

Abstract
Cancer genomic analysis requires accurate identification of somatic variants in sequencing data. Manual review to refine somatic variant calls is required as a final step after automated processing. However, manual variant refinement is time-consuming, costly, poorly standardized, and non-reproducible. Here, we systematized and standardized somatic variant refinement using a machine learning approach. The final model incorporates 41,000 variants from 440 sequencing cases. This model accurately recapitulated manual refinement labels for three independent testing sets (13,579 variants) and accurately predicted somatic variants confirmed by orthogonal validation sequencing data (212,158 variants). The model improves on manual somatic refinement by reducing bias on calls otherwise subject to high inter-reviewer variability.
Somatic variant callers are commonly used to identify somatic variants from aligned sequence reads in cancer genomics studies and in clinical cancer assays1. These callers attempt to statistically model sample purity, sequencing errors, zygosity, ploidy, and other factors. Post-processing of called variants is an approach we term ‘somatic variant refinement’ and is an important, and distinct, next step from variant calling. Somatic variant refinement eliminates false positives from a candidate somatic variant list through heuristic filtering and manual review. Heuristic filtering includes setting project-specific thresholds for sequencing features such as read coverage depth, variant allele fraction (VAF), base quality metrics, and others. Manual review requires direct examination of aligned reads using a genomic viewer such as Integrative Genomic Viewer (IGV)2,3 to identify false positives that are consistently missed by automated somatic variant callers.
Somatic variant refinement remains indispensable for accurate analysis of cancer data, especially as cancer genomics is brought into the clinic, where variants are used to guide therapy4,5. Manual reviewers look for patterns that are neglected or unavailable to standard variant callers to alter confidence in a variant call. For example, confidence is reduced if: all supporting reads are oriented in the same read direction; a variant is supported exclusively by overlapping reads from short DNA fragments; a variant is located in or near homopolymer stretches, short repeats, or other low-complexity sequences; supporting reads indicate multiple mismatches relative to the reference genome; variant support is identified in the normal data track; variant support occurs exclusively at the ends of sequencing reads; in addition to other factors. If the number of problematic variant reads at a locus is high, a reviewer may label a variant identified by a somatic variant caller as a false positive.
In our experience, somatic variant refinement can dramatically improve the quality of final variant calls by eliminating large percentages of false positives from automated callers. However, despite extensive use of somatic variant refinement in clinical and translational genomics, filtering and refinement protocols are usually unstated or only briefly mentioned. Some illustrative examples of this reporting are, “mutations … were called with MuTect and filtered with oxidation and panel of normal samples filters to remove artefacts,”6 or, ‘all indels were manually reviewed in IGV.’7 These excerpts exemplify a prevalent history of under-reporting variant refinement details from our institute and others8–10.
Discrepancies in manual review procedures may result in significant inter- and intra-lab variability and error. To address the issue of reproducibility, our group generated a standard operating procedure for somatic variant refinement through the use of manual review11. However, even with complete conformity of manual review standard operating procedures, the process is time-consuming and expensive. Automated somatic variant callers can identify thousands of variants per cohort, which corresponds to hundreds of hours of manual review by a highly trained staff12. Machine learning could automate somatic variant refinement and essentially eliminate this bottleneck, reducing the required time and expense associated with variant identification.
Current software used for automated somatic variant calling includes VarScan13, SAMtools14, Pindel15, Sniper16, Strelka17, and MuTect18, among others. To improve on these algorithms, researchers have incorporated machine learning models to reduce the false positive rate intrinsic to automated somatic variant callers19,20. These initial attempts show promise for using machine learning approaches for somatic variant refinement; however, use of small training datasets (fewer than 3,000 variants) and limited number of cancer types prevents extrapolation of existing models onto a wide variety of sequencing data21.
Here we present a robust model that automates somatic variant refinement. We show that use of this model could substantially reduce a major bottleneck in cancer genomic analysis while improving reproducibility and inter-lab comparability in genomic studies and in clinical settings. This model is built on a training dataset of 41,000 variants from 21 studies, with 440 cases derived from nine cancer subtypes. All cases include paired tumor and normal samples that have been sequenced, evaluated for somatic variants using automated callers, and manually reviewed by individuals (an estimated 585 hours of manual effort). For each variant, we assembled 71 features to train the model including cancer type, sample type, tumor read depth, normal read depth, tumor VAF, normal VAF, base quality, mapping quality, and so on (Supplementary Table 1). To our knowledge, this is the largest dataset assembled to develop a machine learning approach for somatic variant detection. This dataset includes both solid and hematological malignancies, covers a broad range of average mutation burden, and includes data from multiple different sequencing pipelines (Table 1). This broad representation supports the generalizability of this machine learning approach for somatic variant refinement.
Table 1 |.
The cancer sequence data used to develop machine learning models included a variety of different tumor subtypes, sequencing approaches and manual review calls
| Variants | |||
|---|---|---|---|
| Training set | Hold out test set | Total | |
| Malignancy | |||
| Leukemia (n = 243) | 5,815 | 2,877 | 8,692 |
| Lymphoma (n = 23) | 8,692 | 628 | 1,891 |
| Breast (n = 135) | 8,986 | 4,320 | 13,306 |
| Small-cell lung (n = 18) | 9,177 | 4,601 | 13,778 |
| Glioblastoma (n = 17) | 844 | 412 | 1,256 |
| Melanoma (n = 1) | 185 | 100 | 285 |
| Colorectal (n = 1) | 842 | 419 | 1,261 |
| Gastrointestinal stromal (n = 1) | 70 | 31 | 101 |
| Malignant peripheral nerve sheath (n = 1) | 288 | 142 | 430 |
| Total | 27,470 | 13,530 | 41,000 |
| Sequencing methods | |||
| Capture sequencing | 9,479 | 4,755 | 14,234 |
| Exome sequencing | 9,367 | 4,677 | 14,044 |
| Genome sequencing | 8,264 | 4,098 | 12,722 |
| Variant calls | |||
| Somatic | 12,266 | 6,115 | 18,381 |
| Ambiguous | 7,189 | 3,454 | 10,643 |
| Fail | 5,909 | 2,945 | 8,854 |
| Germline | 2,106 | 1,016 | 3,122 |
The number of cases for each malignancy is given in parentheses.
Results
Data assembly and standardization.
The 41,000 called and reviewed variants used to train the model were derived from 440 individual tumors, which represent nine cancer types. Sequencing methods were evenly split between capture sequencing (14,234 variants), exome sequencing (14,044 variants), and genome sequencing (12,722 variants). Among all manually reviewed variant calls, 18,381 were confirmed as somatic, 10,643 were assessed as ambiguous, 8,854 as failed, and 3,122 as germline. The training data include both hematopoietic (10,583 variants) and solid tumors (30,417 variants), which often have distinct characteristics during manual variant refinement (Table 1).
