

Summary
DNA repair deficiency leads to genome instability and hence human disease. Depletion of the RNA processing factor Y14/RBM8A in cultured cells or Rbm8a haplodeficiency in the developing mouse cortex results in the accumulation of DNA damage. Y14 depletion differentially affected the expression of DNA damage response (DDR) factors and induced R-loops, both of which threaten genomic stability. Immunoprecipitation coupled with mass spectrometry revealed DDR factors as potential Y14-interacting partners. Further results confirmed that Y14 interacts with Ku and several DDR factors in an ATM-dependent manner. Y14 co-fractionated with Ku in chromatin-enriched fractions and further accumulated on chromatin upon DNA damage. Y14 knockdown delayed recruitment of DDR factors to DNA damage sites and formation of γH2AX foci and also led to Ku retention on chromatin. Accordingly, Y14 depletion compromised the efficiency of DNA end joining. Therefore Y14 likely plays a direct role in DNA damage repair via its interaction with DDR factors.
Subject Areas: Biological Sciences, Molecular Biology, Cell Biology
Graphical Abstract
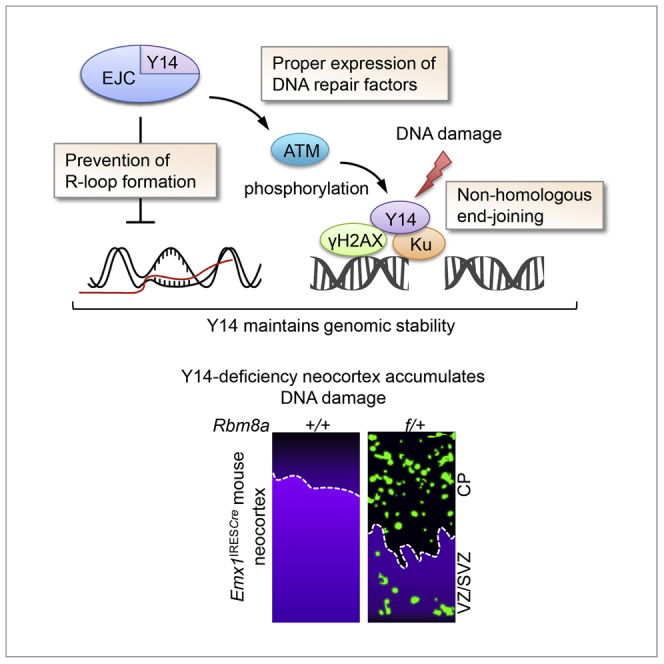
Highlights
-
•
Y14 deficiency leads to DNA damage accumulation
-
•
Y14 depletion disturbs DNA damage response and induces R-loops
-
•
Y14 promotes Ku70/80 recruitment to DNA damage sites
-
•
Y14 participates in DNA damage repair
Biological Sciences; Molecular Biology; Cell Biology
Introduction
Genome stability can be threatened by a variety of DNA lesions, which may result from exogenous DNA-damaging agents or endogenous causes such as DNA repair factor dysfunction or oxidative stress. Failure to repair DNA damage leads to a wide spectrum of human diseases and developmental disorders (Jackson and Bartek, 2009). Cellular response to DNA damage is initiated by damage recognition followed by complex signaling cascades that determine repair mechanisms, and ultimately, DNA repair factors are recruited to damaged sites for DNA repair (Chapman et al., 2012).
Double-strand breaks (DSBs) are one of the most deleterious DNA lesions. DSB repair essentially involves homologous recombination or non-homologous end joining (NHEJ), which require overlapping but distinct sets of DNA repair factors (Chapman et al., 2012). Homologous recombination is more prevalent during cell cycle phases S and G2, whereas NHEJ is active throughout the entire cell cycle and thus constitutes one of the major DNA repair pathways (Chapman et al., 2012). The sensor complexes, Mre11/Rad50/Nbs1 (MRN) and Ku70/Ku80 (abbreviated Ku70/80), recognize DSB ends and recruit respective DNA damage response (DDR) and repair factors. In NHEJ, the complex of Ku70/80 and DNA-dependent protein kinase (DNA-PK) recruits the end-processing ligation complex containing XLF, XRCC4, and DNA ligase IV, which rejoins broken ends. In addition, DDR signaling may activate cell cycle checkpoints or apoptosis.
Accumulating evidence indicates that RNA processing factors contribute both directly and indirectly to genome stability (Naro et al., 2015, Wickramasinghe and Venkitaraman, 2016). Several splicing factors modulate alternative splicing of DDR or apoptosis factors (Shkreta and Chabot, 2015). Depletion of RNA processing factors results in the formation of R-loops that are prone to DNA damage (Santos-Pereira and Aguilera, 2015). Moreover, RNA processing factors may have a direct role in DNA repair via their interactions with DNA repair factors (Naro et al., 2015, Wickramasinghe and Venkitaraman, 2016). Some of these RNA processing factors are recruited to DNA damage sites in a manner that is dependent on DDR-kinase-mediated phosphorylation or poly-ADP ribosylation. Recent reports indicate the intriguing role of RNA molecules in DNA damage repair (Hawley et al., 2017). Therefore a set of RNA processing factors may participate in small RNA biogenesis around DSBs or coordinate with non-coding RNAs in DDR. These findings underscore the importance of RNA-processing-mediated DNA repair.
The RNA processing factor Y14, encoded by Rbm8a, plays multiple roles in mRNA metabolism. Essentially, Y14 acts as a core factor of the exon-junction complex (EJC), which is involved in nonsense-mediated mRNA decay and mRNA translation (Chuang et al., 2015). The EJC components, including Y14, also participate in alternative splicing of apoptosis factors, of which the spliced isoforms have opposing functions in apoptosis (Michelle et al., 2012, Wang et al., 2014, Fukumura et al., 2016). Y14 depletion in general induces a switch to pro-apoptotic isoforms and leads to cell death. Mouse genetic studies show that Y14 is critical for embryonic cortical development (Mao et al., 2015, Mao et al., 2016). Rbm8a haploinsufficiency in mouse embryonic brain causes cell death and reduces the number of neural progenitors and neurons. Depletion of Y14 in cultured cells increases the number of sub-G1 phase cells and ultimately leads to apoptosis (Ishigaki et al., 2013, Lu et al., 2017). Moreover, Y14-depleted cells spontaneously accumulate DSBs and exhibit hypersensitivity to DNA-damaging agents (Lu et al., 2017). Therefore, we attempted to explore the potential role of Y14 in the maintenance of genome integrity. We uncovered the interaction of Y14 with DNA damage repair factors and demonstrated its unprecedented role in DNA damage repair and DDR signaling.
