

Graphical Abstract
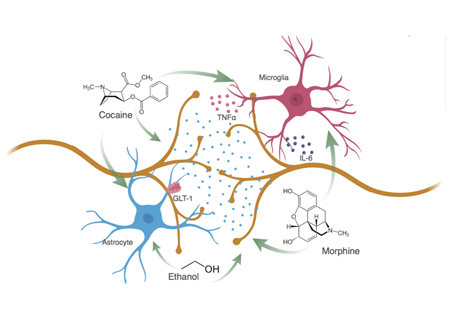
Drugs of abuse have direct actions on both microglia and astrocytes and promote a reactive glial state where pro-inflammatory cytokines, such as IL-6 and TNFα, are released into the synaptic environment. Glia modulate neuronal activity through a variety of receptors including the astrocyte specific glutamate transporter, GLT-1. These drug-induced changes to glia alter neuron-glia interactions, circuit functions and ultimately drug-associated behaviors, and provide a promising therapeutic target for substance use disorders.
Keywords: microglia, astrocytes, addiction, opiates, psychostimulants
Introduction
Addiction is a chronic relapsing disorder, characterized by the compulsion to seek and take drugs despite negative consequences. It is extremely damaging and costly, accounting for $740 billion annually in the United States resulting from healthcare, crime, and lost work productivity (Birnbaum et al., 2011; CDC 2017; Xu et al., 2014). There is also an immeasurable cost in the loss of life and immense individual suffering. In order to find effective therapies for addiction, we must identify effective targets for treatment by understanding the complex mechanisms that are fundamental to this impenetrable disorder. Critical, well-defined changes to the brain that underlie these behavioral stereotypes have been identified (Koob & Volkow, 2010). Although evidence has implicated both neurons and glia in mechanisms that contribute to these addictive behaviors, the largest body of work has focused on understanding neuronal mechanisms of addiction.
Extensive investigations of the pathogenesis of drug abuse have revealed the critical role of two dopamine-producing nuclei in the ventral midbrain, the ventral tegmental area (VTA) and the substantia nigra (SN). These two regions provide dopaminergic innervation to the forebrain, including prefrontal cortex (PFC), amygdala, hippocampus (HPC), dorsal striatum, and nucleus accumbens (NAc). Of these, the VTA to NAc pathway is noteworthy because it is consistently implicated in drug use across species, including humans (Volkow et al., 2017). More specifically, this circuit is critical for motivated instrumental behaviors, such as self-administration and conditioned place preference (CPP) in both mice and rats. Fast-scan cyclic voltammetry studies have shown that natural rewards and drugs of abuse increase the amplitude of phasic dopamine release in the NAc (Phillips et al., 2003; Vander Weele et al., 2014). Although dopamine is a key neurobiological substrate that encodes rewarding properties of drugs of abuse, recent findings have shown other mechanisms that critically influence the reinforcing properties of drugs, including other neurotransmitters, transcription factors, and other substances released from neurons and glia alike. Whereas there is an expansive body of work on neural mechanisms underlying addiction (Hyman et al., 2006; Kauer & Malenka, 2007; Kalivas, 2009), successful therapeutic targets remain elusive. Progress in understanding glial biology has revealed these cells to be potential key regulators in drug abuse. Glia were once viewed as merely immune response cells with no active role in cognitive function. Although microglia and astrocytes are master regulators of the central immune system, mounting evidence demonstrates that they influence processes beyond inflammation and immune surveillance. These cells actively regulate synaptic glutamate spillover, activity-dependent synaptogenesis and elimination, neuronal morphology, neurotransmitter synthesis, synaptic connectivity, and neural circuit function (Guerra-Gomes et al., 2017; York et al., 2017; Scofield & Kalivas, 2014). These functions of glial cells are significantly altered by exposure to abused drugs, and such changes likely contribute to the behavioral outcomes associated with substance abuse (Miguel-Hidalgo, 2009; Coller & Hutchinson, 2012).
In this review, we argue that glia influence the cellular, molecular, and synaptic changes that occur in neurons following drug exposure, and that these neural–glial interactions influence drug-associated behaviors. Here we focus on microglia and astrocytes, rather than oligodendrocytes, endothial, or other glial cells, due to the large body of evidence implicating microglia and astrocytes in addiction. We will evaluate the role of glia in addiction-like brain states and behaviors. First, we will discuss how abused drugs directly affect glia. We will then explore neural-glia communications, and how this affects circuit-glia interactions. Finally, we will review how these molecular, cellular, and circuit changes ultimately produce changes in drug-associated behavior, and discuss the clinical evidence for glia as a therapeutic target for addiction.
Direct Effects of Drugs of Abuse on Microglia and Astrocytes
Microglia and astrocytes are immune competent cells that prevail throughout the central nervous system (CNS). Although microglia are the resident macrophages of the brain, recent research demonstrates that they have functions in addition to biological defense (Wu et al., 2015). Astrocytes are abundant, bushy cells that provide metabolic support for neurons, control synaptic microenvironments, and influence circuit development (Kim et al., 2017). Both microglia and astrocytes exhibit significant changes in morphology, gene expression, and function following drug exposure. Furthermore, there is increasing evidence that microglia and astrocytes can perpetuate the detrimental effects of drugs of abuse on the brain.
Identifying Reactive Glia
Microglia and astrocytes can be identified by a host of general markers that include IBA1, Cd11b, and CX3CR1 for microglia and GFAP, S100b, and GLT-1 for astrocytes (Sofroniew & Vinters, 2010; Kim et al., 2017). In the studies reviewed here, the predominant marker for microglia is IBA1, and for astrocytes, GFAP. It is important to note that IBA1 does not differentiate between central and peripheral macrophages, and that GFAP is only expressed at detectable levels in certain brain regions. However, both GFAP and IBA1 are upregulated after biological insult or during activation. Microglia and astrocytes can become reactive in response to harmful stimuli, including drugs of abuse, leading to stereotyped patterns of alterations in glial morphology and transcriptome. Reactive microglia have an increase in soma size and a simultaneous shrinkage and thickening of processes (Ramirez et al., 2017; Coller & Hutchinson, 2012; Lacagnina et al., 2017). Reactive astrocyte morphology is more difficult to define, although reactive astrocytes do proliferate (Liddelow & Barres, 2017). Reactive microglia and astrocytes can release cytokines and chemokines that are barely detectable in normal conditions, but that are increased following insult (e.g., exposure to drugs of abuse) and critically alter glial and neural function. Although cytokines released by microglia and astrocytes are categorized as either pro- or anti-inflammatory, the microenvironment that a cytokine is released into can alter its properties (Cavaillon, 2001). Generally, cytokines from the tumor necrosis factor (TNF) family, gamma-interferon (IFN-γ) family, and most cytokines from the interleukin (IL) family are considered pro-inflammatory. In contrast, IL-10, IL-13, IFN-α, and TGF-β are considered anti-inflammatory (Cavaillon, 2001; Sofroniew, 2014). Glial cells also release various chemokines, which are chemotactic cytokines that move in response to a chemical stimulus. These are classified into two families: CXC, which attract neutrophils and lymphocytes, or CC, which attract monocytes and T cells (Cavaillon, 2001). However, recent research demonstrates that chemokines participate in many functions in addition to chemotaxis.
Drugs of Abuse Promote Reactive Glia
Both microglia and astrocytes exhibit significant changes in morphology, gene expression, and function following drug exposure (Lacagnina et al., 2017). Furthermore, increasing evidence demonstrates that microglia and astrocytes can perpetuate the detrimental effects of drugs of abuse on the brain. Many drugs of abuse increase expression of microglial and astrocytic markers, increase cytokine and chemokine release, and promote a pro-inflammatory glial phenotype (Table 1; Ramirez et al., 2017; Coller & Hutchinson, 2012; Lacagnina et al., 2017). Indeed, psychostimulants, such as cocaine and methamphetamine, promote a reactive glial phenotype (Coller & Hutchinson, 2012; Lacagnina et al., 2017) and increase IBA1 and GFAP expression (Fattore et al., 2002; Thomas et al., 2004; Liao et al., 2016). The cytokines Il-1β, TNFα, and IL-6, among others, are also increased by psychostimulant exposure (Sriram et al., 2006; Northcutt et al., 2015; Liao et al., 2016). Opioids and alcohol also increase glial markers and pro-inflammatory cytokine release (Alfonso-Loeches et al., 2010; Wang et al., 2012). In contrast, nicotine suppresses reactive microglia in the adult brain (Li et al., 2016; Noda & Kobayashi, 2017), and blocks LPS-induced increases in microglial activation and expression of Toll-like receptor 4 (TLR4, see below; Kim et al., 2014; Li et al., 2016). The anti-inflammatory state produced by nicotine results from activation of the α7 nicotinic acetylcholine receptor (nAChR) (Shytle et al., 2004; Noda & Kobayashi, 2017), possibly in a metabotropic signaling mode (King et al., 2017). Astrocytes have also been shown to express α7 nAChRs (Shen & Yakel, 2012), which modulate synaptic responsiveness by controlling glial release of the N-methyl D-aspartate (NMDA) receptor co-agonist, D-serine (Papouin et al., 2017). Although nicotine has anti-inflammatory effects in adults, we have recently shown that adolescent nicotine exposure increases IBA1 expression and induces a pro-inflammatory microglia state (Linker et al., 2017). Although the direct mechanism for this increase is unknown, this finding suggests that adolescent microglia may have different sensitivities to drugs of abuse. Indeed, adolescent morphine exposure increases subsequent TLR4 expression in adulthood, whereas adult morphine exposure does not (Schwarz & Bilbo, 2013).
Table 1-.
Cytokines and chemokines that contribute to drug use
| Cytokine | Acronym / Receptor | Function |
|---|---|---|
| Interferon Gamma | IFNγ / IFNϒR1,2 | Increases lysosome activity of macrophages; Activates iNOS |
| Interleukin 1 Alpha | IL-1α / IL-1R1,2 | Induces synthesis of proteases; Induces TNFα production |
| Interleukin 1 Beta | IL-1β / IL-1R1,2 | Proinflammatory mediator, involved in apoptosis and proliferation |
| Interleukin 10 | IL-10 / IL-10R1,2 | Block NFkβ activity |
| Interleukin 12 | IL-12 / IL-12R-β1,2 | Stimulates production of TNFα,IFNγ |
| Interleukin 4 | IL-4 / IL-4Rα | Cooperates with TGFβ, drives mitogenesis |
| Interleukin 6 | IL-6 /IL-6R | Drives chronic inflammation and autoimmunity |
| Transforming Growth Factor Beta | TGFβ / TGFβR | Inhibits secretion of TNFα, IFNγ and various interleukins |
| Tumor Necrosis Factor Alpha | TNFα / TNFR1,2 | Apoptosis, inflammation |
| Chemokine | Acronym / Receptor | Function |
| C-C motif chemokine ligand 2 | CCL2 / CCR2 | Recruits immune cells since to sites of inflammation |
| C-X-C motif chemokine ligand 12 | CXCL12 / CXCR4 | Induced by TNFα and IL-1 |
| C-X3-C motif ligand 1 | Fracktalkine / CX3CR1 | Microglial synaptic pruning |
| C-C motif chemokine ligand 4 | CCL4 / CCR5 | Mediate pro-inflammatory signals |
| C-C motif chemokine ligand 17 | CCL17 / CCR4;CCR8 | Induces chemotaxis in T cells |
Timing is Critical for Drug-Induced Glial Activation
Although nearly all drugs of abuse activate glia, it is pertinent to note that the timing of drug exposure is a vital factor determining drug-induced changes in glial responding. For example, 1-day and 7-day exposure to cocaine increases GFAP, whereas 14-day cocaine exposure does not (Fattore et al., 2002). In addition, withdrawal from cocaine after extended self-administration decreases GFAP and astrocytic volume (Scofield et al., 2016b). A similar pattern is observed after alcohol exposure, with increased GFAP expression after 4–12 week alcohol access and decreased GFAP following extended (36 week) access (Franke, 1995). There are also regional differences in inflammatory responses.
Drugs of Abuse Act Directly on Toll-like Receptor 4
The mechanisms by which drugs of abuse activate glia vary. For some drugs of abuse this can occur through direct actions on glial receptors. Microglia and astrocytes express a wide array of receptors that may or may not be glial-specific. These include cytokine and chemokine receptors, numerous neurotransmitter receptors (including dopamine receptors), neuropeptide receptors, interferon receptors, Toll-like receptors, and many others (Pocock & Kettenmann, 2007; Sofroniew, 2014). Microglia and astrocytes also have cell-type specific receptor expression, notably the fracktalkine (CXC3CL1) receptor for microglia and glutamate transporter 1 (GLT-1) for astrocytes (Scofield & Kalivas, 2014; Ransohoff & El Khoury, 2015). Both of these signaling proteins are critical for glial modulation of synapses Toll-like receptors (TLRs), are an important receptor family expressed by both microglia and astrocytes, and they are the most extensively studied immune receptors for biological defense and drug response (Lacagnina et al., 2017). TLR activation promotes an adaptor protein, myeloid differentiation primary response gene 88 (MyD88), to assist translocation of the transcription factor Nf-κβ to the nucleus, which then induces the production of Il-1β, TNFα, IL-6, IL-10, and other cytokines and chemokines (Lee & Kim, 2007; Trotta et al., 2014). These cytokines are released into the extracellular milieu and bind to receptors expressed in microglia, astrocytes, and neurons, causing changes to intracellular signaling and cell function.