Model development.
Three models were developed (logistic regression, random forest, and deep learning) using the 41,000 variant dataset. To guard against overfitting, we randomly selected one-third of the dataset as a hold out test set and used the remaining two-thirds as a training set. Using a tenfold cross-validation strategy, all three models (logistic regression, random forest, and deep learning) achieved better than random performance on variant classification. The logistic regression model demonstrated the worst performance (average area under the curve (AUC) = 0.89) and limited ability to classify ambiguous calls (AUC = 0.79). Both random forest and deep learning models performed well across all classes attaining an average AUC of 0.98 and 0.96, respectively (Fig. 1a). Performance of the hold out test set mirrored the tenfold cross validation (example of deep learning output in Supplementary Fig. 1a). For the hold out test set, decomposition of model performance based on disease, reviewer, and sequencing depth showed no change in model performance for the deep learning and random forest models (example of deep learning cross-tabulation analysis in Supplementary Table 2).
Fig. 1 |. Deep learning and random forest models achieved very high manual review classification performance during tenfold cross-validation.

a, Comparison of performance of three machine learning models via ROC AUC. Performance was parsed by the three classification classes (ambiguous, fail, and somatic) for cross-validation data (n = 27,470 variants). b, Reliability diagrams depict how closely model outputs scale to a probability (between 0 and 1) using cross-validation data (n = 27,470 variants). Bar graphs show 10 equally distributed bins of model output. The bar graphs plot the number of model calls that agree and disagree with the manual review call. The diagonal line indicates a perfectly scaled probabilistic prediction. The colored points display the ratio of predictions that agree with the call to the total number of predictions for a given bin. Binomial proportion confidence intervals were calculated for each bin. Pearson correlation coefficient comparing colored points to the diagonal line was calculated to assess the output of the respective model.
Reliability diagrams were used to determine whether model outputs could be interpreted as a well-scaled probability. Comparing the reliability diagrams for each model indicated that the random forest model and the deep learning model produced outputs that are most closely scaled to a probability. The random forest model and the deep learning model yielded Pearson correlation coefficients (r) of 0.99 and 1.00, respectively (Fig. 1b). The logistic regression model output was most divergent from a well-scaled probability with r = 0.29. When reliability diagrams were plotted independently for each class (somatic, ambiguous, and fail) for the deep learning and random forest models, all classes produced well-scaled outputs (example of deep learning output in Supplementary Fig. 2).
Feature importance.
The feature importance analysis determined which features were important for making model predictions. For the deep learning model, feature importance was ranked using the average change in the AUC after randomly shuffling individual features. For the random forest model, the built-in feature importance metric was used. To assess how manual reviewers rank feature importance, seven experienced manual reviewers at our institute ranked the top 15 (of 71) features that were most important in their manual review decision-making process. Feature ranks were normalized, and average importances across the seven reviewers were used to determine feature importance for manual reviewers. All three lists were rank normalized for comparison. Comparison shows that the models rely on many features that expert manual reviewers also use to make classification decisions (Fig. 2). The random forest feature importance was moderately correlated to the deep learning and manual reviewer feature importance (Pearson r = 0.47 and 0.50, respectively). The deep learning importance was only weakly correlated with manual reviewer survey results (Pearson r = 0.17). Of note, both the random forest model and the deep learning model ranked reviewer identity higher than reviewers themselves ranked this feature. Similarly, cancer type was ranked as an important feature for both models but was not ranked highly by manual reviewers.
Fig. 2 |. Machine learning models and manual reviewers use similar features when making manual review classification decisions.

Features ranked as important by random forest and deep learning models were also ranked highly by experienced manual reviewers (n = 71 features). Human manual reviewer feature importance was determined by asking seven individuals to rank feature importance. Single feature impact for the deep learning model was obtained by training a model on the training set (n = 27,470 variants), shuffling each feature individually, and calculating the mean ROC AUC for all three variant classes. The change in mean ROC AUC for all classes was sorted and plotted. Random forest feature importance was obtained via scikit-learn’s feature importance parameter. All feature importance metrics were ranked normalized. The random forest feature importance is moderately correlated to the deep learning and manual reviewer feature importance (Pearson r = 0.47 and 0.50, respectively). The deep learning importance was weakly correlated with manual reviewer survey results (Pearson r = 0.17). The top 30 (of 71) most important features are shown.
We hypothesized that the cancer type feature was mediated by differences between liquid and solid tumors. Specifically, the concentration of leukemia cells in normal tissue for patients with high circulating counts is higher than in solid tissue malignancies with circulating tumor cells22. This contamination ultimately increases the risk that a somatic variant will be mis-called. To test this hypothesis, we collapsed the cancer type features to a single solid/liquid boolean. Using the deep learning model as an example, the tenfold cross-validation performance for models trained with individual tumor types was similar to models trained with a simplified tumor type (solid/liquid boolean) (Supplementary Fig. 1b).
Inter-reviewer variability.
Reviewer identity was highly ranked by both the deep learning and random forest models, indicating reviewer-specific patterns in manual review. To quantify the variability between manual reviewers, we had three independent reviewers call a random subset of 176 sites from the training dataset. This resulted in three independent review calls for each of the 176 variants. Reviewers achieved fair agreement with a Fleiss’ Kappa statistic of 0.37 (ref. 23). When evaluating all calls in the inter-reviewer variability analysis, 77.3% showed good or acceptable agreement (that is, all three reviewers agreed on the call or reviewers only disagree between ambiguous and somatic or ambiguous and fail calls) (Fig. 3a). Model performance was correlated with reviewer agreement such that when all three reviewers called a variant as somatic, the model produced a high somatic probability (average output > 0.8). Conversely, when all reviewers agreed that a call was fail, the model produced a low somatic probability (average output < 0.2). As expected, in situations where there was inter-reviewer variability, the model produced a wider distribution of somatic probabilities (Fig. 3b,c). Together, these results indicate that there is as much as 22.7% disagreement among reviewers, especially on ambiguous calls.
Fig. 3 |. Model confidence closely parallels reviewer confidence.