Results
Y14 Depletion Results in Cumulative DNA Damage and Reduced Cell Viability and Proliferation Capacity
We previously showed that Y14 depletion increases the level of phosphorylated H2AX (γH2AX) and apoptosis in HeLa cells (Lu et al., 2017). HeLa cells exhibit diminished p53 function, and depletion of Y14 by small interfering RNA (siRNA)-induced p53β, a splice isoform of p53, to a great extent (Lu et al., 2017; Figure 1A, lane 2). We therefore evaluated the aforementioned aspects using human osteosarcoma U2OS cells, which express functional p53 and exhibited only a minimal level of p53β upon Y14 depletion (Figure 1A, lane 4). Y14 depletion consistently increased the level of γH2AX in both cell lines, although U2OS had a lower basal γH2AX level (Figure 1A). This observation was consistent with immunofluorescence, which shows a higher background level of γH2AX foci in HeLa cells than U2OS cells. Y14 depletion, nevertheless, increased the signal of γH2AX foci in both cells (Figure S1). Clonogenic assay revealed that Y14 depletion significantly reduced survival of both cell lines (Figure 1B). Therefore, Y14-depletion-induced DNA damage and cell growth inhibition may be irrespective of p53 status.
Figure 1.

Y14 Deficiency Results in Cumulative DNA Damage, Reduced Cell Viability, and Impaired Neurosphere Formation
(A) HeLa and U2OS cells were transfected with control siRNA (siC) or siY14. Immunoblotting shows γH2AX and p53 in both short and long exposures and Y14 and α-tubulin. Asterisk indicates p53β.
(B) Clonogenic assay was performed in siRNA-transfected HeLa and U2OS cells. The bar graph shows relative colony-forming units (percentage; mean ± SD). N indicates the number of replicates.
(C) E13.5 dorsal neocortices of Emx1IREScre and Emx1IREScre;Rbm8af/+ mice were subjected to immunostaining using antibodies against γH2AX and Pax6 and Hoechst staining. Dashed line indicates the boundary of the ventricular zone/subventricular zone (VZ/SVZ) and the cortical plate (CP). Scale bar, 50 μm.
(D) Primary cells dissociated from the dorsal neocortices as in (C) were subjected to immunostaining using antibodies against Pax6 and γH2AX as well as Hoechst staining (also see Figure S2F). Representative magnified images show Pax6+, γH2AX+, and double-positive cells of Emx1IREScre;Rbm8af/+ without Hoechst staining. Scale bar in, 10 μm in (D and E). Bar graphs show percentage of γH2AX+ cells among Pax6+ cells (mean ± SD). (D–F) The number of cells analyzed is indicated above the bars; cells were obtained from three pairs of littermates.
(E) As in (D), immunostaining was performed using anti-Pax6 and anti-cleaved caspase 3 (CC3) (also see Figure S1G). Representative magnified images show Pax6+, CC3+, and double-positive cells. Bar graphs show percentage of CC3+ cells among Pax6+ cells (mean ± SD).
(F) Neurosphere formation was performed using dissociated cells from E13.5 dorsal neocortices as in (C) (scale bar, 200 μm). Stacked bar graph shows percentage of different sizes (<100 μm, 100–200 μm, and >200 μm) of neurospheres.
In all bar graphs of Figures 1, 2, 3, 4, 5, 6, and 7, p values are as follows: *p < 0.05, **p < 0.01, ***p <0.001.
Meanwhile, we assessed Y14-depletion-induced DNA damage in animal models. It has been reported that Rbm8a haploinsufficiency causes apoptosis of neural progenitor cells in the embryonic cerebral cortex (Mao et al., 2015). We speculated that Y14-deficient neocortex has accumulative DNA damage, which leads to cell death. To test this hypothesis, we generated Rbm8af/+ mice (Supplemental Information) as previously reported (Mao et al., 2015), for which loxP insertion was confirmed by genotyping and sequencing (Figures S2A and S2B). Rbm8af/+ mice were mated with Emx1IREScre mice (Gorski et al., 2002) to generate Emx1IREScre;Rbm8af/+. Immunoblotting of Emx1IREScre;Rbm8af/+ cortical plates at embryonic day (E) 13.5 revealed reduced Y14 expression (Figure S2C). As reported (Mao et al., 2015), the Emx1IREScre;Rbm8af/+ brain at postnatal day 10 exhibited microcephaly compared with their littermate controls (Figure S2D). This Rbm8a haploinsufficiency significantly increased the level of γH2AX and activated caspase 3 (CC3) in the neocortex at E13.5 (Figures 1C and S2E, respectively). Immunostaining of dissociated cell cultures of the E13.5 neocortex showed an ∼50% reduction of Pax6-positive radial glia cells in Emx1IREScre;Rbm8af/+ (Figure S2F), as reported (Mao et al., 2015). Nevertheless, higher levels of γH2AX and CC3 were detected in the remaining Pax6-positive cells (Figures 1D and S2G upper for γH2AX, and Figures 1E and S2G lower for CC3). Accordingly, Rbm8a haploinsufficiency impaired the expansion of neural stem or progenitor cells to form neurospheres (Figure 1F), which might have in part resulted from accumulated DNA damage and cell apoptosis. Together, we provided evidence from both cultured cells and animal models for the role of Y14 in genome stability. Y14 deficiency has a detrimental effect on cell viability and neural stem cell proliferation.
Y14 Depletion Causes DNA Damage, Perturbs the DDR, and Induces R-Loops
To quantify DNA damage in Y14-depleted cells, we performed the neutral comet assay. The result showed that Y14 depletion generated fragmented DNA, i.e., comet tail, in HeLa cells even without ionizing radiation (IR) treatment (Figure 2A). Y14-depleted MCF7 and U2OS cells exhibited different levels of DNA damage (Figure S3A), which may reflect their different repair or tolerance capacities. IR treatment caused an increase in olive tail moment in both control and Y14-depleted HeLa cells, but the latter exhibited a significantly higher level of DNA damage after IR (Figure 2B). Consistently, IR treatment reduced cell viability of both HeLa and U2OS cells (Figure S3B), indicating that Y14 loss increased cell sensitivity to DNA damage.