The Toll-like receptor 4 (TLR4), in particular, is a critical interface for direct and indirect effects of many drugs of abuse on glia (Figure 1). Extensive studies have demonstrated that opioids can act directly on TLR4 as an agonist at a non-classical binding site (Watkins et al., 2009; Coller & Hutchinson, 2012). Opioids bind to the MD-2 binding pocket for lipopolysaccharide (LPS; TLR4 agonist and classical immune activator) to induce TLR4 oligomerization, which triggers downstream signaling to Nf-κβ and the production of pro-inflammatory cytokines (Figure 1; Wang et al., 2012). Furthermore, the (+) isomers of the opioid receptor antagonists, naloxone and naltrexone, act as direct TLR4 antagonists (Wang et al., 2016). Interestingly, Northcutt et al., (2015) demonstrated that cocaine interacts with the same TLR4 binding domain as (+)-naloxone, which blocks cocaine-induced increases in cytokine release (Figure 1). Alcohol also induces a TLR4-dependent release of pro-inflammatory cytokines (Alfonso-Loeches et al., 2010). Knockdown of TLR4 genetically, or with an siRNA, blocks alcohol-induced increases in microglial (Cd11b) and astrocytic (GFAP) pro-inflammatory markers (Alfonso-Loeches et al., 2010). The mechanism by which alcohol enhances activation of TLR4 is unclear, but one possibility is through modulation of lipid rafts in astrocyte membranes, and endocytosis of active TLR4 receptors (Figure 1; Blanco et al., 2008; Pascual-Lucas et al., 2014). Even with these complexities, the above studies demonstrate that most drugs of abuse can produce profound pro-inflammatory states in both microglia and astrocytes, and that TLR4 receptors are a central regulator of their downstream inflammatory signaling.
Figure 1:

Toll-like receptor 4 (TLR4) is a central intermediary between abused drugs and glia. Morphine is a direct agonist of TLR4 and binds to a unique binding site in the myeloid differentiation factor 2 (MD-2) domain of the TLR4 receptor. Cocaine is a direct agonist for TLR4 and binds to a domain in the MD-2 region of the receptor, this binding site is the same site of (+)-naltrexone. Alcohol promotes active endocytosis of TLR4 receptors by modulating lipid rafts. TLR4 activation causes the translocation of the transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (Nf-κβ) to the nucleus via the myeloid differentiation primary response 88 (MyD88) pathway and MyD88-independent pathways.
Distinct Alterations in Neuron-Glial interactions Following Drug Exposure
Cytokines and Chemokines Modulate Synaptic Plasticity and Neurotransmitter Synthesis
As reviewed above, exposure to drugs of abuse increases microglial and astrocytic markers, largely promoting a pro-inflammatory state that triggers glial cells to release an arsenal of cytokines and chemokines, but how do these changes affect neurons? Glial-derived cytokines and chemokines can alter neurotransmitter synthesis and release, neuronal activity, and synaptic plasticity (Cui et al., 2014). Increasing evidence has demonstrated that the pro-inflammatory cytokine, TNFα, regulates synaptic activity via AMPA receptor trafficking and astrocyte glutamate release (Santello & Volterra, 2012). TNFα decreases inhibitory synaptic strength by promoting endocytosis of GABAA receptors through p38 MAPK (Pribiag & Stellwagen, 2013), and also enhances excitatory synaptic efficacy by increasing AMPA receptors (GluR1) at the cell surface (Beattie et al., 2002; Stellwagen et al., 2005). Furthermore, TNFα is required for astrocyte glutamate release (Santello et al., 2011). This glutamatergic change is critical as an imbalance in glutamate synaptic plasticity is a hallmark of addiction (Kalivas, 2009; Scofield & Kalivas, 2014; Scofield et al., 2016a). IL-1β increases excitatory transmission through an increase in surface expression of GluR1, but IL-6 and IL-10 do not produce these receptor changes (Stellwagen et al., 2005). However, IL-6 may downregulate NMDA receptor function or expression (Ali et al., 2000; Pizzi et al., 2004). Increasing evidence has demonstrated that TNFα regulates synaptic activity via AMPA receptor trafficking and astrocyte glutamate release. These cytokine-derived changes to glutamate and GABA transmission may be critical to understanding addiction-like states, as these neurotransmitter systems are significantly altered after repeated drug exposure. Lewitus et al. (2016) demonstrated that semi-chronic cocaine exposure increased TNFα levels which resulted in a significant reduction in AMPA/NMDA receptor ratios in D1-containing medium spiny neurons. This alteration in glutamate receptor signaling is evident 24 hours after repeated cocaine exposure (Kourrich et al., 2007; Lewitus et al., 2016). Furthermore, disruption of cocaine signaling at TLR4, and prevention of subsequent TNFα production, blocks cocaine-induced dopamine release (Northcutt et al., 2015).
Chemokines are also key glial-derived neuronal modulators. CXC motif chemokine 12 (CXCL12) increases dopamine neuron action potential frequency, and switches the firing pattern of depolarized dopamine neurons from tonic to burst firing (Skrzydelski et al., 2007). Chemokine ligand 2 (CCL2) also increases striatal dopamine release (Guyon et al., 2009). The receptor for CCL2, chemokine receptor 2 (CCR2), has distinct expression throughout the brain, with high levels in the cortex, striatum, and basolateral amygdala (BLA) – regions involved in reward and addiction (Banisadr et al., 2002). Importantly, CCR2 knockout mice also exhibit attenuated cocaine-induced immediate early gene activation in the striatum (Trocello et al., 2011). These preliminary studies suggest that CCL2, its receptor CCR2, and CXCL12 may contribute to the rewarding effects of drugs by their modulation of striatal dopaminergic firing.
Cytokines have also been shown to stimulate intra-neural signaling pathways that alter the synthesis of neurotransmitters essential for reward and addiction-like behaviors (Cui et al., 2014). Dopamine is synthesized by a series of chemical reactions where phenylalanine is converted to tyrosine by phenylalanine hydroxylase (PAH) and tyrosine is converted into the precursor of dopamine, L-DOPA, by tyrosine hydroxylase (TH). Tetrahydrobiopterin (BH4) is an essential co-factor for both enzymes, and BH4 production is stimulated by pro-inflammatory cytokine release, suggesting that inflammatory cytokines may promote dopamine synthesis via BH4 (Cui et al., 2014; Figure 2). However, inflammatory cytokines also increase reactive oxygen species that ultimately decrease the oxidation-labile BH4 and, in turn, decrease TH production (Neurauter et al., 2008). Many studies in the depression field have demonstrated that a chronic excess of inflammatory cytokines, associated with sickness and mood disorders, ultimately causes a decrease in dopamine levels (Neurauter et al., 2008; Felger & Miller, 2012). This mechanism is not well understood in the context of addiction, but recent evidence suggests drug-induced changes in cytokines may have different effects on BH4-controlled TH. TLR4 pro-inflammatory signaling has been shown to increase TH production in VTA dopaminergic neurons, which contrasts with cytokine-induced decreases in TH observed in depression (Aurelian et al., 2016). This discrepancy in the impact on TH may reflect differences in the duration or concentration of cytokines. Indeed, acute cytokine exposure increases dopamine whereas extended exposure decreases dopamine and TH levels (Dunn, 2006). Clearly this research area is understudied and should be further investigated in relation to how drug-induced increases in cytokines alter neurotransmitter production.
Figure 2:

Dopamine synthesis relies on the conversion of tyrosine to L-3,4-dihydroxyphenylalanine (L-DOPA) by tyrosine hydroxylase (TH). Phenylalanine is converted to tyrosine by phenylalanine hydroxylase (PAH). Both TH and PAH require tetrahydrobiopterin (BH4) as a cofactor. Although inflammatory cytokines induce GTP-cyclohydrolase I expression, an enzyme necessary for BH4 synthesis, they also increase free radicals. Reactive oxygen species (ROS) contribute to oxidative stress and reduction of BH4. Inflammatory cytokines can both increase and decrease dopamine synthesis through different pathways. Red arrows indicate inhibitory effects and green arrows indicate excitatory effects on enzymes.
Astrocytic GLT-1 is Important for Glutamate Signaling
Astrocytes also modulate synaptic function by tightly controlling synaptic glutamate levels through the astrocyte-specific glutamate transporter GLT-1, which is responsible for ~90% of synaptic glutamate clearance (Rao et al., 2015). GLT-1 is decreased following cocaine, nicotine, and heroin exposure, demonstrating a consistent decrease across drug groups (Smith et al., 2015). Drug-induced downregulation of GLT-1 is associated with glutamate synaptic spillover to activate extrasynaptic NMDARs (Shen et al., 2014), which is critical for drug-induced plasticity and reinstatement.
Microglial and Astrocytic Creation and Elimination of Synapses
Glia also modulate synapse elimination and formation. Specifically, microglia prune synapses via phagocytosis of dendritic spines (Nimmerjahn et al., 2005; Wake et al., 2009, Wu et al., 2015) in an activity-dependent manner (Tremblay et al., 2010; Paolicelli et al., 2011). Several mechanisms have been elucidated for this pruning, most notably the classical complement cascade and fracktalkine-CX3CR1 signaling (Stevens et al., 2007; Paolicelli et al., 2011). Foundational studies in vitro have also demonstrated that astrocytes are key for promoting synapse formation, since cultured neurons develop very few synapses in their absence (Clark et al., 2013). Astrocytes secrete thrombospondins (TSP1 and TSP2) and the matricellular protein, hevin (SC1), to promote synaptic formation (Christopherson et al., 2005; Kucukdereli et al., 2011). Although astrocytes are classically known to promote synapse formation while microglia prune synapses, both cell types contribute to synaptic formation and elimination. Microglia can promote synaptic formation through the anti-inflammatory cytokine IL-10, and astrocytes can promote synaptic elimination through SPARC (Kucukdereli et al., 2011; Miyamoto et al., 2015).
Drugs of Abuse Influence Glial Modulation of Synaptic Plasticity
We highlight the significant role that glia play in synaptic modulation due to the long-lasting effects of drugs of abuse on synaptic density, structure, and function (Lüscher & Malenka, 2011; Gipson et al., 2014). There is minimal, but promising, evidence that glia are also involved in drug-induced changes to synaptic plasticity. For example, alcohol-induced depression of long-term potentiation (LTP) is blocked in the absence of glial-derived chemokine CCL2 (Bray et al., 2013). Adolescent intermittent alcohol exposure also increases thrombospondin-2 and PSD95 protein levels, suggesting that alcohol may increase synapse count through secreted astrocyte chemokines (Risher et al., 2015). These effects are not limited to alcohol, as cocaine self-administration and extinction also decrease astrocyte-synaptic contacts (Scofield et al., 2016b), and morphine decreases microglial NAc levels of fracktalkine, a chemokine involved in microglial synaptic pruning (Schwarz et al., 2013). Taken together, these studies demonstrate that different drug exposure paradigms can increase or decrease synaptic count or contacts through glial-derived factors. Although these few studies are encouraging, this field remains largely unexplored.
Drugs of Abuse Critically Alter Circuit-Glial Network Interactions
Some brain circuits and regions are particularly sensitive to inflammation, and heterogeneous glial populations respond to neural and foreign stimuli differently. This diverse and elaborate circuit-glia intercommunication remains poorly understood, but is an important component of brain function in health and disease. Diversity of microglia and astrocytes throughout the brain is well understood (Jessen et al., 1984; Reynolds & Herschkowitz, 1987; Lawson et al., 1990). However, the extent to which these cells differ functionally and in smaller, discrete brain regions is a more recent discovery. Understanding this heterogeneity is crucial to understanding the role of glia in drug use, as drug exposure has region-specific effects.
Microglia and Astrocytes are Multifarious Cell Types
Mounting evidence shows that microglia are very diverse throughout the CNS, both constitutively and after insult. Systemic LPS, a TLR4 agonist, induces IL-1β and TNFα cytokine production in the HPC, PFC, and other cortical brain regions, with the highest induction in the striatum (Tyagi et al., 2010; Noh et al., 2014). An autoimmune paradigm also significantly increases IL-1α in the striatum as compared to the HPC and amygdala (Gentile et al., 2015). Taken together, these studies demonstrate that the striatum may be particularly sensitive to inflammatory insults. Microglia have distinct cell body densities, morphologies, transcriptomes, and receptor expression in the subnuclei of the basal ganglia (NAc, VTA, SN; De Biase et al., 2017). Heterogeneity within these nuclei suggests that each basal ganglia region possesses a unique population of microglia with a variety of prospective functions (De Biase et al., 2017). Some of the most differentially expressed gene functional groups within these subnuclei are related to vesicle release and secretion, lysosome function, and reactive oxygen species homeostasis. Understanding these spatial differences in microglia is key to interpreting the regional changes in microglia following drug exposure, and how these region-specific changes to glia may modify reward circuitry.
