

Abstract

Synaptic dysfunction is a pathological feature in many neurodegenerative disorders, including Alzheimer’s disease, and synaptic loss correlates closely with cognitive decline. Histone deacetylases (HDACs) are involved in chromatin remodeling and gene expression and have been shown to regulate synaptogenesis and synaptic plasticity, thus providing an attractive drug discovery target for promoting synaptic growth and function. To date, HDAC inhibitor compounds with prosynaptic effects are plagued by known HDAC dose-limiting hematological toxicities, precluding their application to treating chronic neurologic conditions. We have identified a series of novel HDAC inhibitor compounds that selectively inhibit the HDAC–co-repressor of repressor element-1 silencing transcription factor (CoREST) complex while minimizing hematological side effects. HDAC1 and HDAC2 associate with multiple co-repressor complexes including CoREST, which regulates neuronal gene expression. We show that selectively targeting the CoREST co-repressor complex with the representative compound Rodin-A results in increased spine density and synaptic proteins, and improved long-term potentiation in a mouse model at doses that provide a substantial safety margin that would enable chronic treatment. The CoREST-selective HDAC inhibitor Rodin-A thus represents a promising therapeutic strategy in targeting synaptic pathology involved in neurologic disorders.
Keywords: HDAC, CoREST, synaptopathy, prosynaptic, synaptic resilience, neurodegenerative
Introduction
Synaptic dysfunction, or synaptopathy, is a pathological feature in many neurodegenerative disorders, including Alzheimer’s disease, and synaptic loss correlates closely with cognitive decline in both humans and animals.1−7 Emerging evidence implicates epigenetic dysregulation in cognitive and neurodegenerative disorders.8−12 Post-translational epigenetic modifications of histone proteins, such as acetylation, play a central role in regulating neuronal gene expression and brain function. Many studies with HDAC inhibitors (HDACi) have shown increases in histone acetylation and enhanced synaptic plasticity, learning, and memory, providing promise for the use of HDACi in treating synaptopathies.13−15 However, the HDACi tested in these studies target multiple HDAC isoforms and have displayed dose-limiting hematological toxicities typically associated with HDACi.16,17 For conditions that are likely to require chronic dosing, such as neurologic disorders, these hematological toxicities are a significant safety concern and have relegated the use of HDACi only to in vitro and animal model studies rather than clinical treatment of patients afflicted with these debilitating conditions. To harness the potential of HDACi to treat neurologic disorders, much safer compounds are required to enable long-term treatment of patients. More selective inhibitors of HDACs have been explored to test whether improved selectivity could maintain efficacy and minimize the dose-limiting toxicities, but with limited success. Strategies pursued toward this goal have included isoform and kinetic selectivity.18,19
The 18 human HDAC enzymes are divided into four classes based on sequence homology and function. The class I (HDAC1, HDAC2, HDAC3, and HDAC8), class II (HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, and HDAC10), and class IV (HDAC11) members are Zn+2 dependent, while the class III sirtuins (SIRT1–7) are NAD+ dependent.8 Selective targeting of the class I HDAC isoforms HDAC1, HDAC2, and HDAC3 has been achieved using benzamide and other ortho-aminoanilide inhibitors.15,18−20 These class I HDACs exist in multiprotein co-repressor complexes, including the CoREST, NuRD, Sin3, and SMRT/NCoR complexes, through which they differentially engage chromatin and deacetylate specific histone and non-histone sites.8 The pioneering work by Bantscheff et al. demonstrated via chemoproteomics that the benzamides CI-994 and BML-210 were selective for a subset of HDAC co-repressor complexes, binding the CoREST, NuRD, and NCoR complexes, but not the Sin3 complex.21 This was the first demonstration that some measure of HDACi selectivity beyond the isoform level was possible, achieving limited selectivity between the HDAC–co-repressor complexes. Despite this additional level of co-repressor complex selectivity, CI-994 still displayed significant hematological toxicity17 and maintained the prosynaptic effects at both the structural and functional levels.22 Of the different co-repressor complexes, the CoREST complex preferentially associates with HDAC2 in the brain and has been shown to have a key function in repressing neuronal gene expression.8,9,23,24 We hypothesized that selectively targeting HDACs associated with the CoREST complex could limit peripheral toxicity while maintaining beneficial neuronal effects.
Here we describe our design of a series of molecules that specifically targets the HDACs in the CoREST co-repressor complex and maintains prosynaptic effects while minimizing deleterious hematopoietic toxicity.
Results
Rodin Compounds Selectively Inhibit HDAC1 and HDAC2
To test the hypothesis that a selective HDAC–CoREST co-repressor complex inhibitor would maintain prosynaptic effects and improve the safety profile, we embarked on a medicinal chemistry campaign to identify molecules with these properties. We separated our chemistry efforts into two design stages. In the first stage, we designed new HDACi core scaffolds that maintained the inhibitory activity toward HDAC1 and HDAC2, similar to the classic benzamide scaffolds. In the second design stage, we implemented an in vitro predictive safety assay in human bone marrow cells to optimize the structure–activity relationship (SAR) for human hematological safety in this series of compounds while maintaining selective HDAC inhibitory activity.
Encouraged by multiple reports of isoform selectivity toward HDAC1 and HDAC2 achieved with benzamides and other ortho-aminoanilide containing appendages, which occupy the hydrophobic internal cavity “foot pocket” in the HDAC active site,18−20,25−28 we devised a strategy to modify the aniline core and Zn+2-chelating pharmacophore of class I selective compounds such as CI-994 and Cpd-60 (Figure 1A) as a means of honing the selectivity profile. By replacing the core diamino-aniline with other diamino-heterocycles, we tuned down the Zn+2 chelating ability while simultaneously occupying the “foot pocket”, with the aim of achieving the right balance of electrostatic properties. To test this hypothesis, a range of different heterocyclic replacements for the diamino-aniline core were explored, and most modifications resulted in a decrease in HDAC inhibitory activity (data not shown), but the 3-amino-pyridine-2-urea chemotype emerged with the best balance of HDAC potency and drug-like properties. Iterative design, synthesis, and compound optimization efforts resulted in a series of compounds represented by Rodin-A, Rodin-B, Rodin-C, and Rodin-D, which are all novel class I HDACi featuring the 3-amino-pyridine-2-urea core structure (Figure 1A). Key structural features include the central diaminopyridine ring, fused bicyclic pyrrolidine ureas, and the lipophilic “foot pocket” appendage. The four compounds feature four different cap groups (highlighted as the yellow region of Rodin-A in Figure 1A) and three different foot pockets (purple region), while all utilize the same Zn+2 chelating group (blue region).
Figure 1.

Structures of HDACi and crystal structure of Rodin-A with HDAC2. (A) Structures of four Rodin 3-amino-pyridine-2-urea HDACi and three published HDACi. (B) X-ray crystal structure of HDAC2 in complex with Rodin-A. Overall structure of HDAC2 containing the ligand Rodin-A shown as a ribbon diagram. One monomer of HDAC2 is shown as a ribbon representation. The ligand Rodin-A is shown as a stick model, colored according to the chemical atom type (CRodin-A in cyan, N in blue, O in red, and S in yellow). (C) Binding pocket of HDAC2 containing the ligand Rodin-A. The ligand and neighboring protein side chains are shown as a stick model, colored according to the chemical atom type (CRodin-A in cyan, CHDAC2 in gray, N in blue, O in red, and S in yellow). The ligand molecule is superimposed with the refined 2Fo – Fc electron density map contoured at 1.0σ. Hydrogen bonds are indicated as green dotted lines. For details of data collection, see SI (Tables S2 and S3).
In profiling against isolated recombinant HDAC isoforms for inhibition of enzymatic activity, these compounds selectively inhibited the HDAC1 and HDAC2 isoforms (Table 1). Modest inhibitory activity toward HDAC11 was exhibited by Rodin-C and Rodin-D. The well-studied HDACi CI-99422,29 and Cpd-6020,25 were also profiled as key comparators. The benzamide CI-994 inhibited HDAC1, HDAC2, and HDAC3, with modest inhibition of HDAC10. The more elaborated Cpd-60, containing the 2-thiophenyl substituent on the CI-994 benzamide core, potently inhibited HDAC1 and HDAC2 and showed modest inhibition of HDAC3 and HDAC11.
Table 1. In Vitro Inhibitory Profile of Rodin Compounds, CI-994, and Cpd-60a.
| IC50 (μM) |
||||||
|---|---|---|---|---|---|---|
| target | Rodin-A | Rodin-B | Rodin-C | Rodin-D | CI-994 | Cpd-60 |
| HDAC1 | 0.15 | 0.27 | 0.059 | 0.12 | 0.62 | 0.008 |
| HDAC2 | 0.43 | 0.28 | 0.18 | 0.28 | 0.54 | 0.031 |
| HDAC3 | >30 | 27.6 | 20.2 | >30 | 0.28 | 2.59 |
| HDAC4 | >30 | >30 | >30 | >30 | >30 | >30 |
| HDAC5 | >30 | >30 | >30 | >30 | >30 | >30 |
| HDAC6 | >30 | >30 | >30 | >30 | >30 | >30 |
| HDAC7 | >30 | >30 | >30 | >30 | >30 | >30 |
| HDAC8 | >30 | >30 | >30 | >30 | >30 | >30 |
| HDAC9 | >30 | >30 | >30 | >30 | >30 | >30 |
| HDAC10 | >30 | >30 | >30 | >30 | 8.95 | >30 |
| HDAC11 | >30 | >30 | 5.39 | 11.8 | >30 | 5.79 |
Inhibitory activity of Rodin-A, Rodin-B, Rodin-C, Rodin-D, CI-994, and Cpd-60 toward deacetylase activity of HDAC isoforms 1–11 (N ≥ 3).