Figure 2.

Y14 Depletion Causes DNA Damage, Induces R-Loops, and Perturbs DDR
(A) The comet assay of siRNA-transfected HeLa cells. Immunofluorescence shows SYBR Green staining of DNA. (A, C, and E) Immunoblotting shows Y14 and α-tubulin.
(B) siRNA-transfected HeLa cells as in (A) were either not treated (−) or treated with 2 Gy IR followed by 16-h recovery. Box and whiskers plot shows the comet tail moment; mean ± SD were from ∼300 cells in three independent experiments.
(C) Immunoblotting of siC- or siY14-transfected HeLa cell lysates was performed using antibodies against the indicated proteins.
(D) siRNA-transfected HeLa cells were treated with 10 Gy IR followed by recovery for 30 min. Immunofluorescence was performed using anti-53BP1 and anti-Y14. Arrowheads, Y14-depleted cells. Scale bar, 10 μm. The number (mean ± SD) of 53BP1 foci per cell is shown in the bar graph; ∼200 cells were counted in three independent experiments.
(E) siRNA-transfected HeLa cells were subjected to immunofluorescence using antibody S9.6; the images were merged with DAPI staining (scale bar, 10 μm). Box and whiskers plot shows quantification of nuclear (left) and mock or actinomycin D-treated nucleolus-excluding nucleoplasmic (right) S9.6 signals. For each transfection, ∼100 cells were scored from three independent experiments.
Y14 depletion differentially affects gene expression (Michelle et al., 2012, Wang et al., 2014, Fukumura et al., 2016, Lu et al., 2017). Therefore we examined the expression of a panel of DDR signaling factors in Y14-depleted HeLa cells. Immunoblotting showed that Y14 depletion induced p53β and γH2AX, as previously reported (Lu et al., 2017) and reduced the level of three major upstream DDR kinases, namely, ATM, ATR, and DNA-PK (Figure 2C). A similar result was obtained in Y14-depleted MCF7 and U2OS cells (Figure S4A). Moreover, a reduction in the phosphorylation level of the two checkpoint kinases, Chk1 and Chk2, in Y14-depleted HeLa cells supported defective DDR signaling (Figure 2C). In addition, we observed that the level of 53BP1, but not Rad51, was also reduced in Y14-depleted cells (Figures 2C and S4A). Immunofluorescence further indicated the reduction in IR-induced 53BP1 foci in Y14-depleted HeLa cells (Figure 2D).
R-loops are RNA-DNA hybrids that form naturally during transcription and can act as a source of genomic instability (Santos-Pereira and Aguilera, 2015). Using immunofluorescence with antibody S9.6, we observed that Y14 depletion enhanced R-loop signals in both the nucleus and the cytoplasm (Figure 2E, HeLa; Figure S5A, MCF7 and U2OS cells). Dot blot analysis confirmed that the R-loop signal was increased by ∼1.7-fold upon Y14 depletion and diminished after treatment with RNase H (Figure S5B). Although R-loops were abundant in the nucleoli, the nucleoplasmic R-loop signal was still visibly increased (Figures 2E and S5C for the images without DAPI staining). Such a signal was reduced by actinomycin D treatment (Figures S5C, and 2E, bar graph at the right), suggesting that R-loops form in a transcription-dependent manner. Overexpression of RNase H1 reduced γH2AX foci in Y14-depleted HeLa cells (Figure S5D), as observed in SRSF2-depleted cells (Sridhara et al., 2017), suggesting that R-loops contribute to Y14-deficiency-induced DNA damage. A DNA-RNA immunoprecipitation (DRIP)-coupled quantitative PCR (qPCR) analysis has previously shown that silencing of BRCA2 or THOC1 promotes R-loop accumulation at actively transcribed genes (Bhatia et al., 2014). Using DRIP-qPCR, we observed that Y14 depletion similarly increased R-loops in several transcriptionally active genes tested, including Egr1, Apoe, and Rpl13A, but not significantly in Btdb19 and an intergenic negative control (Figure S5E). Our results thus indicated a potential role for Y14 in preventing co-transcriptional R-loop formation. Together, Y14 deficiency likely causes DNA damage, compromises DDR, and induces R-loops, all of which can lead to genome instability.
As Y14 is a part of the EJC, we also evaluated the effect of eIF4A3. Indeed, depletion of eIF4A3 caused DNA damage, downregulated several DDR proteins, and induced R-loops, as observed in Y14 depletion (Figures S3C, S4B, and S5F). This result was not unexpected, because it is well recognized that RNA processing factors contribute to genome stability via different mechanisms (Naro et al., 2015, Shkreta and Chabot, 2015, Wickramasinghe and Venkitaraman, 2016). However, whether Y14 and eIF4A3 function independently or inter-dependently in such events remains to be determined.
Y14 Interacts with DNA Damage Repair Factors
To explore the mechanism of Y14 depletion-induced DNA damage, we performed immunoprecipitation in combination with mass spectrometry using overexpressed FLAG-Y14 in HEK293 cells. Y14 co-precipitated proteins included the components of the EJC, the methylosome, the transcription factor/nuclear export (THOC) complexes, splicing factors, and ribosomal proteins, consistent with previous reports (reviewed in Chuang et al., 2015) (Table S1). Moreover, we identified a set of proteins implicated in DNA repair, including Ku70/80, Trim28, and Rad23a (Table 1). We first focused on the heterodimeric Ku complex owing to its prominent function in DSB repair and the maintenance of genomic stability (Jackson and Bartek, 2009). Immunoprecipitation of FLAG-Y14 followed by immunoblotting confirmed the interaction between Y14 with Ku70/80 and Trim28 as well as the DNA damage sensor and signaling factors (such as the MRN complex, MDC1, and γH2AX) and Ku-associated NHEJ factors (XRCC4, XLF, and DNA ligase IV) (Figure 3A, lane 5). We also demonstrated the interaction between endogenous Y14 and Ku80 (Figure 3B). However, eIF4A3, TAP, or Upf3b could not interact with Ku (Figures 3A, lane 6; S6A), emphasizing the specific interaction between Y14 and Ku.
