Emery K. Nat. Energy 2016, 1, 16152. 10.1038/nenergy.2016.152. [DOI] [Google Scholar]
- Yang W. S.; Park B.-W.; Jung E. H.; Jeon N. J.; Kim Y. C.; Lee D. U.; Shin S. S.; Seo J.; Kim E. K.; Noh J. H.; Seok S. I. Science 2017, 356, 1376–1379. 10.1126/science.aan2301. [DOI] [PubMed] [Google Scholar]
- Etgar L.; Gao P.; Xue Z.; Peng Q.; Chandiran A. K.; Liu B.; Nazeeruddin M. K.; Grätzel M. J. Am. Chem. Soc. 2012, 134, 17396–17399. 10.1021/ja307789s. [DOI] [PubMed] [Google Scholar]
- Schmidt L. C.; Pertegas A.; Gonzalez-Carrero S.; Malinkiewicz O.; Agouram S.; Minguez Espallargas G.; Bolink H. J.; Galian R. E.; Perez-Prieto J. J. Am. Chem. Soc. 2014, 136, 850–853. 10.1021/ja4109209. [DOI] [PubMed] [Google Scholar]
- Protesescu L.; Yakunin S.; Bodnarchuk M. I.; Krieg F.; Caputo R.; Hendon C. H.; Yang R. X.; Walsh A.; Kovalenko M. V. Nano Lett. 2015, 15, 3692–3696. 10.1021/nl5048779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akkerman Q. A.; Rainò G.; Kovalenko M. V.; Manna L. Nat. Mater. 2018, 17, 394–405. 10.1038/s41563-018-0018-4. [DOI] [PubMed] [Google Scholar]
- Kovalenko M. V.; Protesescu L.; Bodnarchuk M. I. Science 2017, 358, 745–750. 10.1126/science.aam7093. [DOI] [PubMed] [Google Scholar]
- Imran M.; Caligiuri V.; Wang M.; Goldoni L.; Prato M.; Krahne R.; De Trizio L.; Manna L. J. Am. Chem. Soc. 2018, 140, 2656–2664. 10.1021/jacs.7b13477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning C.-Z.; Dou L.; Yang P. Nat. Rev. Mater. 2017, 2, 17070. 10.1038/natrevmats.2017.70. [DOI] [Google Scholar]
- Dong Y.; Qiao T.; Kim D.; Parobek D.; Rossi D.; Son D. H. Nano Lett. 2018, 18, 3716–3722. 10.1021/acs.nanolett.8b00861. [DOI] [PubMed] [Google Scholar]
- Liu F.; Zhang Y.; Ding C.; Kobayashi S.; Izuishi T.; Nakazawa N.; Toyoda T.; Ohta T.; Hayase S.; Minemoto T.; Yoshino K.; Dai S.; Shen Q. ACS Nano 2017, 11, 10373–10383. 10.1021/acsnano.7b05442. [DOI] [PubMed] [Google Scholar]
- Bekenstein Y.; Koscher B. A.; Eaton S. W.; Yang P.; Alivisatos A. P. J. Am. Chem. Soc. 2015, 137, 16008–16011. 10.1021/jacs.5b11199. [DOI] [PubMed] [Google Scholar]
- Akkerman Q. A.; Motti S. G.; Srimath Kandada A. R.; Mosconi E.; D’Innocenzo V.; Bertoni G.; Marras S.; Kamino B. A.; Miranda L.; De Angelis F.; Prato M.; Manna L.; Petrozza A. J. Am. Chem. Soc. 2016, 138, 1010–1016. 10.1021/jacs.5b12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.; Eaton S. W.; Yu Y.; Dou L.; Yang P. J. Am. Chem. Soc. 2015, 137, 9230–9233. 10.1021/jacs.5b05404. [DOI] [PubMed] [Google Scholar]
- Imran M.; Di Stasio F.; Dang Z.; Canale C.; Khan A. H.; Shamsi J.; Brescia R.; Prato M.; Manna L. Chem. Mater. 2016, 28, 6450–6454. 10.1021/acs.chemmater.6b03081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J.; Xu L.; Li J.; Xue J.; Dong Y.; Li X.; Zeng H. Adv. Mater. 2016, 28, 4861–4869. 10.1002/adma.201600225. [DOI] [PubMed] [Google Scholar]