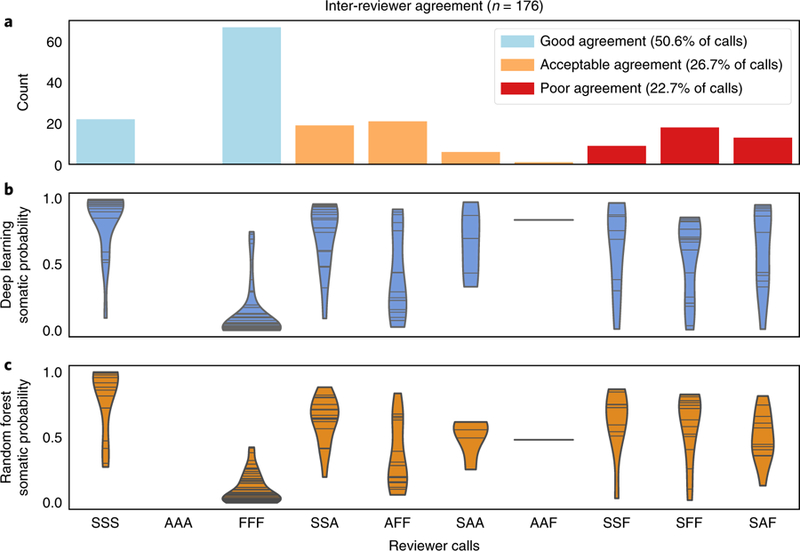
When reviewers exhibit strong agreement on a variant call, the model outputs confident probabilities (>0.8 or <0.2), whereas when reviewers exhibit inter-reviewer variability for a variant call, the model outputs inconclusive probabilities (0.2> and <0.8). a, Bar graphs show binned agreement of three reviewers for 176 variants. The x axis outlines all possible permutations of agreement among three reviewers. The y axis outlines the frequency of each permutation. ‘S’ denotes a somatic call, ‘A’ denotes an ambiguous call, and ‘F’ denotes a fail call. ‘SSS’ is the case where all three reviewers call the same variant somatic and the other permutations follow a similar pattern (for example, ‘SAF’ = somatic, ambiguous, fail). It is considered good agreement when all three reviewers agree, acceptable agreement when reviewers only disagree between ambiguous and somatic or ambiguous and fail calls, and poor agreement when one reviewer calls a variant somatic while another calls a variant fail. b, Violin plots of deep learning somatic probability whereby the horizontal lines indicate the occurrence of a probability and the width indicates the distribution of probabilities (n = 528 variants (176 variants for each of the three reviewers)). c, Violin plots of random forest somatic probability whereby the horizontal lines indicate the occurrence of a probability and the width indicates the distribution of probabilities (n = 528 variants (176 variants for each of the three reviewers)).
Model outputs that do not depend on reviewer identity are most desirable to reduce the impact of idiosyncratic criteria on ultimate calls. Therefore, new models were developed after removing the reviewer feature from the training data to assess performance in situations when the reviewer is unknown. Using the deep learning model as an example, tenfold cross-validation with all 71 features resulted in an average AUC of 0.960, whereas tenfold cross-validation without the reviewer feature resulted in an average AUC of 0.956. This experiment illustrates expected performance on de novo data that does not include a reviewer feature (Supplementary Fig. 1c).
Independent sequencing data with orthogonal validation.
To validate model performance on unfiltered ‘raw’ variant calls, deep learning and random forest models were used to predict manual review labels for 192,241 putative somatic variants in the acute myeloid leukemia case (AML31) described by Griffith et al.24 This case study had deep (312×) genome sequencing data as well as ultra-deep (1,000×) orthogonal custom capture validation for all 192,241 predicted variant sites. Variants validated by the custom capture data were considered true positives and those that failed validation were considered false positives (Supplementary Table 3). When comparing somatic model predictions to validation sequencing results, the deep learning model and the random forest model achieved receiver operating characteristic (ROC) AUCs of 0.95 and 0.96, respectively (Fig. 4a).
Fig. 4 |. Machine learning models accurately predict orthogonal validation sequencing results.
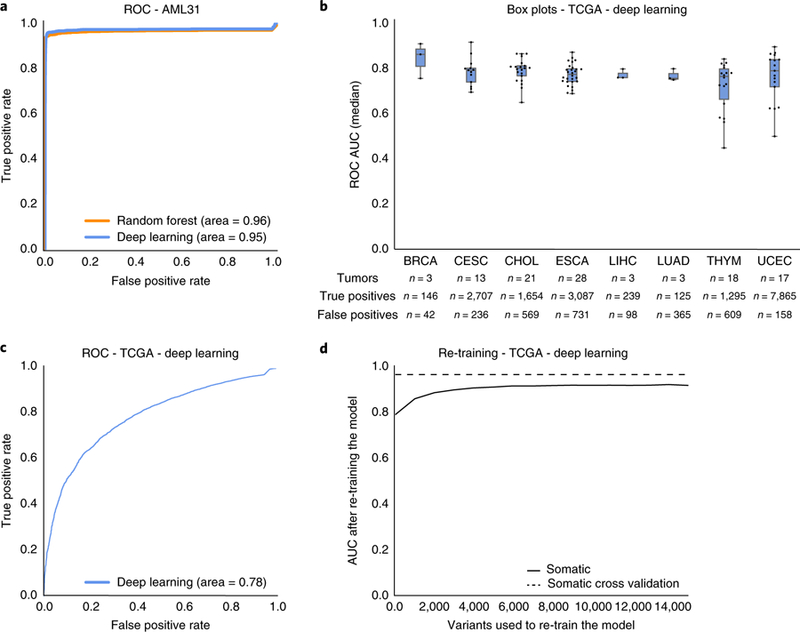
a, A single AML case with 312× genome sequencing had seven automated somatic variant callers identify 192,241 putative somatic variants. Orthogonal sequencing at ~1,000× was performed for all 192,241 variants to identify true positives and false positives. The random forest and deep learning models predicted labels for all variants using the 312× genome sequencing data as input. Model accuracy was determined by comparing model predictions to orthogonal sequencing labels. b, Box plots describe the median ROC AUC for each of eight TCGA cancer types (n = 106 tumor/normal pairs (see Supplementary Table 3 for abbreviations; n = 19,917 variants)). Each dot represents a single TCGA tumor/normal pair, the center represents the 50th percentile, the lower and upper limits of the box represent 25th and 75th percentiles, respectively, and whiskers represent data minimum and maximum. The table below the boxplots shows information on the total number of samples assayed and the distribution of true positive and false positive calls for each cancer type. c, ROC AUC for all TCGA data (n = 19,917 variants) using the deep learning classifier trained on the 41,000 variants described in Table 1. d, Change in ROC AUC after re-training the deep learning model with increments of the TCGA data. TCGA data was partitioned in random stratified increments of 5% (from 0–75%) and used to train a new model (increments = 1,327 variants). The x axis outlines the number of test variants included in re-training. The y axis plots the resulting model’s ROC AUC.
