

Abstract
Opioids are the most commonly used and effective analgesic treatments for severe pain, but they have recently come under scrutiny owing to epidemic levels of abuse and overdose. These compounds act on the endogenous opioid system, which comprises four G protein-coupled receptors (mu, delta, kappa, and nociceptin) and four major peptide families (β-endorphin, enkephalins, dynorphins, and nociceptin/orphanin FQ). In this review, we first describe the functional organization and pharmacology of the endogenous opioid system. We then summarize current knowledge on the signaling mechanisms by which opioids regulate neuronal function and neurotransmission. Finally, we discuss the loci of opioid analgesic action along peripheral and central pain pathways, emphasizing the pain-relieving properties of opioids against the affective dimension of the pain experience.
Keywords: opioid, analgesia, pain, signaling, neuroanatomy, perception
INTRODUCTION
Throughout human history, opioids have been used medicinally as an analgesic and recreationally as a euphorigenic. In the 1970s and 1980s, the efficacy of opioids to treat illness was vastly improved by advancements in modern medicinal chemistry and neuroscience. Specifically, the identification of the endogenous opioid peptides and receptors, accompanied by the development of new hyperselective and potent opioid drugs such as fentanyl and heroin, contributed both beneficial and detrimental effects on society and medicine. Today, opioids remain the mainstay analgesic treatment for severe acute, perioperative, and chronic pain. Paralleling the outstanding magnitude of pain in the United States (Institute of Medicine (US) Committee on Advancing Pain Research, Care, and Education 2011), the use of opioids for pain management has increased dramatically in the past two decades such that hydrocodone topped all prescriptions in 2011 (CDC 2013, Manchikanti et al. 2012). Unfortunately, opioids cause numerous detrimental effects, including analgesic tolerance, paradoxical hyperalgesia, nausea and vomiting, constipation, respiratory depression, and transition to addiction (Inturrisi 2002, Streicher & Bilsky 2017, Volkow & McLellan 2016). These side effects dramatically impact the quality of life of patients, and the number of deaths from opioid overdose now exceeds that of car accidents (CDC 2013). Elucidation of the neural mechanisms underlying opioid effects is urgently needed to develop innovative adjuvant therapies that dissociate opioid analgesia from side effects. In this review, we discuss the recent advancements made in understanding opioid mechanisms of function.
ENDOGENOUS OPIOID SYSTEM
Opioid Receptors
The endogenous opioid system comprises four seven-transmembrane G protein-coupled receptors (GPCRs): mu, delta, kappa, and nociceptin (MOPR, DOPR, KOPR, NOPR). Each receptor is encoded by a unique gene (Oprm1, Oprd1, Oprk1, Oprl1) but shares upward of 60% of its amino acid composition (Al-Hasani & Bruchas 2011, Kieffer & Evans 2009, Toll et al. 2016). Importantly, each receptor has a distinct expression pattern throughout the nervous system (Mansour et al. 1994, Neal et al. 1999). The recent crystal structures of all four receptors illustrate with unprecedented detail several similar molecular characteristics that may open new avenues for novel drug design (Granier et al. 2012, Manglik et al. 2012, Thompson et al. 2012, Wu et al. 2012). In particular, the crystal structures for the inactive state of each receptor have been identified (Figure 1a). These studies provided the first glimpse into atomic-level details of the receptors necessary for pinpointing the unique opioid binding pockets that maintain ligand preferences. For example, the active state of MOPR has been crystalized with nanobodies to stabilize the structure; comparisons of the active and inactive states can identify potential sites of action for different molecules. A recent computational docking and drug design study, based on the active MOPR structure, was used to identify novel biased opioid analgesics (e.g., PZM21) that preferentially promote unique active-state conformations and signaling pathways (Manglik et al. 2016). In the case of NOPR, the least well understood of the opioid receptors, structural crystallization has indicated the lack of a salt bridge, which is common to the other receptors, resulting in an overall shift in the conformation of the fifth and sixth helices. This shift may be relevant for NOPR’s lack of extracellular domain interactions with the other endogenous opioid ligands, which may be relevant for the development of receptor-specific drugs. Collectively, these results provide insight into how different agonists distinctly alter receptor conformations to direct downstream intracellular cascades, which may ultimately lead to more effective pharmacological treatments. Additionally, other mechanisms including alternative splicing and receptor interactions may contribute to the diversity of analgesic responses mediated by opioids (Fujita et al. 2015, Pasternak 2018, Samoshkin et al. 2015, Wieskopf et al. 2014).
Figure 1.
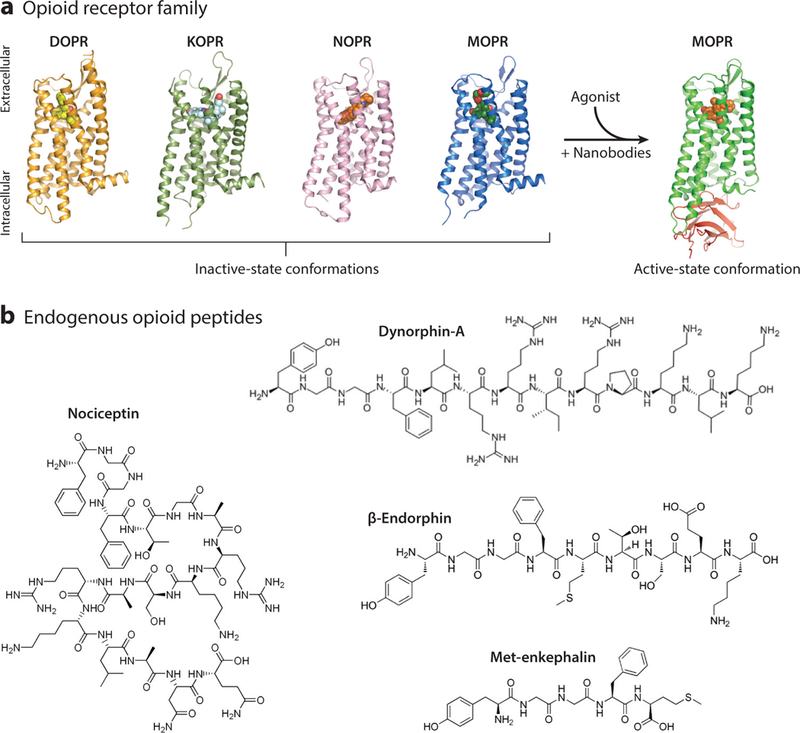
The endogenous opioid system. (a) Crystal structures of the inactive state of all four opioid receptors (DOPR, KOPR, NOPR, and MOPR). When an opioid agonist enters the binding pocket of its cognate receptor, a conformational change in the transmembrane domains allows for intracellular effector molecules to bind and activate signaling cascades that modulate neural function. The addition of stabilizing nanobodies to the crystal preparation has elucidated the active state of MOPR. Images courtesy of Dr. Aashish Manglik (UCSF) and used with his permission. (b) Chemical structures of the four main classes of opioid peptides: met-enkephalin, dynorphin-A, nociceptin, and β -endorphin. Abbreviations: DOPR, delta opioid receptor; KOPR, kappa opioid receptor; MOPR, mu opioid receptor; NOPR, nociceptin opioid receptor.
Opioid Ligands
There are four major families of endogenous opioid ligands: β-endorphins, enkephalins, dynorphins, and nociceptin/orphanin FQ (Figure 1b). These opioid peptides along with their cognate receptors are widely expressed across the neuraxis and, in particular, pain pathways. In contrast to the amino acid or monoamine neurotransmitters, the opioid peptides are packaged into dense core vesicles in the soma and transported down to axon terminals. During this process, enzymatic splicing of the prepropeptides results in the formation of the diverse, receptor-specific peptide transmitters. The classic example of this process involves β-endorphin, the canonical mu-preferring ligand. β-Endorphin is cleaved from the parent molecule proopiomelanocortin (POMC), which is expressed in the arcuate nucleus and the nucleus of the solitary tract (Bloom et al. 1978, Lazarus et al. 1976). After packaging, POMC is cleaved into either proopiocorticotropin or adrenocorticotropin molecules, which are then again broken down into β-endorphin, α-melanocyte-stimulating hormone, and corticotropin-releasing hormone. These peptides act on MOPR, melanocortin, and corticotropin receptors, respectively. Additionally, 3-endorphin can be further cleaved into met-enkephalin, a nonselective agonist with affinity for both DOPR and MOPR.
Similar to β-endorphin, enkephalins and dynorphins arise from larger molecules that are broken down into more specific peptide transmitters. Preproenkephalin is cleaved into either met-or leuenkephalin (Bower et al. 1976). Prodynorphin can be cleaved into several KOPR-selective ligands, includingdynorphin-A[1–17], dynorphin-B[1–13], and α-neoendorphin. Further complicating the relationship between opioid receptors and their ligands, dynorphin can also be cleaved into less-opioid-selective leu-enkephalin or dynorphin-A[1–8], essentially making dynorphin a potential agonist for MOPRs, DOPRs, and KOPRs (Chavkin 2013, Goldstein et al. 1979). Last, nociceptin is derived from prepronociceptin and has a significantly higher affinity for NOPR than for the other opioid receptors (Meunier et al. 1995). This selectivity is likely due to the Phe amino acid in the first position of the nociceptin peptide sequence (James et al. 1982).
Contrasting with the tight, spatially controlled synaptic transmission of small-molecule transmitters such as glutamate or dopamine, opioids are thought to rely on volumetric release into synaptic and extrasynaptic spaces and diffuse toward their receptors (Banghart & Sabatini 2012, Duggan 2000). Indeed, electron microscopy illustrates that most MOPRs are extrasynaptic, being hundreds of microns away from release sites (Glass et al. 2009, Mansour et al. 1988, Svingos et al. 1996). That is, they are not found in the bed of symmetric or asymmetric synapses but rather shifted over, next to the synapse. Similarly, dynorphin release has been suggested to travel up to nearly 100 μm from the released terminal (Chavkin 2013, Drake et al. 1994), implying that opioid synapses may include a much broader area than typical fast transmitter synapses. The mechanisms that command the spatial and temporal dynamics of opioid release, and that direct peptides to these distant receptors, remain some of the biggest and exciting mysteries in the field.