Binding Pose of Rodin-A Confirmed by X-ray Cocrystal Structure with HDAC2
An X-ray cocrystal structure of Rodin-A with HDAC2 at a resolution of 2.25 Å confirmed the predicted binding pose in the active site, with the 3-amino-pyridine-2-urea pharmacophore chelating the Zn+2 and the 2-thiophenyl group occupying the 14 Å hydrophobic “foot pocket”, a feature that has previously been described to result in selective inhibition of HDAC1/2 over HDAC319,20,25−28 (Figure 1B,C). The bicyclic urea cap group of Rodin-A participates in π-stacking interactions with Phe210 and Phe155 of HDAC2. The urea NH participates in a backbone interaction with Gly154, and the NH2 participates in a hydrogen bond network with His145 and His146, in addition to binding the Zn+2. This binding mode is consistent with the observed binding mode of Cpd-60 with HDAC2.27
Rodin Compounds Selectively Inhibit the Deacetylase Activity of the HDAC–CoREST Complex
Since HDAC1, HDAC2, and HDAC3 are components of multiprotein co-repressor complexes, inhibition of the deacetylase activity of the enzymes as members of complexes would be a more physiologically relevant measure of the HDAC activity of the compounds.21,30,31 To explore the ability of the 3-amino-pyridine-2-urea compound series to inhibit specific HDAC co-repressor complexes, we implemented co-immunoprecipitation (co-IP) activity assays to measure the ability of the Rodin compounds, CI-994, Cpd-60, and SAHA to inhibit the HDAC activity within CoREST, Sin3, NuRD, and NCoR co-repressor complexes when purified and isolated from a cell lysate. Of the complexes investigated, Rodin-A, Rodin-B, Rodin-C, and Rodin-D primarily inhibit only the HDAC–CoREST complex activity (Table 2, Figure 2). The co-IP assay confirmed the earlier published findings that CI-994 inhibits the HDAC activity of CoREST, NuRD, and NCoR but not Sin3, while SAHA inhibits all the isolated HDAC complexes.21,30 Our studies also demonstrate that Cpd-60, which adds the 2-thiophenyl group to the CI-994 scaffold to extend into the “foot pocket” cavity, inhibits the CoREST and NCoR complexes.
Table 2. HDAC Co-repressor Complex Deacetylase Activity Inhibition Profile of Rodin Compounds and of CI-994, Cpd-60, and SAHAa.
| IC50 (μM) |
|||||||
|---|---|---|---|---|---|---|---|
| target | Rodin-A | Rodin-B | Rodin-C | Rodin-D | CI-994 | Cpd-60 | SAHA |
| CoREST | 1.80 | 0.50 | 0.38 | 0.49 | 1.82 | 0.16 | 0.10 |
| Sin3 | >250 | >250 | >250 | >250 | >250 | 191 | 0.11 |
| NuRD | 222 | >250 | 202 | >250 | 8.43 | 107 | 0.076 |
| NCoR | 160 | 88 | 52 | 209 | 0.86 | 4.03 | 0.14 |
| Fold CoREST selectivity | |||||||
| vs Sin3 | >138 | >499 | >649 | >510 | >137 | 1224 | 1.1 |
| vs NuRD | 123 | >499 | 524 | >510 | 4.6 | 686 | 0.8 |
| vs NCoR | 88 | 175 | 135 | 426 | 0.5 | 26 | 1.3 |
Inhibitory activity of Rodin-A, Rodin-B, Rodin-C, Rodin-D, CI-994, Cpd-60, and SAHA toward deacetylase activity of co-immunoprecipitated co-repressor complexes CoREST, Sin3A, NuRD, and NCoR. IC50 values for co-repressor complex co-immunoprecipitation activity assays are reported as the average of two experimental replicates. Each experiment includes technical replicates. See Methods for details.
Figure 2.
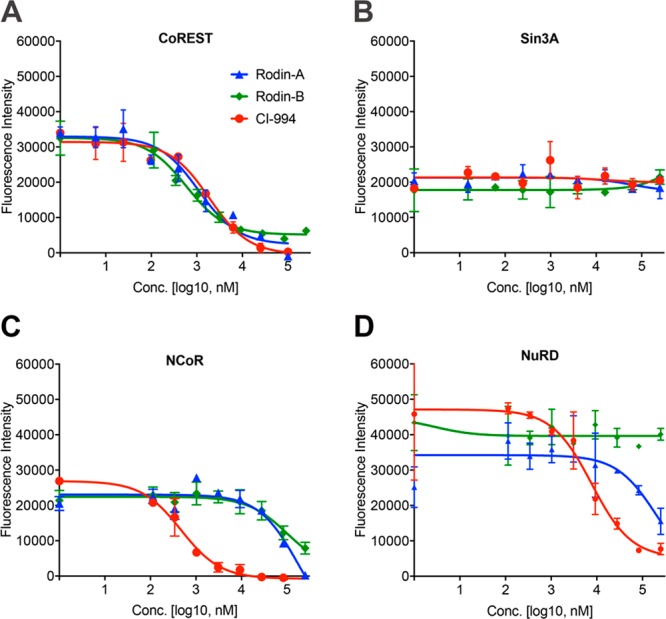
Rodin-A and Rodin-B selectively inhibit activity of the HDAC–CoREST complex. Overlaid inhibition curves of Rodin-A, Rodin-B, and CI-994 for the HDAC-containing isolated CoREST (A), Sin3A (B), NCoR (C), and NuRD (D) complexes in the co-immunoprecipitation complex deacetylase activity assays (technical duplicates; error bars are SD).
The HDAC–CoREST co-repressor complex also contains the lysine-specific histone demethylase 1 (LSD1). While strategies for targeting the LSD1–CoREST complex32 or the LSD1–HDAC–CoREST complex have been reported via dual LSD1/HDACi,33,34 these make no mention of selectivity over other HDAC complexes. Rodin compounds were screened against LSD1 and LSD1–CoREST and showed no inhibition of the LSD1 activity, confirming that they target the HDACs in the CoREST complex and do not have an effect on LSD1 (SI; Table S4).
Rodin Compounds Show Less Hematological Toxicity in Vitro than CI-994 and Cpd-60
To explore the impact of complex selectivity on hematological toxicity, in vitro assays were run in human bone marrow cells to predict hematological effects in humans. The potential effects on human erythroid and myeloid progenitors of the CoREST selective inhibitors (Rodin-A, Rodin-B, Rodin-C, and Rodin-D), CI-994, and Cpd-60 were assessed in a colony forming unit (CFU) cell assay system, which has been shown to be predictive for clinical neutropenia and anemia.35 Compound treated cells were cultured for 14 days, whereupon the relative safety of compounds was assessed by counting cell colonies and comparing between control and compound treated plates. After 14 days at 10 μM compound treatment, the in vitro hematological effects of the CoREST selective inhibitors Rodin-A, Rodin-B, and Rodin-D were mild, with all having greater than 59% of control remaining in the myeloid progenitors (Table 3). Rodin-C showed more toxicity, with 30% of control remaining in the myeloid progenitors. The erythroid progenitors were more sensitive to compound treatment in all cases. The class I selective HDACi CI-994 (HDAC1, 2, 3) and Cpd-60 (HDAC1, 2) had severe toxic effects on both the myeloid and erythroid progenitor cells, with 0% control remaining after 10 μM compound treatment.
Table 3. In Vitro Hematological Safety Profiling of Rodin Compounds and of CI-994 and Cpd-60a.
| target | Rodin-A | Rodin-B | Rodin-C | Rodin-D | CI-994 | Cpd-60 |
|---|---|---|---|---|---|---|
| myeloid progenitors: % control at 10 μM | 59 | 63 | 30 | 59 | 0 | 0 |
| erythroid progenitors: % control at 10 μM | 27 | 40 | 16 | 23 | 0 | 0 |
In vitro hematological safety assays: effects on human myeloid and erythroid progenitors using colony forming cell (CFU) assays, assessed in a semi-solid, methylcellulose-based culture system. The mean and SEM of three replicate cultures were calculated for progenitors of each category (myeloid and erythroid). Compounds were dosed at 0.4, 2, and 10 μM. See Methods for details.
Because Rodin-A demonstrated a similar CoREST complex IC50 to CI-994 and a clean isoform inhibition profile (HDAC1 and HDAC2 at similar IC50 to CI-994), it represented the best comparator compound to CI-994 to explore the effects of selective CoREST inhibition on synapses. Based on this similarity, in vitro ADME and safety profiles, and pharmacokinetic profile, a thorough in vitro and in vivo characterization was conducted on this novel and representative HDAC-CoREST co-repressor selective compound.
Rodin-A Increases H3K9 Acetylation
The most proximal effect of HDACi is an increase in histone acetylation at multiple sites in the genome (typically tracked as global changes in the acetylation status of specific histone lysine side chains). Traditionally, several key histone lysines linked to gene regulation are monitored during HDACi treatment;36 H3K9 is a commonly analyzed position, and we report effects of Rodin-A on this position in K562 cells. Rodin-A treatment of K562 cells stimulated histone acetylation with a similar potency as the less selective CI-994 (Figure 3A,B; Figure S1). Both CI-994 and Rodin-A increase the levels of acetylation at position lysine 9 on histone H3 (Ac-H3K9), relative to treatment with DMSO, in K562 cells treated in vitro for 24 h. The more complex-selective Rodin-A has a lower magnitude of effect on global Ac-H3K9 acetylation than the less selective CI-994 when each are dosed over the same dose range up to 100 μM in a dose-response treatment.