Table 1.
Identification of Y14-interacting Proteins Implicated in DNA Damage Response, Repair, and Metabolism
| Protein Name | Accession No. | Peptide No. | Function |
|---|---|---|---|
| Ku80 | P13010 | 5 | DSB repair |
| Ku70 | P12956 | 3 | DSB repair |
| BRCA2 | P51587 | 3 | DNA damage repair |
| Trim28 | Q13263 | 3 | Transcriptional regulation DNA damage repair |
| RFC2 | P35250 | 3 | Replication factor |
| MCM8 | Q9UJA3 | 2 | DNA replication licensing factor |
| MCM10 | Q7L590 | 2 | DNA replication licensing factor |
| Rad23a | P54725 | 1 | Nucleotide excision repair |
| RFC3 | P40938 | 1 | Replication factor |
Figure 3.

Y14 Interacts with DNA Damage Repair Factors
(A) HEK293 cells were transfected with empty vector (vec) or vector encoding FLAG-Y14 or FLAG-eIF4A3. Cell lysates were subjected to immunoprecipitation with anti-FLAG, followed by immunoblotting using antibodies against FLAG or indicated proteins.
(B) HEK293 cell lysates were subjected to immunoprecipitation using control IgG or anti-Ku or anti-Y14, followed by immunoblotting. Asterisk indicates the immunoglobulin light chain.
(C) HEK293 cells were transfected with the empty or FLAG-Y14 vector. Immunoprecipitation was performed using anti-FLAG in the cell lysates in the absence (−) or presence (+) of RNase A, followed by immunoblotting.
(D) Transfected cells as in (C) were left untreated or were treated with the ATM inhibitor KU55933, followed by 10 Gy irradiation. The cell lysates were each subjected to immunoprecipitation using anti-FLAG, followed by immunoblotting.
(E) In vitro pull-down assay was performed by incubating GST or GST-Y14 (full-length, ΔN, ΔC, or phosphorylated) with the HeLa cell lysate. Bound proteins were analyzed by immunoblotting using anti-Ku70/80. GST fusions were detected by Coomassie blue staining.
(F) HEK293 cells were transfected with the empty (vec) or FLAG-Y14 vector. FLAG-Y14 transfectants were mock treated or treated with the RNA polymerase II inhibitor DRB. Anti-FLAG immunoprecipitates were subjected to immunoblotting.
(G) Model shows the role of Y14 in a positive circuit of DDR signaling. Y14 promotes the expression of several DDR factors including ATM, which is likely required for Y14 function in DNA damage repair.
Next, we characterized the nature of the interaction between Y14 and Ku. Intriguingly, the Y14-Ku interaction was completely disrupted by RNase treatment regardless of DNA damage (Figures 3C and S6B). Moreover, DNA damage did not particularly enhance their interaction (Figure 3D, lane 11). Inhibition of the DDR kinase ATM by KU55933, however, abolished the interaction of Y14 with Ku and γH2AX (Figure 3D, lane 12). Similar result was observed by depletion of ATM using siRNA (Figure S6C). ATM inactivation appeared to abolish the phosphorylation of overexpressed Y14 (Figure S6D). The C-terminal arginine/serine dipeptides are the major phosphorylation of Y14 in cells (Hsu Ia et al., 2005). To evaluate the effect of this C-terminal phosphorylation on the Y14-Ku interaction, we performed an in vitro pull-down assay using recombinant glutathione-S-transferase-Y14 fusion as bait. The result showed that full-length unphosphorylated Y14 could interact with Ku80, albeit barely, and, nevertheless, the C-terminal region was critical for the interaction (Figure 3E, lanes 4 and 5). Strikingly, phosphorylation of Y14 (see Transparent Methods) greatly enhanced its interaction with Ku70/80 (lane 6), supporting the observation in cells. Finally, we evaluated the interaction between Y14 and Trim28, which functions in both transcription and DNA repair (Bunch and Calderwood, 2015). We observed that the transcription inhibitor DRB disrupted the interaction of Y14 with RNA polymerase II (RNA pol II) and Trim28, but not γH2AX (Figure 3F), suggesting that the Y14-Trim28 interaction is more likely involved in transcriptional regulation.
Together, our results revealed an EJC-independent but RNA-dependent interaction of Y14 with Ku, and, moreover, suggested a positive feedback loop between Y14 and DDR signaling factors, which likely promotes genome stability (Figure 3G and see Discussion).
Y14 Associates with Chromatin and Further Accumulates on Chromatin after DNA Damage
Next, we accessed subnuclear localization of Y14 and Ku by fractionation of HeLa cell lysates (Brown et al., 2015). The result showed that Y14, but not eIF4A3 or Magoh, co-fractionated with Ku70/80 in the chromatin-enriched fraction (Figure 4A, lane 2). After RNase treatment, the vast majority of Ku70/80 was derived from chromatin, but Y14 still remained (lane 3). A similar result was observed in MCF7 and HEK293 cells (Figure S7A). Therefore, RNA-independent chromatin association of Y14 suggested that it may directly interact with chromatin factor(s) of unknown identity (Figure S7B, upper). Nevertheless, we observed that IR or phleomycin treatment, which also causes DSBs, increased the level of Y14, Ku, and γH2AX on chromatin even after RNase treatment (Figures 4B for IR; S7C for phleomycin; Figure S7B, bottom). The ATM inhibitor KU55933 significantly reduced γH2AX signals on chromatin and slightly reduced the level of chromatin-bound Y14 (Figure 4C). Therefore, ATM-dependent Ku interaction and chromatin association of Y14 suggested its direct role in DNA repair.
Figure 4.

Y14 Associates with Chromatin and Accumulates on Chromatin after DNA Damage
(A) Immunoblotting was performed using the whole-cell extract (WCE) and RNase A-treated (+) and non-treated (−) chromatin fractions (CSK) of HeLa cells.
(B) HeLa cells were mock-irradiated (−) or irradiated with 10 Gy followed by recovery for 0, 1, or 2 h. Immunoblotting shows the indicated proteins in the WCE and RNase-treated (CSK + R) and non-treated (CSK) chromatin fractions.
