

Abstract
We present the first large-scale methylome-wide association studies (MWAS) for major depressive disorder (MDD) to identify sites of potential importance for MDD etiology. Using a sequencing-based approach that provides near-complete coverage of all 28 million common CpGs in the human genome, we assay methylation in MDD cases and controls from both blood (N=1,132) and postmortem brain tissues (N=61 samples from Brodmann Area 10, BA10). The MWAS for blood identified several loci with P 1.91×10−8-4.39×10−8 and a resampling approach showed that the cumulative association was significant (P=4.03×10−10) with the signal coming from the top 25,000 MWAS markers. Furthermore, a permutation based analysis showed significant overlap (P=5.4×10−3) between the MWAS findings in blood and brain (BA10). This overlap was significantly enriched for a number of features including being in eQTLs in blood and frontal cortex, CpG islands and shores, and exons. The overlapping sites were also enriched for active chromatin states in brain including genic enhancers and active transcription start sites. Furthermore, three loci located in GABBR2, RUFY3 and in an intergenic region on chromosome 2 replicated with the same direction of effect in the second brain tissue (BA25, N=60) from the same individuals and in two independent brain collections (BA10, N=81 and 64). GABBR2 inhibits neuronal activity through G protein-coupled second-messenger systems and RUFY3 is implicated in the establishment of neuronal polarity and axon elongation. In conclusion, we identified and replicated methylated loci associated with MDD that are involved in biological functions of likely importance to MDD etiology.
Background
Major depressive disorder (MDD) is characterized by marked and persistent dysphoria that is often accompanied by considerable morbidity1 and mortality.2 Because MDD has a lifetime prevalence of almost 15%,3 tends to start early in life,4 and is often chronic,5 it is the leading contributor to disability worldwide6 with associated costs expected to further double by 2030.7 In comparison to other (psychiatric) disorders, discerning the biological basis of MDD has been difficult. Only recently, large GWAS meta-analyses have identified and replicated genetic variants.8–10 However, these variants explained only a very small proportion of the disease risk.
DNA methylation is an epigenetic modification that provides stability and diversity to the cellular phenotype. Methylome-wide association studies (MWAS) are a promising complement to GWAS. For example, DNA methylation can be dynamic in post-mitotic tissues11 and altered by environmental factors. Therefore methylation can potentially account for key clinical features of MDD such as its episodic nature or mediate the effects of the environmental risk factors (e.g., stress).12, 13 Methylation studies of MDD are ideally performed in brain where the majority of pathogenic processes likely occur. However, brain tissue cannot be obtained from living patients. It can be collected postmortem, but it remains challenging to obtain the sample sizes needed for adequate statistical power. Studies have found correlations of 0.6–0.7 between methylation profiles in human blood and brain14 as well as correlated (psychotropic drug induced) methylation changes in rodent blood and brain.15 Multiple factors may contribute to this overlap. Peripheral tissues such as blood may reveal methylation marks predating or resulting from the epigenetic reprogramming events affecting the germ line and embryogenesis,16 sequence variants can affect methylation levels and will be identical across tissues,17, 18 systemic MDD disease processes such as stress and inflammation will affect both blood and brain, and blood interacts with brain during perfusion. This observed overlap suggests that one approach to improve statistical power of studies of postmortem brain, is to combine results with studies in (antemortem) blood samples that are easier to collect in large sample sizes.
Here we present the largest and most comprehensive CpG methylome study of MDD to date. It uses the convergence of evidence across data sets to identify sites of potential importance for MDD etiology using both blood and brain tissues. To avoid missing sites of potential etiological importance we used a sequencing-based approach that provides near-complete coverage of all 28 million common CpGs in the human genome.19, 20 The design involves three phases. First, the discovery phase includes a methylome-wide association study (MWAS) of blood samples from 1,132 individuals. Next, in the overlap phase we identify sites that are in the top of both the MWAS in blood and MWASs in postmortem brain samples from two brain regions from 61 individuals. Finally, in the replication phase the overlapping sites are further followed-up in two independent postmortem brain collections from 81 and 64 individuals, respectively.
Materials and Methods
Our three-phase, multi-tissue design improves statistical power and helps to avoid potential tissue/study specific confounders. An overview of the study design is given in Figure 1.
Figure 1:
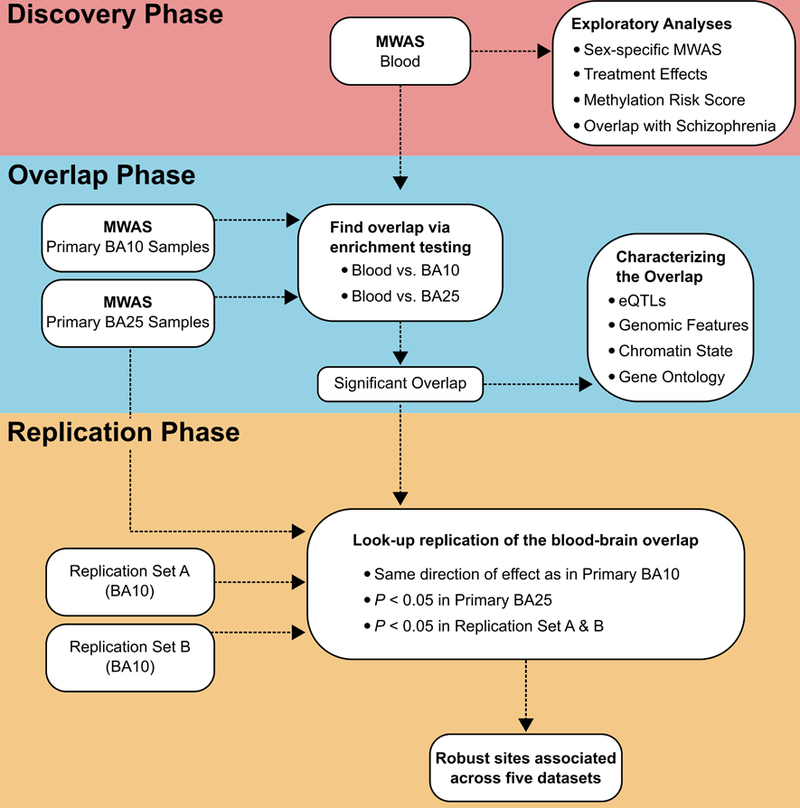
In this study we present a three-phase, multi-tissue design: Top) a Discovery Phase involving a MWAS in a large set of blood samples as well as a number of exploratory analysis; Middle) an Overlap Phase where we test for enrichment of MWAS overlap between blood and brain; Bottom) a Replication Phase where all sites in the significant overlap are followed up with “look-up replication” in two independent brain collections from BA10 and in the second brain tissue (BA25) from the original brain MWAS sample.