Figure 3.
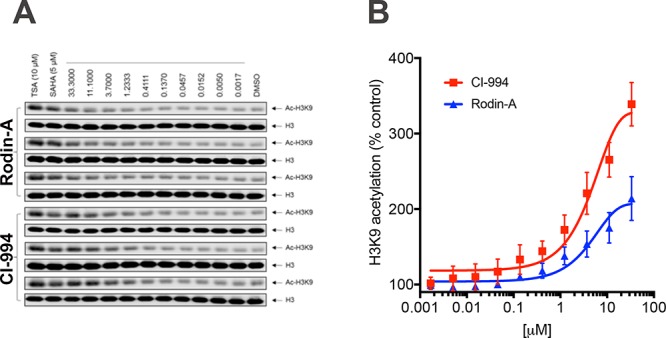
Inhibition of histone deacetylation at site H3K9 in vitro in a dose-dependent manner by Rodin-A and CI-994. CI-994 and Rodin-A treatment of K562 cells results in increased levels of acetylation at position lysine 9 on histone H3 (Ac-H3K9), relative to treatment with DMSO after 24 h. (Western blot image in left panel, dose-response effect in right panel, n = 3 independent treatments; for all figures, error bars are SEM).
Rodin-A Increases Density of Total and Thin Spines
HDAC inhibition has been shown to enhance synaptic structure and function.22 To investigate whether chronic treatment with Rodin-A and CI-994, with different co-repressor complex selectivity profiles, affects structural plasticity to a similar degree, we measured dendritic spines in the hippocampus following 14 days of daily administration. Rodin-A (0.1, 0.3, 1, 3, and 6 mg/kg/day), CI-994 (1 and 3 mg/kg/day), or vehicle (20% HPβCD) was dosed orally to wild-type (WT) mice for 14 days, and the mice were sacrificed 24 h after the final dose. Dendritic spine density and morphology were evaluated in the CA1 region of the dorsal hippocampus, using ballistic dye labeling. The 6 mg/kg dose of Rodin-A was chosen to match the predicted free brain exposures of CI-994 at the efficacious dose of 1 mg/kg based on PK experiments and in vitro protein binding data (SI; Table S5). Rodin-A treatment resulted in a significant increase in dendritic spine density when compared to vehicle-treated controls (Figure 4A,B). No significant changes were observed at the lower doses of 0.1 or 0.3 mg/kg (data not shown). The strong prosynaptic effects at the 1 and 3 mg/kg doses can be explained by high concentration of free drug in brain as demonstrated in a separate microdialysis experiment (Figure 4C; SI, Table S6). Mice treated with CI-994 at 1 and 3 mg/kg displayed significant increases in spine density compared to vehicle-treated animals, which were similar to Rodin-A (Figure 4B).
Figure 4.
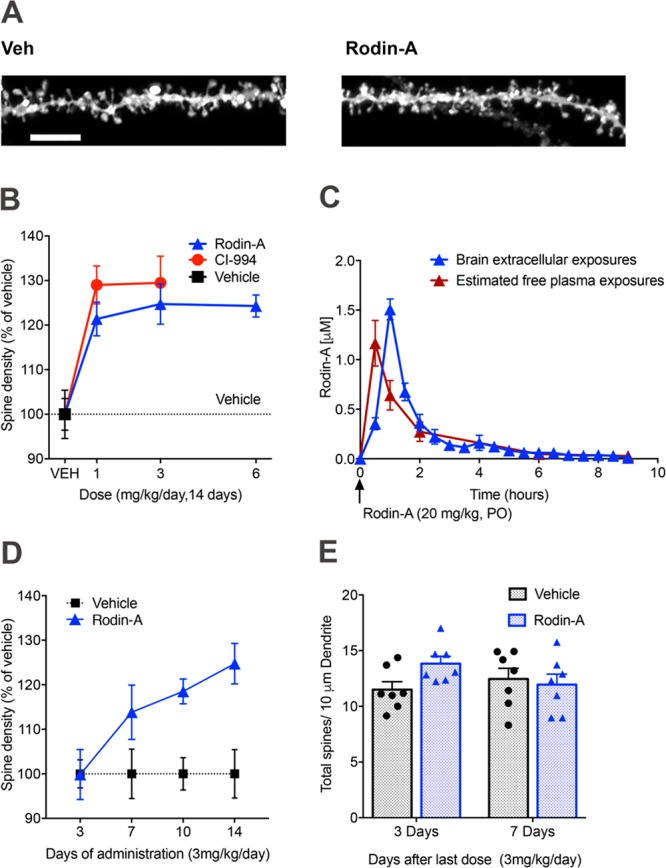
Rodin-A increases synaptic density in mice. (A) Representative three-dimensional images of dendritic spines after ballistic dye labeling (scale bar, 5 μm). (B) Quantification of total dendritic spine density in hippocampal pyramidal neurons in WT mice after 14 days of treatment with Rodin-A (n = 7) or CI-994 (n = 5). Significant increases occur in spine density following treatment of mice with different doses of Rodin-A (mean ± SEM, one-way ANOVA and Dunnett’s post hoc analysis; 1 mg/kg, p = 0.004; 3 mg/kg p = 0.001; and 6 mg/kg, p = 0.0012) and CI-994 (1 mg/kg, p = 0.02; 3 mg/kg, p = 0.018). (C) PK curves from mouse microdialysis experiment with Rodin-A dosed at 20 mg/kg. Exposure curves for brain dialysate showing free exposure in brain at different time points overlaid with estimated free plasma exposures calculated using in vitro plasma protein binding data (fu = 25.1% in mouse plasma in vitro). Free brain-to-plasma ratio (Kpuu) at brain Cmax = 2.4. (D) Total spine density quantification after different duration of daily treatment with Rodin-A (n = 7 animals each). Spine density does not significantly change after 3 or 7 days of treatment but significantly increased after 10 days of dosing (mean ± SEM, unpaired t test, p = 0.0016). (E) Quantification of spine density at different days after last dose (n = 7 animals each) indicates a reversible effect of the drug after 7 days from the last dose. Error bars are SEM.
When spine morphology is analyzed, chronic treatment with Rodin-A produced a dose-dependent increase in thin spines, with significant effects measured at all tested doses (SI; Figure S2A). In contrast, there were no significant changes in mushroom and stubby spine density (SI; Figure S2B,C). To further characterize the Rodin-A mediated effects on spine density, we also investigated both the duration of dosing required to mediate the increase in spine density and the duration of increase following 14 days of dosing. Chronic treatment with Rodin-A (3 mg/kg/day) produced a time-dependent increase in spines, with no changes in spine density after 3 or 7 days of treatment but a statistically significant increase in total spine density after 10 days of dosing (Figure 4D). Following termination of dosing (14 days, 3 mg/kg/day), the spine density gradually decreased over a 7 day period and returned to vehicle-treated levels at day 7 post-treatment (Figure 4E). Taken together, our findings demonstrate that chronic treatment with Rodin-A resulted in increased synaptic density that gradually developed over a 10 day period and has returned to baseline 7 days following termination of dosing. This confirms our hypothesis that chronic treatment is likely required for a selective HDACi to achieve desired prosynaptic effects, thus increasing the safety requirements for a neurotherapeutic HDACi. The findings also suggest that after chronic treatment, those structural changes are sustained for a period of time postdosing.
Rodin-A Increases Coincidence of Synaptic Proteins
To further characterize the effects of chronic administration of Rodin-A on the structure of excitatory synapses, presynaptic vesicle protein 2A (SV2A) and postsynaptic density protein 95 (PSD95) proteins were quantified by immunofluorescence in the dorsal hippocampus of WT mice that had been treated with Rodin-A daily for 14 days.37,38 Structurally mature synapses were identified by colocalization of pre- and postsynaptic SV2A and PSD95 puncta. Treatment with Rodin-A led to a dose-dependent increase in the coincidence of SV2A and PSD95 (Figure 5C). The low dose produced a 20% increase that did not reach significance, while the 3 and 6 mg/kg doses led to a significant increase in SV2A–PSD95 coincidence (∼27%) that matches the increase in spine density (Figure 4B). There was, however, no effect on total PSD95 and SV2A elements, separately (Figure 5A,B).
Figure 5.

Immunohistochemical analysis of pre- and postsynaptic elements. Multiple doses of Rodin-A in WT mice do not change the total number of synaptic PSD95 (A) and SV2A (B) proteins but significantly increase coincidence of synaptic proteins SV2A and PSD95 (C, n = 7), in the CA1 dorsal hippocampus (scale bar, 2 μm). Similarly, treatment with Rodin-A significantly increases pTrk elements and coincidence of pTrK (D, n = 5) and PSD95 (E, F, n = 5) in the CA1 dorsal hippocampus (mean ± SEM, one-way ANOVA and Dunnett’s post hoc analysis. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001).
A key mediator of structural and functional synaptic changes is the brain-derived neurotrophic factor (BDNF), which acts predominantly via TrkB receptors in hippocampal neurons.39,40 Increases in BDNF binding to and signaling through TrkB should be accompanied by increases in phosphorylated TrkB (pTrkB) at postsynaptic dendrites of excitatory synapses in hippocampus.40 We therefore investigated whether chronic treatment with Rodin-A affected pTrkB protein in the CA1 region of the dorsal hippocampus in WT mice. Excitatory synapses were identified by PSD95, which is highly enriched at glutamatergic synapses.41 Rodin-A treatment produced a significant increase in both total pTrk levels (Figure 5D) and in the coincidence of pTrk and PSD95 (Figure 5F). There was no effect of treatment with Rodin-A on total PSD95 levels (Figure 5E).