SIGNALING
General Principles
Here, we briefly summarize the basic signaling properties of the four opioid receptors (Figure 2). Extensive reviews of opioid receptor signaling can be found elsewhere (Al-Hasani & Bruchas 2011, Lamberts & Traynor 2013, Toll et al. 2016, Williams et al. 2013). All four opioid receptors couple to the inhibitory G proteins (Gαi and Gαo). Upon activation by agonists, either endogenous or exogenous, the Gα and Gβγ subunits dissociate from one another and subsequently engage a variety of effectors and intracellular signaling cascades that typically depress neural functions. Note that MOPR, DOPR, and KOPR have been shown to signal through an agonist-independent mechanism called constitutive activity, including during persistent pain and stress (Corder et al. 2013, Polter et al. 2017, Yao et al. 2016). Although further in vivo studies are needed to understand the initiation mechanisms, constitutive activity of MOPR and DOPR is also observed after prolonged exogenous opioid stimulation (Liu & Prather 2001, Meye et al. 2012, Shoblock & Maidment 2006) and likely involves lowering the energy barrier to assume the active conformation, as predicted by the crystal structure (Manglik et al. 2012). Such activity might result from a variety of mechanisms, including changes in receptor density, changes in receptor phosphorylation, modulation of allosteric binding sites, or changes in interactions with accessory proteins such as β-arrestin and Src (Kenakin 2001, Walwyn et al. 2007).
Figure 2.

Opioid modulation of signaling and synaptic transmission. (a) Presynaptic and postsynaptic effects of opioids on nociception. (Left) Noxious stimuli trigger action potential firing along DRG nociceptors. Upon reaching the synaptic terminal, VGCCs (yellow) open, facilitating neurotransmitter release. These neurotransmitters (e.g., glutamate) then open postsynaptic AMPA and NMDA receptors, which continue the nociceptive signals along pain circuits. (Right) Activation of opioid receptors promotes dissociation of inhibitory Gα and Gβγ protein subunits. Gα subunits suppress adenylate cyclase, and Gβγ subunits presynaptically inhibit VGCC opening and postsynaptically activate GIRK channels, resulting in reduced neurotransmitter release and membrane hyperpolarization, respectively. (b) Biased signaling pathways. Agonist binding to opioid receptors causes conformational changes that promote distinct recruitment of G protein and arrestin effector signaling cascades. While G proteins mediate the inhibitory action of opioid signaling on neurotransmission, arrestin signaling is required both for internalization of opioid receptors and for kinase activities. The balance between G protein and arrestin signaling is thought, in part, to determine the analgesic versus detrimental effects of opioids. (c) Within pain circuits opioid receptors are activated by opioid analgesics such as enkephalin (endogenous) or morphine (exogenous). Endogenous opioids, such as enkephalins, can be released from infiltrating immune cells at the site of injuries and from neurons in the central nervous system. Abbreviations: AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; DRG, dorsal root ganglion; EPSC, excitatory postsynaptic current; ERK, extracellular signal regulated kinase; GIRK, G protein gated inwardly rectifying potassium; JNK, c-Jun N-terminal kinase; NMDA, N-methyl-D-aspartate; RVM, rostral ventromedial medulla; VGCC, voltage-gated calcium channel.
Opioid receptor activity inhibits adenylate cyclase (AC), thereby reducing cyclic AMP production (Minneman & Iversen 1976), as evidence of pertussis toxin sensitivity was established in later experiments. Further studies revealed that guanine nucleotides such as GTP modulate agonist binding to opioid receptors in membrane preparations from brain tissue and that opioids stimulate GTPase activity (Barchfeld & Medzihradsky 1984, Childers & Snyder 1978). Beyond coupling to Gi and Go proteins, all four opioid receptors engage other G proteins that modulate a multitude of effectors in addition to AC (Al-Hasani & Bruchas 2011, Toll et al. 2016, Williams et al. 2013).
Ion Channel Mechanisms
One of the most highly conserved pathways that opioid receptors use to alter neuronal function is the modulation of ion channels (Figure 2a). All four opioid receptors inhibit in N-, P/Q-, and L-type voltage-gated calcium channels (Rusin et al. 1997). This process, which occurs via the Gβγ subunit inhibition of the channel, decreases the presynaptic calcium-dependent fusion of synaptic vesicles with the membrane terminal and subsequent neurotransmitter release. In dorsal root ganglion (DRG) neurons, N-type calcium channels along with opioid receptors can be co-internalized following prolonged agonist exposure, which may further reduce neurotransmitter release and the transmission of pain signals to the central nervous system (CNS) (Altier et al. 2006). Postsynaptically, opioids also cause a Gβγ-mediated activation of G protein gated inwardly rectifying potassium (GIRK) channels (Torrecilla et al. 2002). This process is particularly important in postsynaptic compartments where dendritic hyperpolarization filters synaptic input. Mutant mice lacking GIRK channels, or expressing dysfunctional channels, show reduced opioid antinociception, establishing the importance of G protein-mediated potassium conductance modulation for opioid analgesia (Lujan et al. 2014, Nagi & Pineyro 2014).
Although the acute action of opioids on calcium and potassium channels typically reduces neurotransmission within seconds to minutes, chronic (hours to days) or abruptly interrupted opioid signaling can also facilitate excitatory synaptic plasticity. For example, withdrawal of exogenous opioids can elicit long-term potentiation (LTP) of synaptic transmission between primary afferent DRG nociceptors and second-order spinal cord neurons (Drdla et al. 2009, Zhou et al. 2010). This form of spinal LTP is considered a major substrate for opioid-induced hyperalgesia (OIH), a paradoxical decrease in pain threshold following opioid administration, and might also contribute to analgesic tolerance. The detailed molecular mechanisms underlying OIH and analgesic tolerance are not fully resolved, but they require presynaptic MOPRs in nociceptors (Corder et al. 2017) and involve the activation of microglia and molecules, including pannexin1, P2X4, and Toll-like receptors, that differentially contribute to OIH and tolerance in these cells (Burma et al. 2017, Trang et al. 2015). Finally, spinal LTP is also induced by peripheral injuries and represents a major mechanism of pathological pain. In this setting, a high dose of MOPR agonist can depotentiate synaptic transmission and erase spinal pain memory (Drdla-Schutting et al. 2012, Ruscheweyh et al. 2011).
Desensitization and Trafficking
Following activation, opioid receptors are phosphorylated by GPCR kinases, leading to β-arrestin 2 or 3 recruitment (Figure 2b). Arrestin molecules are key proteins that bind to phosphorylated GPCRs to regulate their G protein signaling through desensitization and internalization. The interaction of an opioid receptor with arrestin is thought to depend on the cellular context, agonist type, and model system studied. Importantly, mice that lack β-arrestin 2 show enhanced morphine antinociception and increased conditioned place preference (Bohn et al. 1999, 2003). Additionally, studies examining the aversive qualities of KOPR stimulation have shown that GRK3 knockout mice show no conditioned place aversion to KOPR agonists, and that phosphorylation of the receptor is required for these effects, implicating arrestin signaling in behavioral function (Bruchas et al. 2007a, 2011). Remarkably, and contrary to previous models, internalized GPCRs are not inactive but may still signal, including from endosomal compartments (Eichel et al. 2016, Irannejad et al. 2013). These observations suggest, on the basis of the intracellular fate and signaling of internalized receptors (Bahouth & Nooh 2017, Irannejad & von Zastrow 2014), an additional level of complexity through which distinct ligands acting on the same opioid receptor can produce different cellular effects.
Arrestin Signaling
Whereas arrestin and opioid-receptor interactions were originally defined by their ability to regulate receptors, more recent studies have shown that arrestin is in fact a key signal effector at these receptors, mediating an array of cellular and behavioral responses. Phosphorylated arrestin-bound GPCR complexes recruit alternate, critically important downstream signaling cascades, including the mitogen-activated protein kinase (MAPK) cascade (Figure 2b). These MAPKs, which consist ofthree major proteins [extracellular signal regulated kinase 1 and2 (ERK1/2), c-Jun N-terminal kinase 1–3 (JNK 1–3), and p38], notably modulate cell proliferation, differentiation, apoptosis, transcription factor regulation, ion channel regulation, neurotransporter regulation, and protein scaffolding (Raman et al. 2007). MAPKs can regulate these effects over either short or long temporal domains to affect intra-and extracellular functions. All the opioid receptor subtypes stimulate phosphorylation of ERK 1/2, as well as JNK and p38 (Al-Hasani & Bruchas 2011, Bruchas et al. 2006, Chen et al. 2008, Eisinger & Ammer 2008, Macey et al. 2006). However, recent studies have reported that JNK phosphorylation by MOPR and KOPR can additionally engage noncanonical, arrestin-independent signaling pathways that inhibit G protein signaling at these receptors for long periods (Bruchas et al. 2007b, Melief et al. 2010, Schattauer et al. 2017).
Recent efforts have aimed to take advantage of the G protein versus arrestin signaling pathways by creating biased opioid receptor ligands. G protein-biased ligands could have fewer adverse effects, including constipation, respiratory depression, and even abuse liability (Brust et al. 2016, Manglik et al. 2016, Raehal et al. 2011, Schmid et al. 2017, Spangler & Bruchas 2017). However, the utility of biased agonists toward mitigating complex side effects, such as analgesic tolerance and OIH, remains controversial as numerous alternate signaling pathways and compensatory mechanisms are likely to be involved (Chen et al. 2016, Roeckel et al. 2016). Finally, how these biased agonists work in vivo, within selected circuits, remains to be dissected.