Additional sequencing data were obtained from The Cancer Genome Atlas (TCGA) dataset. Specifically, we obtained a cohort of 106 TCGA tumor-normal pairs that had original exome sequencing and subsequent targeted orthogonal validation25. This cohort comprised eight cancer types and 19,917 total variants, whereby 17,109 were true positives and 2,808 were false positives (Supplementary Table 3). When employing the deep learning model on this dataset, average ROC AUC for each cancer type ranged from 0.724–0.878, and average ROC AUC for all variants was 0.78 (Fig. 4b and c). To overcome apparent batch effect, orthogonal validation calls were randomly selected in increments of 5% (from 0%–75%) to include in re-training the model. The newly trained model was then used to predict calls for remaining variants in the TCGA dataset. When retraining the model using incremental amounts of the testing set, the total AUC improved. After incorporating 20% of the TCGA data, the model attained a ROC AUC of 0.90 and when incorporating 75% of the TCGA data, the model attained a ROC AUC of 0.93 (Fig. 4d).
Independent sequencing data with manual review validation.
To test model performance on external manual review data, three independent datasets were obtained whose characteristics differed from the training set. These datasets included 4 small-cell lung cancer (SCLC) cases with 2,686 variants, 14 follicular lymphoma (FL) cases with 1,723 variants, and 19 head and neck squamous cell carcinoma (HNSCC) cases with 9,170 variants (Supplementary Table 4). The SCLC cases were sequenced independently from the training set SCLC cases, utilized different methods for automated somatic variant calling, and were reviewed by new manual reviewers. The FL cases had a unique distribution of call classes (50.2% somatic, 49.8% fail, and 0% ambiguous) when compared to the training set (44.8% somatic, 29.2% fail, and 26% ambiguous). The HNSCC cases represented a new tumor type and were aligned to a different version of the human reference genome (GRCh38).
For the deep learning model, ROC AUC for independent test sets (n = 37 cases) ranged from 0.78–0.92 for somatic variants, 0.74–0.92 for failed variants, and 0.43–0.47 for ambiguous variants (Fig. 5). When re-training the model using incremental amounts of the testing set, as described above, model performance improved. For the deep learning model, inclusion of approximately 250 manual review calls restored performance to levels observed in cross-validation (Fig. 5). Initial model performance for the deep learning model outperformed the random forest model, especially for somatic and fail variants (Supplementary Fig. 3).
Fig. 5 |. The deep learning model performance on three independent test sets validated with manual review and subsequent correction for batch effect using re-training with 5% increments of the test data.

a, ROC curves outlining model performance on four SCLC cases with 2,686 variants and independent test set correction through model re-training to overcome batch effects associated with new manual reviewers, new sequencers, and a new alignment strategy. b, ROC curves outlining model performance on 14 FL cases with 1,723 variants and independent test set correction through model re-training to overcome batch effects associated with different frequencies of manual review classes. c, ROC curves outlining model performance on 19 HNSCC cases with 9,170 variants and independent test set correction through model re-training to overcome batch effects associated with alignment to a different reference genome (GRCh38).
Analysis of clinically relevant variants.
The deep learning model was used to assess whether machine learning algorithms for variant analysis could improve detection of clinically actionable variants mislabeled by manual refinement strategies. Of the 21,100 variants identified as somatic by either the deep learning model or by manual review, there were 16,722 variants that were called as somatic by both methods, 1,659 manual review (MR)-specific variants, and 2,719 classifier-specific variants (Fig. 6).
Fig. 6 |. Manual review misclassifications recovered by the deep learning model.
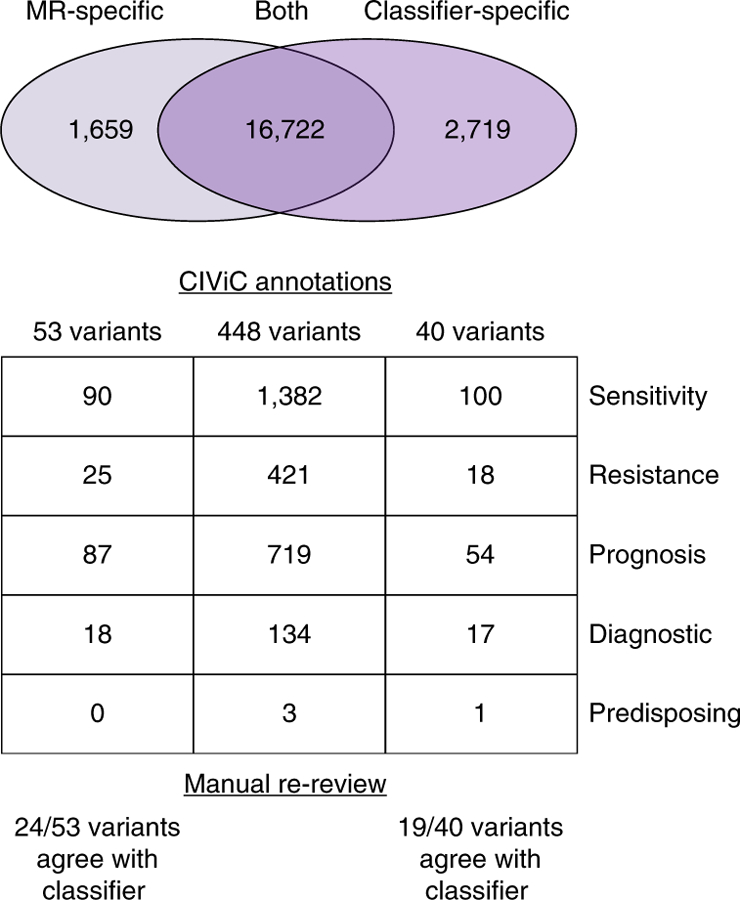
The Venn diagram illustrates variants identified as somatic by manual review (MR-specific), by both pipelines (Both), and by the deep learning classifier (Classifier-specific). For these three groups, the number of variants that have direct overlap with CIViC annotations and the total number of evidence items associated with all variants within each group are shown. These evidence items are parsed by those that convey variant sensitivity to a drug, variant resistance to a drug, variant that confers better or worse prognosis, variant that confers disease diagnosis, and variant that shows predisposing evidence for disease. The manual re-review panel shows the number of clinically relevant variants that agreed with the classifier call upon re-review by seven individuals.
Discordant variants (MR- or classifier-specific) were evaluated for clinical relevance using the Clinical Interpretations of Variants in Cancer database (CIViC)26. Each annotation within CIViC is based on evidence summaries that detail therapeutic, prognostic, predisposing, or diagnostic implications in cancer. After filtering extraneous evidence summaries (see Methods), there were 425 clinically relevant CIViC annotations. Using these CIViC annotations, 40 classifier-specific variants were identified that were clinically actionable. These 40 variants were associated with 100 evidence items related to therapeutic sensitivity, 18 evidence items related to therapeutic resistance, 54 evidence items that detailed prognostic information, 17 evidence items that indicated diagnostic information, and one evidence item that supported predisposition to cancer (Supplementary Table 5). If we assume that the classifier more accurately predicts the true variant label, this would represent an 8.9% increase in detection of clinically relevant variants. Using relevant CIViC annotations, 53 manual review-specific variants were identified as clinically actionable. Of these manual review-specific variants, 90 evidence items related to therapeutic sensitivity, 25 evidence items related to therapeutic resistance, 87 evidence items detailed prognostic information, and 18 items illustrated diagnostic information (Supplementary Table 5). If we again assume that the classifier call is more accurate relative to the original manual review call, this would represent an 11.8% reduction in mislabeled, clinically relevant calls.