Rodin-A Improves Impaired LTP in a 5xFAD Mouse Model
Synaptic plasticity encompasses structural changes, dendritic spine plasticity, and the resulting physiological plasticity, including long-term potentiation (LTP), which together are considered the foundation of learning and memory. LTP is a persistent strengthening of synapses, with a long-lasting increase in synaptic efficacy and in signal transmission between neurons, which has been shown to be related to dendritic spine size and numbers.42,43
To evaluate the effect of Rodin-A on LTP, we employed the 5xFAD transgenic mouse model of amyloid pathology, in which mice begin to display deficits in LTP by the age of 4–7 months.44 Aged 5xFAD mice were treated with vehicle or three doses of Rodin-A and compared to an age-matched cohort of WT animals treated with vehicle (Figure 6A,B). The 5xFAD animals display an impaired LTP relative to WT animals, and in this model, treatment with Rodin-A (3, 6, and 20 mg/kg; Figure 6B) for 14 days stimulated a dose-related improvement of the impaired LTP similar to the results seen with CI-994 at 1 mg/kg (Figure 6C).
Figure 6.

Compound effects of Rodin-A and CI-994 on impaired hippocampal LTP. (A) Effects of Rodin-A treatment on normalized last 10 min fEPSP slope % in hippocampus of transgenic 5xFAD mice. Average fEPSP change for aged, 10–12 months old, 5xFAD vehicle treated mice (n = 8), matched WT vehicle treated mice (n = 9), and aged, 10–12 months old, 5xFAD mice treated with 6 mg/kg Rodin-A, po, for 14 days (n = 6). (B) Treatment with 3 mg/kg, 6 mg/kg, and 20 mg/kg doses of Rodin-A improves impaired LTP in aged 5xFAD mice relative to vehicle treated animals (combination of two experiments; n = 16, 16, 6, and 8 for WT Veh, 5xFAD Veh, 6 mg/kg, and 20 mg/kg treated animals, respectively). (C) Dosing aged, 10–12 months old, 5xFAD mice with 1 mg/kg of CI-994, po (all groups n = 8), for 14 days improves impaired LTP in treated mice relative to vehicle. For all figures, error bars are SEM. For panels B and C, statistical analysis was by one-way ANOVA followed by a Holm–Sidak post hoc test (*p < 0.05, **p < 0.01, ***p < 0.005).
In Vivo Safety Profile of Rodin-A in Rats and Dogs
In order to confirm the in vitro findings that suggest this series of CoREST-selective HDACi possess an improved hematological safety profile compared to less selective compounds with respect to co-repressor complexes in vivo, we profiled Rodin-A in toxicology studies in male Sprague–Dawley rats (n = 20) and male beagle dogs (n = 15), with daily oral dosing for 14 days. There were no Rodin-A related observations in male Sprague–Dawley rats after 14 days daily oral dosing at 30 mg/kg or 45 mg/kg. The no-observed-adverse-effect level (NOAEL) was considered to be 45 mg/kg (day 14, AUC 13 200 ng·h/mL, Cmax 3780 ng/mL; Table 4). The 60 mg/kg dose group of Rodin-A in rats showed hematological and bone marrow effects and microscopic findings in the testes. In male beagle dogs, after daily oral dosing for 14 days at 45 mg/kg, 60 mg/kg, and 120 mg/kg, there were no target organ effects observed at any dose levels, and the NOAEL was considered to be 120 mg/kg (day 14, AUC of 25 000 ng·h/mL, Cmax of 18 600 ng·h/mL; Table 4).
Table 4. Rodin-A Toxicokinetic Parameters in Rats and Dogs Following Repeated Daily Oral Administration for 14 Days.
| SD rata |
beagle dogb |
|||||
|---|---|---|---|---|---|---|
| 30 mg/kg | 45 mg/kg | 60 mg/kg | 45 mg/kg | 60 mg/kg | 120 mg/kg | |
| Cmax (ng/mL) | ||||||
| day 14 | 1670 ± 184 | 3780 ± 1450 | 3760 ± 1160 | 6283 ± 1510 | 10100 ± 4730 | 18600 ± 7410 |
| AUC (ng·h/mL) | ||||||
| day 14 | 8240 ± 1940 | 13200 ± 4760 | 28700 ± 7570 | 11487 ± 1918 | 13000 ± 5790 | 25000 ± 14300 |
Two animals per group; value reported is the mean ± SD.
Three animals per group; value reported is the mean ± SD.
Discussion
As a result of our drug discovery program focused on identifying safer HDACi to treat neurologic disorders, we have identified a series of 3-amino-pyridine-2-urea compounds that selectively inhibit the activity of the HDAC–CoREST co-repressor complex. In comparing inhibition of individual HDAC isoforms, the Rodin compounds all exhibit more potent inhibition of HDAC1 and HDAC2 than does CI-994, a class I HDACi (HDAC1, 2, 3) that had previously been reported to have prosynaptic effects22 (Figure 1, Table 1). Since the Rodin compounds also feature a foot pocket group as a key structural feature, we also profiled Cpd-60,20,25,27 a very potent inhibitor of HDAC1 and HDAC2, which has been reported to show effects on mood and behavior at relatively high doses (45 mg/kg, ip).45 Both CI-99417 and Cpd-6025 have demonstrated safety and tolerability issues reported at doses where the CNS efficacy has been reported. The key differentiating profile of the 3-amino-pyridine-2-urea compounds was confirmed in HDAC co-repressor complex activity assays: Rodin-A, Rodin-B, Rodin-C, and Rodin-D primarily inhibit only the CoREST complex activity (Table 2, Figure 2). This is in contrast to CI-994, which inhibits the activity of the CoREST, NuRD, and NCoR complexes but not Sin3, as had been shown previously in the Bantscheff et al. chemoproteomics studies.21,30 The co-IP assay also confirmed the earlier findings that SAHA inhibits all the studied HDAC complexes. By adding a foot pocket to CI-994, Cpd-60 gains additional complex selectivity, targeting the CoREST and NCoR complexes. It is interesting to note that Cpd-60 shows inhibition of the NCoR complex, which contains only HDAC3, despite a high degree of selectivity for HDAC1 (>320-fold) and HDAC2 (>80-fold) over HDAC3 in recombinant enzymatic assays (Table 1). This suggests that recombinant enzymatic assays are not a good surrogate for the cellular context where HDACs are involved in multiprotein complexes. The comparison of binding poses of Rodin-A (Figure 1B,C) with Cpd-6027 does not offer any apparent explanation for differences in complex selectivity. While protein crystal structures of HDAC3/SMRT (NCoR)46 and HDAC1/MTA1 (NuRD)47 have been published, to the best of our knowledge there are no known ligand bound cocrystal structures of any HDAC multiprotein complexes. Future work in the structure of the HDAC–CoREST co-repressor complex would help elucidate reasons for the observed CoREST complex selectivity profile of these novel molecules. The high level of HDAC–CoREST co-repressor complex selectivity that we observed for the series of 3-amino-pyridine-2-urea compounds toward the tested HDAC complexes is unprecedented.
The in vitro safety profiling of the CoREST selective compounds shows they are clearly differentiated from known prosynaptic HDACi like CI-994. The series of Rodin compounds all exhibit significantly improved in vitro hematological safety relative to the class I selective CI-994, despite the fact that they are more potent inhibitors of HDAC1 and HDAC2 activity than CI-994 (Table 1) and equally or more potent when associated with CoREST (Table 2). The hematological effects of Rodin compounds in vitro were mild compared to the severe effects of the class I selective HDACi CI-994 on both the myeloid and erythroid progenitor cells at the same concentrations after 14 days in culture (Table 3). This data suggests that the more complex-selective HDACi will have an improved hematological profile in humans.35 Whether the inhibition of the additional complexes is responsible for the additional toxicity is unknown. Cpd-60, which inhibits only CoREST and NCoR, also shows severe hematological toxicity in vitro, similar to CI-994. However, Cpd-60 is also much more potent on HDAC1, HDAC2, and HDAC3 than Rodin compounds. The comparison between the Rodin series compounds and CI-994 suggests that the enhanced selectivity might play a role in the differentiated safety profile. Effects of complex selectivity are also suggested when comparing the effects of Rodin-A and CI-994 on global histone H3K9 acetylation in K562 cells treated at concentrations up to 100 μM (Figure 3). Rodin-A treated cells show a lower magnitude of effect despite the fact that the inhibitory potencies of both compounds toward the HDAC–CoREST complex are similar and Rodin-A is more potent toward HDAC1 and HDAC2. While saturation of effect may not have been reached in these experiments, the acetylation differential observed at these high concentrations in vitro should offer a fair estimation of what might be observed at therapeutically relevant doses. The lower magnitude of acetylation of H3K9 could be a consequence of complex selectivity or simply differences in acetylation profile, and this is currently under investigation.
Because of the similarity in CoREST IC50 to CI-994, the representative CoREST selective compound Rodin-A was extensively profiled in vivo to interrogate the effects of CoREST complex selective HDACi treatment. Rodin-A was shown to dose-dependently increase dendritic spine density in mouse hippocampus (Figure 4) and functionally improve the impaired LTP in the 5xFAD transgenic mouse model of amyloid pathology (Figure 6) at similar doses to CI-994. Rodin-A treatment also caused the associated increase in both SV2A/PSD95 coincidence and TrkB phosphorylation (Figure 5). Importantly, the prosynaptic effects of Rodin-A were achieved at doses and exposures far below the NOAEL exposures identified in rats and dogs (Table 4). Although it is not ideal to compare across species, if we look at the efficacious and toxic doses of Rodin-A from different studies, we can obtain a general assessment of the safety margin for the compound. Based on the 1 mg/kg dose of Rodin-A administered daily for 14 days to WT mice that stimulated a 21% increase in total spine density (Figure 4), a projected human equivalent dose (HED) based on body surface area48 is 4.8 mg/kg. The NOAEL in rats of 45 mg/kg correlates to a HED of 432 mg/kg, and the NOAEL in dogs of 120 mg/kg projects a HED of 3888 mg/kg. Thus, even when basing calculations on rat as the most sensitive species, the efficacious HED of 4.8 mg/kg provides a safety margin of 90-fold for Rodin-A at the NOAEL of 45 mg/kg.