Astrocytes are also a remarkably diverse cell type, which exhibit regionally distinct transcriptomes, electrophysiological properties, morphologies, calcium signaling, and synaptic proximity (Chai et al., 2017). Astrocytes are sparse in regions with a high density of neuronal cell bodies, but are dense in regions occupied by dendrites and axons (Khakh & Sofroniew, 2015). As such, there are regional differences in astrocyte-to-neuron ratios throughout the brain. For example, there is a lower number of astrocytes per neuron in the striatum as compared to the HPC (Chai et al., 2017). Although the functional outcomes of this differential distribution remain poorly understood, sparsely populated striatal astrocytes have larger calcium responses to excitatory and inhibitory designer receptors exclusively activated by designer drugs (DREADD) modulation as compared to the HPC (Chai et al., 2017).
Different Drugs of Abuse Alter Glia in a Region-Specific Manner
Drugs of abuse produce varied regional changes to microglia. As discussed earlier, opioids activate TLR4 receptors and, therefore, promote the production of several pro-inflammatory (IL-1β, TNFα, IL-6) and anti-inflammatory (IL-10) cytokines through downstream activation of the glial transcription factor Nf-κβ (Figure 1). However, this cytokine induction is not homogeneously distributed throughout the brain (Table 2). Chronic morphine increases IL-1β in the HPC and dorsal periaqueductal grey (dPAG), while it increases IL-6 in the SN and IFN-γ in the NAc. FACS sorted tissue in the NAc has shown that morphine induces production of chemokines CCL4, CCL17, and chemokine receptor CCR4, as well as the anti-inflammatory cytokine IL-10, by microglia, but not astrocytes (Schwarz et al., 2013). Opioids also induce reactive phenotypes in microglia and astrocytes broadly and differentially throughout the CNS. Following chronic morphine exposure, astrocytic GFAP is increased in the locus coeruleus, nucleus of the solitary tract, caudate putamen (CPu), NAc, dentate gyrus (DG), dPAG, SN, PFC, and other midbrain nuclei, but not the dorsal raphe nucleus (DRN) (Marie-Claire et al., 2004; Alonso et al., 2007; Hutchinson et al., 2009). Morphine also promotes morphological changes in astrocytes in the NAc and lateral septal nucleus (Lazriev et al., 2001). Furthermore, microglial IBA1 expression is increased by morphine in the VTA, SN, NAc, DG, and CPu, but not the mPFC or DRN (Hutchinson et al., 2009).
Table 2-.
Brain regional differences in glial response to drugs of abuse
| Brain Region | Drug of Abuse | Chemokine/Cytokine | Effect | References |
|---|---|---|---|---|
| Nucleus accumbens | Morphine | IFN-γ | Shwartz et al. 2014 | |
| CCL4 / CCR4 | ||||
| CCL17 | ||||
| IL-10 | ||||
| GFAP | Marie-Claire et al. 2004 | |||
| IBA1 | Hutchinson et al. 2009 | |||
| Cocaine | IBA1 | Lewitus et al. 2016 | ||
| IL-1β | Cearley et al. 2011; Northcutt et al. 2015 | |||
| TNFα | Cearley et al. 2011; Northcutt et al. 2015 | |||
| Amphetamine | GFAP | = | Armstrong et al. 2004 | |
| Caudate Putamen | Morphine | GFAP | Marie-Claire et al. 2004 | |
| IBA1 | ||||
| Cortex | Morphine | IBA1 | Hutchinson et al. 2009 | |
| Alcohol | GFAP | Udomuksorn et al. 2011; Kane et al. 2014 | ||
| CCL2 | Kane et al. 2014 | |||
| IL-6 | = | |||
| TNFα | ||||
| IBA1 | Qin and Crews 2012 | |||
| Cocaine | IL-1β | Cearley et al. 2011; Northcutt et al. 2015 | ||
| TNFα | ||||
| Amphetamine | GFAP | = | Armstrong et al. 2004 | |
| Methamphetamine | TNFα | Goncalves et al. 2008 | ||
| IL-6 | ||||
| Hippocampus | Morphine | IL-1β | Shwartz et al. 2014 | |
| IBA1 | Hutchinson et al. 2009 | |||
| Alcohol | GFAP | Tagliaferro et al. 2002; Kane et al. 2014 | ||
| CCL2 | Kane et al. 2014 | |||
| IL-6 | = | |||
| TNFα | ||||
| IBA1 | Qin and Crews 2012 | |||
| Cocaine | GFAP | Fattore et al. 2002 | ||
| Methamphetamine | TNFα | Goncalves et al. 2008 | ||
| IL-6 | ||||
| Ventral Tegmental Area | Morphine | IBA1 | Hutchinson et al. 2009 | |
| Cocaine | IL-1β | Cearley et al. 2011; Northcutt et al. 2015 | ||
| TNFα | ||||
| Periaqueductal Gray | Morphine | IL-1β | Shwartz et al. 2014 | |
| Substantia Nigra | Morphine | IL-6 | Shwartz et al. 2014 | |
| IBA1 | Hutchinson et al. 2009 | |||
| Locus coeruleus | Morphine | GFAP | Alonso et al. 2007 | |
| Nucleus of the Solitary Tract | Morphine | GFAP | Alonso et al. 2007 | |
| Dorsal Raphe | Morphine | IBA1 | = | Hutchinson et al. 2009 |
IFN-γ = interferon gamma; CCL4 = chemokine ligand 4; CCR4 = chemokine receptor type 4; CCL17 = chemokine ligand 17; IL-10 = interleukin 10; GFAP = glial fibrillary acidic protein; IBA1 = ionized calcium binding adaptor molecule 1; CCL2 = chemokine ligand 2; IL-6 = interleukin 6; TNFα = tumor necrosis factor alpha; IL-1β = interleukin 1 beta
Alcohol also induces unique patterns of glial activation throughout the brain. Chronic alcohol exposure for a period of 3–6 months increases GFAP, astrocyte volume, and S100b expression in the CA1 of the HPC, and increases GFAP+ astrocytes in frontal, parietal, and temporal cortex (Tagliaferro et al., 2002; Udomuksorn et al., 2011). Semi-chronic alcohol exposure (10 days) increases GFAP and the chemokine CCL2 in the adult rat cortex, cerebellum, and HPC, but not the cytokines TNFα or IL-6 (Kane et al., 2014). This alcohol exposure paradigm also increases IBA1 expression in the cortex and HPC, along with increasing Nf-κβ expression (Qin & Crews, 2012). The NAc, BLA, and PFC all express different glial-enriched transcript profiles following alcohol exposure (Osterndorff-Kahanek et al., 2015). Importantly a genetic knockout of TLR4 blocks chronic alcohol-induced changes to glial networks (Alfonso-Loeches et al., 2010).
Psychostimulants also promote region-specific changes to microglia and astrocytes. Repeated amphetamine exposure increases GFAP in the dorsal CPu but not the NAc or PFC (Armstrong et al., 2004). Cocaine increases IBA1 and microglial reactivity in the NAc, and increases GFAP in the HPC (Fattore et al., 2002; Lewitus et al., 2016). Psychostimulants also induce circuit-specific functional consequences. Acute cocaine increases IL-1B and TNFα in the NAc, PFC, and VTA (Cearley et al., 2011; Northcutt et al., 2015), whereas methamphetamine increases TNFα and IL-6 in the HPC and PFC (Gonçalves et al., 2008). Together, these data highlight important drug-induced alterations in glial markers and cytokine levels that vary based on drug class, as well as by region and glial cell type.
Drugs of Abuse Alter Glial Regulation of Circuit Communication
Drug-induced changes to glia also affect how brain regions communicate with each other. Astrocytes in the striatum respond to D1-receptive or D2-receptive cells in a mutually exclusive manner. D1-responsive astrocytes increase calcium levels in response to D1 receptor stimulation and not to D2 receptor stimulation, and vice versa (Martin et al., 2015). Similarly, increased calcium levels trigger astrocytes to release glutamate to modulate excitability of neurons in a selective manner, with calcium waves in D2-responsive astrocytes only increasing excitatory post synaptic currents in D2 responsive, but not D1 responsive, neurons (Martin et al., 2015). This foundational work demonstrates that astrocytes modulate neurons in a circuit specific (D1 vs. D2) manner. Future work should investigate how astrocyte circuit-specific modulations affect drug-associated behaviors.
LTP is a persistent increase in synaptic strength, and nearly all drugs of abuse induce LTP within the VTA that is associated with drug intake. Increasing evidence demonstrates that cytokines and chemokines influence LTP in a cell-type specific manner. Hippocampal LTP induces IL-1β and IL-6 release, and blocking their signaling inhibits or supports LTP maintenance, respectively (Del Rey et al., 2013). Bath application of IL-10 in the VTA increases firing of dopaminergic, but not GABAergic, neurons (Williams et al., 2017). Signaling of TGF-β, an anti-inflammatory astrocyte-derived chemokine, also regulates excitatory and inhibitory synaptic balance of dopamine neurons (Luo et al., 2016). Action potential-independent vesicular GABA release in a subset of mouse central amygdala neurons is decreased by IL-1β, an effect that is reversed by co-administration of alcohol (Bajo et al., 2015). Although these initial findings are promising, there needs to be greater analysis of how glial activation, and subsequent release of cytokines and chemokines, alter complex circuit dynamics and the communication between reward-associated regions.
Glial mechanisms underlying behavioral changes after drug exposure
A rapidly growing body of literature has highlighted the potential role of glia in modulating behaviors associated with a variety of drugs, including opioids, alcohol, and stimulants. Animal behavioral models can provide critical information on the neurobiological underpinnings of drug dependence with high face and construct validity, although a full description of these paradigms is beyond the scope of this review, but see (Koob & Le Moal, 2008; Crabbe et al., 2011). Importantly, the knowledge gained from these preclinical models has the potential to guide the development of glial-targeted therapeutics in humans.
Since exposure to alcohol, opioids, and psychostimulants activates the neuroimmune system, preventing glial activation may prove to be a valuable therapeutic approach. Indeed, as shown in Table 3, the predominant effect of microglial inhibitors is to attenuate the rewarding and reinforcing properties of drugs of abuse. Minocycline is a blood-brain barrier permeable tetracycline antibiotic and microglial inhibitor that has been shown to have many inhibitory effects on opioid-associated behaviors. Minocycline and ibudilast, another glial inhibitor, inhibit morphine-induced dopamine release and the development of morphine CPP (Hutchinson et al., 2008; Bland et al., 2009). Naloxone-precipitated withdrawal in mice chronically treated with morphine is also attenuated by minocycline and ibudilast, and spontaneous withdrawal-induced weight loss and hyperactivity is reduced by glial inhibition. These changes are accompanied by reductions in morphine-induced CD11b and GFAP expression, measures of microglial and astrocytic activation, respectively. Interestingly, the analgesic effects of morphine and oxycodone are potentiated by ibudilast or minocycline, without any change in plasma morphine levels (Hutchinson et al., 2008; Hutchinson et al., 2009), suggesting that ibudilast may extend the intended analgesic effects of morphine, while dampening the unwanted rewarding effects. Alcohol consumption is also reduced by minocycline and ibudilast in the two-bottle choice, drinking-in-the-dark, and chronic intermittent access paradigms (Agrawal et al., 2011; Bell et al., 2015; Syapin et al., 2016). Ibudilast’s inhibitory effect on alcohol consumption is seen in multiple strains of mice and rats, including strains selectively bred for high alcohol consumption (Bell et al., 2015). Minocycline has also been shown to protect against ethanol-induced damage in the early postnatal brain, decreasing or blocking caspase-3 activation and expression of the pro-inflammatory markers monocyte chemoattractant protein 1 (MCP-1) and CCR2. Minocycline also protects against ethanol-induced activation of microglia, as measured by IBA1 and CD11b, and lysosomal phagocytic marker CD68 expression (Wang et al., 2017).
Table 3-.