NEUROANATOMICAL SUBSTRATES FOR OPIOID ANALGESIA
Somatosensory Neurons of the Dorsal Root Ganglia
A remarkable feature of opioid receptors is that they are present at virtually all neural loci contributing to the pain experience. Neurons of the DRG and trigeminal ganglia innervate peripheral organs and relay somatosensory information, including pain, to the spinal cord and medulla (Basbaum et al. 2009). All four opioid receptors are expressed by DRG somatosensory neurons (Arvidsson et al. 1995a,b; Zhu et al. 1998), and their activation by intradermal or intrathecal agonists produces antinociception (Chan et al. 2017, Gunther et al. 2017, Stein et al. 2009) (Figure 3a). Opioid receptor activation depresses glutamate and neuropeptide release from somatosensory afferents onto CNS neurons. Initial studies had suggested that the different types of opioid receptors, particularly MOPR and DOPR, were coexpressed by the same class of DRG neurons, namely unmyelinated peptidergic nociceptors. These neurons detect noxious stimuli in skin and internal organs and express the neuropeptides substance P and calcitonin gene-related protein (CGRP) and the heat-and capsaicin-sensitive transient receptor potential cation channel subfamily V member 1 (TRPV1) (Chen & Pan 2008, Ueda 2006, Vetter et al. 2006). MOPR expression in these cells is thought to contribute to the remarkable utility of mu agonists for perioperative pain management (Figure 2c). In recent years, this coexpression model has been reappraised following the emergence of novel techniques to investigate opioid receptor expression, particularly reporter mice expressing fluorescent opioid receptors and single-cell RNA sequencing (scRNA-seq) (Erbs et al. 2015, Scherrer et al. 2006, Usoskin et al. 2015). These studies suggest that each opioid receptor is differentially distributed among different DRG neuron classes, implying that receptor classes preferentially control distinct types of pain and somatosensory modalities. For example, delta opioid receptor-green fluorescent protein (DOR-GFP) knockin mouse line and scRNA-seq indicate that DOPR is enriched in myelinated mechanosensory neurons that project to the skin and that have been implicated in tactile hypersensitivity (allodynia) in the setting of chronic inflammatory or neuropathic pain (Bardoni et al. 2014, Scherrer et al. 2009, Usoskin et al. 2015). Note, however, that the expression pattern and function of DOPR in DRG remain debated and differ between species (Francois & Scherrer 2017, Gendron et al. 2015). MOPRs in DRG can be targeted by peripherally restricted agonists (i.e., limited blood-brain barrier permeability) to produce analgesia without CNS-derived side effects (DeHaven-Hudkins & Dolle 2004, Vadivelu et al. 2011). Recently, Spahn et al. (2017) refined this approach and developed an opioid analgesic with a low acid dissociation constant, such that this compound selectively activates MOPRs at acidic inflammation sites. Interestingly, however, studies using conditional knockout mice with a selective deletion of MOPRs in DRG nociceptors, but intact receptor expression in the CNS, showed that these MOPRs in DRG are not necessary for the antinociception resulting from systemic morphine (Corder et al. 2017, Weibel et al. 2013). Instead, MOPRs in DRG are important contributors to two of the adverse side effects associated with chronic MOPR agonist treatments, tolerance and OIH (Araldi et al. 2018, Corder et al. 2017; but see also Weibel et al. 2013). Other brain regions, including the periaqueductal gray and rostral ventromedial medulla, contribute to opioid analgesia, tolerance, and OIH (Connor et al. 2015, Eidson et al. 2013, Gaspari et al. 2018, Lane et al. 2005, Morgan et al. 2006, Vanderah et al. 2001, Wilson-Poe et al. 2017). However, activation of MOPR in peripheral nociceptor populations appears to be the key molecular event that initiates pathological plasticity within CNS pain circuits, thereby facilitating the onset of opioid antinociceptive tolerance, physical dependence, and the pronociceptive effects of opioids (Chu et al. 2008, Joseph et al. 2010, Kandasamy & Price 2015, Ossipov et al. 2005).
Figure 3.
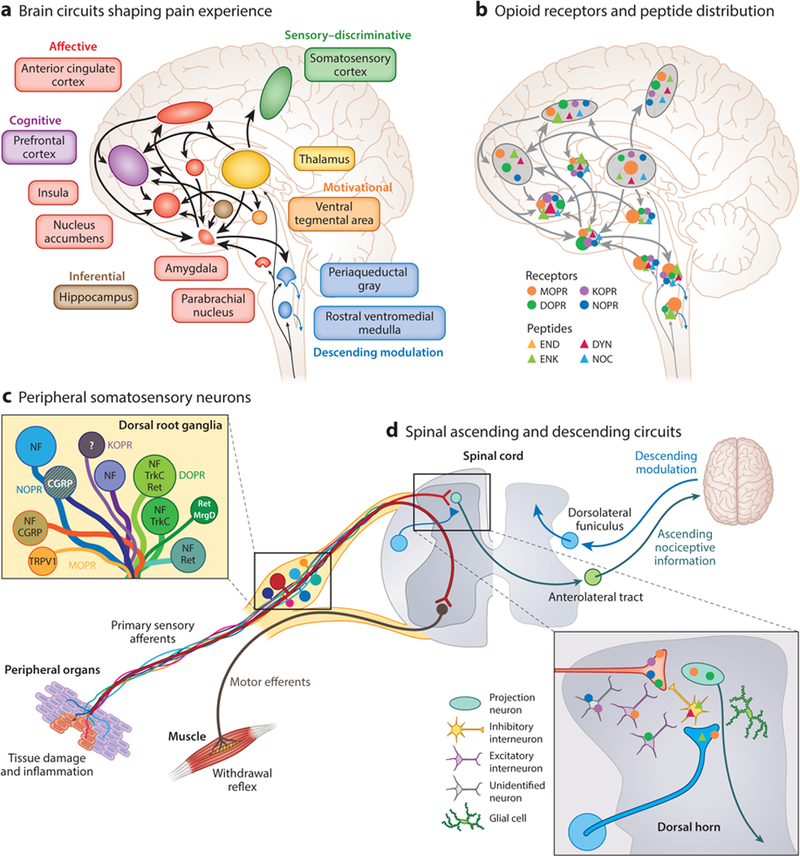
Neuroanatomical substrates of pain perception and remodeling by opioids. (a) A large interconnected neural network of supraspinal brain circuits transforms nociceptive information ascending from the spinal cord into an aversive, painful experience. (b) The opioid system is well positioned within this brain network to modify the perception of pain. The different opioid receptors and peptides are distinctively, though broadly, expressed in different sites, the function of which is under intense investigation. Relative opioid receptor (circles) and peptide (triangles) expression levels are denoted by the size of the shapes. (c,d) Opioid receptor types and peptides are also distributed in distinct subpopulations of (c) DRG neurons, identified with the indicated markers such as TRPV1, and (d) second-order spinal cord dorsal horn neurons. NF marks large-diameter DRG neurons with myelinated axons. Striped neurons coexpress different opioid receptor types. Abbreviations: CGRP, calcitonin gene-related peptide; DRG, dorsal root ganglion; DOPR, delta opioid receptor; DYN, dynorphin; END, p-endorphin; ENK, enkephalin; KOPR, kappa opioid receptor; MOPR, mu opioid receptor; MrgD, Mas-related G protein-coupled receptor member D; NF, neurofilament; NOC, nociceptin/orphanin FQ; NOPR, nociceptin opioid receptor; Ret, Ret proto-oncogene; TrkC, tropomyosin receptor kinase C; TRPV1, transient receptor potential cation channel subfamily V member 1.
KOPR expression and function in DRG can now also be investigated with reporter mice (Cai et al. 2016, Liu-Chen 2017). Multiple preclinical studies provided evidence that KOPR in DRG may control visceral pain and suggested the use of peripherally restricted kappa agonists for these types of pain (Kivell & Prisinzano 2010, Vanderah 2010). The function of NOPR in DRG is not well understood, but the recent generation of a NOPR-enhanced GFP (eGFP) receptor revealed a broad distribution of NOPR in DRG neurons, including in unmyelinated peptidergic nociceptors, and in several populations of myelinated neurons that may include cutaneous mechanoreceptors and proprioceptors (Ozawa et al. 2015).
Spinal Cord Dorsal Horn Circuits
Opioid receptors are expressed by second-order neurons of pain pathways (Figure 3b). MOPR has long been known to be expressed by nociceptive dorsal horn neurons, including excitatory interneurons and lamina I projection neurons of the anterolateral tract that relay nociceptive information to the lateral parabrachial nucleus, thalamus, and periaqueductal gray matter (Aicher et al. 2000, Spike et al. 2002). Immunohistochemical studies suggested that DOPR expression in the dorsal horn was restricted to primary afferent terminals (Dado et al. 1993), whereas DOR-GFP mice, as well as in situ hybridization and electrophysiological recordings in wild-type mice, support the idea that DOPR is expressed by multiple classes of spinal neurons (Wang et al. 2018). Specifically, DOPR expression in somatostatin-positive excitatory interneurons that gate mechanosensory inputs (Duan et al. 2014) contributes to the analgesic properties of DOR agonists. Additionally, DOPR and MOPR coexpression in projection neurons of the anterolateral tract (Wang et al. 2018) suggests that these two receptors may cooperate postsynaptically in cells receiving convergent inputs from segregated delta-positive and mu-positive afferents. The use of an antibody against the phosphorylated form of KOPR suggested expression of this receptor in inhibitory interneurons and spinal astrocytes (Xu et al. 2007), and electrophysiological recordings documented KOPR-selective, agonist U50488H-responsive neurons in the dorsal horn (Eckert & Light 2002). The development of reporter mice for KOPR, along with transcriptomic approaches, will enable the definitive identification of these neurons.