As a point of comparison, a previous report on the toxicity of CI-994 in rats and dogs after 2 weeks of dosing showed principal signals in tissues with rapidly dividing cell populations (bone marrow, testes), with similar effects in rats and dogs.17 Rats were dosed daily at 1.5 mg/kg, 5 mg/kg, and 15 mg/kg orally (exposures not reported), and dogs were given daily doses of 0.5 mg/kg, 2 mg/kg, or 5 mg/kg (day 12 AUC of 9.91 ng·h/mL at 2 mg/kg in males). The dose-limiting toxicity was primarily bone marrow suppression, but nonreversible testicular effects were also observed (rat 5 mg/kg, dog 2 mg/kg). Using the 1 mg/kg dose of CI-994 to WT mice that stimulated a similar increase in total spine density (29%, Figure 4) to Rodin-A (21%) after 14 days of dosing, the projected HED for CI-994 is also 4.8 mg/kg. The reported NOAEL in dogs of 0.5 mg/kg translates to a HED of 16.2 mg/kg (10 mg/m2).17 The 16.2 mg/kg HED therefore correlates to a safety margin of 3.4-fold for CI-994. Thus, the CoREST selective Rodin-A has maintained the prosynaptic effects of CI-994 while improving the safety margin from 3.4-fold to 90-fold based on HED and NOAEL in the most sensitive species. This suggests that the prosynaptic efficacy has been dissociated from classic HDAC-related toxicity in this new series of compounds. The in vivo studies performed with Rodin-A also show that the predictive in vitro hematological safety assays (Table 3) translate well to in vivo rat and dog safety studies for this series of compounds.
Taken together, our data suggests that CoREST inhibition is sufficient to achieve prosynaptic effects and that the CoREST co-repressor complex is a key controller of synaptic growth and function, based on the exquisite selectivity of Rodin-A for CoREST over the Sin3, NCoR, and NuRD co-repressor complexes. Since we have not yet identified compounds that selectively inhibit Sin3, NCoR, or NuRD, we cannot say if selective inhibition of one of these HDAC co-repressor complexes will lead to prosynaptic benefits, nor can we say that selective inhibition of one of these other complexes will lead to an improved safety profile. It is also important to note that HDAC1 and HDAC2 are involved in more multiprotein complexes than just the four complexes that were investigated in this study. These include the less studied multiprotein MiDAC complex and complexes involving the proteins MIER1, MIER2, and MIER3.21,30 Since we did not evaluate experimentally the ability to inhibit any of these complexes, we cannot comment on whether the series of 3-amino-pyridine-2-urea compounds inhibit these complexes or whether these complexes are involved in the prosynaptic or improved safety effects observed. Our data allow us to say that of the four HDAC complexes studied, inhibition of CoREST alone is sufficient for maintaining prosynaptic effects and can also result in an improved safety profile in vitro and in vivo.
Achieving improved hematological safety with compounds such as Rodin-A enables the application of HDACi to long-term dosing of patients. Thus, the novel, CoREST co-repressor complex-selective HDACi, Rodin-A, represents a promising new potential therapeutic in synaptic pathology driven neurologic disorders such as Alzheimer’s disease. Further studies with this and related compounds, including compounds with different complex inhibition profiles, are ongoing and will be reported in due course.
Methods
The data that support the findings reported in this paper are available upon reasonable request.
All animal studies were conducted in accordance with local guidelines for the humane treatment of animals and were approved by the local Ethics Committees.
Microdialysis (Brains-on-Line) experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee of Brains On-Line, LLC. LTP, spine density, and PK (ChemPartner) experimental protocols were approved by the Institutional Animal Care and Use Committee of ChemPartner, Shanghai, China. Safety studies (rat, dog) were conducted at Charles River Massachusetts and were reviewed and approved by Charles River Laboratories Institutional Animal Care and Use Committee (IACUC).
CI-994 was purchased from Sigma-Aldrich (C0621 SIGMA). SAHA was purchased from Sigma-Aldrich (SML0061). Cpd-60 was synthesized according to literature procedures.20 For the synthesis of Rodin-A, Rodin-B, Rodin-C, and Rodin-D, see the Supporting Information.
Study Design
For all in vivo studies, pilot studies were utilized to determine the variability in the readout of interest, and this information was used to determine the appropriate sample size. Rules for stopping data collection, data inclusion/exclusion criteria, and handling of outliers is described in the respective experimental sections below, where applicable. The end points and number of replicates are also described.
The research objectives, research subjects, and experimental design are outlined in the respective sections for each experiment, where applicable. Details on the statistical analyses used are provided in the experimental design and data analysis description or in the figure or table legends containing the data.
Code Availability
Dendritic spine analysis was done using Afraxis ESP software (Afraxis Inc., San Diego, CA). Queries regarding access to the analysis software code should be addressed to Afraxis Inc.
Recombinant HDAC Isoform Biochemical Assay (Nanosyn)
Screening of HDAC Co-repressor Complex Inhibition via Activity Determination of Co-immunoprecipitated HDAC Complexes (OmicScouts, Inc.)
Cell Culture and Extraction of Nuclear Proteins
K562 cells were obtained from ATCC (ATCC CCL-243) and cultured according to the manufacturer’s recommendations. Cells were harvested by centrifugation and subsequently washed three times in phosphate buffered saline without calcium and magnesium (1000 rpm, 5 min, room temperature). Cell pellets were frozen at −80 °C until further use.
Screening of HDAC Co-repressor Complex Inhibition via Activity Determination of Co-immunoprecipitated HDAC Complexes
Nuclear protein extraction from K562 cell pellets and co-immunoprecipitation (Co-IP) directed against HDAC complexes were performed using Active Motif’s Nuclear complex Co-IP kit (no. 54001). Reagents and conditions for the different HDAC complex Co-IPs are summarized in Table 5. The following antibodies have been used: LSD1 (C69G12) rabbit mAb (no. 2148, Cell Signaling Technology), mSin3A rabbit pABb (no. ab3479, abcam), HDAC3 rabbit pAb (np. ab7030, abcam), MTA3 (428C2a) sc-81325 mouse mAb (no. sc-81325, Santa Cruz Biotechnology). In brief, the extract has been incubated with the specific antibody overnight at 4 °C followed by incubation with 50 μL of Protein G Agarose (no. 11243233001, Sigma; 1 h, 4 °C). One Co-IP (∼260 μL total volume) was distributed into 10 wells (20 μL per well) and incubated with the test compound at 9 concentrations; the 10th aliquot was incubated with 100 μM SAHA as positive control. Two replicate Co-IP and activity assay procedures were performed per compound. HDAC activity was measured after 120 min compound incubation using Active Motif’s Fluorescent HDAC Assay Kit according to the manufacturer’s instructions. Fluorescence was read-out using a FLUOstar microplate reader (excitation wavelength 355 nm, emission wavelength 460 nm).
Table 5. Co-repressor Complex Immunoprecipitation Parameters.
| complex | antibody | antibody dilution | nuclear extract amount [mg] |
|---|---|---|---|
| CoREST | LSD1 | 1:25 | 0.7 |
| Sin3 | mSin3A | 1:50 | 0.7 |
| NCoR | HDAC3 | 1:625 | 3 |
| NuRD | MTA3 | 1:5 | 3 |
LC-MS/MS Based Identification of Co-immunoprecipitated Proteins
An aliquot of the Co-IPs directed against each HDAC complex was subjected to LC-MS/MS based protein identification to confirm the integrity of the co-immunoprecipitated HDAC complex and the absence of other HDAC complexes and their activities. To this end, bound proteins were eluted from the beads via incubation in LDS buffer for 5 min at 95 °C. Eluted proteins were processed for quantitative proteomics using standard in-gel digestion procedure.49,50
Nanoflow LC-MS/MS analysis of peptide samples was performed on an UltiMate 3000 RSLCnano System (Thermo Scientific, Dreieich, Germany) coupled to a Q-Exactive Plus quadrupole orbitrap mass spectrometer (Thermo Scientific, Bremen, Germany). Peptides were delivered to a trap column (100 μm inner diameter × 2 cm, packed with 5-μm C18 resin (Reprosil GOLD, Dr. Maisch, Germany) at a flow rate of 5 μL/min for 10 min in 0.1% FA in HPLC-grade water. After loading and washing, peptides were transferred to an analytical column (75 μm ID × 55 cm C18 column; Reprosil GOLD, 3 μm, Dr. Maisch, Germany) and separated using a linear gradient of 51 min ramping from 4% to 32% solvent B (0.1% FA, 5% DMSO in ACN) at a flow rate of 300 nL/min; 5% (v/v) DMSO was used in solvents A (0.1% FA, 5% DMSO in HPLC-grad water) and B to boost the nanoelectrospray response. Peptides were ionized using a 2.2 kV electrospray voltage and a capillary temperature of 275 °C. The mass spectrometer was operated in data-dependent acquisition mode, automatically switching between MS and MS2 scans. Full-scan MS spectra (m/z 360–1300) were acquired in the Orbitrap at 70 000 (m/z 200) resolution with an automatic gain control (AGC) target value of 3e6. Fragment mass (MS2) spectra were generated for up to 20 precursors with a normalized collision energy of 28% using higher energy collision-induced dissociation (HCD). The fixed first mass was set to 100 m/z for fragment mass spectra, and fragment ions were read out in the Orbitrap mass analyzer at a resolving power of 17 500 at m/z 200. The isolation window was set to 1.7 Th, and a MS2 AGC target value of 1e5 was used. Sequenced precursor ions were dynamically excluded for 35 s.