(C) HeLa cells were mock-treated or treated with KU55933 followed by IR irradiation and subsequent recovery. The WCE and RNase-treated chromatin fraction (CSK + R) were subjected to immunoblotting. Bar graph shows the relative level of chromatin-bound Y14 without (lanes 9–12) or after KU55933 treatment (lanes 13–16); lane 9 was set as 1.
Y14 Depletion Delays the Formation of DNA Damage Foci and Recruitment of DNA Repair Factors
Next, we evaluated the formation of DNA damage foci in Y14- or eIF4A3-depleted HeLa cells. Immunofluorescence using anti-Y14 or anti-eIF4A3 was able to distinguish transfected from non-transfected cells (Figure 5A; dashed circle for Y14- or eIF4A3-depleted cells and arrowhead for non-transfected cells). IR exposure induced γH2AX foci in non-transfected cells within 5 min (Figure 5A, arrowheads). γH2AX foci also formed in eIF4A3-depleted but not in Y14-depleted cells (Figure 5A, dashed circles, and Figure 5B, 3 min), suggesting that Y14 depletion specifically affects the formation of γH2AX foci. We performed a time course experiment and observed that the focus number was raised in Y14-depleted cells between 15 and 30 min after exposure (Figure 5B), indicating that Y14 depletion delayed, but did not completely block, γH2AX foci formation. This delay may have partially resulted from defective DDR signaling in Y14-deficient cells, although there still could be a direct role for Y14 in recruiting DNA repair factors to damaged sites.
Figure 5.

Y14 Depletion Delays the Formation of DNA Damage Foci and Recruitment of DNA Repair Factors
(A) HeLa cells were transfected with an siRNA targeting Y14 or eIF4A3 for 48 h and then left non-irradiated or irradiated at 10 Gy. Cells were fixed 3 min to 4 h (see B) after IR treatment, followed by immunofluorescence using antibodies against Y14, eIF4A3, and γH2AX and Hoechst staining. Representative images show 3-min post-IR. Dashed circle indicate Y14- or eIF4A3-depleted cells, and arrowheads indicate non-transfected cells; three selected cells are indicated. Scale bar, 20 μm.
(B) The experiment was as described in (A). The fluorescence intensities of γH2AX were scored using ImageJ from at least 30 cells per condition. Bar graph shows the relative γH2AX intensity (mean ± SD) of Y14-depleted cells versus non-depleted cells at the indicated time points.
(C–E) U2OS cells that stably expressed the GFP fusion with (C) MDC1, (D) Ku70, or (E) Ku80 were transiently transfected with the Y14 shRNA-mCherry-expressing vectors. Cells were subjected to laser microirradiation (405 nm) followed by live-cell imaging using confocal microscopy. Representative confocal images show accumulation of GFP fusion proteins at sites (dashed lines) of laser microirradiation at the indicated time points. NT indicates samples before microirradiation. The intensity (high, arrowhead; low, empty triangle) of mCherry represents the expression level of shY14. Curve graphs show fluorescence intensities of GFP-fusion proteins at the irradiated region that were quantified periodically, normalized, and presented as mean ± SD for at least 10 cells in each experiment. Scale bar, 10 μm.
To more precisely assess the molecular mechanism by which Y14 modulates the formation of foci, we used live-cell imaging to track the dynamic mobilization of MDC1 and Ku70/80 after laser-induced DNA damage. MDC1 is a key mediator of the DDR that recognizes γH2AX (Chapman et al., 2012). GFP-tagged MDC1 steadily accumulated at sites of DNA damage, consistent with a previous report (Chang et al., 2015), and this accumulation significantly slowed upon transfection of a Y14 small hairpin RNA-expressing vector (Figure 5C). Analogously, the recruitment of GFP-tagged Ku70 and Ku80 was also delayed upon Y14 depletion (Figures 5D and 5E, lower panels), supporting the notion that Y14 is required for the early assembly of DSB repair complexes.
Y14 Depletion Disrupts the Ubiquitination of Ku and Impairs DNA Repair
Y14 accumulated on chromatin after DNA damage and may facilitate Ku recruitment to DNA lesions (Figures 4 and 5). Intriguingly, we observed that Y14 depletion, however, resulted in Ku accumulation on chromatin following phleomycin treatment (Figure 6A). Depletion of eIF4A3 had no such effect compared with control (Figure S8). It has been reported that ubiquitination modulates the release of Ku70/80 from DNA lesions after repair (Brown et al., 2015), so we examined whether Y14 may influence Ku ubiquitination. Ubiquitination of overexpressed FLAG-Ku70 was enhanced upon phleomycin treatment (Figure 6B, lane 3), as reported (Brown et al., 2015). Y14 depletion diminished Ku ubiquitination by ∼60% (lane 4 and bar graph). This result suggested that Y14 may modulate the ubiquitination of Ku and provided an explanation for our observed persistence of Ku70/80 on chromatin in Y14-depleted cells.
Figure 6.

Y14 Depletion Disrupts Ku Ubiquitination and Impairs DNA Repair
(A) siC or siY14-transfected HeLa cells were mock treated (−) or treated with phleomycin, followed by recovery for 0, 1, or 2 h. WCE and CSK + R (see Figure 4B) were subjected to immunoblotting. Bar graph shows the relative level of chromatin-bound Ku70/80 in siC (lanes 9–12) and siY14 cells (lanes 13–16); lane 9 was set as 1.
(B) HEK293 cells were transfected with vectors or siRNA (HA-ubiquitin, lanes 1–4; FLAG-Ku70, lanes 2–4; siC, lanes 1–3; siY14, lane 4), and then mock treated (lane 2) or treated with phleomycin (lanes 1, 3, 4). Immunoprecipitation was performed using anti-FLAG, followed by immunoblotting. Bar graph shows the level of Ku ubiquitination in Y14-depleted cells (lane 5) versus control cells (lane 4) after phleomycin treatment.
(C) Diagram shows the pEGFP-Pem1-Ad2 vector as the NHEJ reporter (Seluanov et al., 2004). U2OS cells were co-transfected with uncut (Un), HindIII (H)-, or I-SceI (I)-digested NHEJ reporter and siRNA as indicated. The number of GFP-expressing cells was determined by fluorescence-activated cell sorting and normalized by the transfection rate (i.e., the level of GFP PCR product). (C–E) Bar graphs show end joining efficiency (mean ± SD) relative to siC. Immunoblotting shows knockdown or overexpressed proteins; PCR of a GFP fragment (horizontal line in the diagram; primers in Table S2) was used as the transfection control.