Dynorphin and enkephalin are expressed by distinct classes of dorsal horn interneurons (Boyle et al. 2017, Francois et al. 2017) and are upregulated in the spinal cord following peripheral injury to modulate chronic pain (Lai et al. 2008, Podvin et al. 2016, Xu et al. 2004). Additionally, recent evidence suggests that dynorphin, released by dorsal horn inhibitory interneurons, is an essential mediator of itch (Kardon et al. 2014). The NOPR-eGFP diffuse fluorescence signal throughout laminae I−III strongly suggests that NOPR may be expressed by dorsal horn neurons in addition to primary afferents (Ozawa et al. 2015), but the precise identity of these neurons, as well as the endogenous source of nociceptin peptide that acts on NOPR in laminae I−III, remains to be established. This identification of NOPR-expressing DRG and spinal neurons is likely to clarify the mechanisms by which NOPR agonists can facilitate or counteract mu-mediated antinociception (Toll et al. 2016).
Opioid Action in Brain Circuits for Pain Affect: Remodeling of Pain Percept
Painful experiences are both personal and complex; they are not linearly correlated to noxious input but rather are constructed from neural information relating sensory, emotional, interoceptive, inferential, and cognitive information, which coalesce into a unified perception of pain (Craig 2003, Wiech 2016).
A major site of action of mu opioid analgesics is the descending pain modulatory system, which includes the ventrolateral periaqueductal gray (vlPAG), rostral ventromedial medulla (RVM), and spinal cord (Basbaum & Fields 1984). Microinjection of mu opioids into the vlPAG, or the RVM, is sufficient to produce antinociception (al-Rodhan et al. 1992, Rossi et al. 1994). RVM neurons receive monosynaptic inputs from the vlPAG and have been categorized as on, off, or neutral cells on the basis of their action potential firing pattern, pronociceptive or antinociceptive properties, and response to opioids (Basbaum & Fields 1984, Cheng et al. 1986, Fang et al. 1989, Morgan et al. 1992). Mu opioids can inhibit on cells, and indirectly disinhibit off cells, to produce antinociception. Using endogenous opioids, genetic approaches have begun to molecularly identify RVM neuron subpopulations and clarify the synaptic mechanisms by which these neurons regulate pain thresholds at the spinal level. These studies showed that at least two populations of RVM GABAergic neurons project to the spinal cord and modulate pain (Figures2c and 3b). The first population coexpresses preproenkephalin (Penk) and projects directly onto nociceptor terminals in the dorsal horns to inhibit pain (Zhang et al. 2015); they functionally correspond to off cells. In contrast, the second population, which expresses MOPRs, projects onto Penk-positive dorsal horn interneurons that then presynaptically inhibit mechanosensory neurons to facilitate mechanical pain (Francois et al. 2017).
Furthermore, rostral, subcortical, and cortical sites appear to be especially important for affective processing of pain, as well as the affective and rewarding aspects of pain analgesia (Cahill et al. 2013, Fields & Margolis 2015, Hummel et al. 2008, Kupers et al. 1991, Price et al. 1985) (Figure 3c). Clinical studies suggest that opioids produce pain relief by altering affective and somatic responses. For example, patient self-reports of morphine analgesia reveal that the sensation of pain is still present but affective aversive qualities are reduced (Price et al. 1985). Interestingly, this experience appears to be a dose-dependent pharmacological phenomenon, whereby progressively increasing doses of opioids diminishes first pain affect, then pain sensation (Cobos et al. 2012, LaGraize et al. 2006, Navratilova et al. 2015). Consistent with this, human functional MRI (fMRI) studies showed that much higher doses of opioids are required to reduce blood-oxygen-level-dependent activity in sensory brain regions than in limbic regions (Oertel et al. 2008).
Human positron emission tomography (PET) binding and fMRI studies of the anterior cingulate cortex (ACC) reveal that endogenous opioid release occurs during sustained pain experiences and largely correlates with analgesia against pain affect (Borras et al. 2004, Zubieta et al. 2005). This finding is also true for placebo analgesia (Bingel et al. 2006, Wager et al. 2007, Zubieta et al. 2005). Rodent models have further pinpointed the role of MOPR signaling in the ACC toward the relief of pain-induced aversion (LaGraize et al. 2006, Navratilova et al. 2015). Injection of naloxone, an opioid antagonist, into the ACC reduces the positive affect associated with pain relief, including by nonopioid analgesics, suggesting that endogenous opioids not only reduce nociceptive processes but also facilitate the reinforcing features of exogenous analgesia (Remeniuk et al. 2015). This feature of the endogenous opioid system is further supported by the result thatMOPR blockade reduces dopamine release in the nucleus accumbens (NAc) that accompanies pain relief (Navratilova et al. 2012). Opioid analgesics thus act on multiple cortical and subcortical sites to influence dopaminergic neurotransmission between the ventral tegmental area (VTA) and NAc to reduce pain aversion. Adding to this complexity, chronic pain is accompanied by changes in plasticity in the mesolimbic dopaminergic system. Inflammatory pain desensitizes MOPR in the VTA, promoting opioid consumption (Hipolito et al. 2015, Narita et al. 2005), and neuropathic pain is accompanied by decreased NAc dopamine release, an effect that involves microglial activation in the VTA (Taylor et al. 2015), as well as other negative regulators of dopamine transmission. Additionally, in the amygdala, a crucial node in affective brain circuits, MOPR is expressed by GABAergic neurons of the central nucleus and intercalated cell masses (Winters et al. 2017). Inhibition of these neurons by mu agonists may reduce aversive behavior and reduce amygdala inhibitory input onto descending brainstem pain pathway responses (Han et al. 2015, Namburi et al. 2015). Despite this progress, the precise aspects of the pain experience that are encoded in the NAc and amygdala (salience, valence, motivation, analgesia), and the identity ofMOPR-expressing neurons that modulate pain in the ACC, NAc, amygdala, and VTA, remain to be determined.
KOPRs, DOPRs, and NOPRs also modulate pain supraspinally (Miaskowski et al. 1991, Yamamoto et al. 2001). KOPR activation in the dorsal raphe nucleus mediates descending antinociception (Land et al. 2009, Zhao et al. 2007). Additionally, the KOPR system gates affective information relating to stress and anxiety from the basolateral amygdala to the bed nucleus of the stria terminalis, as well as from inputs from the locus coeruleus (Crowley et al. 2016, McCall et al. 2017, Nygard et al. 2016). Although it is not yet fully understood for pain perception, the KOPR system is well positioned within the NAc circuitry to modify the hedonic value of nociceptive events and shape motivational behaviors in response to painful experiences (Al-Hasani et al. 2015, Castro & Berridge 2014, Negrete et al. 2017, Park et al. 2015). The dynorphin-kappa system regulates stress, aversion, mood, and relapse to drug-seeking for all major classes of abused drugs (Bruchas et al. 2010; Land et al. 2008, 2009) and may also contribute to shaping pain-induced negative affect (Massaly et al. 2017) and to driving comorbid depression and addiction. Interestingly, a recent study supports the idea that KOPR antagonists could be used to prevent stress-induced migraine (Xie et al. 2017). DOPRs and NOPRs are broadly expressed in pain affect and descending control circuits and are particularly enriched in the amygdala and ACC (Goody et al. 2002, Mansour et al. 1994, Ozawa et al. 2015, Scherrer et al. 2006, Toll et al. 2016); however, how these different receptor populations alter the different dimensions of pain experience requires further clarification.
CONCLUSIONS: DISSOCIATING DELETERIOUS SIDE EFFECTS FROM ANALGESIA
There are currently two main research paths to battle the opioid epidemic: discovering nonopioid analgesic therapies that could replace opioids or improving current opioid analgesics. For both paths, the complete resolution of opioid analgesics’ mechanism of action, at the circuit, neural ensemble, synaptic, and molecular levels, will be a decisive step. For instance, the identification of MOPR-expressing neuronal populations in affective circuits that mediate opioid-induced reductions in pain affect will enable transcriptional and proteomic studies to uncover novel nonopioid analgesic targets. These studies are facilitated by the development of genetically engineered mouse lines for visualizing and manipulating opioid receptor-expressing neurons (Cai et al. 2016, Erbs et al. 2015, Scherrer et al. 2006). Similar tools can now be used in vivo to study the cells that endogenously release enkephalins, dynorphins, endorphins, and nociceptin (Al-Hasani et al. 2015, Cowley et al. 2001, Francois et al. 2017).
By contrast, improving current opioid treatments requires an understanding of the mechanisms that underlie their deleterious side effects. At the cellular level, the development of conditional knockout mice lacking opioid receptors in defined cell types will greatly facilitate an understanding of the CNS structures that mediate OIH, antinociceptive tolerance, respiratory depression, and transition to addiction (Convertino et al. 2015, Corder et al. 2017, Gaveriaux-Ruff et al. 2011, Nygard et al. 2016, Weibel et al. 2013). At the level of signaling, biased agonists will clarify which signaling pathways need to be engaged to facilitate analgesia and limit deleterious effects such as respiratory depression, addiction, and constipation (Bohn & Aube 2017, Manglik et al. 2016, Schmid et al. 2017, Siuda et al. 2017, Spangler & Bruchas 2017). Collectively, this suite of novel genetic and pharmacological tools, together with the development of new behavioral paradigms for evaluating the pain experience and opioid analgesia in animal models (see the sidebar titled Translational Hurdles in Pain and Opioid Research), will likely yield insights into previously unanswerable questions. These advances are likely to lead to the development of more effective and safer analgesic treatments.
TRANSLATIONAL HURDLES IN PAIN AND OPIOID RESEARCH.