Study samples from blood and brain
The three phases of our study include both DNA from blood and from three postmortem brain collections. Table 1 summarizes the characteristics of the participants and full descriptions are given in the Supplement. The blood samples were obtained from 1,200 individuals from the Netherlands Study of Depression and Anxiety (NESDA). MDD was diagnosed using the DSM-IV based Composite International Diagnostic Interview (CIDI version 2.1).21 In addition to a current MDD diagnosis, cases had a symptom score > 14 on the IDS-SR30.22 Controls had no lifetime psychiatric disorders and an IDS-SR30 score < 14. Thus, by using this criteria, individuals that report symptoms without having received a psychiatric diagnosis were prevented from inclusion in the study, thereby maximizing the contrast between the two groups in terms of depression status. This study was approved by the ethical committees of all participating locations, and participants provided written informed consent.
Table 1.
Characteristics of study participants
| Controls | MDD | Test | |||
|---|---|---|---|---|---|
| Mean | SD | Mean | SD | P value | |
| a) Discovery phase | |||||
| Blood MWAS Sample | n = 320 | n = 812 | |||
| Sex | 0.591 | 0.492 | 0.667 | 0.471 | 0.018 |
| Age | 41.61 | 14.64 | 41.53 | 12.25 | 0.912 |
| EDU | 13.13 | 3.158 | 11.46 | 3.198 | <0.001 |
| IDS | 5.022 | 3.533 | 33.79 | 10.94 | <0.001 |
| TCA | 0.000 | 0.000 | 0.048 | 0.214 | <0.001 |
| SSRI | 0.003 | 0.056 | 0.299 | 0.458 | <0.001 |
| Other AD | 0.000 | 0.000 | 0.112 | 0.315 | <0.001 |
| CD3 | 0.296 | 0.086 | 0.281 | 0.087 | 0.008 |
| CD14 | 0.113 | 0.028 | 0.109 | 0.028 | 0.049 |
| CD15 | 0.509 | 0.115 | 0.536 | 0.114 | <0.001 |
| CD19 | 0.082 | 0.034 | 0.074 | 0.033 | <0.001 |
| b) Overlap phase | |||||
| Brain MWAS Sample* | n = 31 | n = 30 | |||
| Sex | 0.581 | 0.502 | 0.567 | 0.504 | 1 |
| Age | 51.77 | 17.66 | 51.43 | 18.59 | 0.942 |
| PMI | 46.3 | 14.97 | 41.49 | 15.65 | 0.224 |
| pH | 6.304 | 0.217 | 6.482 | 0.273 | 0.009 |
| c) Replication phase | |||||
| BA10 Brain Collection A | n = 37 | n = 44 | |||
| Sex | 0.297 | 0.463 | 0.409 | 0.497 | 0.416 |
| Age | 61 | 18.64 | 55 | 20.57 | 0.176 |
| PMI | 21.77 | 16.78 | 32.08 | 25.07 | 0.047 |
| pH | 6.605 | 0.223 | 6.595 | 0.183 | 0.822 |
| BA10 Brain Collection B | n = 25 | n = 39 | |||
| Sex | 0.36 | 0.49 | 0.308 | 0.468 | 0.871 |
| Age | 61.48 | 15.02 | 47.9 | 16.94 | 0.002 |
| PMI | 39.22 | 25.57 | 48.58 | 26.39 | 0.166 |
| pH | 6.534 | 0.238 | 6.648 | 0.315 | 0.212 |
| Validation in BA25 included the participants described in section b (see above). | |||||
Note: n is number of samples left after quality control. Sex indicates the proportion of males, age is measures in years. EDU years of education. The IDS (Inventory of Depressive Symptomatology) is a self-report measure of symptom severity. The usage of antidepressants is indicated for TCA (tricyclic antidepressant), SSRI (serotonin reuptake inhibitor) and other AD (antidepressant). The estimated blood cell type proportions are indicated by CD3 (T-lymphocytes), CD14 (monocytes), CD15 (granulocytes) and CD19 (B-lymphocytes). PMI (postmortem interval) is measured in hours. * The numbers given are for the participants contributing BA10 tissue. For BA25, while the number of cases remained the same, the number of control participants was 30.
The postmortem brain collections were obtained from several repositories. Diagnosing disease in subjects providing postmortem brain samples can be challenging.23 In most cases psychiatrists determine the diagnosis by using information obtained from a family member who is well acquainted with the deceased. This technique has been validated for axis I and II diagnoses24, 25 and has shown to have high inter-rater agreement.26
Methylome-wide association testing of MBD-seq data
We assayed the methylomes using an optimized protocol for methyl-CG binding domain sequencing (MBD-seq) that provides near-complete coverage of all 28 million common CpGs in the genome.19 Samples were performed in a randomized order and all lab technical procedures were performed blind to any phenotype information. The sequence reads were aligned to the human reference genome (hg19/GRCh37). Data was processed and analyzed using the RaMWAS Bioconductor package,27 which is specifically designed for large-scale methylation studies. A summary of the quality control procedure for the different datasets, the CpG score calculation and further details about the procedures are given in the Supplement.
To test each CpG for association with MDD, we performed multiple regression analyses while controlling for covariates. First, we regressed out 19 potential technical artifacts (i.e., the quantity of methylation-enriched DNA captured, sample batch, and peak location20), age and sex. Next, to avoid confounding due to cell type heterogeneity, we regressed out blood cell type proportions as estimated from the methylation data28 using “reference methylomes” specifically generated for this purpose.29 Finally, principle component analysis was used to capture unmeasured remaining sources of variation. Specifically, using a scree-test we selected the first principle component, explaining 2.61% of the remaining methylation variation. To perform an (exploratory) MWAS to detect methylation sites that have different effects on MDD in males and females we included the product MDD × sex to estimate the interaction effects in the model that also allowed for main effects of MDD and sex.
Furthermore, to ensure that the detected MWAS findings were not a direct confounding effect of antidepressant treatment we performed two additional (exploratory) MWAS. First, we performed an MWAS of all individuals (cases and controls) where we regressed out a variable indicating if the individual were currently using any antidepressant. It should be noted that as antidepressant use is highly confounded by MDD status (i.e., it is rarely present among controls) simply controlling for use of antidepressants by regressing out this effect may also eliminate part of the effects that may be related to MDD etiology (and not caused by the treatment). In the second MWAS we limited the analysis to cases only and studied whether antidepressant treatment was associated with methylation changes.