Effects of pharmacological manipulation of glia or TLRs on drug-associated behaviors
| Drug of Abuse | Inhibitor | Measure - Rodents | Effect | Measure – Humans | Effect | References |
|---|---|---|---|---|---|---|
| Opioids | Minocycline | CPP | Hutchinson et al. 2008; 2009 | |||
| Spontaneous somatic withdrawal | ||||||
| Plasma drug levels | = | |||||
| Analgesia | ||||||
| Ibudilast | NAc dopamine release | Ratings of ‘restless’, ‘anxious’, ‘perspiring’, and ‘stomach cramps’ | Bland et al. 2009; Cooper et al. 2016; Metz et al. 2017 | |||
| Naloxone-precipitated somatic withdrawal | Craving | |||||
| Spontaneous somatic withdrawal | Subjective ‘liking’ | Hutchinson et al. 2008; 2009; Cooper et al. 2016; Metz et al. 2017 | ||||
| Plasma drug levels | = | Reinforcing effects | ||||
| Analgesia | Analgesia | |||||
| Positive subjective effects | = | |||||
| (+)-naloxone | NAc dopamine | Hutchinson et al. 2012; Tanda et al. 2016 | ||||
| CPP | Hutchinson et al. 2012 | |||||
| Self-administration | ||||||
| Analgesia | ||||||
| LPS-RS | Development of CPP | Chen et al. 2017 | ||||
| Expression of CPP | = | |||||
| (+)-naltrexone | NAc dopamine release | = | Tanda et al. 2016 | |||
| Cue-induced reinstatement | Theberge et al. 2013 | |||||
| Ceftriaxone | Hyperthermia | Rawls et al. 2007 | ||||
| Naloxone-precipitated somatic withdrawal | Rawls et al. 2010 | |||||
| Cue-induced reinstatement | Shen et al. 2014 | |||||
| N-acetylcysteine | Cue-induced reinstatement | Zhou and Kalivas 2008 | ||||
| Drug-induced reinstatement | Agrawal et al. 2011 | |||||
| Alcohol | Minocycline | Ethanol intake | Syapin et al. 2016 | |||
| Ibudilast | Ethanol intake | Cue-induced reinstatement of craving | Bell et al. 2015; Ray et al. 2017 | |||
| Stress-induced reinstatement of craving | ||||||
| Positive subjective effects | ||||||
| (+)-naloxone | Ethanol intake | = | Blednov et al. 2017; Harris et al. 2017 | |||
| LORR | Wu et al. 2012 | |||||
| T5342126 | Ethanol intake | Bajo et al. 2016 | ||||
| Ceftriaxone | Ethanol intake | Sari et al. 2011; 2013a; 2013b; Rao and Sari 2014; Das et al. 2015 | ||||
| CPP | Das et al. 2015 | |||||
| ‘Relapse-like’ drinking | Qrunfleh et al. 2013; Alhaddad et al. 2014 | |||||
| Cocaine | Minocycline | NAc dopamine release | Northcutt et al. 2015 | |||
| CPP | Northcutt et al. 2015 | |||||
| Ibudilast | Locomotor sensitization | Craving | Poland et al. 2016; Metz et al. 2017 | |||
| (+)-naloxone (+)-naltrexone | NAc dopamine release | Northcutt et al. 2015 | ||||
| Tanda et al. 2016 | ||||||
| CPP | Northcutt et al. 2015; Tanda et al. 2016 | |||||
| Self-administration | Northcutt et al. 2015; Tanda et al. 2016 | |||||
| Ceftriaxone | Cue-induced reinstatement | Sari et al. 2009; Knackstedt et al. 2010 | ||||
| Drug-induced reinstatement | Knackstedt et al. 2010 | |||||
| N-acetylcysteine | Drug-induced reinstatement | Craving | La Rowe et al. 2006; Madayag et al. 2007; Amen et al. 2007; Moussawi et al. 2011 | |||
| Cue-induced reinstatement | Moussawi et al. 2011; Reissner et al. 2015 | |||||
| Methamphetamine | Minocycline | Self-administration | Snider et al. 2013 | |||
| NAc dopamine release | Zhang et al. 2006 | |||||
| Locomotor sensitization | Zhang et al. 2006; Mizoguchi et al. 2008 | |||||
| = | Mizoguchi et al. 2008 | |||||
| Ibudilast | Hyperlocomotion | Positive subjective effects | Snider et al. 2012; Worley et al. 2016 | |||
| Locomotor sensitization | Snider et al. 2012 | |||||
| Self-administration | Snider et al. 2013 | |||||
| Drug-induced reinstatement | Beardsley et al. 2010 | |||||
| Stress-induced reinstatement | ||||||
| (+)-naltrexone | Cue-induced reinstatement | = | Theberge et al. 2013 | |||
| Ceftriaxone | Drug-induced reinstatement of CPP | Abulseoud et al. 2012 | ||||
| d-Amphetamine | Minocycline | Positive subjective effects | Soguoglu et al. 2011 | |||
| Self-administration | = | |||||
| Nicotine/Tobacco | Minocycline | Smoking behavior | = | Sofuoglu et al. 2009 | ||
| Metabolism | ||||||
| Physiological effects | ||||||
| Ratings of ‘depressed mood’ during withdrawal | ||||||
| Cigarette craving | ||||||
| Ibudilast | Craving | Cooper et al. 2017; Metz et al. 2017 | ||||
| Ceftriaxone | Reinstatement of CPP | Alajaji et al. 2013 | ||||
| Acquisition of CPP | = | |||||
| N-acetylcysteine | Self-administration | Craving | Schmaal et al. 2011; Gipson et al. 2013; Froeliger et al. 2015 | |||
| Reinstatement | = | |||||
| Positive affect | Froeliger et al. 2015 | |||||
| Withdrawal symptoms | = |
References in italics are human clinical studies, all others are preclinical animal studies. CPP = conditioned place preference; DID = drinking-in-the-dark; CIE = chronic intermittent ethanol; LPS-RS = lipopolysaccharide from Rhodobacter sphaeroides; 2BC = two-bottle choice; CIE-2BC = two-bottle choice after chronic intermittent ethanol exposure
Cocaine-associated behaviors are also impacted by minocycline and ibudilast. Minocycline attenuates cocaine-induced dopamine release and CPP (Northcutt et al., 2015), whereas ibudilast decreases cocaine-induced locomotor sensitization in both male and female rats (Poland et al., 2016). Both minocycline and ibudilast decrease methamphetamine self-administration, locomotor sensitization, reinstatement of drug-seeking, and the neurotoxic effects of methamphetamine. This effect is strongest when minocycline is administered before methamphetamine exposure, which may suggest that the timing of glial inhibition plays an important role in its effect (Zhang et al., 2006; Mizoguchi et al., 2008; Beardsley et al., 2010; Snider et al., 2012; Snider et al., 2013).
Manipulations of TLR4 function Modulate Drug-Associated Behaviors
TLRs are also a critical interface between drugs and glial activation that promotes drug consumption. TLRs are expressed on both astrocytes and microglia, but have higher expression on microglia. The TLR4, in particular, has garnered much interest in the addiction research field in the past decade (Figure 1). Blockade of TLR4s appears to generally decrease opioid-associated behaviors and accompanying neurochemical responses (Table 3). TLR4 antagonists prevent morphine-induced dopamine release in the NAc, a hallmark neurochemical signal of drug reward, as well as interfere with the acquisition and maintenance of morphine CPP and reduce self-administration of the opioid remifentanil (Hutchinson et al., 2012; Chen et al., 2017). This may be due to TLR4-mediated activation of the signaling pathway Signal Transducers and Transcription Activator 3 (STAT3) within the VTA, which has been implicated in regulating reward and motivated behavior (Chen et al., 2017). Similarly, chronic, but not acute, TLR4 blockade prevents incubation of heroin craving during withdrawal, suggesting that TLR4 antagonism does not directly alter the reinforcing properties of heroin but modulates withdrawal-associated synaptic remodeling (Theberge et al., 2013). Tanda et al. (2016) have reported that the TLR4 antagonists, (+)-naloxone and (+)-naltrexone, have no effect on heroin-induced dopamine release, suggesting that TLR4 may alter behavior through mechanisms outside of dopamine release.
Pharmacological and genetic manipulations of TLR4s have also implicated these receptors in alcohol-associated behaviors (Table 4). However, TLR4 signaling may have opposing modulatory roles on ethanol consumption as compared to ethanol-associated behaviors. Indeed, alcohol self-administration increases expression of the TLR4 protein in the VTA of alcohol-preferring rats (June et al., 2015). Mice lacking the TLR4 protein or the TLR4 adapter protein, MyD88, display lower sensitivity to the sedative and motor-impairing effects of alcohol compared to wild-type mice (Wu et al., 2012; Harris et al., 2017), whereas MYD88 knockout increases ethanol consumption (Blednov et al., 2017). TLR4 activation in the VTA and central amygdala, in particular, may be an important signal for binge alcohol consumption, as site-selective knockdown of TLR4 in these regions using siRNA reduces binge drinking in alcohol-preferring rats (Liu et al., 2011; June et al., 2015). Furthermore, genetic knockout of TLR2 or CD14, suppresses ethanol intake (Blednov et al., 2017). In contrast, some have reported no effect of pharmacological or genetic manipulation of TLR4 on excessive alcohol consumption in the two-bottle choice, drinking-in-the-dark, or chronic intermittent access paradigms (Blednov et al., 2017; Harris et al., 2017). Blednov et al. (2017) demonstrated that TLR4 mutant mice have unaltered alcohol consumption behavior, However, Blednov et al. (2017) reported significant effects of TLR4 knockout on total fluid consumption. This may suggest that TLR4 activity modulates general consummatory behavior. Similarly, although the TLR4 inhibitor, T5342126, decreased ethanol drinking in dependent mice, it also had non-specific effects on saccharin intake and locomotor activity (Bajo et al., 2016).. IL-1 receptor antagonists also reduce binge-like consumption of alcohol, suggesting that cytokine expression modulates the reinforcing effects of alcohol (Marshall et al., 2016). Taken together these studies demonstrate that immune-derived factors and receptors modulate alcohol consumption in certain paradigms, but the mechanisms underlying this are unclear.
Table 4-.
Effects of genetic manipulation of TLR4 signaling on drug-associated behaviors
| Drug of Abuse | Genotype | Measure | Effect | Notes | References |
|---|---|---|---|---|---|
| Alcohol | TLR4 −/− | LORR | Wu et al. 2012; Harris et al. 2017 | ||
| Self-administration | = | In dependent and non-dependent rats | Harris et al. 2017 | ||
| 2BC | = | Both sexes; reduced total fluid intake in females more | Blednov et al. 2017 | ||
| DID | Males; reduced total fluid intake | ||||
| CIE-2BC | Males and females | Harris et al. 2017 | |||
| Site specific TLR4 knockdown | 2BC | = | NAc knockdown | Harris et al. 2017 | |
| DID | VTA knockdown in P rats | June et al. 2015 | |||
| CeA knockdown in P rats | Liu et al. 2011; June et al. 2015 | ||||
| MYD88 −/− | LORR | Wu et al. 2012 | |||
| 2BC | Males; accompanied by reduction in saccharin preference | Blednov et al. 2017 | |||
| Females drinking 20% EtOH; reduced total fluid intake | |||||
| Ethanol preference | = | 2BC or DID; males and females | |||
| TLR2 −/− | 2BC | ||||
| DID | |||||
| CD14 −/− | 2BC | Increased total fluid intake | |||
| Cocaine | TLR4 −/− | CPP | Lower sensitivity; no persistent CPP | Kashima and Grueter 2017 | |
| TLR4 point mutation (C3H/HeJ) | Self-administration | Acquisition and progressive-ratio testing | Northcutt et al. 2015 |
LORR = loss of righting reflex; TLR4 = Toll-like receptor 4; 2BC = two-bottle choice; CIE-2BC; two-bottle choice after chronic intermittent ethanol exposure; DID = drinking-in-the-dark; NAc = nucleus accumbens; VTA = ventral tegmental area; CeA = central amygdala; VP = ventral pallidum; MYD88 = myelin differentiation primary response gene 88; P rats = alcohol preferring rats; TLR2 = Toll-like receptor 2; CD14 = cluster of differentiation 14; C3H/HeJ = mouse strain with point mutation preventing TLR-NFκB signaling
The exact role of TLR4 activation in modulating cocaine-associated behaviors is ambiguous, but TLR4 receptors are clearly important for cocaine reinforcement. The TLR4 antagonists, (+)-naloxone and (+)-naltrexone, have been shown to inhibit cocaine-induced dopamine release in the NAc and attenuate cocaine CPP and self-administration (Northcutt et al. 2015). Similarly, pharmacological blockade of TLR4s in the VTA reduces drug-primed reinstatement of cocaine-seeking, while activation of VTA TLR4s is sufficient to produce modest reinstatement of cocaine-seeking (Brown et al., 2018). However, another study using parameters similar to those in Northcutt et al. (2015) reported an opposite effect of (+)-naltrexone and (+)-naloxone on cocaine-induced dopamine release (Tanda et al., 2016). Interestingly, both groups reported that TLR4 blockade attenuated cocaine CPP and self-administration, although Tanda et al. (2016) reported that the ability of (+)-naltrexone and (+)-naloxone to attenuate cocaine self-administration required high doses that were accompanied by alterations in responding for food reward, which may suggest nonspecific effects of these antagonists. In contrast, genetic manipulation of TLR4 signaling lowered cocaine responding. In C3H/HEJ mice that have impaired TLR4-Nf-κβ signaling, cocaine reinforcement and motivation is lower compared to wild-type mice, without any change in sucrose self-administration (Northcutt et al., 2015). Complete knockout of TLR4 decreases sensitivity to the rewarding effects of cocaine, requiring higher doses to develop significant place preference compared to wild-type animals, and TLR4 knockout prevents the persistent enhancement of cocaine preference that is seen in wild-type controls (Kashima & Grueter, 2017). The authors suggest that alterations in cocaine reward learning in TLR4 knockouts is due to deficits in NMDAR-dependent long-term depression in the NAc core. TLR4 signaling may play a less prominent role in methamphetamine addiction, as TLR4 blockade has no effect on incubation of methamphetamine craving (Theberge et al., 2013). However, a growing body of literature suggests that glial activation and subsequent TLR4 signaling positively regulates opioid-, alcohol-, and cocaine-associated behaviors.