Current preclinical models of pain have elucidated detailed mechanisms for sensory detection and spinal encoding of nociceptive information. Unfortunately, a disconnect exists between clinical and preclinical assessments of pain: Human studies primarily use patient self-reports, whereas animal models typically use withdrawal reflexes or other indirect measures of pain. This raises the concern that animal models do not capture the holistic (i.e., sensory and affective) experience of pain in patients. This limitation has likely hampered the discovery of novel analgesic strategies to dampen pain negative affect in the clinic. Looking forward, efforts need to be directed toward dissecting the brain circuits of pain and require the development of measures of pain in animal models that more accurately reflect the in-the-moment and perceptual qualities of what it is like to experience pain. Tight modulation of neural circuits in vivo (e.g., optogenetic holography), paired with high-resolution, mesoscale monitoring of brain activity, may hold tremendous promise for determining how neural networks encode various dimensions of pain. Indeed, the combination of human functional imaging, behavior, and machine learning has already led to important advances in linking dynamic brain states to pain, thus paving a new avenue for preclinical research to follow in kind.
ACKNOWLEDGMENTS
We apologize to all investigators whose work could not be appropriately cited owing to space and citation limitations of this journal. This work was supported by National Institutes of Health grants R01DA044481 (G.S.), R01DA033396 (M.R.B), K99DA043609 (G.C.), andF32DA043999 (D.C.C). G.S. is a New York Stem Cell Foundation - Robertson Investigator. We thank Dr. Aashish Manglik for providing images of opioid receptor crystal structures (Figure 1).
Footnotes
DISCLOSURE STATEMENT
G.S. is cofounder of Epiodyne, an opioid analgesic discovery company. M.R.B. is a cofounder of Neurolux, a neuroscience technology company.
LITERATURE CITED
- Aicher SA, Punnoose A, Goldberg A. 2000. μ.-Opioid receptors often colocalize with the substance P receptor (NK1) in the trigeminal dorsal horn. J. Neurosci. 20:4345–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hasani R, Bruchas MR. 2011. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 115:1363–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hasani R, McCall JG, Shin G, Gomez AM, Schmitz GP, et al. 2015. Distinct subpopulations of nucleus accumbens dynorphin neurons drive aversion and reward. Neuron 87:1063–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- al-Rodhan NR, Yaksh TL, Kelly PJ. 1992. Comparison of the neurochemistry of the endogenous opioid systems in two brainstem pain-processing centers. Stereotact. Funct. Neurosurg. 59:15–19 [DOI] [PubMed] [Google Scholar]
- Altier C, Khosravani H, Evans RM, Hameed S, Peloquin JB, et al. 2006. ORL1 receptor-mediated internalization of N-type calcium channels. Nat. Neurosci. 9:31–40 [DOI] [PubMed] [Google Scholar]
- Araldi D, Khomula EV, Ferrari LF, Levine JD. 2018. Fentanyl induces rapid onset hyperalgesic priming: type I at peripheral and type II at central nociceptor terminals. J. Neurosci. 38:2226–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvidsson U, Dado RJ, Riedl M, Lee JH, Law PY, et al. 1995a. delta-Opioid receptor immunoreactivity: distribution in brainstem and spinal cord, and relationship to biogenic amines and enkephalin. J. Neurosci. 15:1215–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvidsson U, Riedl M, Chakrabarti S, Lee JH, Nakano AH, et al. 1995b. Distribution and targeting of a mu-opioid receptor (MOR1) in brain and spinal cord. J. Neurosci. 15(5 Pt. 1):3328–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahouth SW, Nooh MM. 2017. Barcoding of GPCR trafficking and signaling through the various trafficking roadmaps by compartmentalized signaling networks. Cell. Signal. 36:42–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banghart MR, Sabatini BL. 2012. Photoactivatable neuropeptides for spatiotemporally precise delivery of opioids in neural tissue. Neuron 73:249–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barchfeld CC, Medzihradsky F. 1984. Receptor-mediated stimulation of brain GTPase by opiates in normal and dependent rats. Biochem. Biophys. Res. Commun. 121:641–48 [DOI] [PubMed] [Google Scholar]
- Bardoni R, Tawfik VL, Wang D, François A, Solorzano C, et al. 2014. Delta opioid receptors presynaptically regulate cutaneous mechanosensory neuron input to the spinal cord dorsal horn. Neuron 81:1312–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. 2009. Cellular and molecular mechanisms of pain. Cell 139:267–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Fields HL. 1984. Endogenous pain control systems: brainstem spinal pathways and endorphin circuitry. Annu. Rev. Neurosci. 7:309–38 [DOI] [PubMed] [Google Scholar]
- Bingel U, Lorenz J, Schoell E, Weiller C, Büchel C. 2006. Mechanisms of placebo analgesia: rACC recruitment of a subcortical antinociceptive network. Pain 120:8–15 [DOI] [PubMed] [Google Scholar]
- Bloom FE, Rossier J, Battenberg EL, Bayon A, French E, et al. 1978. beta-endorphin: cellular localization, electrophysiological and behavioral effects. Adv. Biochem. Psychopharmacol. 18:89–109 [PubMed] [Google Scholar]
- Bohn LM, Aubé J. 2017. Seeking (and finding) biased ligands of the kappa opioid receptor. ACS Med. Chem. Lett. 8:694–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Sotnikova TD, Medvedev IO, Lefkowitz RJ, et al. 2003. Enhanced rewarding properties of morphine, but not cocaine, in βarrestin-2 knock-out mice. J. Neurosci. 23:10265–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. 1999. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science 286:2495–98 [DOI] [PubMed] [Google Scholar]
- Borras MC, Becerra L, Ploghaus A, Gostic JM, DaSilva A, et al. 2004. fMRI measurement of CNS responses to naloxone infusion and subsequent mild noxious thermal stimuli in healthy volunteers. J. Neurophysiol. 91:2723–33 [DOI] [PubMed] [Google Scholar]
- Bower JD, Guest KP, Morgan BA. 1976. Enkephalin. Synthesis of two pentapeptides isolated from porcine brain with receptor-mediated opiate agonist activity. J. Chem. Soc. Perkin Trans. 1 (23):2488–92 [PubMed] [Google Scholar]
- Boyle KA, Gutierrez-Mecinas M, Polgár E, Mooney N, O’Connor E, et al. 2017. A quantitative study of neurochemically defined populations of inhibitory interneurons in the superficial dorsal horn of the mouse spinal cord. Neuroscience 363:120–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Aita M, Xu M, Barot SK, et al. 2007a. Stress-induced p38 mitogen-activated protein kinase activation mediates K-opioid-dependent dysphoria. J. Neurosci. 27:11614–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Chavkin C. 2010. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 1314:44–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Macey TA, Lowe JD, Chavkin C. 2006. Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J. Biol. Chem. 281:18081–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Schindler AG, Shankar H, Messinger DI, Miyatake M, et al. 2011. Selective p38a MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron 71:498–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Yang T, Schreiber S, Defino M, Kwan SC, et al. 2007b. Long-acting к opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating c-Jun N-terminal kinase. J. Biol. Chem. 282:29803–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brust TF, Morgenweck J, Kim SA, Rose JH, Locke JL, et al. 2016. Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria. Sci. Signal. 9:ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burma NE, Bonin RP, Leduc-Pessah H, Baimel C, Cairncross ZF, et al. 2017. Blocking microglial pannexin-1 channels alleviates morphine withdrawal in rodents. Nat. Med. 23:355–60 [DOI] [PubMed] [Google Scholar]
- Cahill CM, Xue L, Grenier P, Magnussen C, Lecour S, Olmstead MC. 2013. Changes in morphine reward in a model of neuropathic pain. Behav. Pharmacol. 24:207–13 [DOI] [PubMed] [Google Scholar]
- Cai X, Huang H, Kuzirian MS, Snyder LM, Matsushita M, et al. 2016. Generation of a KOR-Cre knockin mouse strain to study cells involved in kappa opioid signaling. Genesis 54:29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro DC, Berridge KC. 2014. Opioid hedonic hotspot in nucleus accumbens shell: mu, delta, and kappa maps for enhancement of sweetness “liking” and “wanting.” J. Neurosci. 34:4239–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC (Cent. Dis. Control Prev.). 2013. Vital signs: overdoses of prescription opioid pain relievers and other drugs among women-United States, 1999–2010. MMWRMorb. Mortal Wkly. Rep. 62:537–42 [PMC free article] [PubMed] [Google Scholar]
- Chan HCS, McCarthy D, Li J, Palczewski K, Yuan S. 2017. Designing safer analgesics via μ-opioid receptor pathways. Trends Pharmacol. Sci. 38:1016–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C 2013. Dynorphin—still an extraordinarily potent opioid peptide. Mol. Pharmacol. 83:729–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Xie R-G, Gao Y-J, Xu Z-Z, Zhao L-X, et al. 2016. β-arrestin-2 regulates NMDA receptor function in spinal lamina II neurons and duration of persistent pain. Nat. Commun. 7:12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L-Y, Huang J-X, Yu L-C. 2008. Involvement of ORL1 receptor and ERK kinase in the orphanin FQ-induced nociception in the nucleus accumbens of rats. Regul. Pept. 151:43–47 [DOI] [PubMed] [Google Scholar]
- Chen S-R, Pan H-L. 2008. Removing TRPV1-expressing primary afferent neurons potentiates the spinal analgesic effect of delta-opioid agonists on mechano-nociception. Neuropharmacology 55:215–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng ZF, Fields HL, Heinricher MM. 1986. Morphine microinjected into the periaqueductal gray has differential effects on 3 classes of medullary neurons. Brain Res. 375:57–65 [DOI] [PubMed] [Google Scholar]