Data processing and peptide and protein identification and quantification was performed using MaxQuant (version 1.5.3.8) (including its built-in iBAQ calculations).51 Briefly, raw MS data were processed by MaxQuant for peak detection and quantification. MS/MS spectra were searched against the current version of the human UniProt database (download date 21.08.2017) supplemented with common contaminants using the Andromeda search engine with the following search parameters: full tryptic specificity, up to two missed cleavage sites, carbamidomethylation of cysteine residues set as a fixed modification, and N-terminal protein acetylation and methionine oxidation as variable modifications. Mass spectra were recalibrated within MaxQuant (first search, 20 ppm precursor tolerance) and subsequently researched with a mass tolerance of 6 ppm. Fragment ion mass tolerance was set to 20 ppm. Search results were filtered to a maximum false discovery rate (FDR) of 1% for proteins and 1% for peptide spectrum matches, and a minimum peptide length of at least 7 amino acids was required.
Effects of Rodin-A, Rodin-B, Rodin-C, Rodin-D, and CI-994 on Human Myeloid and Erythroid Progenitors Using Colony Forming Cell Assays (ReachBio)
Compounds were tested to evaluate the potential effects on human erythroid and myeloid progenitors using colony forming cell assays. Clonogenic progenitors of human erythroid (CFU-E, BFU-E), granulocyte-monocyte (CFU-GM), and multipotential (CFU-GEMM) lineages were assessed in a semisolid methylcellulose-based media formulation containing recombinant human interleukin 3 (rhIL-3, 10 ng/mL), recombinant human granulocyte-macrophage colony-stimulating factor (rhGM-CSF, 10 ng/mL), recombinant human stem cell factor (rhSCF, 50 ng/mL) and erythropoietin (Epo, 3 U/mL).
Cells
Normal human bone marrow light density cells derived from normal bone marrow (NorCal Biologics, California) and qualified at ReachBio were stored in the gaseous phase of liquid nitrogen (−152 °C) until required for the assay. On the day of the experiment, the cells were thawed rapidly, the contents of each vial was diluted in 10 mL of Iscove’s modified Dulbecco’s medium containing 10% fetal bovine serum (IMDM + 10% FBS) and washed by centrifugation (approximately 1200 rpm for 10 min, room temperature). The supernatant was discarded, and the cell pellets resuspended in a known volume of IMDM + 10% FBS. A cell count (3% glacial acetic acid) and viability assessment (trypan blue exclusion test) was performed for the bone marrow sample.
Compounds
On the day of the experiment, the compounds were dissolved in DMSO to a stock concentration of 10 mM. Serial dilutions were prepared from the stock concentration to achieve concentrations of 2 and 0.4 mM. When added to the methylcellulose-based media at 1:1000 (v/v), the final test concentrations of 10, 2, and 0.4 μM were achieved. Additionally, 5-FU was evaluated at 1.0, 0.1, and 0.01 μg/mL.
Method Summary
Clonogenic progenitors of the human erythroid (CFU-E and BFU-E) and myeloid (CFU-GM) lineages were set up in the methylcellulose-based media formulations described above. All compounds were added to the medium to give the final desired concentrations (10, 2, and 0.4 μM). 5-Fluorouracil (Sigma-Aldrich) was used as a positive control for progenitor proliferation (inhibition of colony growth) and was introduced to the human bone marrow cultures at 1.0, 0.1, and 0.01 μg/mL. Solvent control cultures (containing no compound but 0.1% DMSO) as well as standard controls (containing no compound and no DMSO) were also initiated. Human myeloid and erythroid progenitor assays were initiated at 2.0 × 104 cells per culture. Following 14 days in culture, myeloid and erythroid colonies were assessed microscopically and scored by trained personnel. The colonies were divided into the following categories based on size and morphology: CFU-E, BFU-E, CFU-GM, and CFU-GEMM.
Statistical Analyses of CFC Numbers
The mean ± SEM of three replicate cultures was calculated for progenitors of each category (CFU-E, BFU-E, etc.). Two-tailed t tests were performed to assess if there was a difference in the number of colonies generated between solvent control and treated cultures. Due to the potential subjectivity of colony enumeration, a p value of less than 0.01 is deemed significant. To calculate the concentration of 50% inhibition of colony growth (IC50) for each compound, a dose response curve was generated plotting the log of the compound concentration versus the percentage of control colony growth using XLfit software (IDBS). The concentration of 50% inhibition of colony growth (IC50) was calculated based on the sigmoid curve fit using dose–response, one-site model formula: y = A + [(B – A)/(1 + (C/x)D)], where A = the initial value (baseline response), B = maximum response, C = center (drug concentration that provokes a response halfway between A and B), and D = slope of the curve at midpoint. Further plots and additional dose response curves were generated using GraphPad Prism 7.0.
Morphological Assessment of Colonies
Photographs were taken of representative hematopoietic progenitor-derived colonies from various lineages, illustrating colonies in the presence of the solvent control as well as colonies in the presence of the test compounds.
Erythroid (CFU-E and BFU-E), myeloid (CFU-GM), and multipotential (CFU-GEMM) colony enumeration was performed by trained personnel. The distribution of colony types as well as general colony and cellular morphology was analyzed. For statistical analysis, colony numbers in compound treated cultures were compared to the solvent control cultures. 5-FU was used as a positive control for toxicity in these assays, and the inhibitory effects obtained for this compound were exactly as expected. The experiment was used to evaluate the potential effect of test compounds on human erythroid and myeloid progenitor proliferation in a methylcellulose-based medium. The IC50 values were calculated from XLfit. Dose response curves for erythroid and myeloid toxicity were generated by XLfit. Finally, nonlinear regression curve fitting and IC50 values ± 95% CI, were calculated by Prism 7.0.
Measuring the Effects of Rodin-A and CI-994 on Histone Acetylation
K562 cells were purchased from ATCC (Virginia, USA) and stored in liquid nitrogen until use (cultured for 10 days after thaw). K562 cells were cultured in IMDM medium plus 10% FBS and pen/strep, in a cell culture incubator at 37 °C with 5% CO2. Anti-Ac-H3K9 rabbit mAb (no. 9649) and anti-H3 mouse mAb (no. 3638) were purchased from Cell Signaling Technology. Anti-rabbit IgG IRDye 680RD and anti-mouse IgG IRDye 800CW secondary antibodies were purchased from LI-COR. Bis-Tris gel (12%) and nitrocellulose membrane were purchased from Thermo Fisher Scientific. For the assays, 3 × 105 cells/well of K562 cells in 1 mL of complete culture medium was seeded to the wells of 24-well plates overnight. The cells were treated with compounds in triplicate (starting at 33.3 μM, 10-doses with 3-fold dilution) for 24 h. Cells treated with 5 μM SAHA and 10 μM TSA were used as the positive control. Cells treated with 0.1% DMSO were used as the negative control. After treatment, the K562 cells were washed once with PBS and lysed with 100 μL of 1× SDS sample buffer containing 50 mM DTT. The lysate samples were sonicated and then heated at 90 °C for 5 min. Cell lysate samples (13 μL) were subjected to SDS-PAGE with 12% Bis-Tris gel and transferred onto nitrocellulose membrane by iBlot dry blotting system. The membrane was blocked with 3% nonfat milk for 1 h and probed with anti-Ac-H3K9 and anti-H3 antibodies. Anti-rabbit IgG IRDye 680RD and anti-mouse IgG IRDye 800CW secondary antibodies were used to detect the primary antibodies. The membranes were scanned with LI-COR Odyssey Fc Imaging System. The specific bands of interest were quantified by LI-COR Image Studio Lite software. Change in acetylation status of H3K9 was determined by normalizing Ac-H3K9 bands to H3 bands (Figure 2C, Figure S1). The percent acetylated H3K9 relative control EC50 curves were plotted, and EC50 values were calculated using the GraphPad Prism 7 program based on a 4-parameter sigmoidal dose–response equation.
Evaluation of the Free Drug Concentration of Rodin-A in Mouse Hippocampus by MetaQuant Microdialysis and Plasma Concentration
The biotechnical and bioanalytical experiments described in the present report were performed at Brains On-Line, LLC (South San Francisco, USA).
Animals
Eight adult male C57Bl/6 mice (20–30 g) were used for this study. Upon arrival, the animals were group-housed in plastic cages on a 12/12 h light/dark cycle. Animals had access to food and water ad libitum. Experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee of Brains On-Line, LLC.
Test Substance and Formulation
Test compound (Rodin-A) was prepared in 20% HPβCD in sterile water and sonicated at room temperature for 90 min. Solutions were prepared fresh daily for dosing.