Blinded retrospective review of these mislabeled variants in IGV confirmed confidence in model predictions. Four examples of manual review miscalls that were originally labeled as somatic but were failed by the classifier are shown in Supplementary Fig. 4. Two examples of manual review miscalls that were originally labeled as ambiguous or fail by manual reviewers but were identified as somatic by the classifier are shown in Supplementary Fig. 5. In Supplementary Fig. 5a, two clinically relevant PIK3CA variants were missed due to the manual reviewer assuming that two adjacent variants on the same strand were considered multiple mismatches. In Supplementary Fig. 5b, a TP53 variant was missed in an AML case due to the manual reviewer’s lack of awareness that hematologic cancers can have tumor cell contamination in normal tissue.
Analysis of discrepant calls.
The classifier agrees with manual review in 89.3% (35,622/41,000) of calls; however, there were 4,378 variants (10.7%) for which the original manual review call was discrepant with the classifier call. To understand features influencing discrepant calls, unbiased manual re-review was performed on 179 discordant variants. Seven individuals proficient in manual review re-reviewed IGV snapshots of the 179 variants (Supplementary Table 6). For each variant, a consensus call was determined (see Methods) (Supplementary Table 7). When comparing the original manual review and classifier calls to the consensus call, 51 variants (28.5%) showed call-agreement between the consensus call and classifier call and 53 variants (29.6%) showed call-agreement between consensus call and the original manual review. Additionally, 34 variants (19.0%) showed disagreement between the consensus call, the classifier call, and the original manual review call, and 41 variants (22.9%) had no consensus (Supplementary Fig. 6). Of the 93 discrepant clinical variants evaluated during rereview, 50 variants were classified as either ‘no agreement’ or ‘no consensus’. Therefore, we estimate that approximately 5.8% of all clinically relevant variant calls are fundamentally ambiguous, even on re-review.
Discussion
The random forest and deep learning models achieved high (average AUCs > 0.95) classification performance across all variant refinement classes (somatic, ambiguous, and fail), whereas the logistic regression model demonstrated reduced performance (average AUC = 0.89), particularly with the ambiguous class. High performance of model predictions confirms that an automated strategy can reduce the need for manual variant refinement. In addition, maintenance of performance after elimination of the manual reviewer feature further demonstrated that the trained model can be used on de novo data without reviewer information (Supplementary Fig. 1c).
The deep learning and random forest models also showed high accuracy (AUCs > 0.95) when classifying independent sequencing data with orthogonal validation. The AML31 case outlined by Griffith et al.24 had two unique features that made it optimal for assessing model performance. First, variants had manual review calls for both the original genome sequencing and the ultra-deep orthogonal sequencing. Second, the ultra-deep orthogonal sequencing was performed on all variants (false positives and true positives) called by automated somatic variant callers, allowing for quantification of both sensitivity and specificity.
With regards to the TCGA orthogonal validation datasets, the deep learning model showed initial reduction in average AUC (AUC = 0.78) relative to cross-validation performance. We hypothesized that reduction in accuracy was attributable to methods used for classifying TCGA false positives. Specifically, TCGA false positives were filtered by eliminating variants caused by 8-Oxoguanine (OxoG) DNA lesions using the DetOxoG tool27, eliminating variants with strand bias, and eliminating germline variants using a panel of normals. Since these features are not typically available to manual reviewers, they were not incorporated into the original model. Re-training with TCGA false positives allowed the model to learn new sequencing features that improve its ability to recognize false positives, ultimately restoring model accuracy (AUC = 0.93). Given these findings, we are hopeful that development of a model that incorporates these data will improve somatic variant refinement and eventually reduce the need for orthogonal validation sequencing.
For the independent sequencing data with manual review validation, we also observed a decrease in model performance. However, when re-training the model with as few as 250 calls, model performance was restored (Fig. 5). Therefore, when employing the classifier on new datasets, we recommend manually reviewing or performing validation sequencing for a small subset of variants called via statistical variant callers (for example, 5% of all data) to re-train the classifier and improve performance. Our group has provided a command line interface to allow individuals to train a custom deep learning classifier, prepare data, and classify variants (see URLs). The deep learning model was selected as the optimal method for somatic variants refinement due to its increased accuracy when employed on validation sets.
These results together show that a machine learning model can effectively automate somatic variant refinement. Standardization and systematization of this process decreases the human variability associated with manual refinement and increases the reproducibility of variant calling. In addition, automation of variant refinement eliminates a labor bottleneck, and its associated costs, allowing any number of somatic variant calls to be evaluated in a negligible amount of time. Finally, since the model offers probabilistic output, an economic framework can be used to set thresholds for confirmatory follow up testing, allowing investigators to optimize experimental design to improve accuracy within budgetary constraints28.
To illustrate the extent of this advance, we compared the manual review burden in a standard cancer genomics workflow with a workflow that utilizes the machine learning classifier. In a previously conducted breast cancer study12, 10,112 variants were identified via automated somatic variant callers. In this example, 1,066 variants were filtered using heuristic cutoffs, and 9,046 variants required manual review. Given that experienced reviewers can evaluate 70–100 variants per hour, manual review for this study would have taken ~90–130 hours. Using the machine learning approach, 5% of the data (~500 variants) would require manual review. This manual review data would be used to re-train the model and correct for associated batch effects. This manual review would require approximately 5 hours. In this example, the manual review burden would be reduced from ~100 hours to ~5 hours, detailing the considerable improvement in efficiency.
Through the re-review analysis, we showed that inter-reviewer variability affects variant detection, which can ultimately impact patient care. Many of the variants with high inter-reviewer variability and/or no consensus call had clinical significance. We believe that in these cases, an automated model can provide an unbiased and probabilistic output for variant classification, thereby eliminating reproducibility issues associated with manual refinement. In instances where the model makes an ambiguous call for a variant of clinical relevance, we recommend manually reviewing these variants to make a definitive call.