- Childers SR, Snyder SH. 1978. Guanine nucleotides differentiate agonist and antagonist interactions with opiate receptors. Life Sci. 23:759–61 [DOI] [PubMed] [Google Scholar]
- Chu LF, Angst MS, Clark D. 2008. Opioid-induced hyperalgesia in humans: molecular mechanisms and clinical considerations. Clin. J. Pain 24:479–96 [DOI] [PubMed] [Google Scholar]
- Cobos EJ, Ghasemlou N, Araldi D, Segal D, Duong K, Woolf CJ. 2012. Inflammation-induced decrease in voluntary wheel running in mice: a nonreflexive test for evaluating inflammatory pain and analgesia. Pain 153:876–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor M, Bagley EE, Chieng BC, Christie MJ. 2015. β-Arrestin-2 knockout prevents development of cellular μ-opioid receptor tolerance but does not affect opioid-withdrawal-related adaptations in single PAG neurons. Br. J. Pharmacol. 172:492–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Convertino M, Samoshkin A, Gauthier J, Gold MS, Maixner W, et al. 2015. μ-Opioid receptor 6-transmembrane isoform: a potential therapeutic target for new effective opioids. Prog. Neuropsychopharmacol. Biol. Psychiatry 62:61–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder G, Doolen S, Donahue RR, Winter MK, Jutras BL, et al. 2013. Constitutive μ-opioid receptor activity leads to long-term endogenous analgesia and dependence. Science 341:1394–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder G, Tawfik VL, Wang D, Sypek EI, Low SA, et al. 2017. Loss of μ opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat. Med. 23:164–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, et al. 2001. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411:480–84 [DOI] [PubMed] [Google Scholar]
- Craig AD. 2003. A new view of pain as a homeostatic emotion. Trends Neurosci. 26:303–7 [DOI] [PubMed] [Google Scholar]
- Crowley NA, Bloodgood DW, Hardaway JA, Kendra AM, McCall JG, et al. 2016. Dynorphin controls the gain of an amygdalar anxiety circuit. Cell Rep. 14:2774–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dado RJ, Law PY, Loh HH, Elde R. 1993. Immunofluorescent identification of a delta (delta)-opioid receptor on primary afferent nerve terminals. Neuroreport 5:341–44 [DOI] [PubMed] [Google Scholar]
- DeHaven-Hudkins DL, Dolle RE. 2004. Peripherally restricted opioid agonists as novel analgesic agents. Curr. Pharm. Des. 10:743–57 [DOI] [PubMed] [Google Scholar]
- Drake CT, Terman GW, Simmons ML, Milner TA, Kunkel DD, et al. 1994. Dynorphin opioids present in dentate granule cells may function as retrograde inhibitory neurotransmitters. J. Neurosci. 14:3736–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drdla R, Gassner M, Gingl E, Sandkühler J. 2009. Induction of synaptic long-term potentiation after opioid withdrawal. Science 325:207–10 [DOI] [PubMed] [Google Scholar]
- Drdla-Schutting R, Benrath J, Wunderbaldinger G, Sandkühler J. 2012. Erasure of a spinal memory trace of pain by a brief, high-dose opioid administration. Science 335:235–38 [DOI] [PubMed] [Google Scholar]
- Duan B, Cheng L, Bourane S, Britz O, Padilla C, et al. 2014. Identification of spinal circuits transmitting and gating mechanical pain. Cell 159:1417–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggan AW. 2000. Neuropeptide spread in the brain and spinal cord. Prog. Brain Res. 125:369–80 [DOI] [PubMed] [Google Scholar]
- Eckert WA, Light AR. 2002. Hyperpolarization of substantia gelatinosa neurons evoked by μ-, κ-, δ1-, and δ2-selective opioids. J. Pain 3:115–25 [DOI] [PubMed] [Google Scholar]
- Eichel K, Jullie D, von Zastrow M. 2016. ß-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat. Cell Biol. 18:303–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisinger DA, Ammer H. 2008. δ-Opioid receptors activate ERK/MAP kinase via integrin-stimulated receptor tyrosine kinases. Cell. Signal. 20:2324–31 [DOI] [PubMed] [Google Scholar]
- Erbs E, Faget L, Scherrer G, Matifas A, Filliol D, et al. 2015. A mu-delta opioid receptor brain atlas reveals neuronal co-occurrence in subcortical networks. Brain Struct. Funct. 220:677–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang FG, Haws CM, Drasner K, Williamson A, Fields HL. 1989. Opioid peptides (DAGO-enkephalin, dynorphin A(1–13), BAM 22P) microinjected into the rat brainstem: comparison of their antinociceptive effect and their effect on neuronal firing in the rostral ventromedial medulla. Brain Res. 501:116–28 [DOI] [PubMed] [Google Scholar]
- Fields HL, Margolis EB. 2015. Understanding opioid reward. Trends Neurosci. 38:217–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- François A, Low SA, Sypek EI, Christensen AJ, Sotoudeh C, et al. 2017. A brainstem-spinal cord inhibitory circuit for mechanical pain modulation by GABA and enkephalins. Neuron 93:822–839.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- François A, Scherrer G. 2017. Delta opioid receptor expression and function in primary afferent somatosensory neurons. Handb. Exp. Pharmacol. doi: 10.1007/164_2017_58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita W, Gomes I, Devi LA. 2015. Heteromers of μ-δ opioid receptors: new pharmacology and novel therapeutic possibilities. Br. J. Pharmacol 172:375–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspari S, Purushothaman I, Cogliani V, Sakloth F, Neve RL, et al. 2018. Suppression of RGSz1 function optimizes the actions of opioid analgesics by mechanisms that involve the Wnt/ β-catenin pathway. PNAS 115:E2085–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaveriaux-Ruff C, Nozaki C, Nadal X, Hever XC, Weibel R, et al. 2011. Genetic ablation of delta opioid receptors in nociceptive sensory neurons increases chronic pain and abolishes opioid analgesia. Pain 152:1238–48 [DOI] [PubMed] [Google Scholar]
- Gendron L, Mittal N, Beaudry H, Walwyn W. 2015. Recent advances on the δ opioid receptor: from trafficking to function. Br. J. Pharmacol. 172:403–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass MJ, Vanyo L, Quimson L, Pickel VM. 2009. Ultrastructural relationship between N-methyl-D-aspartate-NR1 receptor subunit and mu-opioid receptor in the mouse central nucleus of the amygdala. Neuroscience 163:857–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein A, Tachibana S, Lowney LI, Hunkapiller M, Hood L. 1979. Dynorphin-(1–13), an extraordinarily potent opioid peptide. PNAS 76:6666–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goody RJ, Oakley SM, Filliol D, Kieffer BL, Kitchen I. 2002. Quantitative autoradiographic mapping of opioid receptors in the brain of δ-opioid receptor gene knockout mice. Brain Res. 945:9–19 [DOI] [PubMed] [Google Scholar]
- Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, et al. 2012. Structure of the δ-opioid receptor bound to naltrindole. Nature 485:400–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther T, Dasgupta P, Mann A, Miess E, Kliewer A, et al. 2017. Targeting multiple opioid receptors— improved analgesics with reduced side effects? Br. J. Pharmacol. doi: 10.1111/bph.13809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Soleiman MT, Soden ME, Zweifel LS, Palmiter RD. 2015. Elucidating an affective pain circuit that creates a threat memory. Cell 162:363–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipólito L, Wilson-Poe A, Campos-Jurado Y, Zhong E, Gonzalez-Romero J, et al. 2015. Inflammatory pain promotes increased opioid self-administration: role of dysregulated ventral tegmental area μ opioid receptors. J. Neurosci. 35:12217–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel M, Lu P, Cummons TA, Whiteside GT. 2008. The persistence of a long-term negative affective state following the induction of either acute or chronic pain. Pain 140:436–45 [DOI] [PubMed] [Google Scholar]
- Institute of Medicine (US) Committee on Advancing Pain Research, Care, and Education. 2011. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. Washington, DC: National Academies Press; [PubMed] [Google Scholar]
- Inturrisi CE. 2002. Clinical pharmacology of opioids for pain. Clin. J. Pain 18(4 Suppl):S3–13 [DOI] [PubMed] [Google Scholar]
- Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, et al. 2013. Conformational biosensors reveal GPCR signalling from endosomes. Nature 495:534–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irannejad R, von Zastrow M. 2014. GPCR signaling along the endocytic pathway. Curr. Opin. Cell Biol. 27:109–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James IF, Chavkin C, Goldstein A. 1982. Preparation of brain membranes containing a single type of opioid receptor highly selective for dynorphin. PNAS 79:7570–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph EK, Reichling DB, Levine JD. 2010. Shared mechanisms for opioid tolerance and a transition to chronic pain. J. Neurosci. 30:4660–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandasamy R, Price TJ. 2015. The pharmacology of nociceptor priming. Handb. Exp. Pharmacol. 227:15–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardon AP, Polgar E, Hachisuka J, Snyder LM, Cameron D, et al. 2014. Dynorphin acts as a neuromodulator to inhibit itch in the dorsal horn of the spinal cord. Neuron 82:573–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T 2001. Inverse, protean, and ligand-selective agonist: matters of receptor conformation. FASEB J. 3:593–611 [DOI] [PubMed] [Google Scholar]
- Kieffer BL, Evans CJ. 2009. Opioid receptors: from binding sites to visible molecules in vivo. Neuropharmacology 56(Suppl. 1):205–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivell B, Prisinzano TE. 2010. Kappa opioids and the modulation of pain. Psychopharmacology 210:109–19 [DOI] [PubMed] [Google Scholar]
- Kupers RC, Konings H, Adriaensen H, Gybels JM. 1991. Morphine differentially affects the sensory and affective pain ratings in neurogenic and idiopathic forms of pain. Pain 47:5–12 [DOI] [PubMed] [Google Scholar]