(D) U2OS cells were transfected with the reporter as in (C), siRNA (siC or siY14), and expression vector (empty or FLAG-Y14R encoding siRNA-resistant Y14). Asterisk indicates FLAG-tagged proteins (D–G).
(E) U2OS cells were transfected with the reporter as in (C) and expression vector (empty or FLAG-Y14 or FLAG-Y14SA).
(F) Diagram shows the DR-GFP construct that is genomically integrated in U2OS cells (U2OS-HDR; Gunn et al., 2011). U2OS-HDR cells were co-transfected with the empty or vector encoding FLAG-Y14 or FLAG-Ku70 and the I-SceI (white bar) expression vector. (F and G) Bar graphs show relative fold-enrichment of precipitated DNA determined by qPCR; mock transfection was set as 1. Immunoblotting shows knockdown or overexpressed proteins as in (C–E).
(G) U2OS-HDR cells were co-transfected with siRNA (mock, siC or siY14) and the empty or FLAG-Ku70 expression vector and the I-SceI vector.
Next, we tested whether Y14 depletion could affect DNA repair using a previously characterized NHEJ reporter, pEGFP-Pem1-Ad2 (Seluanov et al., 2004). The I-SceI- or HindIII-digested reporter plasmid was transfected into U2OS cells, as per the instruction of Seluanov et al. (2004). Unlike complementary ends that can be joined via simple ligation, end repair of DSB-mimicking I-SceI-generated inverted overhangs involves nucleolytic end processing. Depletion of Y14 reduced the rejoining efficiency of the I-SceI-linearized plasmid (Figure 6C). Overexpression of the siY14-resistent FLAG-Y14 (Y14R) in Y14-depleted cells restored end joining (Figure 6D), supporting a role for Y14 in DNA repair. Because inhibition of ATM reduced the phosphorylation of Y14 (Figure S6D), we tested the effect of a non-phosphorylatable Y14 mutant (SA) on DNA repair. We found that Y14-SA had a dominant-negative effect on DNA repair, even though it was expressed poorly (Figure 6E). As observed in U2OS cells, knockdown of Y14 or overexpression of Y14-SA also reduced the end joining efficiency in HeLa cells (Figures S9A and S9B), supporting the role of Y14 in DNA repair. Knockdown of eIF4A3, although had no significant effect in U2OS cells (Figure 6C), suppressed end joining to an extent equivalent to Y14 depletion in HeLa cells (Figure S9B); whether eIF4A3 modulates DSB repair via a mechanism different from that of Y14 remains to be investigated.
Finally, to examine whether Y14 indeed associates with DSBs, we took advantage of a line of U2OS cells that have a stably integrated GFP reporter with an I-SceI recognition site (Gunn et al., 2011). Chromatin immunoprecipitation and qPCR revealed that FLAG-Y14 associated with the GFP reporter upon cleavage by transiently expressed I-SceI; a similar result was obtained with FLAG-Ku70 (Figure 6F). The association of FLAG-Ku70 with the cleaved reporter was diminished by ∼20% in Y14-depleted cells (Figure 6G), suggesting that Y14 facilitates or stabilizes Ku binding to DNA ends. Together, our results indicated two possible direct roles for Y14 in DNA repair, i.e., recruiting Ku to DNA lesions and modulating its ubiquitination, both of which influence DNA repair efficiency.
In conclusion, Y14 acts through multiple pathways to maintain genome stability. First, Y14 is essential for the expression of several DDR factors, which maintains cellular capacity for DNA damage repair, and also functions to prevent R-loop accumulation during transcription (Figure 7, left). Moreover, upon DNA damage, Y14 promotes the recruitment of DDR factors to DNA lesions and perhaps also orchestrates their functions in DNA repair (Figure 7). Depletion of Y14 reduces DNA repair efficiency. Hence Y14 deficiency results in DNA damage accumulation in cultured cells and in the developing neocortex.
Figure 7.

Model of both Direct and Indirect Roles of Y14 in Genome Stability
Y14 maintains proper expression of DDR factors and prevents the formation of co-transcriptional R-loops likely through its role as an EJC factor (left). Y14 may directly participate in DNA damage repair via its RNA-mediated interaction with Ku and other DDR factors. Upon DNA damage, Y14 promotes Ku and DDR factors to DNA lesion and modulates Ku ubiquitination (ubi) and release after repair (right). Y14 deficiency impairs NHEJ.
Discussion
We have previously reported that Y14 depletion resulted in the accumulation of a DNA damage marker and sensitized cells to irradiation (Lu et al., 2017). This study reveals that Y14 not only indirectly but also directly contributes to genome stability. We provide evidence for the functions of Y14 in DDR signaling and DSB repair.
Previous studies have reported that depletion of Y14 alters the splicing pattern of apoptotic and cell-cycle-related genes (Michelle et al., 2012, Wang et al., 2014, Fukumura et al., 2016, Lu et al., 2017), which likely threatens cell viability. Here, we show that depletion of Y14 downregulated several DDR factors, including three critical DDR kinases (Figure 2), leading to attenuated DDR signaling and inefficient DSB repair. Moreover, depletion of Y14 induced R-loops (Figure 2). Thus, as many other splicing factors and the THOC (Naro et al., 2015, Wickramasinghe and Venkitaraman, 2016), Y14 prevents co-transcriptional aberrant R-loop formation. It is not surprising to observe that depletion of eIF4A3 also compromised the expression of DDR factors and induced R-loops (Figures S4 and S5). Therefore, Y14, eIF4A3, and perhaps other EJC factors likely via their role in RNA processing maintain proper expression of DDR factors and prevent R-loop accumulation, and thereby contribute to genome stability. Nevertheless, in this regard, whether they act independently or in the EJC context is not addressed in this study.