This model does have some limitations. Given the identified inter-reviewer variability associated with manual variant refinement calls, the training data likely contain a substantial amount of noise that might impact model performance. Moreover, there are sources of data that can be used to build better models. In an ideal scenario, highly accurate orthogonal validation sequencing would be performed to determine somatic variant status. Unfortunately, validation sequencing has a large monetary and tissue material expense, limiting our ability to use these types of data in the training set. Lastly, while the training data were produced using a varied array of capture strategies, libraries, Illumina sequencing instruments, and somatic variant callers, the model will likely require evaluation and some amount of retraining for non-Illumina sequencing instruments and divergent somatic variant analysis pipelines. It is also possible that the model has learned various other institutional batch-effects from our sequencing and analysis workflows. However, our results suggest that retraining with a small amount of supplemental calls from an independent dataset may be sufficient to overcome these effects. We anticipate improving this model by adding genomic and sequencing features such as proximal sequence complexity (for example, presence of repeat regions), functional prediction (for example, conservation based variant impact scores), and other indicators associated with false positives29,30.
In conclusion, persistent weaknesses in variant calling pipelines remain, especially in an era of constantly changing and variable sequencing data quality. Sophisticated context-specific pattern matching abilities of humans are still needed to refine and confirm somatic variant calls, which is expensive and laborious. We show that with a relatively small amount of project-specific review for model retraining that most manual review can be replaced with an automated classifier approach, providing more reproducible and refined calls for clinically relevant variants.
URLs.
Clinical Interpretation of Variants in Cancer database, http://www.civicdb.org/; GitHub, https://github.com/; Convert_zero_one_based, https://github.com/griffithlab/convert_zero_one_based; DeepSVR GitHub Repo, https://github.com/griffithlab/DeepSVR; Bam-Readcount, https://github.com/genome/bam-read-count; Keras Library, https://github.com/fchollet/keras; DeepSVR GitHub Wiki, https://github.com/griffithlab/DeepSVR/wiki; CIViC interface public API, http://griffithlab.org/civic-api-docs/.
Methods
Training data.
We assembled manual variant refinement data from 21 different cancer genomic studies conducted at the McDonnell Genome Institute (MGI), including 11 genomic discovery cohorts, 1 clinical trial, and 9 case studies8,24,31–40. Samples present in multiple studies were eliminated by removing all sample pairs with more than 70% co-occurrence of genomic mutations. In total, 440 sample pairs were evaluated, with 266 samples derived from hematologic malignancies and 174 samples derived from solid tumors (Table 1). Samples were only included if paired tumor/normal sequencing data and manual somatic variant refinement calls were available. Sequencing data from this cohort were analyzed using standard cancer genome pipelines at the McDonnell Genome Institute over a period of several years1. Briefly, sequencing data were produced using genome, exome, or custom capture sequencing. Reads were aligned to reference genome hg19/ GRCh37 using Burrows–Wheeler aligner (BWA)41 or BWA-MEM (maximal exact matches)42, duplicates were marked by Picard43, and variants were called with SAMtools14 or (predominantly) the union of SAMtools and VarScan13. Variants identified by automated callers were annotated and subjected to false positive filtering strategies such as removal of variants with low VAF (for example, < 5%) or low coverage (for example, < 20×). Much of the raw sequencing data from these 21 cancer genomic studies is publicly available (Supplementary Table 8), and all variant calls, manual review data, and associated features required for model development are provided in a publicly available GitHub repository (see URLs).
Manual variant refinement for all projects was performed by individuals at the MGI, who recently described a standard operating procedure for this process11. In this operating procedure, reviewers manually refine variants using four distinct classes: ‘somatic’—a variant that has sufficient sequence read data support in the tumor in the absence of obvious sequencing artifacts; ‘ambiguous’ —a variant with insufficient sequence read data support to definitively classify the variant; ‘germline’—a variant that has sufficient support in the normal sample beyond what might be considered attributable to tumor contamination of the normal; and ‘fail’—a variant with low variant sequence read data support and/or reads that indicate sequencing artifacts, yet has acceptable variant coverage. In accordance with the standard operating procedure, as reviewers call variants, they often provide additional notes or tags describing the reason for each call.
Germline and fail calls represent two distinct types of failure for somatic variant calling. However, since germline and fail calls rarely invoke different downstream analysis procedures, they were merged into one class called ‘failed’. Therefore, the machine learning model was developed for ‘somatic’, ‘ambiguous’, and ‘fail’ classes. All manual variant refinement results were standardized to a one-based coordinate system using the convert_zero_one_based Python tool. Relevant metrics were extracted from the bam files using bam-readcount. Bam file metrics were merged with cancer type and reviewer information. All continuous features were normalized to fall between 0 and 1 using Scikit-learn’s MinMaxScaler44. All categorical variables were one-hot boolean indexed to split any feature with n categories into an n column boolean array. Following processing, the training dataset included 71 features (Supplementary Table 1).
Model development and analysis.
Logistic regression, random forest, and deep learning were tested as alternative models for somatic variant refinement. A logistic regression model was implemented using the keras library. Scikit-learn was used to implement the random forest model44,45. The random forest was trained using the parameters n_estimators = 1,000 and trees max_features = 8. The deep learning model was implemented using the keras library as a feed-forward neural network with the input layer equaling the number of features, four hidden layers with 20-node hidden layers, and an output layer equaling the three outputs (somatic, ambiguous, fail). The input and hidden layers used a hyperbolic tangent (tanh) activation function, the output layer used a softmax activation function. Categorical cross-entropy was used as a loss function and the Adam optimizer was used over 700 epochs with a batch size of 2,000. L2 regularization was used with a weight of 0.001.
To compare model performance, one-versus-all receiver operator characteristic curves were generated, and area under the curve metrics (AUC) were quantified using scikit-learn44. We used multiple out-of-sample model validation strategies on the 41,000 variant dataset. We randomly selected two thirds of the data to serve as a training set and the remaining one-third served as the hold out test set. On the training set, we performed tenfold cross-validation for model selection and hyperparameter tuning. When models and hyper parameters were selected, a model was trained on the training set and evaluated against the hold out test set to understand model performance44,45. Model performance was decomposed by performing a cross-tabulation analysis on data features including: reviewer, disease type, and sequencing depth (Supplementary Table 2).
Reliability diagrams were used to determine if model outputs could be interpreted as the probability of a manual variant refinement call. Model output, which was a continuous value, was plotted for 10 equally distributed bins that were separated by whether the model’s output matched or did not match the manual variant refinement call. For each bin, we calculated the ratio between the number of sites where the model agreed with the call and the total number of sites in the bin. It is expected that if the model output estimates a well-scaled probability, then the calculated ratio will be correlated to an identity line (x = y). Pearson correlation coefficient was used to test for a well-scaled probability using the scipy.stats. pearsonr function46.