Surgery
Mice were anesthetized using isoflurane (2%, 800 mL/min O2). Bupivacaine/epinephrine was used for local anesthesia, and carprofen was used for peri-/postoperative analgesia. First, a jugular vein (JV) cannula was implanted for serial blood collection (BrainLink, The Netherlands). Then, the animals were placed in a stereotaxic frame (Kopf Instruments, USA) and a MetaQuant (MQ) microdialysis probe (4 mm PAN membrane, BrainLink, The Netherlands) was implanted into the right hippocampus. A second microdialysis probe (3 mm PEE membrane, BrainLink, The Netherlands) was inserted into the left hippocampus. Coordinates for the tips of the probes into the hippocampus were as follows: anterior–posterior (AP) = −3.1 mm from bregma, lateral (L) = ±2.8 mm from midline, and ventral (V) = −4.5 mm from dura; the toothbar set at 0.0 mm. After surgery, animals were housed individually in cages and provided food and water ad libitum.
Experiments
Experiments were performed 1 day after surgery. On experimental day 1, the MetaQuant microdialysis probe was connected with flexible PEEK tubing to a microperfusion pump (Harvard PHD 2000 Syringe pump, Holliston, MA). Microdialysis probes were perfused with aCSF containing 147 mM NaCl, 3.0 mM KCl, 1.2 mM CaCl2, 1.2 mM MgCl2 + 0.2% β-CD, at a slow flow rate of 0.15 μL/min and a carrier flow (UP + 0.2% β-CD) at a rate of 0.8 μL/min. Microdialysis samples were collected for 30 min periods by an automated fraction collector (820 Microsampler, Univentor, Malta) into 300 μL polypropylene minivials. After stabilization, one baseline sample was collected, and test compounds were given PO. Samples were collected for an additional 9 h. All the samples were stored at −80 °C awaiting analysis by Brains On-Line. Blood samples were collected via the JV cannula at 0, 30, 60, 120, 360, and 540 min after dosing into EDTA anticoagulant vials (∼25 μL blood/sample). Plasma was harvested within 15 min after collection and aliquoted (∼10 μL/sample) into a separate vial and stored at −80 °C awaiting analysis by Brains On-Line. On the following day (day 2), the push/pull microdialysis probes were connected with FEP tubing to a microperfusion pump (Harvard PHD 2000 Syringe pump, Holliston, MA or similar). Microdialysis probes were perfused with aCSF containing 147 mM NaCl, 3.0 mM KCl, 1.2 mM CaCl2 and 1.2 mM MgCl2, and 0.15% BSA at a flow rate of 0.75 μL/min. Microdialysis samples were collected for 60 min periods by an automated fraction collector (820 Microsampler, Univentor, Malta) into polypropylene (300 μL) minivials. Samples were collected for a total of 8 h after dosing. At the end of day 2 of dialysis, mice were euthanized and brain was collected for verification of probe placement by Brains On-Line. Push/pull dialysis samples were stored at −80 °C before shipping.
Bioanalysis
Concentrations of Rodin-A in dialysates and plasma were determined by HPLC coupled to a tandem mass spectrometer (LC/MS/MS).
Quantification of Rodin-A
Plasma samples were first mixed for protein precipitation with a solution of acetonitrile/ultrapure water (80:20). After 5 min incubation at room temperature, the samples were centrifuged for 5 min (13000 rpm, 4 °C) and the supernatant was diluted 10-fold in ultrapure water/acetonitrile (80:20). Dialysate samples and diluted plasma supernatants were injected (5 μL) into an Infinity 1290 LC system (Agilent, USA) by an automated sample injector (SIL-20AD, Shimadzu, Japan). Analytes were separated by liquid chromatography using a linear gradient of mobile phase B at a flow rate of 0.300 mL/min on a reversed phase Phenomenex Luna C18(2)-HST column (3.0 mm × 100 mm, 2.5 μm particle size; Phenomenex, USA) held at a temperature of 35 °C. Mobile phase A consisted of UP H2O with 5 mM ammonium acetate. Mobile phase B was MeOH with 5 mM ammonium acetate. Acquisitions were achieved in positive ionization mode using a QTrap 5500 (Applied Biosystems, USA) equipped with a Turbo Ion Spray interface. The ion spray voltage was set at 5.0 kV, and the probe temperature was 500 °C. The collision gas (nitrogen) pressure was kept at the Medium setting level. The following MRM transitions were used for quantification: 353.3/218.0 for Rodin-A. Data were calibrated and quantified using the Analyst data system (Applied Biosystems, version 1.5.2).
Hippocampal Dendritic Spine Density Analysis
Research Objectives
To measure the effects on dendritic spines in hippocampus after oral dosing of compound daily for 3,7, 10, or 14 days.
Research Subjects
Male C57BL/6J mice (from Shanghai Lingchang Biotech, Shanghai, China) were dosed orally with CI-994 (daily with 1 and 3 mg/kg for 3 to 14 days, n = 5 per group) or with Rodin-A (daily with 1, 3, and 6 mg/kg, for 3, 7, 10, and 14 days, n = 7 per group) or vehicle (20% HPβCD, daily for 3, 7, 10, and 14 days, n = 5 or 7 per group) at ChemPartner, Shanghai, China. Male C57BL/6J mice were dosed orally with CI-994 (daily with 0.3 and 3 mg/kg for 14 days) or vehicle (0.5% MC + 0.1% Tween80 in ddH2O, daily for 14 days, n = 7 per group). Animals were sacrificed 24 h after the last dose of CI-994 and 24 h, 3 days, and 7 days after last dose of Rodin-A and vehicle. Ballistic dye labeling was performed according to protocols developed by Afraxis to label target neurons (Afraxis, SanDiego, CA).52 Laser-scanning confocal microscopy (Olympus FV1000) was performed using a 63× objective (1.42 NA) to scan individually labeled neurons at high resolution (0.103 × 0.103 × 0.33 μm3 voxels). Target neurons were identified in the brain region of interest by anatomical location and cell morphology. A minimum of 5 samples per mouse were measured for each segment. Blind deconvolution was applied to raw three-dimensional digital images, which were then analyzed for spine density and morphology by trained analysts. Individual spines were measured manually for (a) head diameter, (b) length, and (c) neck thickness. Each dendrite was analyzed by 3 (on average) independent analysts. Automated image assignment software distributed images to analysts in a randomized manner and ensured that each analyst performed measurements of near equal numbers of dendrites per group. Analysts were blinded to all experimental conditions (including treatment, brain region, and cell type).
Statistical analysis of interanalyst variability for each dendrite was examined online and used to eliminate dendrites that did not meet interanalyst reliability criteria. For spine density and spine morphological classification, data across analysts were averaged to report data for each dendrite. Data for each dendrite were then averaged to report data for each mouse.
Means and SEM for spine density were calculated using animal’s individual values. A one-way ANOVA was performed followed by a Dunnett’s post hoc analysis.
Immunofluorescence and Microscopy Measurements of SV2A, pTrkB, and PSD-95
Research Objectives
To measure SV2A, pTrkB, and PSD-95 in brain slices from mice dosed orally with compound.
Research Subjects
From male C57BL/6J mice, 20 μm slices from the brain region of interest were incubated for 24 h (4 °C) in blocking solution (0.1 M PB, 4% bovine serum albumin, 0.3% Triton-X-100) with the following primary antibodies: rabbit anti-SV2A (1:500; Abcam ab32942), mouse anti-PSD95 (1:1000, Affinity Bioreagents, no. MA1-045), rabbit anti-phosphorylated Trk (Tyr490; 1:200; Cell Signaling no. 9141). Sections were incubated for 1 h (RT) in secondary antisera with Alexa594 anti-rabbit IgG and Alexa488 anti-mouse IgG (Invitrogen). Although anti-pTrkY490 recognizes a site conserved across TrkA, -B, and -C, TrkB is predominantly expressed by hippocampal neurons, and both TrkC and TrkA are therefore unlikely to contribute to results. Super-resolution laser-scanning confocal microscopy (Zeiss LSM880, Airyscan)52,53 was performed using a 63× objective (1.4 NA) to scan regions of interest (20 × 20 × 5 μm3, xyz) at high resolution (scan resolution = 0.07 μm/pixel; axial resolution = 0.07 μm/focal step). Target laminae were identified in the brain region of interest by epifluorescence navigation (DAPI) using anatomical landmarks. Microscopy was performed blind to experimental conditions. A minimum of 5 mice were tested in each experimental condition. A minimum of 5 samples per mouse were measured for each target lamina. Micrographs were analyzed using image segmentation analysis based on established protocols.53,54 Intensity normalization to a target background level (15% of maximum) was performed on image subsections (5 × 5 × 1 μm3) to standardize object quantification across sections and batches of tissue. A one-way ANOVA was performed followed by a Dunnett’s post hoc analysis.
Measurement of LTP in 5xFAD Mice
Research Objectives
To measure the effects on long-term potentiation upon daily dosing for 14 days to mice with cognitive deficits.