Manipulating Astrocytic Function Critically Alters Drug-Seeking
Astrocytes may also be important regulators of alcohol consumption. Disruption of astrocytic networks with gap-junction hemichannel blockers increases the motivation for alcohol self-administration, whereas activation of astrocytes using Gq-coupled DREADDs under the control of a GFAP promoter reduces alcohol self-administration (Bull et al., 2014). Similarly, stimulation of Gq-coupled DREADDSs on astrocytes in the NAc normalizes extracellular glutamate levels during cocaine abstinence through stimulation of release-regulating group II metabotropic glutamate autoreceptors to decrease cue-induced reinstatement of cocaine seeking (Scofield et al., 2015).
As discussed previously, astrocyte control of glutamate spillover is critically impaired following drug exposure (Scofield & Kalivas, 2014; Roberts-Wolfe & Kalivas, 2015; Scofield et al., 2016a). Psychostimulants, alcohol, and opioids all decrease expression of GLT-1, an astrocyte-specific glutamate transporter that clears glutamate from the synapse (Smith et al., 2015). Ceftriaxone, a β-lactam antibody that restores expression of GLT-1, decreases morphine tolerance and heroin-induced reinstatement of drug seeking (Rawls et al., 2007; Rawls et al., 2010; Shen et al., 2014), and has been found to reduce alcohol consumption and reduce ‘relapse-like drinking’ (Qrunfleh et al., 2013; Sari et al., 2013a; Sari et al., 2013b; Alhaddad et al., 2014; Rao & Sari, 2014; Das et al., 2015). Ceftriaxone can also inhibit cocaine and methamphetamine seeking (Sari et al., 2009; Knackstedt et al., 2010; Abulseoud et al., 2012) and prevents the reinstatement, but not the acquisition, of nicotine CPP (Alajaji et al., 2013). N-acetylcysteine (NAC), which restores glutamate homeostasis, also blocks drug-induced decreases in GLT-1, decreases nicotine self-administration (Gipson et al., 2013), and blocks reinstatement of nicotine, cocaine, and heroin seeking (Madayag et al., 2007; Zhou & Kalivas, 2008; Amen et al., 2011; Moussawi et al., 2011; Gipson et al., 2013; Reissner et al., 2015). In addition, mice expressing a dominant-negative SNARE protein selectively in astrocytes do not exhibit cocaine-induced reinstatement of CPP or cue-induced reinstatement of cocaine-seeking (Turner et al., 2013). Taken together, these studies demonstrate that astrocytes, and GLT-1 in particular, may be critical for reinstatement of drug seeking for multiple drugs of abuse.
Clinical implications and potential therapeutic targets
Neuroimmune activation has been found in individuals with substance use disorders, and the immune system represents a promising target for addiction therapeutics. Importantly, genome-wide association studies have demonstrated significant correlations between single nucleotide polymorphisms in cytokine genes, in particular IL-1β and IL-10, with alcohol, opioid, and cocaine use disorders (Pastor et al., 2005; Marcos et al., 2008; Liu et al., 2009; Smith & Humphries, 2009; Coller & Hutchinson, 2012). These genetic correlations suggest that glia may contribute to human addiction. In addition, the patterns of drug-associated alterations in glia that are observed in rodent models are also seen in humans. Indeed, chronic psychostimulant use can lead to astrogliosis and microgliosis, as measured by positron emission tomography or histopathological assessment at autopsy (Sekine et al., 2008; Büttner, 2011). However, some reports have found smaller or minimal effects of chronic methamphetamine use on astrocytes and microglia (Kitamura et al., 2010; Clark et al., 2013). Alcohol-dependent individuals have increased number of glial-associated markers, including IBA1, TLRs, MCP-1, and IL-1β (He & Crews, 2008). These glial changes are highly correlated with lifetime alcohol consumption and the age of drinking onset, with earlier initiation or greater use being associated with higher expression of TLR4s (Crews & Vetreno, 2016). In contrast to microglial-associated makers, alcohol-dependent individuals also display significant decreases in astrocyte number and GFAP intensity in the hippocampus and dorsolateral PFC (Korbo, 1999; Miguel-Hidalgo et al., 2002). Together, these data highlight critical interactions between drug use and the neuroimmune system in humans, and create an important backdrop for the use of glial inhibitors as potential cessation aids. Indeed, a growing body of clinical data has demonstrated the effectiveness of glial-targeted therapeutics in substance abuse (Table 3). These studies are small in number and typically have small sample sizes (i.e., <50 participants), but the results are promising.
One of the earliest clinical studies examining the influence of glial inhibitors on drug-associated behaviors showed that minocycline reduces the positive subjective effects of d-amphetamine, but has no effect on self-administration of d-amphetamine (Sofuoglu et al., 2011). However, this was tested in healthy non-users and may not have broader applicability to dependent users. It is not known whether minocycline influences consumption and/or craving and relapse in dependent cocaine or amphetamine users. Ibudilast’s effects on psychostimulant use is similarly understudied, although it has been shown to reduce the positive subjective effects of methamphetamine in dependent individuals (Worley et al., 2016) and to decrease cocaine craving in non-treatment seeking opioid-dependent individuals (Metz et al., 2017). Phase 2 clinical trials for 100 mg dosing of ibudilast in methamphetamine-dependent individuals are expected to begin in 2018 (NCT01860807).
Ibudilast is the most studied therapeutic to decrease opioid use, and shows promise as a pharmacotherapy to ameliorate withdrawal symptoms in opioid users. Indeed, in non-treatment seeking heroin-dependent volunteers, ibudilast lowered ratings of ‘anxious’, ‘perspiring’, ‘restless’, and ‘stomach cramps’ (Cooper et al., 2016), and also produced a slight reduction in the subjective ‘liking’, reinforcing effects, and ratings of craving for oxycodone in non-treatment seeking opioid-dependent volunteers (Metz et al., 2017). In parallel with what has been seen in animal models, ibudilast potentiated the analgesic effects of oxycodone in opioid-dependent volunteers (Cooper et al., 2017; Metz et al., 2017). These effects suggest that ibudilast may be a useful alongside opioid treatment to prevent dependence and aversive side effects. Ibudilasts’ effect on the subjective properties of oxycodone was inconsistent, but there was a general trend for ibudilast treatment to decrease craving for heroin, cocaine, and tobacco (Cooper et al., 2017; Metz et al., 2017). As such, Ibudilast may be useful as a maintenance therapy to prevent craving for a variety of drugs. Ibudilast may also decrease cue- and stress-induced reinstatement of alcohol craving, as well as decrease the subjective effects of alcohol in individuals with alcohol use disorder (Ray et al., 2017).
To date, only one study has examined minocycline’s effect on smoking behavior. In a small randomized, double-blind crossover study of non-treatment seeking smokers, Sofuoglu et al. (2009) found that minocycline does not affect smoking behavior, nicotine metabolism, or nicotine’s physiological effects, but it did decrease ratings of ‘depressed mood’ during withdrawal and reduced cigarette craving during intravenous nicotine self-administration and sample cigarette smoking. However, rather than being mediated by a direct glial mechanism, the authors suggest that minocycline effects on smoking behavior may be due to inhibition of the neuronal nitric oxide (NO) synthase enzyme, ultimately reducing NO production. A recent study by Cooper et al. (2017) found a trend for ibudilast to decrease tobacco craving in opioid-dependent individuals, but this was a secondary endpoint and a small effect. Metz et al. (2017) also showed a significant and dose-dependent decrease in tobacco craving by ibudilast treatment in opioid-dependent individuals, but the sample size was small (n=11).
Although relatively few studies have directly examined neuroimmune therapeutics in the treatment of substance use disorders, current data on ibudilast, in particular, show some promise. Future research should focus on how immune modulators can affect withdrawal, long-term abstinence, and relapse. Research that identifies markers and develops tools to accurately measure glial functioning in humans is also needed, as it is currently difficult to attribute the effectiveness of glial-targeted medications such as ibudilast or minocycline to direct alterations in glia.
Discussion
Interactions between neurons and glia are gaining increasing attention due to their involvement in brain function during health and disease. A growing body of literature demonstrates that glia may be key therapeutic targets that perpetuate addiction. Microglia and astrocytes have distinct and consequential changes following exposure to a wide range of drugs of abuse. The vast majority of drugs of abuse (opioids, alcohol, cocaine, and amphetamines) increase glial markers and promote a pro-inflammatory phenotype. However, It is important to note that drug-induced changes to glia are dependent on drug exposure timing, brain region, animal age, and type of drug. Many of the changes to glia induced by abused drugs are dependent on the immune receptor, TLR4, which identifies TLR4 and its downstream targets as potential therapeutic avenues. However, there are other mechanisms by which drugs of abuse alter glia that are not discussed in detail here, and it is reasonable to predict that more mechanisms underlying drug-induced glial changes will be discovered in the future.
It is paramount that we further investigate the role of glia during the different phases of drug use, from acquisition, to dependence, withdrawal, and reinstatement. This is of particular importance as it has been demonstrated for both alcohol and cocaine that timing of exposure has distinct effects on microglia and astrocytes. These differential responses may contribute to the divergent molecular, cellular, circuit, and behavioral changes observed in these different phases of drug use.
In addition, the relationship between glia and complex circuit dynamics is just beginning to be recognized. Foundational studies have shown glial-released cytokines alter synaptic number, connectivity, neurotransmitter release, receptor abundance, and neural response to stimuli. However, the consequence of these cytokine-induced changes to addiction behavior is exceedingly understudied. More recent development of viral, chemogenetic, and optogentic tools to specifically target and modulate glia will aid in answering key questions as to the role of glial mechanisms underlying addiction. Advancing our understanding of glial cells, and our ability to control them, should provide novel therapies for addiction.
Acknowledgements
The national institute of drugs of abuse (NIDA) is acknowledged for funding this work (R01DA040440). FL also receives UCI institutional support while serving as Dean of the Graduate Division.
Footnotes
Conflict of Interest
KL, SC, and FL have no competing interests to declare.
Data Accessibility
All original figures are available upon reasonable request from corresponding authors.