Feature importance for the deep learning model was calculated by using the cross-validation dataset. Each of the 71 features was independently shuffled and change in average AUC was determined by comparing baseline performance to shuffled performance. The random forest feature importance metric was obtained from scikit-learn’s built in feature_importances_ parameter on a trained random forest model.
Validation of model performance by independent sequencing data with orthogonal validation.
To assess model performance on orthogonal sequencing data, we evaluated variant calls from a single AML case, AML31, that had extensive orthogonal validation sequencing. Genome sequencing data (average coverage = 312×) were previously produced for AML31 and evaluated using seven different variant callers. Orthogonal custom capture validation sequencing (average coverage = 1,000×) was used to validate the 192,241 variants identified by any of the seven variant callers (MuTect, Seurat, Shimmer, Sniper, Strelka, VarScan, Bassovac)24. Variants identified as somatic by orthogonal sequencing (the ‘Platinum SNV List’) were considered true positives (n = 1,343). Variants that were identified by only one of the seven callers, but not validated by orthogonal custom capture sequencing, were considered false positives (n = 190,898). Features were obtained from genome sequencing bam files for every site that was called by at least one of seven variant callers in the original study and had been selected for targeted resequencing (n = 192,241). The random forest and deep learning models were used to predict calls for each of the sites in the AML31 dataset and ROC figures were used to illustrate model performance.
Validation data were also obtained from TCGA exome sequencing data that had orthogonal validation25. Using the minor allele frequency (MAF) file (mc3. v0.2.9.CONTROLLED_lt3_b.maf) described by Ellrot et al.25, we identified a cohort of 19,917 variants from 106 tumor/normal pairs for model validation (Supplementary Table 3). This cohort was identified by removing un-powered validation, non-exonic variants, and potential germline calls from the original MAF file. Additionally, eligible variants required original identification via exome sequencing and orthogonal validation via targeted capture (target_status ≠ ‘NaN’). Variants were labeled as true positives if they passed the Broad Institute’s initial quality check (that is, ‘FILTER’ =‘PASS’) and were statistically powered (that is, ‘target_status’ =‘_powered’). Variants were labeled as false positives using the following tools: DetOxoG, strand bias, The Broad Institute’s Panel of Normals, and ExAC47. Any TCGA sample that had at least 20 false positives and 20 false negatives validated on TCGA exome data via targeted capture was eligible for classifier validation. To test model performance on these data, we trained a deep learning model on the entire training dataset and made predictions for all variants in the independent test samples. We assessed the model performance using ROC curves as outlined above. To overcome batch effects associated with new data, we re-trained the model 15 times using incremental amounts of the test data (0%–75% with 5% increments) and employed the new model on the remaining variants.
Validation of model performance by independent sequencing data with manual review.
To assess model robustness when employed on external data, an independent test dataset was assembled from 37 additional paired tumor/normal cases (13,579 variants) that were not included in the training set (Supplementary Table 4). Model development, variant predictions, and accuracy metrics were employed as described in the orthogonal validation analysis.
Annotations of clinical relevance.
All variants identified as somatic by either manual somatic refinement or by the deep learning classifier (n = 21,100) were evaluated for clinical significance. MR-specific calls were defined as variants identified as somatic by the manual review pipeline but labeled as ambiguous or fail by the classifier. Classifier-specific calls were defined as variants identified as ambiguous or fail during manual review but identified as somatic by the classifier. Variants were annotated using the CIViC database26. To evaluate overlap with the CIViC database, coordinates were queried from the CIViC interface using the public API (see URLs). Given that not all variants within CIViC can be analyzed using whole genome or whole exome sequencing, we used the provided Sequence Ontology IDs to filter out variants that cannot be analyzed using DNA-sequencing, such as ‘increased expression’ or ‘methylation’. (Supplementary Table 9). Using coordinates queried from the CIViC interface, we determined overlap between discrepant calls and CIViC annotations.
Re-review of conflicting calls.
A subset of variants whereby the original manual review call disagreed with the classifier call were re-reviewed using IGV. Using a standard operating procedure for manual review setup and execution11, we created IGV snapshots for 40 clinically relevant MR-specific calls, 53 clinically relevant classifier-specific calls, 43 non-clinically relevant MR-specific calls, and 43 non-clinically relevant classifier-specific calls. These 179 variants were manually re-reviewed by seven individuals who were blinded from original manual review calls. To analyze the 179 discordant variants, a consensus call was established as the ‘true label’ by aggregating the seven calls provided by blinded individuals. To be considered a consensus, the most common choice had to exceed any other choice by at least two votes. Any other distribution of votes resulted in that variant being classified as ‘no consensus’ (Supplementary Table 7).
Statistical tests used.
All plots were produced using the MatPlotlib library48. ROC curves were generated and AUC metrics were calculated using scikit-learn44. To assess reviewer agreement, we used Fleiss’ Kappa statistic, which is a statistic that lies between −1 and 1 where a Kappa statistic at or below 0 indicates poor agreement and above 0 indicates good agreement. For reliability diagrams, the binomial proportion confidence intervals were calculated for each bin. Pearson correlation coefficient comparing colored points to the diagonal line was calculated to assess the output of the respective model. Pearson correlation coefficient was used to test for a well-scaled probability using the scipy.stats.pearsonr function46.
Supplementary Material
Acknowledgements
The authors thank A. Petti, G. Chang, T. Li, C. Miller, L. Trani, R. Lesurf, Z. Skidmore, K. Krysiak, A. Ramu, and F. Gomez for assisting in data assembly. We also acknowledge L. Trani for performing manual review and for valuable discussion on the project. We gratefully acknowledge L. Wartman, J. DiPersio, M. Jacoby, B. Van Tine, R. Fields, B. Tan, S. Chi, D. Gutmann, and T. Ley for sharing genomic data that made this project possible. The authors also thank the patients and their families for their selfless contribution to the advancement of science. Part of this work was performed as part of the Washington University School of Medicine Genomics Tumor Board, which was funded with private research support from the Division of Oncology and the McDonnell Genome Institute. E.K.B. was supported by the National Cancer Institute (T32GM007200 and U01CA209936). T.E.R. received support from the National Institutes of Health/ National Cancer Institute (NIH/NCI) (R01CA142942) and the Breast Cancer Research Foundation. Select sample data was funded by the Genomics of AML PPG (T. Ley, PI, P01 CA101937). A.H.W. was supported by the NCI (NIH NCI F32CA206247). B.J.A. was supported by the Siteman Cancer Center. S. Swamidass is funded by the National Library of Medicine (NIH NLM R01LM012222 and NIH NLM R01LM012482) and acknowledges support from the Institute for Informatics at Washington University School of Medicine. M.G. is funded by the National Human Genome Research Institute (NIH NHGRI R00HG007940). O.L.G. is funded by the National Cancer Institute (NIH NCI K22CA188163 and NIH NCI U01CA209936).