- Shamsi J.; Dang Z.; Bianchini P.; Canale C.; Di Stasio F.; Brescia R.; Prato M.; Manna L. J. Am. Chem. Soc. 2016, 138, 7240–7243. 10.1021/jacs.6b03166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S.; Yuan D.; Xu Y.; Wang A.; Deng Z. ACS Nano 2016, 10, 3648–3657. 10.1021/acsnano.5b08193. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Yu Y.; Bekenstein Y.; Wong A. B.; Alivisatos A. P.; Yang P. J. Am. Chem. Soc. 2016, 138, 13155–13158. 10.1021/jacs.6b08373. [DOI] [PubMed] [Google Scholar]
- Bohn B. J.; Tong Y.; Gramlich M.; Lai M. L.; Döblinger M.; Wang K.; Hoye R. L.; Müller-Buschbaum P.; Stranks S. D.; Urban A.; Polavarapu L.; Feldmann J. Nano Lett. 2018, 18, 5231–5238. 10.1021/acs.nanolett.8b02190. [DOI] [PubMed] [Google Scholar]
- Almeida G.; Goldoni L.; Akkerman Q.; Dang Z.; Khan A. H.; Marras S.; Moreels I.; Manna L. ACS Nano 2018, 12, 1704–1711. 10.1021/acsnano.7b08357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi V. K.; Santra P. K.; Joshi N.; Chugh J.; Singh S. K.; Rensmo H.; Ghosh P.; Nag A. J. Phys. Chem. Lett. 2017, 8, 4988–4994. 10.1021/acs.jpclett.7b02192. [DOI] [PubMed] [Google Scholar]
- Pan A.; He B.; Fan X.; Liu Z.; Urban J. J.; Alivisatos A. P.; He L.; Liu Y. ACS Nano 2016, 10, 7943–7954. 10.1021/acsnano.6b03863. [DOI] [PubMed] [Google Scholar]
- Akkerman Q. A.; Park S.; Radicchi E.; Nunzi F.; Mosconi E.; De Angelis F.; Brescia R.; Rastogi P.; Prato M.; Manna L. Nano Lett. 2017, 17, 1924–1930. 10.1021/acs.nanolett.6b05262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saidaminov M. I.; Almutlaq J.; Sarmah S.; Dursun I.; Zhumekenov A. A.; Begum R.; Pan J.; Cho N.; Mohammed O. F.; Bakr O. M. ACS Energy Lett. 2016, 1, 840–845. 10.1021/acsenergylett.6b00396. [DOI] [Google Scholar]
- Yassitepe E.; Yang Z.; Voznyy O.; Kim Y.; Walters G.; Castañeda J. A.; Kanjanaboos P.; Yuan M.; Gong X.; Fan F.; Pan J.; Hoogland S.; Comin R.; Bakr O. M.; Padilha L. A.; Nogueira A. F.; Sargent E. H. Adv. Funct. Mater. 2016, 26, 8757–8763. 10.1002/adfm.201604580. [DOI] [Google Scholar]
- Krieg F.; Ochsenbein S. T.; Yakunin S.; Ten Brinck S.; Aellen P.; Süess A.; Clerc B.; Guggisberg D.; Nazarenko O.; Shynkarenko Y.; Kumar S.; Shih C. J.; Infante I.; Kovalenko M. V. ACS Energy Lett. 2018, 3, 641–646. 10.1021/acsenergylett.8b00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidman M. C.; Goodman A. J.; Tisdale W. A. Chem. Mater. 2017, 29, 5019–5030. 10.1021/acs.chemmater.7b01384. [DOI] [Google Scholar]
- Dohner E. R.; Hoke E. T.; Karunadasa H. I. J. Am. Chem. Soc. 2014, 136, 1718–1721. 10.1021/ja411045r. [DOI] [PubMed] [Google Scholar]
- Dou L. J. Mater. Chem. C 2017, 5, 11165–11173. 10.1039/C7TC02863F. [DOI] [Google Scholar]