Determining the significance of the cumulative MWAS signal by resampling
To study the significance of the cumulative MWAS signals we use a resampling approach. For this purpose RaMWAS27 fits elastic-nets30–32 and employs k-fold cross-validation33 to avoid overfitting and obtain an unbiased estimate of the cumulative effect. Elastic-nets were fitted by using ridge regression (alpha parameter=0). For cross-validation the sample was randomly partitioned into k=10 equal sized subsamples. Of the k subsamples, k−1 were used as a “training set” to fit the elastic-net and obtain weights for each CpG. The weights were then used to pestimate MDD status from methylation in the remaining “test set”. By alternating the samples used in the training and test sets,estimates are obtained for all samples. RaMWAS repeats the entire cycle of MWAS based CpG selection followed by estimation of weights using elastic-nets for each of the k folds. Because both the selection of CpGs and estimation of their weights are not affected by the participants in the test set, we obtained unbiased estimates of the MDD status for each subject. By testing whether these estimates were significantly correlated with actual MDD status, we performed an “in sample replication” of the cumulative MWAS signal.
Similarly, as a proof-of-concept, we created a multi-site biomarker using estimates from one dataset to predict in another. That is, we calculated MDD estimates in blood using CpGs selected based on their performance in brain. As the selection of sites is performed in a different dataset than the prediction itself, this demonstrates the concept that potentially etiologically relevant CpGs detected in one tissue have predictive value, and may potentially function as a multi-site biomarker, in another tissue.
Permutation based enrichment test
To perform enrichment tests of the overlap between datasets we used the shiftR R-package with 1 million permutations. ShiftR first maps the two datasets to each other based on genomic location. Next, the MWAS P values are used to cross-classify each mapped marker in the two datasets as being in the top or bottom. We used two thresholds (1% and 5%) to define “top findings”. Based on the resulting 2 by 2 tables, shiftR tests the null hypothesis that the enrichment odds ratio (OR) equals one. To perform these tests, shiftR uses circular permutations34 to generate the empirical test statistic distribution under the null hypothesis while preserving the correlational structure of the data. To account for “multiple testing” (i.e., the multiple thresholds used), the same thresholds are used in the permutations by selecting the most significant threshold combination. ShiftR is also used to test the overlap for enrichment of a panel of biological features (Supplement).
Look-up replication in independent postmortem brain collections
All sites in the significant blood-brain overlap (detected in the overlap phase) were followed up using look-up replication in association results of MBD-seq data from two additional independent brain collections (Table 1c). Robust findings were identified with a stringent set of criteria. First, we required the effects to be in the same direction as in the original BA10 MWAS that detected the overlap. Second, we required both independent replication samples to show P<0.05. Third, we required the results from BA25, from the same subjects as the original BA10 MWAS, to show P<0.05. Only sites that fulfilled all three criteria were considered robust findings.
Results
Methylome-wide association study in blood
We used MBD-seq to generate methylation profiles from blood for the NESDA discovery sample.35–37 After assessing all methylation profiles and excluding those that failed quality control criteria, methylomes from 1,132 individuals (Table 1a) remained. Akin to filtering SNPs with low minor allele frequency, we excluded rarely methylated sites. This left 21,869,561 CpGs for MWAS, which corresponds to 78.3% of all common CpGs in the human genome. Each methylation profile was sequenced with an average of 59.4 million (SD=11.2 million) reads per sample and obtained an average CpG score38 of 6.029 (SD=1.398) with an average nonCpG-to-CpG score ratio27 of 0.010 (SD=0.005). Thus, the average signal at the tested CpGs is sufficiently strong and the background noise level is exceptionally low.
The QQ-plot (Figure 2a, lambda=1.124) suggests that many associated CpGs with modest effects. MWAS of permuted case-control status for this dataset yields an average lambda that is not significantly different from 1 (Supplement). Thus the observed lambda in the discovery MWAS likely reflect true associations rather than statistical artifacts. The Manhattan plot (Figure 2b) shows that associated loci are spread across the genome. However, in most cases several CpGs support the signal within loci. For example, Table S1 shows the five most significant CpGs associated with MDD (P ranging from 1.91×10−8 to 4.39×10−8, q<0.20), which are located in three distinct loci. All findings with P<1×10−5 are given in Table S2. Although none of the three highly significant loci are directly overlapping with known regulator elements or genes, one of the loci is located within 275 bp of HTR3C (serotonin receptor ionotropic 3, subunit c). We used the resampling approach to study the significance of the cumulative MWAS signal. Although this approach do not evaluate site specific significance it evaluates the collective effect of many markers. Results showed that the cumulative association to MDD was significant (P=4.03×10−10) with the signal coming from the top 25,000 blood markers.
Figure 2:
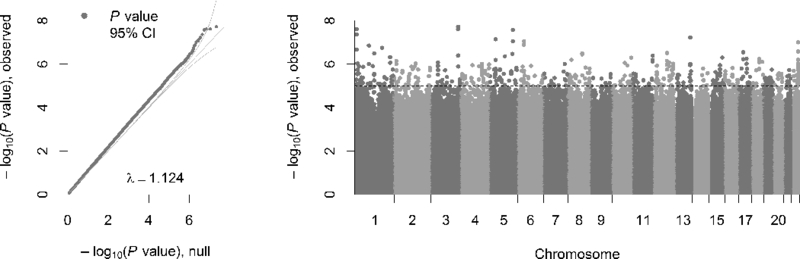
Left. Quantile-Quantile plot of the MWAS in blood. The observed P values, on a –log10 scale, are plotted against their expected values (grey main diagonal line) under the null hypothesis assuming none of the CpGs have an effect. Yellow lines indicate the 95% confidence intervals (CI). The deviation of P values from the main diagonal indicates that there are potentially many markers associated with MDD. The lambda (λ) is close to one, indicating that markers that are not associated behave as expected under the null hypothesis. Right. Manhattan plot of the MWAS in blood. The plot shows the MWAS P values on a –log10 scale (y-axis) by their chromosomal location (x-axis). The dashed line marks the threshold for suggestive significance (P = 1×10−5).