Footnotes
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All analysis, preprocessing code, readcount training data, manual review calls, and trained deep learning and random forest models are available on the DeepSVR GitHub repository (https://github.com/griffithlab/DeepSVR). The raw sequencing data are publicly available for most projects included in this study (Supplementary Table 8). Users can access the classifier command line interface via our open-sourced GitHub repository and can install the package through Bioconda49. After installation, the tool can be used to (1) train and save a deep learning classifier, (2) prepare data for training a classifier or classification, and (3) classify data using either the provided deep learning model or a custom model. A walkthrough of this process is available on the DeepSVR GitHub Wiki.
Competing interests
R.G. consults for Eli Lilly and Genentech. R.G. is on the board/honorarium for EMD Serono, Bristol-Myers Squibb, Genentech, Pfizer, Nektar, Merck, Celgene, Adaptimmune, GlaxoSmithKline, Phillips Gilmore. All remaining authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41588–018-0257-y.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of data availability and associated accession codes are available at https://doi.org/10.1038/s41588–018-0257-y.
References
- 1.Griffith M et al. Genome modeling system: a knowledge management platform for genomics. PLoS Comput. Biol 11, e1004274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robinson JT et al. Integrative genomics viewer. Nat. Biotechnol 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson JT, Thorvaldsdóttir H, Wenger AM, Zehir A & Mesirov JP Variant review with the integrative genomics viewer. Cancer Res 77, e31–e34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li MM et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn 19, 4–23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roy S et al. Standards and guidelines for validating next-generation sequencing bioinformatics pipelines: a joint recommendation of the Association for Molecular Pathology and the College of American Pathologists. J. Mol. Diagn 20, 4–27 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Rheinbay E et al. Recurrent and functional regulatory mutations in breast cancer. Nature 547, 55–60 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ott PA et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma CX et al. A phase I trial of BKM120 (Buparlisib) in combination with fulvestrant in postmenopausal women with estrogen receptor-positive metastatic breast cancer. Clin. Cancer Res 22, 1583–1591 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.The Cancer Genome Atlas Research Network. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N. Engl. J. Med 372, 2481–2498 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rasche L et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun 8, 268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barnell EK et al. Standard operating procedure for somatic variant refinement of sequencing data with paired tumor and normal samples. Genet. Med 10.1038/s41436-018-0278-z(2018) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Griffith OL et al. Truncating prolactin receptor mutations promote tumor growth in murine estrogen receptor-alpha mammary carcinomas. Cell Rep 17, 249–260 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koboldt DC et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25, 2283–2285 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye K, Schulz MH, Long Q, Apweiler R & Ning Z Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simola DF & Kim J Sniper: improved SNP discovery by multiply mapping deep sequenced reads. Genome. Biol 12, R55 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saunders CT et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Cibulskis K et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol 31, 213–219 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding J et al. Feature-based classifiers for somatic mutation detection in tumour-normal paired sequencing data. Bioinformatics 28, 167–175 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spinella J-F et al. SNooPer: a machine learning-based method for somatic variant identification from low-pass next-generation sequencing. BMC Genomics 17, 912 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strom SP Current practices and guidelines for clinical next-generation sequencing oncology testing. Cancer Biol. Med 13, 3–11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bettegowda C et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med 6, 224ra24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McHugh ML Interrater reliability: the kappa statistic. Biochem. Med 22, 276–282 (2012). [PMC free article] [PubMed] [Google Scholar]
- 24.Griffith M et al. Optimizing cancer genome sequencing and analysis. Cell Syst 1, 210–223 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ellrott K et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst 6, 271–281.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Griffith M et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat. Genet 49, 170–174 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Costello M et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res 41, e67 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swamidass SJ, Bittker JA, Bodycombe NE, Ryder SP & Clemons PA An economic framework to prioritize confirmatory tests after a high-throughput screen. J. Biomol. Screen 15, 680–686 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Settles B & Craven M An analysis of active learning strategies for sequence labeling tasks. In Proceedings of the Conference on Empirical Methods in Natural Language Processing - EMNLP ‘08, 10.3115/1613715.1613855 (Association for Computational Linguistics, Stroudsburg, PA, USA; 2008). [DOI] [Google Scholar]
- 30.Settles B in Synthesis Lectures on Artificial Intelligence and Machine Learning Vol. 6 1–114 (Morgan & Claypool, San Rafael, 2012). [Google Scholar]
- 31.Griffith M et al. Comprehensive genomic analysis reveals FLT3 activation and a therapeutic strategy for a patient with relapsed adult B-lymphoblastic leukemia. Exp. Hematol 44, 603–613 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krysiak K et al. Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma. Blood 129, 473–483 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klco JM et al. Association between mutation clearance after induction therapy and outcomes in acute myeloid leukemia. JAMA 314, 811–822 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uy GL et al. Dynamic changes in the clonal structure of MDS and AML in response to epigenetic therapy. Leukemia 31, 872–881 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lesurf R et al. Genomic characterization of HER2-positive breast cancer and response to neoadjuvant trastuzumab and chemotherapy-results from the ACOSOG Z1041 (Alliance) trial. Ann. Oncol 28, 1070–1077 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Welch JS et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N. Engl. J. Med 375, 2023–2036 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rohan TE et al. Somatic mutations in benign breast disease tissue and risk of subsequent invasive breast cancer. Br. J. Cancer 118, 1662–1664 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mahlokozera T et al. Biological and therapeutic implications of multisector sequencing in newly diagnosed glioblastoma. Neuro. Oncol 20, 472–483 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wagner AH et al. Recurrent WNT pathway alterations are frequent in relapsed small cell lung cancer. Nat. Commun 9, 3787 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duncavage EJ et al. Mutation clearance after transplantation for myelodysplastic syndrome. N. Engl. J. Med 379, 1028–1041 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H & Durbin R Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM Preprint at https://arxiv.org/abs/1303.3997(2013) . [Google Scholar]
- 43.Picard Tools (Broad Institute, 2018); http://broadinstitute.github.io/picard/ [Google Scholar]
- 44.Varoquaux G et al. Scikit-learn: machine learning without learning the machinery. GetMobile 19, 29–33 (2015). [Google Scholar]
- 45.Nelli F Machine Learning with scikit-learn. In Python Data Analytics 2nd edn, Ch. 7 237–264 (Apress, New York, 2015). [Google Scholar]
- 46.Oliphant TE Python for scientific computing. Comput. Sci. Eng 9, 10–20 (2007). [Google Scholar]
- 47.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hunter JD Matplotlib: a 2D graphics environment. Comput. Sci. Eng 9, 90–95 (2007). [Google Scholar]
- 49.Grüning B et al. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat. Methods 15, 475–476 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.