- LaGraize SC, Borzan J, Peng YB, Fuchs PN. 2006. Selective regulation of pain affect following activation of the opioid anterior cingulate cortex system. Exp. Neurol. 197:22–30 [DOI] [PubMed] [Google Scholar]
- Lai J, Luo M, Chen Q, Porreca F. 2008. Pronociceptive actions of dynorphin via bradykinin receptors. Neurosci. Lett. 437:175–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberts JT, Traynor JR. 2013. Opioid receptor interacting proteins and the control of opioid signaling. Curr. Pharm. Des. 19:7333–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. 2008. The dysphoric component of stress is encoded by activation of the dynorphin K-opioid system. J. Neurosci. 28:407–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, et al. 2009. Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. PNAS 106:19168–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DA, Patel PA, Morgan MM. 2005. Evidence for an intrinsic mechanism of antinociceptive tolerance within the ventrolateral periaqueductal gray of rats. Neuroscience 135:227–34 [DOI] [PubMed] [Google Scholar]
- Lazarus LH, Ling N, Guillemin R. 1976. beta-Lipotropin as a prohormone for the morphinomimetic peptides endorphins and enkephalins. PNAS 73:2156–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JG, Prather PL. 2001. Chronic exposure to mu-opioid agonists produces constitutive activation of mu-opioid receptors in direct proportion to the efficacy of the agonist used for pretreatment. Mol. Pharmacol. 60:53–62 [DOI] [PubMed] [Google Scholar]
- Liu-Chen L-Y. 2017. Characterization of a mutant mouse line expressing a fusion protein of kappa opioid receptor and tdTomato. Presented at the International Narcotics Research Conference, Chicago, July 9–14 [Google Scholar]
- Luján R, Marron Fernandez de Velasco E, Aguado C, Wickman K. 2014. New insights into the therapeutic potential of GIRK channels. Trends Neurosci. 37:20–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macey TA, Lowe JD, Chavkin C. 2006. Mu opioid receptor activation of ERK1/2 is GRK3 and arrestin dependent in striatal neurons. J. Biol. Chem. 281:34515–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchikanti L, Helm S, Fellows B, Janata JW, Pampati V, et al. 2012. Opioid epidemic in the United States. Pain Phys. 15(3 Suppl):ES9–38 [PubMed] [Google Scholar]
- Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, et al. 2012. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 485:321–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, et al. 2016. Structure-based discovery of opioid analgesics with reduced side effects. Nature 537:185–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Burke S, Meng F, Thompson RC, et al. 1994. Mu, delta, and kappa opioid receptor mRNA expression in the rat CNS: an in situ hybridization study. J. Comp. Neurol. 350:412–38 [DOI] [PubMed] [Google Scholar]
- Mansour A, Khachaturian H, Lewis ME, Akil H, Watson SJ. 1988. Anatomy of CNS opioid receptors. Trends Neurosci. 11:308–14 [DOI] [PubMed] [Google Scholar]
- Massaly N, Wilson-Poe A, Hipolito L, Markovic T, Bruchas MR, Moron J. 2017. Pain recruits accumbal kappa opioid system and alters opioid consumption. Presented at the International Narcotics Research Conference, Chicago, July 9–14 [Google Scholar]
- McCall JG, Siuda ER, Bhatti DL, Lawson LA, McElligott ZA, et al. 2017. Locus coeruleus to basolateral amygdala noradrenergic projections promote anxiety-like behavior. eLife 6:e18247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melief EJ, Miyatake M, Bruchas MR, Chavkin C. 2010. Ligand-directed c-Jun N-terminal kinase activation disrupts opioid receptor signaling. PNAS 107:11608–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C,et al. 1995. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature 377:532–35 [DOI] [PubMed] [Google Scholar]
- Meye FJ, van Zessen R, Smidt MP, Adan RA, Ramakers GM. 2012. Morphine withdrawal enhances constitutive μ-opioid receptor activity in the ventral tegmental area. J. Neurosci. 32:16120–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miaskowski C,Taiwo YO, Levine JD. 1991. Contribution of supraspinal μ-and δ-opioidreceptors to antinociception in the rat. Eur. J. Pharmacol. 205:247–52 [DOI] [PubMed] [Google Scholar]
- Minneman KP, Iversen IL. 1976. Enkephalin and opiate narcotics increase cyclic GMP accumulation in slices of rat neostriatum. Nature 262:313–14 [DOI] [PubMed] [Google Scholar]
- Morgan MM, Fossum EN, Levine CS, Ingram SL. 2006. Antinociceptive tolerance revealed by cumulative intracranial microinjections of morphine into the periaqueductal gray in the rat. Pharmacol. Biochem. Behav. 85:214–19 [DOI] [PubMed] [Google Scholar]
- Morgan MM, Heinricher MM, Fields HL. 1992. Circuitry linking opioid-sensitive nociceptive modulatory systems in periaqueductal gray and spinal cord with rostral ventromedial medulla. Neuroscience 47:863–71 [DOI] [PubMed] [Google Scholar]
- Nagi K, Pineyro G. 2014. Kir3 channel signaling complexes: focus on opioid receptor signaling. Front. Cell Neurosci. 8:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namburi P, Beyeler A, Yorozu S, Calhoon GG, Halbert SA, et al. 2015A circuit mechanism for differentiating positive and negative associations. Nature 520:675–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Kishimoto Y, Ise Y, Yajima Y, Misawa K, Suzuki T. 2005. Direct evidence for the involvement of the mesolimbic κ-opioid system in the morphine-induced rewarding effect under an inflammatory pain-like state. Neuropsychopharmacology 30:111–18 [DOI] [PubMed] [Google Scholar]
- Navratilova E, Xie JY, Meske D, Qu C, Morimura K, et al. 2015. Endogenous opioid activity in the anterior cingulate cortex is required for relief of pain. J. Neurosci. 35:7264–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navratilova E, Xie JY, Okun A, Qu C, Eyde N, et al. 2012. Pain relief produces negative reinforcement through activation of mesolimbic reward-valuation circuitry. PNAS 109:20709–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal CR, Mansour A, Reinscheid R, Nothacker HP, Civelli O, et al. 1999. Opioid receptor-like (ORL1) receptor distribution in the rat central nervous system: comparison of ORL1 receptor mRNA expression with 125I-[14Tyr]-orphanin FQ binding. J. Comp. Neurol. 412:563–605 [PubMed] [Google Scholar]
- Negrete R, García Gutierrez MS, Manzanares J, Maldonado R. 2017. Involvement of the dynorphin/KOR system on the nociceptive, emotional and cognitive manifestations ofjoint pain in mice. Neuropharmacology 116:315–27 [DOI] [PubMed] [Google Scholar]
- Nygard SK, Hourguettes NJ, Sobczak GG, Carlezon WA, Bruchas MR. 2016. Stress-induced reinstatement of nicotine preference requires dynorphin/kappa opioid activity in the basolateral amygdala. J. Neurosci. 36:9937–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertel BG, Preibisch C, Wallenhorst T, Hummel T, Geisslinger G, et al. 2008. Differential opioid action on sensory and affective cerebral pain processing. Clin. Pharmacol. Ther. 83:577–88 [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, King T, Vanderah TW, Porreca F. 2005. Underlying mechanisms of pronociceptive consequences of prolonged morphine exposure. Biopolymers 80:319–24 [DOI] [PubMed] [Google Scholar]
- Ozawa A, Brunori G, Mercatelli D, Wu J, Cippitelli A, et al. 2015. Knock-in mice with NOP-eGFP receptors identify receptor cellular and regional localization. J. Neurosci. 35:11682–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park PE, Schlosburg JE, Vendruscolo LF, Schulteis G, Edwards S, Koob GF. 2015. Chronic CRF1 receptor blockade reduces heroin intake escalation and dependence-induced hyperalgesia. Addict. Biol. 20:275–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak GW. 2018. Mu opioid pharmacology: 40 years to the promised land. Adv. Pharmacol. 82:261–91 [DOI] [PubMed] [Google Scholar]
- Podvin S, Yaksh T, Hook V. 2016. The emerging role of spinal dynorphin in chronic pain: a therapeutic perspective. Annu. Rev. Pharmacol. Toxicol. 56:511–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polter AM, Barcomb K, Chen RW, Dingess PM, Graziane NM, et al. 2017. Constitutive activation of kappa opioid receptors at ventral tegmental area inhibitory synapses following acute stress. eLife 6:e23785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DD, Von der Gruen A, Miller J, Rafii A, Price C. 1985. A psychophysical analysis of morphine analgesia. Pain 22:261–69 [DOI] [PubMed] [Google Scholar]
- Raehal KM, Schmid CL, Groer CE, Bohn LM. 2011. Functional selectivity at the μ-opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacol. Rev. 63:1001–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman M, Chen W, Cobb MH. 2007. Differential regulation and properties of MAPKs. Oncogene 26:3100–12 [DOI] [PubMed] [Google Scholar]
- Remeniuk B, Sukhtankar D, Okun A, Navratilova E, Xie JY, et al. 2015. Behavioral and neurochemical analysis of ongoing bone cancer pain in rats. Pain 156:1864–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeckel L-A, Le Coz G-M, Gaveriaux-Ruff C, Simonin F. 2016. Opioid-induced hyperalgesia: cellular and molecular mechanisms. Neuroscience 338:160–82 [DOI] [PubMed] [Google Scholar]
- Rossi GC, Pasternak GW, Bodnar RJ. 1994. |a and 6 opioid synergy between the periaqueductal gray and the rostro-ventral medulla. Brain Res. 665:85–93 [DOI] [PubMed] [Google Scholar]