Prevalence rates of MDD are substantially higher in females compared to males39–41. Although such an analysis has lower statistical power, we performed an exploratory MWAS to detect methylation sites showing different effects on MDD in males and females (Figure S1 and Table S3a). The top finding (P=5.01×10−8) was located in FBLN2 (fibulin 2). The standardized effect (“beta” value) is −0.330 for the MDD × sex interaction term, meaning that the case-control difference is 0.330 standard devations smaller in females compared to males. Although this finding should be interpreted with caution, FBLN2 is a very interesting candiate because of its connection to sex hormone42 levels in blood43 and the robust link of these hormones to sex differences in depression.44–46
Analysis of MDD case-control status, while controlling for the use of antidepressant treatment, revealed very similar results as those obtained in the main analysis (Figure S2a and Table S3b). For example, all five of the most significant CpGs associated with MDD were still among the top MWAS results with P<1×10−5 and permutation based enrichment testing showed that the similarity of the top 1% of findings were highly consistent (P<1.0×10−5, OR=239.05). On the contrary, there was no evidence of enriched overlap between the top results from the main MWAS with the MWAS of antidepressant treatment effects using cases only (P=0.75 OR=1.00) Figure S2b and Table S3c). Thus, these analyses suggested that the MDD associated findings in the main analyses were not a confounding effect of antidepressant treatment.
Overlap between blood and brain
In the overlap phase, we used MBD-seq to study the methylome of postmortem brain tissues from two brain regions (BA10 and BA25) per individual, obtained from the Victorian Brain Bank Network.47 After quality control and filtering out sites that were rarely methylated, 17,536,447 CpGs remained from 61/60 individuals for BA10/BA25. The Manhattan plots (Figure S3) show that loci with P<1×10−5 are distributed across the genome for BA10/BA25 (Tables S4-S5). To study the potential overlap between top MWAS findings in blood and brain, we performed permutation-based enrichment tests of the overlap (Table S6). For BA10, after correcting for multiple testing, the top 5% of findings from the blood MWAS were significantly enriched for CpGs in the top 1% of the brain MWAS (P=5.4×10−3, OR=1.04). Although we observed a significant enrichment in overlap between the top 5% of MWAS findings between BA10 and BA25 (P<1.0×10−5, OR=1.05), there was no significant enrichment of overlap between the blood and BA25 MWAS for the tested thresholds. This suggests that some of the MDD associated sites detected in blood and BA10 were not among the top sites in BA25.
Focusing on blood and BA10, the significant overlap included 9,085 CpGs (Table S7). These overlapping sites showed enrichment for eQTLs (GTEx v7) in both blood (P=0.03669, OR=1.11) and frontal cortex (P=0.04625, OR=1.16), but not in anterior cingulate cortex (P=0.69171, OR=0.95). Overlapping sites were also significantly enriched for being in CpG islands (P=0.0195, OR=1.26) CpG island shores (P=0.01217, OR=1.17) and exons (P=0.0049, OR=1.14). Furthermore, the overlapping sites were enriched for active chromatin states in brain including genic enhancers (P=0.01052, OR=1.36) and active transcription start sites (P= 0.0264, OR=1.22). Results from all tracks tested for enrichment are given in Table S8. Of the 2,223 genes implicated in the overlap, 2,061 were present in Gene Ontology (GO). Using the over-representation analysis provided in the Consensus Path Database,48 we found significant (P <0.0001) enrichment of 32 level-5 GO terms (Table S9), which were further clustered based on their similarity in gene content (Figure 3). Several of these clusters included GO terms related to neuronal development and second messenger signaling, with the most significant GO term being “neurogenesis” (GO:0022008, P=2.24×10−19).
Figure 3:
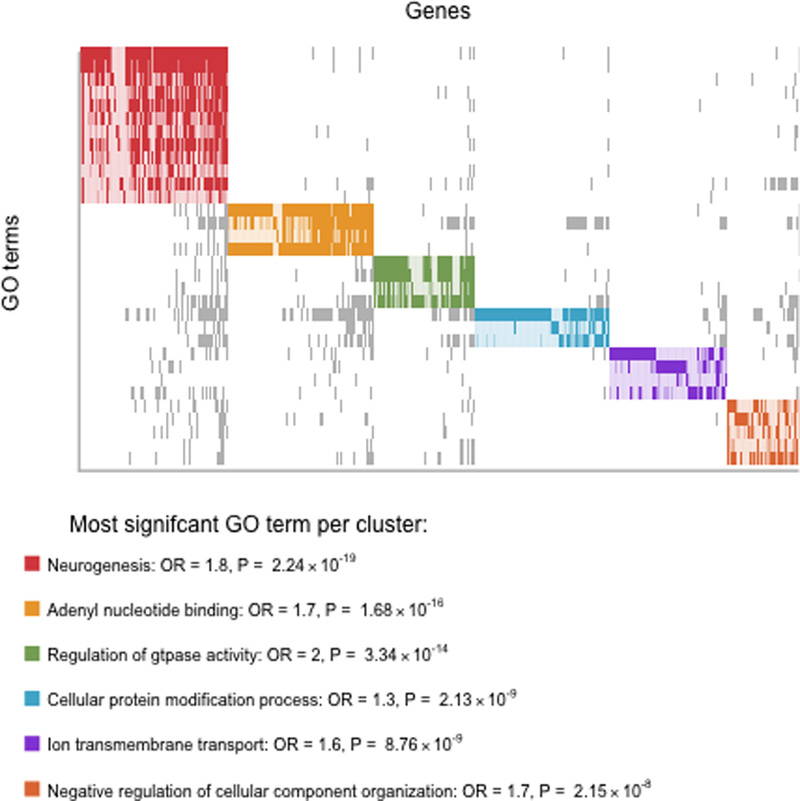
Top. Plot showing level-5 Gene Ontology (GO) terms significantly (P < 0.0001) enriched for genes present in top MWAS results that overlap between blood and brain. Solid rectangles (grey or colored) indicate overlapping genes present in the significant GO terms. GO terms were grouped in color-coded clusters based on the similarity in gene content. Bottom. For each cluster, information (name, odds ratio (OR) and enrichment P value) of the most significant GO term is given. An extended list of all level-5 GO terms with P <0.01 are given in Table S8.
Look-up replication in independent brain collections
As shown in Table 2, we identified three sites that fulfilled our stringent criteria for replication across datasets. The first site was in an intergenic region on chromosome 2 more than 90 kb from any known transcript. The second site was located in GABBR2 (Gamma-aminobutyric acid B receptor 2) and the third site was located in RUFY3 (RUN and FYVE domain containing 3). The results from all replicated sites are reported in Table S10.
Table 2.