References
- Abulseoud OA, Miller JD, Wu J, Choi D-S & Holschneider DP (2012) Ceftriaxone upregulates the glutamate transporter in medial prefrontal cortex and blocks reinstatement of methamphetamine seeking in a condition place preference paradigm. Brain Res, 1456, 14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal RG, Hewetson A, George CM, Syapin PJ & Bergeson SE (2011) Minocycline reduces ethanol drinking. Brain Behav Immun, 25 Suppl 1, S165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alajaji M, Bowers MS, Knackstedt L & Damaj MI (2013) Effects of the beta-lactam antibiotic ceftriaxone on nicotine withdrawal and nicotine-induced reinstatement of preference in mice. Psychopharmacology (Berl), 228, 419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, Sanchez-Vera I & Guerri C (2010) Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J Neurosci, 30, 8285–8295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhaddad H, Das SC & Sari Y (2014) Effects of ceftriaxone on ethanol intake: a possible role for xCT and GLT-1 isoforms modulation of glutamate levels in P rats. Psychopharmacology (Berl), 231, 4049–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali C, Nicole O, Docagne F, Lesne S, MacKenzie ET, Nouvelot A, Buisson A & Vivien D (2000) Ischemia-induced interleukin-6 as a potential endogenous neuroprotective cytokine against NMDA receptor-mediated excitotoxicity in the brain. J Cereb Blood Flow Metab, 20, 956–966. [DOI] [PubMed] [Google Scholar]
- Alonso E, Garrido E, Díez-Fernández C, Pérez-García C, Herradón G, Ezquerra L, Deuel TF & Alguacil LF (2007) Yohimbine prevents morphine-induced changes of glial fibrillary acidic protein in brainstem and alpha2-adrenoceptor gene expression in hippocampus. Neurosci Lett, 412, 163–167. [DOI] [PubMed] [Google Scholar]
- Amen SL, Piacentine LB, Ahmad ME, Li S-J, Mantsch JR, Risinger RC & Baker DA (2011) Repeated N-acetyl cysteine reduces cocaine seeking in rodents and craving in cocaine-dependent humans. Neuropsychopharmacology, 36, 871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong V, Reichel CM, Doti JF, Crawford CA & McDougall SA (2004) Repeated amphetamine treatment causes a persistent elevation of glial fibrillary acidic protein in the caudate-putamen. Eur J Pharmacol, 488, 111–115. [DOI] [PubMed] [Google Scholar]
- Aurelian L, Warnock KT, Balan I, Puche A & June H (2016) TLR4 signaling in VTA dopaminergic neurons regulates impulsivity through tyrosine hydroxylase modulation. Transl Psychiatry, 6, e815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajo M, Montgomery SE, Cates LN, Nadav T, Delucchi AM, Cheng K, Yin H, Crawford EF, Roberts AJ & Roberto M (2016) Evaluation of TLR4 Inhibitor, T5342126, in Modulation of Ethanol-Drinking Behavior in Alcohol-Dependent Mice. Alcohol Alcohol, 51, 541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banisadr G, Quéraud-Lesaux F, Boutterin MC, Pélaprat D, Zalc B, Rostène W, Haour F & Parsadaniantz SM (2002) Distribution, cellular localization and functional role of CCR2 chemokine receptors in adult rat brain. J Neurochem, 81, 257–269. [DOI] [PubMed] [Google Scholar]
- Beardsley PM, Shelton KL, Hendrick E & Johnson KW (2010) The glial cell modulator and phosphodiesterase inhibitor, AV411 (ibudilast), attenuates prime- and stress-induced methamphetamine relapse. Eur J Pharmacol, 637, 102–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS & Malenka RC (2002) Control of synaptic strength by glial TNFalpha. Science, 295, 2282–2285. [DOI] [PubMed] [Google Scholar]
- Bell RL, Lopez MF, Cui C, Egli M, Johnson KW, Franklin KM & Becker HC (2015) Ibudilast reduces alcohol drinking in multiple animal models of alcohol dependence. Addict Biol, 20, 38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum HG, White AG, Schiller M, Waldman T, Cleveland JM, & Roland CL (2011) Societal costs of prescription opioid abuse, dependence, and misuse in the United States. Pain Med, 12, 657–667. [DOI] [PubMed] [Google Scholar]
- Blanco AM, Perez-Arago A, Fernandez-Lizarbe S & Guerri C (2008) Ethanol mimics ligand-mediated activation and endocytosis of IL-1RI/TLR4 receptors via lipid rafts caveolae in astroglial cells. J Neurochem, 106, 625–639. [DOI] [PubMed] [Google Scholar]
- Bland ST, Hutchinson MR, Maier SF, Watkins LR & Johnson KW (2009) The glial activation inhibitor AV411 reduces morphine-induced nucleus accumbens dopamine release. Brain Behav Immun, 23, 492–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Black M, Chernis J, Da Costa A, Mayfield J & Harris RA (2017) Ethanol consumption in mice lacking CD14, TLR2, TLR4, or myd88. Alcohol Clin Exp Res, 41, 516–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray JG, Reyes KC, Roberts AJ, Ransohoff RM & Gruol DL (2013) Synaptic plasticity in the hippocampus shows resistance to acute ethanol exposure in transgenic mice with astrocyte-targeted enhanced CCL2 expression. Neuropharmacology, 67, 115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KT, Levis SC, O’Neill CE, Northcutt AL, Fabisiak TJ, Watkins LR, & Bachtell RK (2018) Innate immune signaling in the ventral tegmental area contributes to drug-primed reinstatement of cocaine seeking. Brain Behav Immun, 67, 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull C, Freitas KCC, Zou S, Poland RS, Syed WA, Urban DJ, Minter SC, Shelton KL, Hauser KF, Negus SS, Knapp PE & Bowers MS (2014) Rat nucleus accumbens core astrocytes modulate reward and the motivation to self-administer ethanol after abstinence. Neuropsychopharmacology, 39, 2835–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büttner A (2011) Review: The neuropathology of drug abuse. Neuropathol Appl Neurobiol, 37, 118–134. [DOI] [PubMed] [Google Scholar]
- Cavaillon JM (2001) Pro- versus anti-inflammatory cytokines: myth or reality. Cell Mol Biol (Noisy-le-grand), 47, 695–702. [PubMed] [Google Scholar]
- Cearley CN, Blindheim K, Sorg BA, Krueger JM & Churchill L (2011) Acute cocaine increases interleukin-1β mRNA and immunoreactive cells in the cortex and nucleus accumbens. Neurochem Res, 36, 686–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (2016) Excessive Drinking is Draining the U.S. Economy Available at: https://www.cdc.gov/features/costsofdrinking/ Accessed March 6, 2018.
- Chai H, Diaz-Castro B, Shigetomi E, Monte E, Octeau JC, Yu X, Cohn W, Rajendran PS, Vondriska TM, Whitelegge JP, Coppola G & Khakh BS (2017) Neural Circuit-Specialized Astrocytes: Transcriptomic, Proteomic, Morphological, and Functional Evidence. Neuron, 95, 531–549.e539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J-X, Huang K-M, Liu M, Jiang J-X, Liu J-P, Zhang Y-X, Yang C, Xin W-J & Zhang X-Q (2017) Activation of TLR4/STAT3 signaling in VTA contributes to the acquisition and maintenance of morphine-induced conditioned place preference. Behav Brain Res, 335, 151–157. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CCA, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P & Barres BA (2005) Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell, 120, 421–433. [DOI] [PubMed] [Google Scholar]
- Clark KH, Wiley CA & Bradberry CW (2013) Psychostimulant abuse and neuroinflammation: emerging evidence of their interconnection. Neurotoxicity Research, 23, 174–188. [DOI] [PubMed] [Google Scholar]
- Coller JK & Hutchinson MR (2012) Implications of central immune signaling caused by drugs of abuse: mechanisms, mediators and new therapeutic approaches for prediction and treatment of drug dependence. Pharmacol Ther, 134, 219–245. [DOI] [PubMed] [Google Scholar]
- Cooper ZD, Johnson KW, Pavlicova M, Glass A, Vosburg SK, Sullivan MA, Manubay JM, Martinez DM, Jones JD, Saccone PA & Comer SD (2016) The effects of ibudilast, a glial activation inhibitor, on opioid withdrawal symptoms in opioid-dependent volunteers. Addict Biol, 21, 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper ZD, Johnson KW, Vosburg SK, Sullivan MA, Manubay J, Martinez D, Jones JD, Saccone PA & Comer SD (2017) Effects of ibudilast on oxycodone-induced analgesia and subjective effects in opioid-dependent volunteers. Drug Alcohol Depend, 178, 340–347. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Harris RA & Koob GF (2011) Preclinical studies of alcohol binge drinking. Ann N Y Acad Sci, 1216, 24–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT & Vetreno RP (2016) Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology (Berl), 233, 1543–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C, Shurtleff D & Harris RA (2014) Neuroimmune mechanisms of alcohol and drug addiction. Int Rev Neurobiol, 118, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SC, Yamamoto BK, Hristov AM & Sari Y (2015) Ceftriaxone attenuates ethanol drinking and restores extracellular glutamate concentration through normalization of GLT-1 in nucleus accumbens of male alcohol-preferring rats. Neuropharmacology, 97, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Biase LM, Schuebel KE, Fusfeld ZH, Jair K, Hawes IA, Cimbro R, Zhang HY, Liu QR, Shen H, Xi ZX, Goldman D & Bonci A (2017) Local cues establish and maintain region-specific phenotypes of basal ganglia microglia. Neuron, 95, 341–356.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Rey A, Balschun D, Wetzel W, Randolf A & Besedovsky HO (2013) A cytokine network involving brain-borne IL-1β, IL-1ra, IL-18, IL-6, and TNFα operates during long-term potentiation and learning. Brain Behav Immun, 33, 15–23. [DOI] [PubMed] [Google Scholar]
- Dunn AJ (2006) Effects of cytokines and infections on brain neurochemistry. Clin Neurosci Res, 6, 52–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattore L, Puddu MC, Picciau S, Cappai A, Fratta W, Serra GP & Spiga S (2002) Astroglial in vivo response to cocaine in mouse dentate gyrus: a quantitative and qualitative analysis by confocal microscopy. Neuroscience, 110, 1–6. [DOI] [PubMed] [Google Scholar]
- Felger JC & Miller AH (2012) Cytokine effects on the basal ganglia and dopamine function: the subcortical source of inflammatory malaise. Front Neuroendocrinol, 33, 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke H (1995) Influence of chronic alcohol treatment on the GFAP-immunoreactivity in astrocytes of the hippocampus in rats. Acta Histochem, 97, 263–271. [DOI] [PubMed] [Google Scholar]
- Gipson CD, Reissner KJ, Kupchik YM, Smith ACW, Stankeviciute N, Hensley-Simon ME & Kalivas PW (2013) Reinstatement of nicotine seeking is mediated by glutamatergic plasticity. Proc Natl Acad Sci U S A, 110, 9124–9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gipson CD, Kupchik YM & Kalivas PW (2014) Rapid, transient synaptic plasticity in addiction. Neuropharmacology, 76 Pt B, 276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves J, Martins T, Ferreira R, Milhazes N, Borges F, Ribeiro CF, Malva JO, Macedo TR & Silva AP (2008) Methamphetamine-induced early increase of IL-6 and TNF-alpha mRNA expression in the mouse brain. Ann N Y Acad Sci, 1139, 103–111. [DOI] [PubMed] [Google Scholar]
- Guerra-Gomes S, Sousa N, Pinto L & Oliveira JF (2017) Functional roles of astrocyte calcium elevations: from synapses to behavior. Front Cell Neurosci, 11, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyon A, Skrzydelski D, De Giry I, Rovère C, Conductier G, Trocello JM, Daugé V, Kitabgi P, Rostène W, Nahon JL & Mélik Parsadaniantz S (2009) Long term exposure to the chemokine CCL2 activates the nigrostriatal dopamine system: a novel mechanism for the control of dopamine release. Neuroscience, 162, 1072–1080. [DOI] [PubMed] [Google Scholar]
- Harris RA, Bajo M, Bell RL, Blednov YA, Varodayan FP, Truitt JM, de Guglielmo G, Lasek AW, Logrip ML, Vendruscolo LF, Roberts AJ, Roberts E, George O, Mayfield J, Billiar TR, Hackam DJ, Mayfield RD, Koob GF, Roberto M & Homanics GE (2017) Genetic and pharmacologic manipulation of TLR4 has minimal impact on ethanol consumption in rodents. J Neurosci, 37, 1139–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J & Crews FT (2008) Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp Neurol, 210, 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzerling K Trial of ibudilast for methamphetamine dependence Identification No. NCT01860807. Available at: https://clinicaltrials.gov/show/NCT01860807. Accessed February 4, 2018.