We observed that Y14 depletion delayed DDR kinetics, i.e., recruitment of DDR factors and formation of γH2AX foci (Figure 5). Although this may in part result from downregulated DDR signaling, our results argue for a direct role for Y14 in DNA signaling and repair. Most importantly, Y14 interacted with DNA damage-sensing, signaling, and repair factors in an EJC-independent manner (Figure 3) and was detected in EJC-deficient chromatin (Figure 4). Moreover, Y14 may possibly associate with some unknown chromatin factor(s) in an RNA-independent manner. DNA damage further enhanced the localization of Y14 to chromatin (Figure 4). However, we were unable to detect Y14 in laser-induced DNA damage sites by live cell imaging (data not shown), perhaps due to its transient or loose association with DNA damage sites. Our observation is in line with previous reports for several RNA processing factors that exhibit dynamic localization at sites of DNA damage (reviewed in Wickramasinghe and Venkitaraman, 2016). Moreover, we found that Y14 depletion diminished ubiquitination of Ku and may hence increase its retention on chromatin (Figure 6). This result is reminiscent of our previous report that Y14 modulates p53 ubiquitination (Lu et al., 2017), but the underlying molecular mechanism remains elusive at present. A recent report indicates that the paraspeckle factor RBM14 also regulates Ku retention during NHEJ-mediated DNA repair (Simon et al., 2017). Therefore a set of RNA-binding proteins may modulate Ku dynamics on chromatin during DNA damage repair. RNA-dependent interaction between Y14 and Ku is not completely unexpected, because non-coding RNAs as well as small RNAs generated at DNA damage loci have been implicated in DDR signaling and DNA repair (d'Adda di Fagagna, 2014). Therefore, whether Y14-mediated Ku recruitment involves RNA and whether Y14 participates in DNA damage-induced RNA processing at damage sites are intriguing topics for future study.
Our results indicate that DDR signaling may modulate Y14 function in DNA repair. Inactivation of ATM reduced the phosphorylation level of Y14 and abolished its interaction with Ku and accumulation on chromatin (Figures 3, 4, and S6). Overexpression of a non-phosphorylatable mutant of Y14 caused a dominant-negative effect on DNA end joining. These results suggest that phosphorylation of Y14 by ATM or as-yet-unidentified kinases is important for DNA repair. It is also notable that Y14 maintains the expression of ATM and several other DDR signaling factors (Figure 2). Therefore, Y14 is possibly engaged in a positive circuit to maintain DNA repair capacity (Figure 3), which emphasizes the importance of Y14 in genome integrity.
In this study, we found a transcription-dependent Y14-Trim28 interaction (Figure 3). Trim28 stabilizes RNA polymerase II pausing at the promoter-proximal region. Upon transcriptional activation, DDR factors associated with topoisomerase II-induced DSBs at promoters phosphorylate Trim28, leading to its release and subsequent transcriptional elongation (Bunch et al., 2015). We indeed observed that Y14 is possibly involved in stimulus-induced transcription through its interaction with Trim28 (data not shown). Notably, a recent report indicates that the nuclear EJC is required for Pol II pausing that prevents premature transcriptional elongation (Akhtar et al., 2018). Therefore whether the individual components of the EJC have any specific role in stimulus-induced pause release remains a topic of future investigation.
In conclusion, Y14 plays a direct role in DNA damage repair via its unprecedented interaction with DNA repair factors and also maintains DDR signaling and prevents co-transcriptional R-loops. Y14 deficiency hampers early brain development at least in part from accumulated DNA damage and genome instability.
Limitations of the Study
This study provides initial evidence for the direct role of Y14 in DNA damage repair. The detailed mechanism by which how Y14 modulates Ku association with DNA damage sites and the possibility of whether any specific RNA molecules are involved in Y14-mediated Ku recruitment during DNA damage repair remain to be investigated.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank J. M. Stark, V. Gorbunova, K. R. Jones, and A. Aguilera for the plasmids, cell lines, mouse lines, antibodies, and experimental suggestions. We also thank the Proteomics and Animal Cores of the Institute of Biomedical Sciences, the Transgenic Facility of the Institute of Molecular Biology, the Imaging and Cell Biology Facility of the Institute of Biological Chemistry for technique assistance, and Timothy C. Taylor for proofreading of the manuscript. We acknowledge Taiwan Protein Project of Academia Sinica grant AS-KPQ-105-TPP to P.-Y.W. This work was primarily supported by the Ministry of Science and Technology grant 106-2311-B-001-019 to W.-Y.T.
Author Contributions
W.-Y.T. oversaw all aspects of the project and wrote the manuscript. Figures were provided by T.-W.C. (Figures 1B, 3A–3F, 4A, 5A, 5B, S1, S3B, S6A–S6C, S7A, and S8), C.-C. Lu (Figures 1A, 2C, 4C, 6C–6G, S1, S3A, S3C, S4, S6D, S7C, S9A, and S9B), C.-H.S. (Figures 1D–1F, S2F, and S2G), P.-Y.W. (Figures 5C–5E), S.E. (Figures 2E, and S5A–S5F), C.-C. Lee (Figures 1C, 2A, 2B, 2D, and S2E), H.-C.K. (Figures 4B, 6A, and 6B), and K.-Y.H. (Figures S2B–S2D). Also, T.-W.C. contributed to the analysis of mass spectrometry data and manuscript preparation, P.-Y.W. provided experimental and manuscript suggestions, K.-Y.H. contributed to mouse breeding and maintenance, K.-M.L. performed immunoprecipitation and mass spectrometry, and C.-Y.T. designed and generated the Rbm8a-loxp mice.
Declaration of Interests
The authors declare no conflicts of interest.
Published: March 29, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.03.005.
Supplemental Information
FLAG-Y14 was immunoprecipitated from HEK293 cells using anti-FLAG; co-immunoprecipitates identified by mass spectrometric analysis are functionally categorized.