Robust loci overlapping between blood and brain (BA10) that is supported by a second brain tissue (BA25) and two independent brain collections.
| Chr. | Position (bp) | Gene | BA10 brain MWAS | BA25 brain | Replication A | Replication B | |
|---|---|---|---|---|---|---|---|
| T | 2-sided P | 1-sided P | 1-sided P | 1-sided P | |||
| 2 | 208,230,169 | intergenic | 2.7064 | 9.18E-03 | 1.68E-02 | 4.24E-02 | 1.02E-02 |
| 9 | 101,119,679 | GABBR2 | -2.6959 | 9.43E-03 | 1.68E-02 | 2.47E-02 | 1.98E-02 |
| 4 | 71,632,888 | RUFY3 | 3.2556 | 1.99E-03 | 2.93E-02 | 1.49E-02 | 4.95E-02 |
Note: Chr. is chromosome; T is t statistic; P is P value. A positive t statistic indicates that more methylation was observed in cases than in controls.
Overlap between MDD and schizophrenia
To study the shared risk for MDD and schizophrenia74, we explored possible overlap in methylation findings between these two disorders74. Using the permutation based enrichment test and results from a previously reported schizophrenia blood MWAS75, we detected a significant enriched overlap of the top 1% of results across the two datasets (P = 5.00 × 10−4, OR = 1.142). This overlap included for example, RERE and RUFY3. These two genes were implicated in MDD by both the blood MWAS and the BA10 MWAS and RUFY3 is one of the three genes that further replicated in the independent MDD brain collections. In total, the 1,564 CpGs implicated in the MDD-schizophrenia overlap, included 462 genes that were present in Gene Ontology (GO). Over-representation analysis showed 24 level-5 terms with P <0.001 and included >20 observed genes (Table S11). The most significant terms were “positive regulation of cell differentiation” (P = 6.49 × 10−5), “small GTPase binding” (P = 6.50 × 10−5), “regulation of nervous system development” (P = 1.91 × 10−4) and “generation of neurons” (P = 2.42 × 10−4). Thus, the results suggest that in addition to a genetic overlap, there may also be an epigenetic overlap in etiology between MDD and schizophrenia.
Exploratory investigation of a multi-site biomarker
To provide a proof-of-concept of that biomarkers exist of potentially etiologically relevant methylation findings, we used MWAS findings in brain to predict MDD status from methylation in blood. In this exploratory analysis the association had a significant Spearman correlation of 0.17 (P=7.84×10−9, r2=0.0289) with case-control status, when using 5,000 CpGs. The use of 5,000 CpGs did not yield the best predictive power but corresponds to the point at which predictive power reaches a stable plateau (Figure S4) suggesting this selection contains the majority of markers with effects in both brain and blood.
Discussion
In the largest methylation study of MDD etiology performed to date, we used a sequencing-based approach that obtains near complete coverage of the entire CpG methylome in both blood and brain samples. Using a resampling approach, we showed that our cumulative blood MWAS signal replicated (P=4.03×10−10). Furthermore, a permutation based enrichment test suggested significant overlap (P=5.4×10−3) of top findings from the MWAS in blood and brain. Three overlapping sites replicated with the same direction of effect in a second brain tissue from the same individuals and in two independent brain collections.
The blood-brain overlap were significantly enriched for eQTLs in both blood and brain, potentially suggesting a mediating role in gene regulation. Interestingly, a significant portion of the genes represented in the overlap were of importance for brain development and function. An example is BDNF (brain-derived neurotrophic factor), a gene involved in induction of synaptic plasticity49, 50 and previously implicated in methylation studies of MDD51, 52 and other psychiatric disorders.53, 54 The overlap also included five genes that recently reached genome-wide significance in GWAS of MDD and related phenotypes: LHPP55 (phospolysine phosphohistidine inorganic pyrophosphate phosphatase), SORCS39 (sortilin-related VPS10 domain containing receptor 3), CELF488 (CUGBP elav-like family member 4), DRD2 (dopamine receptor D2)8 and RERE (arginine-glutamic acid dipeptide (RE) repeats).9 RERE, which plays a role as a transcriptional repressor during development, was the most significant overlapping genic MWAS finding in our BA10 MWAS and was previously associated with schizophrenia.56
However, the most prominent findings in this study involve three loci that received support from all five datasets. The fact that these CpGs were supported across three independent brain collections argues for their likely involvement in MDD etiology. One of these loci was located in an intergenic region on chromosome 2, and the other two were in GABBR2 and RUFY3. GABBR2 encodes a metabotropic GABAB receptor subunit that broadly inhibits neuronal activity including neurotransmitter release and ion channel current. There is substantial support implicating GABAergic dysfunction in MDD57 and a reduced GABBR2 protein level has been observed in brain samples from MDD patients.58 RUFY3 is implicated in the establishment of neuronal polarity59, 60 and is necessary for normal axon elongation.61
A number of methylome-wide association studies for MDD or related phenotypes, have been previously conducted.62–69 With a few exceptions, most of those studies have used approaches that investigated <10% of all CpGs in the genome and have involved relatively few samples. Furthermore, the studies have involved DNA from brain tissues, blood, buccal cells and/or germlines. Thus, given the potentially hampered statistical power in these studies, plus their heterogeneity in studied sites, tissues and phenotypes, the lack of consistent in findings is unsurprising.
This study, as well as the majority of previous methylation studies for MDD, have been performed in bulk tissues that contain multiple cell types. As associations between cell type proportions and MDD status may lead to false positive findings we controlled for possible differences in cell type proportions between cases and controls in our MWAS. If the direction of the differences between cases and controls vary by cell type, effects may be diluted or even cancel out in bulk tissue, resulting in false negative findings. Thus, analyses in bulk tissues will mainly detect loci that are either ubiquitously modulated across all cells or involve a major subset of cell types. This limitation will also affect downstream pathway analyses. It is therefore critical to interpret the results in the context of what cell types/tissues have been studied.
Methylation marks are stable in collected biomaterial and can be measured cost-effectively, which makes them potentially useful biomarkers. Indeed, successful examples of methylation biomarkers already exist for, e.g., smoking related behavior and outcomes.70–75 For a biomarker to be useful in the clinic it is important that biomaterial collection is easy and minimally invasive. Thus, biomarkers detectable only in brain tissue are of limited value. Therefore, methylation sites in blood, potentially reflecting the methylation status of brain, are prospective biomarkers that could improve disease management. Our exploratory analysis in blood using sites selected in brain emphasizes the potential value of such biomarkers. However, a clear limitation of our analysis is the small sample size of the brain collection (N=60) used to select sites, which we observed causes a severe underestimation of the predictive power. Nevertheless, our results show that methylation in blood can track part of the methylation changes that occur in brain and that can be summarized by a single cumulative risk score that would facilitate potential use in the clinic.