- Ruscheweyh R, Wilder-Smith O, Drdla R, Liu X-G, Sandkuhler J. 2011. Long-term potentiation in spinal nociceptive pathways as a novel target for pain therapy. Mol. Pain 7:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusin KI, Giovannucci DR, Stuenkel EL, Moises HC. 1997. K-Opioid receptor activation modulates Ca2+ currents and secretion in isolated neuroendocrine nerve terminals. J. Neurosci. 17:6565–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samoshkin A, Convertino M, Viet CT, Wieskopf JS, Kambur O, et al. 2015. Structural and functional interactions between six-transmembrane μ-opioid receptors and β2-adrenoreceptors modulate opioid signaling. Sci. Rep. 5:18198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattauer SS, Land BB, Reichard KL, Abraham AD, Burgeno LM, et al. 2017. Peroxiredoxin 6 mediates Gαi protein-coupled receptor inactivation by cJun kinase. Nat. Commun. 8:743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherrer G, Imamachi N, Cao Y-Q, Contet C, Mennicken F, et al. 2009. Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell 137:1148–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherrer G, Tryoen-Toth P, Filliol D, Matifas A, Laustriat D, et al. 2006. Knockin mice expressing fluorescent δ-opioid receptors uncover G protein-coupled receptor dynamics in vivo. PNAS 103:9691–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid CL, Kennedy NM, Ross NC, Lovell KM, Yue Z, et al. 2017. Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell 71:1165–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoblock JR, Maidment NT. 2006. Constitutively active micro opioid receptors mediate the enhanced conditioned aversive effect of naloxone in morphine-dependent mice. Neuropsychopharmacology 31:171–77 [DOI] [PubMed] [Google Scholar]
- Siuda ER, Carr R, Rominger DH, Violin JD. 2017. Biased mu-opioid receptor ligands: a promising new generation of pain therapeutics. Curr. Opin. Pharmacol 32:77–84 [DOI] [PubMed] [Google Scholar]
- Spahn V, Del Vecchio G, Labuz D, Rodriguez-Gaztelumendi A, Massaly N, et al. 2017. A nontoxic pain killer designed by modeling of pathological receptor conformations. Science 355:966–69 [DOI] [PubMed] [Google Scholar]
- Spangler SM, Bruchas MR. 2017. Optogenetic approaches for dissecting neuromodulation and GPCR signaling in neural circuits. Curr. Opin. Pharmacol. 32(Suppl. C):56–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spike RC, Puskar Z, Sakamoto H, Stewart W, Watt C, Todd AJ. 2002. MOR-1-immunoreactive neurons in the dorsal horn of the rat spinal cord: evidence for nonsynaptic innervation by substance P-containing primary afferents and for selective activation by noxious thermal stimuli. Eur. J. Neurosci. 15:1306–16 [DOI] [PubMed] [Google Scholar]
- Stein C, Clark JD, Oh U, Vasko MR, Wilcox GL, et al. 2009. Peripheral mechanisms of pain and analgesia. Brain Res. Rev. 60:90–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streicher JM, Bilsky EJ. 2017. Peripherally acting μ-opioid receptor antagonists for the treatment of opioid- related side effects: mechanism of action and clinical implications. J. Pharm. Pract. doi: 10.1177/0897190017732263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svingos AL, Moriwaki A, Wang JB, Uhl GR, Pickel VM. 1996. Ultrastructural immunocytochemical localization of μ-opioid receptors in rat nucleus accumbens: extrasynaptic plasmalemmal distribution and association with Leu5-enkephalin. J. Neurosci. 16:4162–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AMW, Castonguay A, Taylor AJ, Murphy NP, Ghogha A, et al. 2015. Microglia disrupt mesolimbic reward circuitry in chronic pain. J. Neurosci. 35:8442–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AA, Liu W, Chun E, Katritch V, Wu H, et al. 2012. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 485:395–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll L, Bruchas MR, Calo’ G, Cox BM, Zaveri NT. 2016. Nociceptin/orphanin FQ receptor structure, signaling, ligands, functions, and interactions with opioid systems. Pharmacol. Rev. 68:419–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrecilla M, Marker CL, Cintora SC, Stoffel M, Williams JT, Wickman K. 2002. G-protein-gated potassium channels containing Kir3.2 and Kir3.3 subunits mediate the acute inhibitory effects of opioids on locus ceruleus neurons. J. Neurosci. 22:4328–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trang T, Al-Hasani R, Salvemini D, Salter MW, Gutstein H, Cahill CM. 2015. Pain and poppies: the good, the bad, and the ugly of opioid analgesics. J. Neurosci. 35:13879–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H 2006. Molecular mechanisms of neuropathic pain-phenotypic switch and initiation mechanisms. Pharmacol. Ther. 109:57–77 [DOI] [PubMed] [Google Scholar]
- Usoskin D, Furlan A, Islam S, Abdo H, Lonnerberg P, et al. 2015. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 18:145–53 [DOI] [PubMed] [Google Scholar]
- Vadivelu N, Mitra S, Hines RL. 2011. Peripheral opioid receptor agonists for analgesia: a comprehensive review. J. Opioid Manag. 7:55–68 [DOI] [PubMed] [Google Scholar]
- Vanderah TW. 2010. Delta and kappa opioid receptors as suitable drug targets for pain. Clin.J. Pain 26(Suppl. 10):S10–15 [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Suenaga NM, Ossipov MH, Malan TP Jr., et al. 2001. Tonic descending facilitation from the rostral ventromedial medullamediates opioid-induced abnormal pain and antinociceptive tolerance. J. Neurosci. 21:279–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter I, Wyse BD, Monteith GR, Roberts-Thomson SJ, Cabot PJ. 2006. The μ opioid agonist morphine modulates potentiation of capsaicin-evoked TRPV1 responses through a cyclic AMP-dependent protein kinase A pathway. Mol. Pain 2:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, McLellan AT. 2016. Opioid abuse in chronic pain: misconceptions and mitigation strategies. N. Engl. J. Med. 374:1253–63 [DOI] [PubMed] [Google Scholar]
- Wager TD, Scott DJ, Zubieta J-K. 2007. Placebo effects on human μ-opioid activity during pain. PNAS 104:11056–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walwyn W, Evans CJ, Hales TG. 2007. β-Arrestin2 and c-Src regulate the constitutive activity and recycling of μ opioid receptors in dorsal root ganglion neurons. J. Neurosci. 27:5092–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Tawfik VL, Corder G, Low SA, Francois A, et al. 2018. Functional divergence of delta and mu opioid receptor organization in CNS pain circuits. Neuron 98(1):90–108.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel R, Reiss D, Karchewski L, Gardon O, Matifas A, et al. 2013. Mu opioid receptors on primary afferent nav1.8 neurons contribute to opiate-induced analgesia: insight from conditional knockout mice. PLOS ONE 8:e74706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiech K 2016. Deconstructing the sensation of pain: the influence of cognitive processes on pain perception. Science 354:584–87 [DOI] [PubMed] [Google Scholar]
- Wieskopf JS, Pan YX, Marcovitz J, Tuttle AH, Majumdar S, et al. 2014. Broad-spectrum analgesic efficacy of IBNtxAis mediated by exon 11-associated splice variants of the mu-opioid receptor gene. Pain 155:2063–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Ingram SL, Henderson G, Chavkin C, von Zastrow M, et al. 2013. Regulation of μ-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol. Rev. 65:223–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson-Poe AR, Jeong HJ, Vaughan CW. 2017. Chronic morphine reduces the readily releasable pool of GABA, a presynaptic mechanism of opioid tolerance. J. Physiol. 595:6541–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters BL, Gregoriou GC, Kissiwaa SA, Wells OA, Medagoda DI, et al. 2017. Endogenous opioids regulate moment-to-moment neuronal communication and excitability. Nat. Commun. 8:14611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wacker D, Mileni M, Katritch V, Han GW, et al. 2012. Structure of the human к-opioid receptor in complex with JDTic. Nature 485:327–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie JY, De Felice M, Kopruszinski CM, Eyde N, LaVigne J, et al. 2017. Kappa opioid receptor antagonists: a possible new class of therapeutics for migraine prevention. Cephalalgia 37:780–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Bruchas MR, Ippolito DL, Gendron L, Chavkin C. 2007. Sciatic nerve ligation-induced proliferation of spinal cord astrocytes is mediated by K opioid activation of p38 mitogen-activated protein kinase. J. Neurosci. 27:2570–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Petraschka M, McLaughlin JP, Westenbroek RE, Caron MG, et al. 2004. Neuropathic pain activates the endogenous к opioid system in mouse spinal cord and induces opioid receptor tolerance. J. Neurosci. 24:4576–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Sakashita Y, Nozaki-Taguchi N. 2001. Antagonism of ORLI receptor produces an algesic effect in the rat formalin test. Neuroreport 12:1323–27 [DOI] [PubMed] [Google Scholar]
- Yao X-Q, Malik RU, Griggs NW, Skjaerven L, Traynor JR, et al. 2016. Dynamic coupling and allosteric networks in the α subunit of heterotrimeric G proteins. J. Biol. Chem. 291:4742–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhao S, Rodriguez E, Takatoh J, Han B-X, et al. 2015. Identifying local and descending inputs for primary sensory neurons. J. Clin. Investig. 25:3782–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z-Q, Gao Y-J, Sun Y-G, Zhao C-S, Gereau RW, Chen Z-F. 2007. Central serotonergic neurons are differentially required for opioid analgesia but not for morphine tolerance or morphine reward. PNAS 104:14519–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HY, Chen SR, Chen H, Pan HL. 2010. Opioid-induced long-term potentiation in the spinal cord is a presynaptic event. J. Neurosci. 30:4460–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Hsu MS, Pintar JE. 1998. Developmental expression of the μ, к, and δ opioid receptor mRNAs in mouse. J. Neurosci. 18:2538–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubieta J-K, Bueller JA, Jackson LR, Scott DJ, Xu Y, et al. 2005. Placebo effects mediated by endogenous opioid activity on μ-opioid receptors. J. Neurosci. 25:7754–62 [DOI] [PMC free article] [PubMed] [Google Scholar]