References
- Akhtar J., Kreim N., Marini F., Mohana G.K., Brune D., Binder H., Roignant J.-Y. The exon junction complex regulates the release and phosphorylation of paused RNA polymerase II. BioRxiv. 2018 preprint. [Google Scholar]
- Bhatia V., Barroso S.I., Garcia-Rubio M.L., Tumini E., Herrera-Moyano E., Aguilera A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature. 2014;511:362–365. doi: 10.1038/nature13374. [DOI] [PubMed] [Google Scholar]
- Brown J.S., Lukashchuk N., Sczaniecka-Clift M., Britton S., le Sage C., Calsou P., Beli P., Galanty Y., Jackson S.P. Neddylation promotes ubiquitylation and release of Ku from DNA-damage sites. Cell Rep. 2015;11:704–714. doi: 10.1016/j.celrep.2015.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunch H., Calderwood S.K. TRIM28 as a novel transcriptional elongation factor. BMC Mol. Biol. 2015;16:14. doi: 10.1186/s12867-015-0040-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunch H., Lawney B.P., Lin Y.F., Asaithamby A., Murshid A., Wang Y.E., Chen B.P., Calderwood S.K. Transcriptional elongation requires DNA break-induced signalling. Nat. Commun. 2015;6:10191. doi: 10.1038/ncomms10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.F., Chu P.C., Wu P.Y., Yu M.Y., Lee J.Y., Tsai M.D., Chang M.S. PHRF1 promotes genome integrity by modulating non-homologous end-joining. Cell Death Dis. 2015;6:e1716. doi: 10.1038/cddis.2015.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman J.R., Taylor M.R., Boulton S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- Chuang T.W., Lee K.M., Tarn W.Y. Function and pathological implications of exon junction complex factor Y14. Biomolecules. 2015;5:343–355. doi: 10.3390/biom5020343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Adda di Fagagna F. A direct role for small non-coding RNAs in DNA damage response. Trends Cell Biol. 2014;24:171–178. doi: 10.1016/j.tcb.2013.09.008. [DOI] [PubMed] [Google Scholar]
- Fukumura K., Wakabayashi S., Kataoka N., Sakamoto H., Suzuki Y., Nakai K., Mayeda A., Inoue K. The exon junction complex controls the efficient and faithful splicing of a subset of transcripts involved in mitotic cell-cycle progression. Int. J. Mol. Sci. 2016;17:1153. doi: 10.3390/ijms17081153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski J.A., Talley T., Qiu M., Puelles L., Rubenstein J.L., Jones K.R. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn A., Bennardo N., Cheng A., Stark J.M. Correct end use during end joining of multiple chromosomal double strand breaks is influenced by repair protein RAD50, DNA-dependent protein kinase DNA-PKcs, and transcription context. J. Biol. Chem. 2011;286:42470–42482. doi: 10.1074/jbc.M111.309252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley B.R., Lu W.T., Wilczynska A., Bushell M. The emerging role of RNAs in DNA damage repair. Cell Death Differ. 2017;24:1989. doi: 10.1038/cdd.2017.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu Ia W., Hsu M., Li C., Chuang T.W., Lin R.I., Tarn W.Y. Phosphorylation of Y14 modulates its interaction with proteins involved in mRNA metabolism and influences its methylation. J. Biol. Chem. 2005;280:34507–34512. doi: 10.1074/jbc.M507658200. [DOI] [PubMed] [Google Scholar]
- Ishigaki Y., Nakamura Y., Tatsuno T., Hashimoto M., Shimasaki T., Iwabuchi K., Tomosugi N. Depletion of RNA-binding protein RBM8A (Y14) causes cell cycle deficiency and apoptosis in human cells. Exp. Biol. Med. (Maywood) 2013;238:889–897. doi: 10.1177/1535370213494646. [DOI] [PubMed] [Google Scholar]
- Jackson S.P., Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C.C., Lee C.C., Tseng C.T., Tarn W.Y. Y14 governs p53 expression and modulates DNA damage sensitivity. Sci. Rep. 2017;7:45558. doi: 10.1038/srep45558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H., McMahon J.J., Tsai Y.H., Wang Z., Silver D.L. Haploinsufficiency for core exon junction complex components disrupts embryonic neurogenesis and causes p53-mediated microcephaly. PLoS Genet. 2016;12:e1006282. doi: 10.1371/journal.pgen.1006282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H., Pilaz L.J., McMahon J.J., Golzio C., Wu D., Shi L., Katsanis N., Silver D.L. Rbm8a haploinsufficiency disrupts embryonic cortical development resulting in microcephaly. J. Neurosci. 2015;35:7003–7018. doi: 10.1523/JNEUROSCI.0018-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelle L., Cloutier A., Toutant J., Shkreta L., Thibault P., Durand M., Garneau D., Gendron D., Lapointe E., Couture S. Proteins associated with the exon junction complex also control the alternative splicing of apoptotic regulators. Mol. Cell. Biol. 2012;32:954–967. doi: 10.1128/MCB.06130-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naro C., Bielli P., Pagliarini V., Sette C. The interplay between DNA damage response and RNA processing: the unexpected role of splicing factors as gatekeepers of genome stability. Front. Genet. 2015;6:142. doi: 10.3389/fgene.2015.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Pereira J.M., Aguilera A. R loops: new modulators of genome dynamics and function. Nat. Rev. Genet. 2015;16:583–597. doi: 10.1038/nrg3961. [DOI] [PubMed] [Google Scholar]
- Seluanov A., Mittelman D., Pereira-Smith O.M., Wilson J.H., Gorbunova V. DNA end joining becomes less efficient and more error-prone during cellular senescence. Proc. Natl. Acad. Sci. U S A. 2004;101:7624–7629. doi: 10.1073/pnas.0400726101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkreta L., Chabot B. The RNA splicing response to DNA damage. Biomolecules. 2015;5:2935–2977. doi: 10.3390/biom5042935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon N.E., Yuan M., Kai M. RNA-binding protein RBM14 regulates dissociation and association of non-homologous end joining proteins. Cell Cycle. 2017;16:1175–1180. doi: 10.1080/15384101.2017.1317419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhara S.C., Carvalho S., Grosso A.R., Gallego-Paez L.M., Carmo-Fonseca M., de Almeida S.F. Transcription dynamics prevent RNA-mediated genomic instability through SRPK2-dependent DDX23 phosphorylation. Cell Rep. 2017;18:334–343. doi: 10.1016/j.celrep.2016.12.050. [DOI] [PubMed] [Google Scholar]
- Wang Z., Murigneux V., Le Hir H. Transcriptome-wide modulation of splicing by the exon junction complex. Genome Biol. 2014;15:551. doi: 10.1186/s13059-014-0551-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe V.O., Venkitaraman A.R. RNA processing and genome stability: cause and consequence. Mol. Cell. 2016;61:496–505. doi: 10.1016/j.molcel.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FLAG-Y14 was immunoprecipitated from HEK293 cells using anti-FLAG; co-immunoprecipitates identified by mass spectrometric analysis are functionally categorized.