In this study, we present the largest MWAS for MDD etiology performed to date. In addition to a large sample and an approach that assays the majority of all CpGs in the human genome, this study interrogates both blood and brain. For the majority of human tissues, including blood, methylation occurs almost exclusively in the seqence context of CpGs. However, in brain methylation profiles are more complex and also include methylation outside the CpG context as well as hydroxymethylation. Thus, additional studies of these methylation types not covered by the current study would further complement the results.
Our three-phase, multi-tissue design improves statistical power and avoids potential tissue/study specific confounders. Despite this design and controlling for both measured and unmeasured covariates, confounders can never be completely ruled out. One of the potential confounders that we studied more closely is antidepressant treatment. That is, we performed an exploratory analysis where we controlled for current use of antidepressants and also searched for differences in the methylome between MDD patients who were or were not treated with antidepressants. Neither of these analyses raised concerns for that the main MDD MWAS findings would be caused by associations to treatment. In addition, the observed overlap in findings between the MDD MWAS and a previous schizophrenia MWAS further argues that it is unlikely that associated findings are artefacts of specific treatments.
In this study all biosamples were collected after the development of MDD. Thus, some of the associated sites may have been present prior to disease onset, and therefore may act as susceptibility loci, while others may follow disease onset and instead reflect the disease state. To properly disentangle these effects a study design that also includes pre-onset samples would be required.
In conclusion, our most prominent results identified three novel differentially methylated loci associated with MDD that overlapped across blood and brain, and replicated with the same direction of effect across independent datasets and brain regions. These loci are of likely importance for biological functions with potential relevance to MDD. To learn more about the specific biological effects of the methylation marks in these loci, rather than analyzing bulk tissue, it would be valuable to identify the specific cell types where the methylation changes occurred. This would, in turn, allow for proper design of functional follow-up studies with, e.g., epigenetic-editing techniques that may provide great biological context to these methylation loci in MDD etiology.
Supplementary Material
Acknowledgement
The current methylation study was supported by grant R01MH099110 from the National Institute of Mental Health. Tissues were received from five brain banks including: the Victorian Brain Bank, supported by The Florey Institute of Neuroscience and Mental Health, The Alfred and Victorian Forensic Institute of Medicine and funded by Australia’s National Health & Medical Research Council and Parkinson’s Victoria; the Stanley Medical Research Institute; The Netherlands Brain Bank, Netherlands Institute of Neuroscience, Amsterdam; the Harvard Brain Tissue Resource Center; and The Douglas – Bell Canada Brain Bank, Douglas Institute Research Center, Canada. Funding for NESDA was obtained from the Netherlands Organization for Scientific Research (Geestkracht program grant 10–000-1002); the Center for Medical Systems Biology (CSMB, NWO Genomics), Biobanking and Biomolecular Resources Research Infrastructure (BBMRI-NL), VU University’s Institutes for Health and Care Research (EMGO+) and Neuroscience Campus Amsterdam, University Medical Center Groningen, Leiden University Medical Center, National Institutes of Health (NIH, R01D0042157–01A, MH081802, Grand Opportunity grants 1RC2 MH089951 and 1RC2 MH089995). Part of the genotyping and analyses were funded by the Genetic Association Information Network (GAIN) of the Foundation for the National Institutes of Health. Computing was supported by BiG Grid, the Dutch e-Science Grid, which is financially supported by NWO.
Footnotes
Conflict of Interest
B.W.J.H.P. has received research funding (non-related) from Jansen Research and Boehringer Ingelheim. Other authors declare no competing financial interests.
Availability of Data and Materials
Following local IRB approval individual level methylation data will be made available via dbGap (submission in preparation). Extended results are included in the supplementary material of the published article.
Supplementary Information
Supplementary information is available at MP’s website.
References
- 1.Judd LL. The clinical course of unipolar major depressive disorders. Archives of general psychiatry 1997; 54(11): 989–991. [DOI] [PubMed] [Google Scholar]
- 2.Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet 2006; 367(9524): 1747–1757. [DOI] [PubMed] [Google Scholar]
- 3.Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003; 289(23): 3095–3105. [DOI] [PubMed] [Google Scholar]
- 4.Avenevoli S, Swendsen J, He JP, Burstein M, Merikangas KR. Major depression in the national comorbidity survey-adolescent supplement: prevalence, correlates, and treatment. J Am Acad Child Adolesc Psychiatry 2015; 54(1): 37–44 e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mueller TI, Leon AC, Keller MB, Solomon DA, Endicott J, Coryell W et al. Recurrence after recovery from major depressive disorder during 15 years of observational follow-up. The American journal of psychiatry 1999; 156(7): 1000–1006. [DOI] [PubMed] [Google Scholar]
- 6.Depression and Other Common Mental Disorders: Global Health Estimates World Health Organization: Geneva, 2017. [Google Scholar]
- 7.World Health Organization; The global burden of disease: 2004 update Geneva: World Health Organization; 2008. [Google Scholar]
- 8.Okbay A, Baselmans BM, De Neve JE, Turley P, Nivard MG, Fontana MA et al. Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat Genet 2016; 48(6): 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hyde CL, Nagle MW, Tian C, Chen X, Paciga SA, Wendland JR et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet 2016; 48(9): 1031–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.PGC MDDWGot. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression http://www.biorxiv.org/content/early/2017/07/24/1675772017. [DOI] [PMC free article] [PubMed]
- 11.Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron 2007; 53(6): 857–869. [DOI] [PubMed] [Google Scholar]
- 12.Kaffman A, Meaney MJ. Neurodevelopmental sequelae of postnatal maternal care in rodents: clinical and research implications of molecular insights. J Child Psychol Psychiatry 2007; 48(3–4): 224–244. [DOI] [PubMed] [Google Scholar]