- Hutchinson MR, Northcutt AL, Chao LW, Kearney JJ, Zhang Y, Berkelhammer DL, Loram LC, Rozeske RR, Bland ST, Maier SF, Gleeson TT & Watkins LR (2008) Minocycline suppresses morphine-induced respiratory depression, suppresses morphine-induced reward, and enhances systemic morphine-induced analgesia. Brain Behav Immun, 22, 1248–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Lewis SS, Coats BD, Skyba DA, Crysdale NY, Berkelhammer DL, Brzeski A, Northcutt A, Vietz CM, Judd CM, Maier SF, Watkins LR & Johnson KW (2009) Reduction of opioid withdrawal and potentiation of acute opioid analgesia by systemic AV411 (ibudilast). Brain Behav Immun, 23, 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Northcutt AL, Hiranita T, Wang X, Lewis SS, Thomas J, van Steeg K, Kopajtic TA, Loram LC, Sfregola C, Galer E, Miles NE, Bland ST, Amat J, Rozeske RR, Maslanik T, Chapman TR, Strand KA, Fleshner M, Bachtell RK, Somogyi AA, Yin H, Katz JL, Rice KC, Maier SF & Watkins LR (2012) Opioid activation of toll-like receptor 4 contributes to drug reinforcement. J Neurosci, 32, 11187–11200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC & Nestler EJ (2006) Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci, 29, 565–598. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Thorpe R & Mirsky R (1984) Molecular identity, distribution and heterogeneity of glial fibrillary acidic protein: an immunoblotting and immunohistochemical study of Schwann cells, satellite cells, enteric glia and astrocytes. J Neurocytol, 13, 187–200. [DOI] [PubMed] [Google Scholar]
- June HL, Liu J, Warnock KT, Bell KA, Balan I, Bollino D, Puche A & Aurelian L (2015) CRF-amplified neuronal TLR4/MCP-1 signaling regulates alcohol self-administration. Neuropsychopharmacology, 40, 1549–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW (2009) The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci, 10, 561–572. [DOI] [PubMed] [Google Scholar]
- Kane CJM, Phelan KD, Douglas JC, Wagoner G, Johnson JW, Xu J, Phelan PS & Drew PD (2014) Effects of ethanol on immune response in the brain: region-specific changes in adolescent versus adult mice. Alcohol Clin Exp Res, 38, 384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima DT & Grueter BA (2017) Toll-like receptor 4 deficiency alters nucleus accumbens synaptic physiology and drug reward behavior. Proc Natl Acad Sci U S A, 114, 8865–8870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JA & Malenka RC (2007) Synaptic plasticity and addiction. Nat Rev Neurosci, 8, 844–858. [DOI] [PubMed] [Google Scholar]
- Khakh BS & Sofroniew MV (2015) Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci, 18, 942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T-H, Kim S-J & Lee S-M (2014) Stimulation of the α7 nicotinic acetylcholine receptor protects against sepsis by inhibiting Toll-like receptor via phosphoinositide 3-kinase activation. J Infect Dis, 209, 1668–1677. [DOI] [PubMed] [Google Scholar]
- Kim SK, Nabekura J & Koizumi S (2017) Astrocyte-mediated synapse remodeling in the pathological brain. Glia, 65, 1719–1727. [DOI] [PubMed] [Google Scholar]
- King JR, Gillevet TC & Kabbani N (2017) A G protein-coupled α7 nicotinic receptor regulates signaling and TNF-α release in microglia. FEBS Open Bio, 7, 1350–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura O, Takeichi T, Wang EL, Tokunaga I, Ishigami A & Kubo S. i. (2010) Microglial and astrocytic changes in the striatum of methamphetamine abusers. Leg Med (Tokyo), 12, 57–62. [DOI] [PubMed] [Google Scholar]
- Knackstedt LA, Melendez RI & Kalivas PW (2010) Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine seeking. Biol Psychiatry, 67, 81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF & Le Moal M (2008) Addiction and the brain antireward system. Annu Rev Psychol, 59, 29–53. [DOI] [PubMed] [Google Scholar]
- Koob GF & Volkow ND (2010) Neurocircuitry of addiction. Neuropsychopharmacology, 35, 217–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbo L (1999) Glial cell loss in the hippocampus of alcoholics. Alcohol Clin Exp Res, 23, 164–168. [PubMed] [Google Scholar]
- Kourrich S, Rothwell PE, Klug JR & Thomas MJ (2007) Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci, 27, 7921–7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, Chakraborty C, Workman G, Weaver M, Sage EH, Barres BA & Eroglu C (2011) Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci U S A, 108, E440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacagnina MJ, Rivera PD & Bilbo SD (2017) Glial and neuroimmune mechanisms as critical modulators of drug use and abuse. Neuropsychopharmacology, 42, 156–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P & Gordon S (1990) Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience, 39, 151–170. [DOI] [PubMed] [Google Scholar]
- Lazriev IL, Kiknadze GI, Kutateladze II & Nebieridze MI (2001) Effect of morphine on the number and branching of astrocytes in various regions of rat brain. Bull Exp Biol Med, 131, 248–250. [DOI] [PubMed] [Google Scholar]
- Lee MS & Kim Y-J (2007) Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu Rev Biochem, 76, 447–480. [DOI] [PubMed] [Google Scholar]
- Lewitus GM, Konefal SC, Greenhalgh AD, Pribiag H, Augereau K & Stellwagen D (2016) Microglial TNF-α Suppresses Cocaine-Induced Plasticity and Behavioral Sensitization. Neuron, 90, 483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Han X, Bao J, Liu Y, Ye A, Thakur M & Liu H (2016) Nicotine increases eclampsia-like seizure threshold and attenuates microglial activity in rat hippocampus through the α7 nicotinic acetylcholine receptor. Brain Res, 1642, 487–496. [DOI] [PubMed] [Google Scholar]
- Liao K, Guo M, Niu F, Yang L, Callen SE & Buch S (2016) Cocaine-mediated induction of microglial activation involves the ER stress-TLR2 axis. J Neuroinflammation, 13, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA & Barres BA (2017) Reactive astrocytes: production, function, and therapeutic potential. Immunity, 46, 957–967. [DOI] [PubMed] [Google Scholar]
- Linker KE Wood ME, Tawadrous P, Leslie FM (2017) Nicotine increases limbic microglial expression and cocaine self-administration by a D2 receptor mechanism. Poster: Society for Neuroscience
- Liu J, Yang AR, Kelly T, Puche A, Esoga C, June HL, Elnabawi A, Merchenthaler I, Sieghart W, June HL & Aurelian L (2011) Binge alcohol drinking is associated with GABAA alpha2-regulated Toll-like receptor 4 (TLR4) expression in the central amygdala. Proc Natl Acad Sci U S A, 108, 4465–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Hutchinson MR, White JM, Somogyi AA & Coller JK (2009) Association of IL-1B genetic polymorphisms with an increased risk of opioid and alcohol dependence. Pharmacogenet Genomics, 19, 869–876. [DOI] [PubMed] [Google Scholar]
- Luo SX, Timbang L, Kim J-I, Shang Y, Sandoval K, Tang AA, Whistler JL, Ding JB & Huang EJ (2016) TGF-n increased risk of opioid and alcohol dependence. ciated with GABAA atory-Inhibitory Synaptic Balance, and Reversal Learning. Cell Rep, 17, 3233–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C & Malenka RC (2011) Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron, 69, 650–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madayag A, Lobner D, Kau KS, Mantsch JR, Abdulhameed O, Hearing M, Grier MD & Baker DA (2007) Repeated N-acetylcysteine administration alters plasticity-dependent effects of cocaine. J Neurosci, 27, 13968–13976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcos M, Pastor I, González-Sarmiento R & Laso FJ (2008) Interleukin-10 gene polymorphism is associated with alcoholism but not with alcoholic liver disease. Alcohol Alcohol, 43, 523–528. [DOI] [PubMed] [Google Scholar]
- Marie-Claire C, Courtin C, Roques BP & Noble F (2004) Cytoskeletal genes regulation by chronic morphine treatment in rat striatum. Neuropsychopharmacology, 29, 2208–2215. [DOI] [PubMed] [Google Scholar]
- Marshall SA, Casachahua JD, Rinker JA, Blose AK, Lysle DT, & Thiele TE (2016) IL-1 receptor signaling in the basolateral amygdala modulates binge-like ethanol consumption in male C57BL/6J mice. Brain Behav Immun, 51, 258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín R, Bajo-Grañeras R, Moratalla R, Perea G & Araque A (2015) Circuit-specific signaling in astrocyte-neuron networks in basal ganglia pathways. Science, 349, 730–734. [DOI] [PubMed] [Google Scholar]
- Metz VE, Jones JD, Manubay J, Sullivan MA, Mogali S, Segoshi A, Madera G, Johnson KW & Comer SD (2017) Effects of Ibudilast on the Subjective, Reinforcing, and Analgesic Effects of Oxycodone in Recently Detoxified Adults with Opioid Dependence. Neuropsychopharmacology, 42, 1825–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguel-Hidalgo JJ, Wei J, Andrew M, Overholser JC, Jurjus G, Stockmeier CA & Rajkowska G (2002) Glia pathology in the prefrontal cortex in alcohol dependence with and without depressive symptoms. Biol Psychiatry, 52, 1121–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguel-Hidalgo JJ (2009) The role of glial cells in drug abuse. Curr Drug Abuse Rev, 2, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto N, Maki T, Shindo A, Liang AC, Maeda M, Egawa N, Itoh K, Lo EK, Lok J, Ihara M & Arai K (2015) Astrocytes Promote Oligodendrogenesis after White Matter Damage via Brain-Derived Neurotrophic Factor. J Neurosci, 35, 14002–14008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi H, Takuma K, Fukakusa A, Ito Y, Nakatani A, Ibi D, Kim H-C & Yamada K (2008) Improvement by minocycline of methamphetamine-induced impairment of recognition memory in mice. Psychopharmacology (Berl), 196, 233–241. [DOI] [PubMed] [Google Scholar]
- Moussawi K, Zhou W, Shen H, Reichel CM, See RE, Carr DB & Kalivas PW (2011) Reversing cocaine-induced synaptic potentiation provides enduring protection from relapse. Proc Natl Acad Sci U S A, 108, 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurauter G, Schröcksnadel K, Scholl-Bürgi S, Sperner-Unterweger B, Schubert C, Ledochowski M & Fuchs D (2008) Chronic immune stimulation correlates with reduced phenylalanine turnover. Curr Drug Metab, 9, 622–627. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F & Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science, 308, 1314–1318. [DOI] [PubMed] [Google Scholar]
- Noda M & Kobayashi AI (2017) Nicotine inhibits activation of microglial proton currents via interactions with α7 acetylcholine receptors. J Physiol Sci, 67, 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh H, Jeon J & Seo H (2014) Systemic injection of LPS induces region-specific neuroinflammation and mitochondrial dysfunction in normal mouse brain. Neurochem Int, 69, 35–40. [DOI] [PubMed] [Google Scholar]
- Northcutt AL, Hutchinson MR, Wang X, Baratta MV, Hiranita T, Cochran TA, Pomrenze MB, Galer EL, Kopajtic TA, Li CM, Amat J, Larson G, Cooper DC, Huang Y, O’Neill CE, Yin H, Zahniser NR, Katz JL, Rice KC, Maier SF, Bachtell RK & Watkins LR (2015) DAT isn’t all that: cocaine reward and reinforcement require Toll-like receptor 4 signaling. Mol Psychiatry, 20, 1525–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterndorff-Kahanek EA, Becker HC, Lopez MF, Farris SP, Tiwari GR, Nunez YO, Harris RA & Mayfield RD (2015) Chronic ethanol exposure produces time- and brain region-dependent changes in gene coexpression networks. PLoS ONE, 10, e0121522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D & Gross CT (2011) Synaptic pruning by microglia is necessary for normal brain development. Science, 333, 1456–1458. [DOI] [PubMed] [Google Scholar]
- Papouin T, Dunphy JM, Tolman M, Dineley KT & Haydon PG (2017) Septal Cholinergic Neuromodulation Tunes the Astrocyte-Dependent Gating of Hippocampal NMDA Receptors to Wakefulness. Neuron, 94, 840–854.e847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual-Lucas M, Fernandez-Lizarbe S, Montesinos J & Guerri C (2014) LPS or ethanol triggers clathrin- and rafts/caveolae-dependent endocytosis of TLR4 in cortical astrocytes. J Neurochem, 129, 448–462. [DOI] [PubMed] [Google Scholar]
- Pastor IJ, Laso FJ, Romero A & González-Sarmiento R (2005) Interleukin-1 gene cluster polymorphisms and alcoholism in Spanish men. Alcohol Alcohol, 40, 181–186. [DOI] [PubMed] [Google Scholar]
- Phillips PEM, Stuber GD, Heien MLAV, Wightman RM & Carelli RM (2003) Subsecond dopamine release promotes cocaine seeking. Nature, 422, 614–618. [DOI] [PubMed] [Google Scholar]
- Pizzi M, Sarnico I, Boroni F, Benarese M, Dreano M, Garotta G, Valerio A & Spano P (2004) Prevention of neuron and oligodendrocyte degeneration by interleukin-6 (IL-6) and IL-6 receptor/IL-6 fusion protein in organotypic hippocampal slices. Mol Cell Neurosci, 25, 301–311. [DOI] [PubMed] [Google Scholar]
- Pocock JM & Kettenmann H (2007) Neurotransmitter receptors on microglia. Trends Neurosci, 30, 527–535. [DOI] [PubMed] [Google Scholar]
- Poland RS, Hahn Y, Knapp PE, Beardsley PM & Bowers MS (2016) Ibudilast attenuates expression of behavioral sensitization to cocaine in male and female rats. Neuropharmacology, 109, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribiag H & Stellwagen D (2013) TNF-α downregulates inhibitory neurotransmission through protein phosphatase 1-dependent trafficking of GABA(A) receptors. J Neurosci, 33, 15879–15893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L & Crews FT (2012) Chronic ethanol increases systemic TLR3 agonist-induced neuroinflammation and neurodegeneration. J Neuroinflammation, 9, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qrunfleh AM, Alazizi A & Sari Y (2013) Ceftriaxone, a beta-lactam antibiotic, attenuates relapse-like ethanol-drinking behavior in alcohol-preferring rats. J Psychopharmacol (Oxford), 27, 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez K, Fornaguera-Trías J & Sheridan JF (2017) Stress-Induced Microglia Activation and Monocyte Trafficking to the Brain Underlie the Development of Anxiety and Depression. Curr Top Behav Neurosci, 31, 155–172. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM & El Khoury J (2015) Microglia in health and disease. Cold Spring Harb Perspect Biol, 8, a020560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao PSS & Sari Y (2014) Effects of ceftriaxone on chronic ethanol consumption: a potential role for xCT and GLT1 modulation of glutamate levels in male P rats. J Mol Neurosci, 54, 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao PS, Bell RL, Engleman EA, & Sari Y (2015a) Targeting glutamate uptake to treat alcohol use disorder [DOI] [PMC free article] [PubMed]