- Liu Z.; Bekenstein Y.; Ye X.; Nguyen S. C.; Swabeck J.; Zhang D.; Lee S.-T.; Yang P.; Ma W.; Alivisatos A. P. J. Am. Chem. Soc. 2017, 139, 5309–5312. 10.1021/jacs.7b01409. [DOI] [PubMed] [Google Scholar]
- Palazon F.; Almeida G.; Akkerman Q. A.; De Trizio L.; Dang Z.; Prato M.; Manna L. Chem. Mater. 2017, 29, 4167–4171. 10.1021/acs.chemmater.7b00895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udayabhaskararao T.; Houben L.; Cohen H.; Menahem M.; Pinkas I.; Avram L.; Wolf T.; Teitelboim A.; Leskes M.; Yaffe O.; Oron D.; Kazes M. Chem. Mater. 2018, 30, 84–93. 10.1021/acs.chemmater.7b02425. [DOI] [Google Scholar]
- Thirunavukkarasu K.; Thirumoorthy K.; Libuda J.; Gopinath C. S. J. Phys. Chem. B 2005, 109, 13272–13282. 10.1021/jp050478v. [DOI] [PubMed] [Google Scholar]
- De Roo J.; Ibáñez M.; Geiregat P.; Nedelcu G.; Walravens W.; Maes J.; Martins J. C.; Van Driessche I.; Kovalenko M. V.; Hens Z. ACS Nano 2016, 10, 2071–2081. 10.1021/acsnano.5b06295. [DOI] [PubMed] [Google Scholar]
- Kovalenko M. V.; Bodnarchuk M. I. Chimia 2017, 71, 461–470. 10.2533/chimia.2017.461. [DOI] [PubMed] [Google Scholar]
- Rainò G.; Becker M. A.; Bodnarchuk M. I.; Mahrt R. F.; Kovalenko M. V.; Stöferle T.. 2018. arXiv:1804.01873. arXiv.org e-Print archive. https://arxiv.org/abs/1804.01873 (date of access: July 25, 2018).
- Tong Y.; Yao E. P.; Manzi A.; Bladt E.; Wang K.; Doblinger M.; Bals S.; Muller-Buschbaum P.; Urban A. S.; Polavarapu L.; Feldmann J. Adv. Mater. 2018, 30, 1801117. 10.1002/adma.201801117. [DOI] [PubMed] [Google Scholar]
- van der Burgt J. S.; Geuchies J. J.; van der Meer B.; Vanrompay H.; Zanaga D.; Zhang Y.; Albrecht W.; Petukhov A. V.; Filion L.; Bals S.; Swart I.; Vanmaekelbergh D. J. Phys. Chem. C 2018, 122, 15706–15712. 10.1021/acs.jpcc.8b02699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter J.; Iannuzzi M.; Schiffmann F.; VandeVondele J. WIRs. Comput. Mol. Sci. 2014, 4, 15–25. 10.1002/wcms.1159. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Phys. Rev. Lett. 1996, 77, 3865. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- VandeVondele J.; Hutter J. J. Chem. Phys. 2007, 127, 114105. 10.1063/1.2770708. [DOI] [PubMed] [Google Scholar]
- Ten Brinck S.; Infante I. ACS Energy Lett. 2016, 1, 1266–1272. 10.1021/acsenergylett.6b00595. [DOI] [Google Scholar]
- Wu P. S.; Otting G. J. Magn. Reson. 2005, 176, 115–119. 10.1016/j.jmr.2005.05.018. [DOI] [PubMed] [Google Scholar]
- Wagner R.; Berger S. J. Magn. Reson., Ser. A 1996, 123, 119–121. 10.1006/jmra.1996.0222. [DOI] [PubMed] [Google Scholar]
- Boyer R. D.; Johnson R.; Krishnamurthy K. J. Magn. Reson. 2003, 165, 253–259. 10.1016/j.jmr.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Schneider C. A.; Rasband W. S.; Eliceiri K. W. Nat. Methods 2012, 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.