- 13.Szyf M, Weaver IC, Champagne FA, Diorio J, Meaney MJ. Maternal programming of steroid receptor expression and phenotype through DNA methylation in the rat. Front Neuroendocrinol 2005; 26(3–4): 139–162. [DOI] [PubMed] [Google Scholar]
- 14.Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol 2012; 13(6): R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aberg KA, Xie LY, McClay JL, Nerella S, Vunck S, Snider S et al. Testing two models describing how methylome-wide studies in blood are informative for psychiatric conditions. Epigenomics 2013; 5(4): 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Efstratiadis A Parental imprinting of autosomal mammalian genes. Curr Opin Genet Dev 1994; 4(2): 265–280. [DOI] [PubMed] [Google Scholar]
- 17.Sutherland JE, Costa M. Epigenetics and the environment. Ann N Y Acad Sci 2003; 983: 151–160. [DOI] [PubMed] [Google Scholar]
- 18.Kerkel K, Spadola A, Yuan E, Kosek J, Jiang L, Hod E et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat Genet 2008; 40(7): 904–908. [DOI] [PubMed] [Google Scholar]
- 19.Chan RF, Shabalin AA, Xie LY, Adkins DE, Zhao M, Turecki G et al. Enrichment methods provide a feasible approach to comprehensive and adequately powered investigations of the brain methylome. Nucleic Acids Res 2017; 45(11): e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aberg KA, Chan RF, Shabalin AA, Zhao M, Turecki G, Staunstrup NH et al. A MBD-seq protocol for large-scale methylome-wide studies with (very) low amounts of DNA. Epigenetics 2017; 12(9): 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wittchen HU. Reliability and validity studies of the WHO--Composite International Diagnostic Interview (CIDI): a critical review. J Psychiatr Res 1994; 28(1): 57–84. [DOI] [PubMed] [Google Scholar]
- 22.Rush AJ, Giles DE, Schlesser MA, Fulton CL, Weissenburger J, Burns C. The Inventory for Depressive Symptomatology (IDS): preliminary findings. Psychiatry Res 1986; 18(1): 65–87. [DOI] [PubMed] [Google Scholar]
- 23.Deep-Soboslay A, Iglesias B, Hyde TM, Bigelow LB, Imamovic V, Herman MM et al. Evaluation of tissue collection for postmortem studies of bipolar disorder. Bipolar Disord 2008; 10(7): 822–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conner KR, Conwell Y, Duberstein PR. The validity of proxy-based data in suicide research: a study of patients 50 years of age and older who attempted suicide. II. Life events, social support and suicidal behavior. Acta Psychiatr Scand 2001; 104(6): 452–457. [DOI] [PubMed] [Google Scholar]
- 25.Kelly TM, Mann JJ. Validity of DSM-III-R diagnosis by psychological autopsy: a comparison with clinician ante-mortem diagnosis. Acta Psychiatr Scand 1996; 94(5): 337–343. [DOI] [PubMed] [Google Scholar]
- 26.Dumais A, Lesage AD, Alda M, Rouleau G, Dumont M, Chawky N et al. Risk factors for suicide completion in major depression: a case-control study of impulsive and aggressive behaviors in men. Am J Psychiatry 2005; 162(11): 2116–2124. [DOI] [PubMed] [Google Scholar]
- 27.Shabalin AA, Hattab MW, Clark SL, Chan RF, Kumar G, Aberg KA et al. RaMWAS: Fast Methylome-Wide Association Study Pipeline for Enrichment Platforms. Bioinformatics 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 2012; 13: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hattab MW, Shabalin AA, Clark SL, Zhao M, Kumar G, Chan RF et al. Correcting for cell-type effects in DNA methylation studies: reference-based method outperforms latent variable approaches in empirical studies. Genome Biol 2017; 18(1): 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw 2010; 33(1): 1–22. [PMC free article] [PubMed] [Google Scholar]
- 31.Simon N, Friedman J, Hastie T, Tibshirani R. Regularization Paths for Cox’s Proportional Hazards Model via Coordinate Descent. J Stat Softw 2011; 39(5): 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tibshirani R, Bien J, Friedman J, Hastie T, Simon N, Taylor J et al. Strong rules for discarding predictors in lasso-type problems. J R Stat Soc Series B Stat Methodol 2012; 74(2): 245–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hastie T, Tibshirani R, Friedman J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction Springer Verlag: New York, 2001. [Google Scholar]
- 34.Cabrera CP, Navarro P, Huffman JE, Wright AF, Hayward C, Campbell H et al. Uncovering networks from genome-wide association studies via circular genomic permutation. G3 (Bethesda) 2012; 2(9): 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boomsma DI, Willemsen G, Sullivan PF, Heutnik P, Meijer P, Sondervan D et al. Genome-wide association of major depression: Description of samples for the GAIN major depressive disorder study: NTR and NESDA Biobank Projects. European Journal of Human Genetics 2008; 16: 335–342. [DOI] [PubMed] [Google Scholar]
- 36.Penninx B, Beekman A, Smit J. The Netherlands Study of Depression and Anxiety (NESDA): Rationales, Objectives and Methods. International Journal of Methods in Psychiatric Research 2008; 17: 121–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sullivan P, de Geus E, Willemsen G, James MR, Smit JH, Zandbelt T et al. Genomewide association for major depressive disorder: a possible role for the presynaptic protein piccolo. Molecular Psychiatry 2009; 14: 359–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van den Oord EJ, Bukszar J, Rudolf G, Nerella S, McClay JL, Xie LY et al. Estimation of CpG coverage in whole methylome next-generation sequencing studies. BMC Bioinformatics 2013; 14(1): 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weissman MM, Klerman GL. Sex differences and the epidemiology of depression. Archives of General Psychiatry 1977; 34: 98–111. [DOI] [PubMed] [Google Scholar]
- 40.Kessler RC, McGonagle KA, Swartz MS, Blazer DG, Nelson CB. Sex and depression in the National Comorbidity Survey: I. Lifetime prevalence, chronicity and recurrence. Journal of Affective Disorders 1993; 29: 85–96. [DOI] [PubMed] [Google Scholar]
- 41.Bebbington P The origins of sex differences in depressive disorder: Bridging the gap. International Review of Psychiatry 1996; 8: 295–332. [Google Scholar]
- 42.Bouma GJ, Hudson QJ, Washburn LL, Eicher EM. New candidate genes identified for controlling mouse gonadal sex determination and the early stages of granulosa and Sertoli cell differentiation. Biol Reprod 2010; 82(2): 380–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hammond GL. Plasma steroid-binding proteins: primary gatekeepers of steroid hormone action. J Endocrinol 2016; 230(1): R13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soares CN, Zitek B. Reproductive hormone sensitivity and risk for depression across the female life cycle: a continuum of vulnerability? J Psychiatry Neurosci 2008; 33(4): 331–343. [PMC free article] [PubMed] [Google Scholar]