- Rao PSS, Goodwani S, Bell RL, Wei Y, Boddu SHS & Sari Y (2015b) Effects of ampicillin, cefazolin and cefoperazone treatments on GLT-1 expressions in the mesocorticolimbic system and ethanol intake in alcohol-preferring rats. Neuroscience, 295, 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawls SM, Tallarida R, Robinson W & Amin M (2007) The beta-lactam antibiotic, ceftriaxone, attenuates morphine-evoked hyperthermia in rats. Br J Pharmacol, 151, 1095–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawls SM, Baron DA & Kim J (2010) beta-Lactam antibiotic inhibits development of morphine physical dependence in rats. Behav Pharmacol, 21, 161–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray LA, Bujarski S, Shoptaw S, Roche DJ, Heinzerling K & Miotto K (2017) Development of the Neuroimmune Modulator Ibudilast for the Treatment of Alcoholism: A Randomized, Placebo-Controlled, Human Laboratory Trial. Neuropsychopharmacology, 42, 1776–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissner KJ, Gipson CD, Tran PK, Knackstedt LA, Scofield MD & Kalivas PW (2015) Glutamate transporter GLT-1 mediates N-acetylcysteine inhibition of cocaine reinstatement. Addict Biol, 20, 316–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds R & Herschkowitz N (1987) Oligodendroglial and astroglial heterogeneity in mouse primary central nervous system culture as demonstrated by differences in GABA and D-aspartate transport and immunocytochemistry. Brain Res, 433, 13–25. [DOI] [PubMed] [Google Scholar]
- Risher M-L, Sexton HG, Risher WC, Wilson WA, Fleming RL, Madison RD, Moore SD, Eroglu C & Swartzwelder HS (2015) Adolescent Intermittent Alcohol Exposure: Dysregulation of Thrombospondins and Synapse Formation are Associated with Decreased Neuronal Density in the Adult Hippocampus. Alcohol Clin Exp Res, 39, 2403–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts-Wolfe DJ & Kalivas PW (2015) Glutamate Transporter GLT-1 as a Therapeutic Target for Substance Use Disorders. CNS Neurol Disord Drug Targets, 14, 745–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santello M and Volterra A (2012). TNFα in synaptic function: switching gears. Trends in Neurosciences 35(10), 638–647. [DOI] [PubMed] [Google Scholar]
- Santello M, Bezzi P and Volterra A (2011). TNFα controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron 69(5), 988–1001. [DOI] [PubMed] [Google Scholar]
- Sari Y, Smith KD, Ali PK & Rebec GV (2009) Upregulation of GLT1 attenuates cue-induced reinstatement of cocaine-seeking behavior in rats. J Neurosci, 29, 9239–9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sari Y, Franklin KM, Alazizi A, Rao PSS & Bell RL (2013a) Effects of ceftriaxone on the acquisition and maintenance of ethanol drinking in peri-adolescent and adult female alcohol-preferring (P) rats. Neuroscience, 241, 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sari Y, Sreemantula SN, Lee MR & Choi D-S (2013b) Ceftriaxone treatment affects the levels of GLT1 and ENT1 as well as ethanol intake in alcohol-preferring rats. J Mol Neurosci, 51, 779–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM & Bilbo SD (2013) Adolescent morphine exposure affects long-term microglial function and later-life relapse liability in a model of addiction. J Neurosci, 33, 961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Smith SH & Bilbo SD (2013) FACS analysis of neuronal-glial interactions in the nucleus accumbens following morphine administration. Psychopharmacology (Berl), 230, 525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scofield MD & Kalivas PW (2014) Astrocytic dysfunction and addiction: consequences of impaired glutamate homeostasis. Neuroscientist, 20, 610–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scofield MD, Boger HA, Smith RJ, Li H, Haydon PG & Kalivas PW (2015) Gq-DREADD Selectively Initiates Glial Glutamate Release and Inhibits Cue-induced Cocaine Seeking. Biol Psychiatry, 78, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scofield MD, Heinsbroek JA, Gipson CD, Kupchik YM, Spencer S, Smith ACW, Roberts-Wolfe D & Kalivas PW (2016a) The Nucleus Accumbens: Mechanisms of Addiction across Drug Classes Reflect the Importance of Glutamate Homeostasis. Pharmacol Rev, 68, 816–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scofield MD, Li H, Siemsen BM, Healey KL, Tran PK, Woronoff N, Boger HA, Kalivas PW & Reissner KJ (2016b) Cocaine Self-Administration and Extinction Leads to Reduced Glial Fibrillary Acidic Protein Expression and Morphometric Features of Astrocytes in the Nucleus Accumbens Core. Biol Psychiatry, 80, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine Y, Ouchi Y, Sugihara G, Takei N, Yoshikawa E, Nakamura K, Iwata Y, Tsuchiya KJ, Suda S, Suzuki K, Kawai M, Takebayashi K, Yamamoto S, Matsuzaki H, Ueki T, Mori N, Gold MS & Cadet JL (2008) Methamphetamine causes microglial activation in the brains of human abusers. J Neurosci, 28, 5756–5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H. w., Scofield MD, Boger H, Hensley M & Kalivas PW (2014) Synaptic glutamate spillover due to impaired glutamate uptake mediates heroin relapse. J Neurosci, 34, 5649–5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J. x. & Yakel JL (2012) Functional α7 nicotinic ACh receptors on astrocytes in rat hippocampal CA1 slices. J Mol Neurosci, 48, 14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shytle RD, Mori T, Townsend K, Vendrame M, Sun N, Zeng J, Ehrhart J, Silver AA, Sanberg PR & Tan J (2004) Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem, 89, 337–343. [DOI] [PubMed] [Google Scholar]
- Skrzydelski D, Guyon A, Daugé V, Rovère C, Apartis E, Kitabgi P, Nahon JL, Rostène W & Parsadaniantz SM (2007) The chemokine stromal cell-derived factor-1/CXCL12 activates the nigrostriatal dopamine system. J Neurochem, 102, 1175–1183. [DOI] [PubMed] [Google Scholar]
- Smith AJP & Humphries SE (2009) Cytokine and cytokine receptor gene polymorphisms and their functionality. Cytokine Growth Factor Rev, 20, 43–59. [DOI] [PubMed] [Google Scholar]
- Smith ACW, Scofield MD & Kalivas PW (2015) The tetrapartite synapse: Extracellular matrix remodeling contributes to corticoaccumbens plasticity underlying drug addiction. Brain Res, 1628(Pt A), 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider SE, Vunck SA, van den Oord EJCG, Adkins DE, McClay JL & Beardsley PM (2012) The glial cell modulators, ibudilast and its amino analog, AV1013, attenuate methamphetamine locomotor activity and its sensitization in mice. Eur J Pharmacol, 679, 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider SE, Hendrick ES & Beardsley PM (2013) Glial cell modulators attenuate methamphetamine self-administration in the rat. Eur J Pharmacol, 701, 124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV & Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol, 119, 7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV (2014) Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist, 20, 160–172. [DOI] [PubMed] [Google Scholar]
- Sofuoglu M, Waters AJ, Mooney M & O’Malley SS (2009) Minocycline reduced craving for cigarettes but did not affect smoking or intravenous nicotine responses in humans. Pharmacol Biochem Behav, 92, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofuoglu M, Mooney M, Kosten T, Waters A & Hashimoto K (2011) Minocycline attenuates subjective rewarding effects of dextroamphetamine in humans. Psychopharmacology (Berl), 213, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI & O’Callaghan JP (2006) Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: role of TNF-alpha. FASEB J, 20, 670–682. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY & Malenka RC (2005) Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci, 25, 3219–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SWM & Barres BA (2007) The classical complement cascade mediates CNS synapse elimination. Cell, 131, 1164–1178. [DOI] [PubMed] [Google Scholar]
- Syapin PJ, Martinez JM, Curtis DC, Marquardt PC, Allison CL, Groot JA, Baby C, Al-Hasan YM, Segura I, Scheible MJ, Nicholson KT, Redondo JL, Trotter DRM, Edwards DS & Bergeson SE (2016) Effective reduction in high ethanol drinking by semisynthetic tetracycline derivatives. Alcohol Clin Exp Res, 40, 2482–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliaferro P, Vega MD, Evrard SG, Ramos AJ & Brusco A (2002) Alcohol exposure during adulthood induces neuronal and astroglial alterations in the hippocampal CA-1 area. Ann N Y Acad Sci, 965, 334–342. [DOI] [PubMed] [Google Scholar]
- Tanda G, Mereu M, Hiranita T, Quarterman JC, Coggiano M & Katz JL (2016) Lack of Specific Involvement of (+)-Naloxone and (+)-Naltrexone on the Reinforcing and Neurochemical Effects of Cocaine and Opioids. Neuropsychopharmacology, 41, 2772–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theberge FR, Li X, Kambhampati S, Pickens CL, St Laurent R, Bossert JM, Baumann MH, Hutchinson MR, Rice KC, Watkins LR & Shaham Y (2013) Effect of chronic delivery of the Toll-like receptor 4 antagonist (+)-naltrexone on incubation of heroin craving. Biol Psychiatry, 73, 729–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DM, Dowgiert J, Geddes TJ, Francescutti-Verbeem D, Liu X & Kuhn DM (2004) Microglial activation is a pharmacologically specific marker for the neurotoxic amphetamines. Neurosci Lett, 367, 349–354. [DOI] [PubMed] [Google Scholar]
- Tremblay M-È, Lowery RL & Majewska AK (2010) Microglial interactions with synapses are modulated by visual experience. PLoS Biol, 8, e1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trocello JM, Rostene W, Melik-Parsadaniantz S, Godefroy D, Roze E, Kitabgi P, Kuziel WA, Chalon S, Caboche J & Apartis E (2011) Implication of CCR2 chemokine receptor in cocaine-induced sensitization. J Mol Neurosci, 44, 147–151. [DOI] [PubMed] [Google Scholar]
- Trotta T, Porro C, Calvello R & Panaro MA (2014) Biological role of Toll-like receptor-4 in the brain. J Neuroimmunol, 268, 1–12. [DOI] [PubMed] [Google Scholar]
- Turner JR, Ecke LE, Briand LA, Haydon PG & Blendy JA (2013) Cocaine-related behaviors in mice with deficient gliotransmission. Psychopharmacology (Berl), 226, 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi E, Agrawal R, Nath C & Shukla R (2010) Effect of melatonin on neuroinflammation and acetylcholinesterase activity induced by LPS in rat brain. Eur J Pharmacol, 640, 206–210. [DOI] [PubMed] [Google Scholar]
- Udomuksorn W, Mukem S, Kumarnsit E, Vongvatcharanon S & Vongvatcharanon U (2011) Effects of alcohol administration during adulthood on parvalbumin and glial fibrillary acidic protein immunoreactivity in the rat cerebral cortex. Acta Histochem, 113, 283–289. [DOI] [PubMed] [Google Scholar]
- Vander Weele CM, Porter-Stransky KA, Mabrouk OS, Lovic V, Singer BF, Kennedy RT & Aragona BJ (2014) Rapid dopamine transmission within the nucleus accumbens: dramatic difference between morphine and oxycodone delivery. Eur J Neurosci, 40, 3041–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Wise RA, & Baler R (2017) The dopamine motive system: implications for drug and food addiction. Nat Rev Neurosci, 18, 741–752. [DOI] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S & Nabekura J (2009) Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci, 29, 3974–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Loram LC, Ramos K, de Jesus AJ, Thomas J, Cheng K, Reddy A, Somogyi AA, Hutchinson MR, Watkins LR & Yin H (2012) Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc Natl Acad Sci U S A, 109, 6325–6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang Y, Peng Y, Hutchinson MR, Rice KC, Yin H & Watkins LR (2016) Pharmacological characterization of the opioid inactive isomers (+)-naltrexone and (+)-naloxone as antagonists of toll-like receptor 4. Br J Pharmacol, 173, 856–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang K, Yang F, Ren Z, Xu M, Frank JA, Ke Z-J & Luo J (2017) Minocycline protects developing brain against ethanol-induced damage. Neuropharmacology, 129, 84–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Rice KC & Maier SF (2009) The “toll” of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci, 30, 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SB, Pistorius SS, Anderson EQ, Clarke ER, Clark TJ, Hope S, & Steffensen SC (2017) Acute ethanol activates microglia and affects the excitability of ventral tegmental area neurons. Poster: Society for Neuroscience
- Worley MJ, Heinzerling KG, Roche DJO, & Shoptaw S (2016) Ibudilast attenuates subjective effects of methamphetamine in placebo-controlled inpatient study. Drug Alcohol Depend, 162, 245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Lousberg EL, Moldenhauer LM, Hayball JD, Coller JK, Rice KC, Watkins LR, Somogyi AA & Hutchinson MR (2012) Inhibiting the TLR4-MyD88 signalling cascade by genetic or pharmacological strategies reduces acute alcohol-induced sedation and motor impairment in mice. Br J Pharmacol, 165, 1319–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Dissing-Olesen L, MacVicar BA & Stevens B (2015) Microglia: dynamic mediators of synapse development and plasticity. Trends Immunol, 36, 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York EM, Bernier L-P & MacVicar BA (2017) Microglial modulation of neuronal activity in the healthy brain. Dev Neurobiol [DOI] [PubMed]
- Xu X, Bishop EE, Kennedy SM, Simpson SA, Pechacek TF (2014) Annual healthcare spending attributable to cigarette smoking: an update. Am J Prev Med, 48, 326–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Kitaichi K, Fujimoto Y, Nakayama H, Shimizu E, Iyo M & Hashimoto K (2006) Protective effects of minocycline on behavioral changes and neurotoxicity in mice after administration of methamphetamine. Prog Neuropsychopharmacol Biol Psychiatry, 30, 1381–1393. [DOI] [PubMed] [Google Scholar]
- Zhou W & Kalivas PW (2008) N-acetylcysteine reduces extinction responding and induces enduring reductions in cue- and heroin-induced drug-seeking. Biol Psychiatry, 63, 338–340. [DOI] [PMC free article] [PubMed] [Google Scholar]