- 45.Carrier N, Saland SK, Duclot F, He H, Mercer R, Kabbaj M. The Anxiolytic and Antidepressant-like Effects of Testosterone and Estrogen in Gonadectomized Male Rats. Biol Psychiatry 2015; 78(4): 259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McHenry J, Carrier N, Hull E, Kabbaj M. Sex differences in anxiety and depression: role of testosterone. Front Neuroendocrinol 2014; 35(1): 42–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gibbons AS, Brooks L, Scarr E, Dean B. AMPA receptor expression is increased post-mortem samples of the anterior cingulate from subjects with major depressive disorder. J Affect Disord 2012; 136(3): 1232–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kamburov A, Wierling C, Lehrach H, Herwig R. ConsensusPathDB--a database for integrating human functional interaction networks. Nucleic Acids Res 2009; 37(Database issue): D623–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edelmann E, Lessmann V, Brigadski T. Pre- and postsynaptic twists in BDNF secretion and action in synaptic plasticity. Neuropharmacology 2014; 76 Pt C: 610–627. [DOI] [PubMed] [Google Scholar]
- 50.Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci 2013; 14(1): 7–23. [DOI] [PubMed] [Google Scholar]
- 51.Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature 2008; 455(7215): 894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vialou V, Feng J, Robison AJ, Nestler EJ. Epigenetic mechanisms of depression and antidepressant action. Annu Rev Pharmacol Toxicol 2013; 53: 59–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boulle F, van den Hove DL, Jakob SB, Rutten BP, Hamon M, van Os J et al. Epigenetic regulation of the BDNF gene: implications for psychiatric disorders. Mol Psychiatry 2012; 17(6): 584–596. [DOI] [PubMed] [Google Scholar]
- 54.Ikegame T, Bundo M, Murata Y, Kasai K, Kato T, Iwamoto K. DNA methylation of the BDNF gene and its relevance to psychiatric disorders. J Hum Genet 2013; 58(7): 434–438. [DOI] [PubMed] [Google Scholar]
- 55.Consortium Converge. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 2015; 523(7562): 588–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schizophrenia Psychiatric Genome-Wide Association Study Consortium, Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P et al. Genome-wide association study identifies five new schizophrenia loci. Nat Genet 2011; 43(10): 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pehrson AL, Sanchez C. Altered gamma-aminobutyric acid neurotransmission in major depressive disorder: a critical review of the supporting evidence and the influence of serotonergic antidepressants. Drug Des Devel Ther 2015; 9: 603–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fatemi SH, Folsom TD, Thuras PD. Deficits in GABA(B) receptor system in schizophrenia and mood disorders: a postmortem study. Schizophr Res 2011; 128(1–3): 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mori T, Wada T, Suzuki T, Kubota Y, Inagaki N. Singar1, a novel RUN domain-containing protein, suppresses formation of surplus axons for neuronal polarity. J Biol Chem 2007; 282(27): 19884–19893. [DOI] [PubMed] [Google Scholar]
- 60.Honda A, Ito Y, Takahashi-Niki K, Matsushita N, Nozumi M, Tabata H et al. Extracellular Signals Induce Glycoprotein M6a Clustering of Lipid Rafts and Associated Signaling Molecules. J Neurosci 2017; 37(15): 4046–4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wei Z, Sun M, Liu X, Zhang J, Jin Y. Rufy3, a protein specifically expressed in neurons, interacts with actin-bundling protein Fascin to control the growth of axons. J Neurochem 2014; 130(5): 678–692. [DOI] [PubMed] [Google Scholar]
- 62.Uddin M, Koenen KC, Aiello AE, Wildman DE, de los Santos R, Galea S. Epigenetic and inflammatory marker profiles associated with depression in a community-based epidemiologic sample. Psychol Med 2011; 41(5): 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davies MN, Krause L, Bell JT, Gao F, Ward KJ, Wu H et al. Hypermethylation in the ZBTB20 gene is associated with major depressive disorder. Genome Biol 2014; 15(4): R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dempster EL, Wong CC, Lester KJ, Burrage J, Gregory AM, Mill J et al. Genome-wide methylomic analysis of monozygotic twins discordant for adolescent depression. Biol Psychiatry 2014; 76(12): 977–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nagy C, Suderman M, Yang J, Szyf M, Mechawar N, Ernst C et al. Astrocytic abnormalities and global DNA methylation patterns in depression and suicide. Mol Psychiatry 2015; 20(3): 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sabunciyan S, Aryee MJ, Irizarry RA, Rongione M, Webster MJ, Kaufman WE et al. Genome-wide DNA methylation scan in major depressive disorder. PLoS One 2012; 7(4): e34451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murphy TM, Crawford B, Dempster EL, Hannon E, Burrage J, Turecki G et al. Methylomic profiling of cortex samples from completed suicide cases implicates a role for PSORS1C3 in major depression and suicide. Transl Psychiatry 2017; 7(1): e989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haghighi F, Xin Y, Chanrion B, O’Donnell AH, Ge Y, Dwork AJ et al. Increased DNA methylation in the suicide brain. Dialogues Clin Neurosci 2014; 16(3): 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oh G, Wang SC, Pal M, Chen ZF, Khare T, Tochigi M et al. DNA modification study of major depressive disorder: beyond locus-by-locus comparisons. Biol Psychiatry 2015; 77(3): 246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Philibert R, Hollenbeck N, Andersen E, Osborn T, Gerrard M, Gibbons FX et al. A quantitative epigenetic approach for the assessment of cigarette consumption. Front Psychol 2015; 6: 656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Andersen AM, Dogan MV, Beach SR, Philibert RA. Current and Future Prospects for Epigenetic Biomarkers of Substance Use Disorders. Genes (Basel) 2015; 6(4): 991–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang Y, Elgizouli M, Schottker B, Holleczek B, Nieters A, Brenner H. Smoking-associated DNA methylation markers predict lung cancer incidence. Clin Epigenetics 2016; 8: 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baglietto L, Ponzi E, Haycock P, Hodge A, Bianca Assumma M, Jung CH et al. DNA methylation changes measured in pre-diagnostic peripheral blood samples are associated with smoking and lung cancer risk. Int J Cancer 2017; 140(1): 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reynolds LM, Magid HS, Chi GC, Lohman K, Barr RG, Kaufman JD et al. Secondhand Tobacco Smoke Exposure Associations With DNA Methylation of the Aryl Hydrocarbon Receptor Repressor. Nicotine Tob Res 2017; 19(4): 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Philibert R, Hollenbeck N, Andersen E, McElroy S, Wilson S, Vercande K et al. Reversion of AHRR Demethylation Is a Quantitative Biomarker of Smoking Cessation. Front Psychiatry 2016; 7: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.