Research Subjects
The effects of Rodin-A were tested in the 5xFAD mouse model of AD on the properties of LTP at the Schaffer collateral-CA1 hippocampal synaptic layer in 9–12 month old mice (C57BL/6J background strain). Animals were dosed once daily by oral gavage for 14 days. Animals were sacrificed and hippocampal brain slices (350 μm) were cut on the ice-cold stage of a vibrating tissue slicer. The slices were maintained in oxygenated artificial cerebrospinal fluid (aCSF, 124 mM NaCl, 3 mM KCl, 1.2 mM NaH2PO4, 26 mM NaHCO3, 2.4 mM CaCl2, 2 mM MgCl2, 10 mM glucose, pH 7.4) at room temperature for at least 1 h, and then slices were transferred to the multielectrode recording chamber and constantly perfused with oxygenated aCSF at 30 °C for 0.5 h before recording. Electrophysiology was performed with a MED-PG515A 16 electrode array. The basal synaptic transmission was characterized by monitoring the fEPSP slope every second for 30 min before the theta burst stimulus (TBS) was applied, and the 10%–90% slope of input/output (I/O) curves of the field excitatory postsynaptic potential (fEPSP) was determined. The stimulus current was adjusted so that fEPSP stabilized at 50% of maximum. LTP was induced by theta burst stimulus (TBS, 10 bursts with 200 ms interburst interval, each burst consisting of 4 pulses at 100 Hz, delivered at 1.6× stimulation intensity of the baseline stimulation). Test stimulation resumed immediately after the TBS, and recording of the evoked fEPSP responses was monitored for the next 60 min. The plateau of the last 10 min fEPSP slope post-TBS was considered the LTP response. All LTP measurements were performed blinded to treatment. Occasionally, several LTP measures were made in a single slice and/or from multiple slices taken from a single animal; in all cases LTP values were averaged per animal before being used, and the n is the number of animals analyzed per arm. For all experiments, statistical analysis was by ANOVA with post hoc Holm–Sidak analysis corrected for multiple comparisons using statistical hypothesis testing (GraphPad Prism 7 software).
In Vivo 14 Day Safety Evaluation in Rat and Dog
Rat
The objective of this study was to determine the potential toxicity of Rodin-A when given by gavage once daily for 14 days to male Sprague–Dawley rats. In addition, the toxicokinetic characteristics of Rodin-A were determined using a nonvalidated bioanalytical method. In total, 20 male rats were dosed with either Rodin-A (30, 45, or 60 mg/kg/day or vehicle (0.5% methylcellulose/0.1% Tween 80 in RODI (reverse osmosis deionized) water); n = 12 for safety studies and n = 8 for toxicokinetic analysis). End points in the study consisted of mortality checks, clinical observations, body weight (weekly) and food consumption (daily) measurements, clinical pathology (hematology and clinical chemistry), bone marrow smear evaluations, toxicokinetic evaluation, and anatomical pathology evaluations consisting of macroscopic observations, organ weights, and microscopic evaluations.
Dog
The objective of this study was to determine the potential toxicity of Rodin-A when given by gavage once daily for 14 days to male beagle dogs. In addition, the toxicokinetic characteristics of Rodin-A were determined using a nonvalidated bioanalytical method. Naive beagle dogs (15 male animals; 7.8–11.8 kg and 12–13 months old at the time of dosing) were assigned to dose groups and dosed with Rodin-A (45, 60, or 120 mg/kg/day) or vehicle (0.5% methylcellulose/0.1% Tween 80 in RODI water. End points in the study consisted of mortality checks, clinical observations, body weight (weekly) and food consumption (daily) measurements, clinical pathology (hematology and clinical chemistry), bone marrow smear evaluations, toxicokinetic evaluation, and anatomical pathology evaluations consisting of macroscopic observations, organ weights, and microscopic evaluations.
Acknowledgments
We thank Hannes Hahne and colleagues at OmicScouts, Christopher Rex and colleagues at Afraxis, Yingtao Tong, Dongfeng Hou, and colleagues at ChemPartner, Jianghong Wu, Wei Xiao, and colleagues at Reaction Biology, and Emer Clarke and colleagues at ReachBio for experimental support. We thank Stefan Steinbacher and Peter Reinemer of Proteros for the work on the Rodin-A cocrystal structure. We thank John A. Lowe III of JL3 Pharma for useful discussions on medicinal chemistry.
Glossary
Abbreviations
- HDAC
histone deacetylase
- CoREST
co-repressor of Repressor Element1 Silencing Transcription Factor
- HDACi
histone deacetylase inhibitor
- REST
repressor element-1 silencing transcription factor
- NuRD
nucleosome remodeling and deacetylase
- Sin3
switch-independent protein 3
- SMRT
silencing mediator of retinoic acid and thyroid hormone receptor
- NCoR
nuclear receptor co-repressor
- LSD1
lysine-specific demethylase 1
- CFU
colony forming unit
- WT
wild-type
- HPβCD
hydroxypropyl-β-cyclodextrin
- SV2A
presynaptic vesicle glycoprotein 2A
- PSD95
postsynaptic density protein 95
- BDNF
brain-derived neurotrophic factor
- TrkB
tropomyosin receptor kinase B
- pTrkB
phosphorylated tropomyosin receptor kinase B
- LTP
long-term potentiation
- NOAEL
no-obsersved-adverse-effect level
- AUC
area under the curve
- HED
human equivalent dose
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.8b00620.
Experimental procedures for the synthesis of Rodin-A, Rodin-B, Rodin-C, and Rodin-D, experimental details and procedure for recombinant HDAC isoform biochemical assays, data collection and processing statistics for cocrystal structure of Rodin-A bound to HDAC2, experimental procedure and inhibition data for LSD1 and LSD1/CoREST recombinant enzymatic assays, experimental procedure for pharmacokinetic studies, full Western blots of acetylation at histone 3, position lysine 9 for Rodin-A and CI-994, hippocampal dendritic spine density following 14 days of dosing with Rodin-A, pharmacokinetic data for CI-994 and Rodin-A in mice, and pharmacokinetic data from microdialysis experiments with Rodin-A in mice (PDF)
Author Present Address
† M.S.Q.: Takeda Pharmaceuticals, 35 Landsdowne St., Cambridge, MA 02139.
Author Present Address
‡ T.D.M.: Takeda, 75 Sidney St., Cambridge, MA 02139.
Author Contributions
N.O.F. designed compounds and chemistry strategy, analyzed data, and wrote the paper. B.A.L., M.C.H., T.D.M., and M.S.Q. designed and coordinated in vitro and in vivo experiments and analyzed data. A.P. analyzed data and wrote the paper. M.I. contributed to overall strategy and experimental design and wrote the paper.
Part of this work was supported by grants from the NIH (SBIR Grant 1R43AG048651-01A1), Michael J. Fox Foundation (Grant 11581), and the Alzheimer’s Drug Discovery Foundation (Grant No. 20150504.02).
The authors declare the following competing financial interest(s): The authors are, or were, employees of Rodin Therapeutics, Inc., and are stockholders in the company.
Supplementary Material
References
- Coleman P. D.; Yao P. J. (2003) Synaptic slaughter in Alzheimer’s disease. Neurobiol. Aging 24, 1023–1027. 10.1016/j.neurobiolaging.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Imbriani P.; Schirinzi T.; Meringolo M.; Mercuri N. B.; Pisani A. (2018) Centrality of Early Synaptopathy in Parkinson’s Disease. Front. Neurol. 9, 103. 10.3389/fneur.2018.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E.; Mallory M.; Alford M.; DeTeresa R.; Hansen L. A.; McKeel D. W. Jr.; Morris J. C. (2001) Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 56, 127–129. 10.1212/WNL.56.1.127. [DOI] [PubMed] [Google Scholar]
- Scheff S. W.; Neltner J. H.; Nelson P. T. (2014) Is synaptic loss a unique hallmark of Alzheimer’s disease?. Biochem. Pharmacol. 88, 517–528. 10.1016/j.bcp.2013.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry R. D.; Masliah E.; Salmon D. P.; Butters N.; DeTeresa R.; Hill R.; Hansen L. A.; Katzman R. (1991) Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580. 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- de Wilde M. C.; Overk C. R.; Sijben J. W.; Masliah E. (2016) Meta-analysis of synaptic pathology in Alzheimer’s disease reveals selective molecular vesicle machinery vulnerability. Alzheimer's Dementia 12, 633–644. 10.1016/j.jalz.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff S. W.; Price D. A.; Ansari M. A.; Roberts K. N.; Schmitt F. A.; Ikonomovic M. D.; Mufson E. J. (2015) Synaptic change in the posterior cingulate gyrus in the progression of Alzheimer’s disease. J. Alzheimer's Dis. 43, 1073–1090. 10.3233/JAD-141518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ruijter A. J. M.; van Gennip A. H.; Caron H. N.; Kemp S.; van Kuilenburg A. B. P. (2003) Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 370, 737–749. 10.1042/bj20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J.-S.; Haggarty S. J.; Giacometti E.; Dannenberg J.-H.; Joseph N.; Gao J.; Nieland T. J. F.; Zhou Y.; Wang X.; Mazitschek R.; Bradner J. E.; DePinho R. A.; Jaenisch R.; Tsai L.-H. (2009) HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459, 55–60. 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gräff J.; Rei D.; Guan J.-S.; Wang W.-Y.; Seo J.; Hennig K. M.; Nieland T. J. F.; Fass D. M.; Kao P. F.; Kahn M.; Su S. C.; Samiei A.; Joseph N.; Haggarty S. J.; Delalle I.; Tsai L.-H. (2012) An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483, 222–206. 10.1038/nature10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penney J.; Tsai L.-H. (2014) Histone deacetylases in memory and cognition. Sci. Signaling 7, re12. 10.1126/scisignal.aaa0069. [DOI] [PubMed] [Google Scholar]
- Nativio R.; Donahue G.; Berson A.; Lan Y.; Amlie-Wolf A.; Tuzer F.; Toledo J. B.; Gosai S. J.; Gregory B. D.; Torres C.; Trojanowski J. Q.; Wang L.-S.; Johnson F. B.; Bonini N. M.; Berger S. L. (2018) Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat. Neurosci. 21, 497–505. 10.1038/s41593-018-0101-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A.; Sananbenesi F.; Wang X.; Dobbin M.; Tsai L.-H. (2007) Recovery of learning and memory is associated with chromatin remodeling. Nature 447, 178–182. 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Akhtar M. W.; Raingo J.; Nelson E. D.; Montgomery R. L.; Olson E. N.; Kavalali E. T.; Monteggia L. M. (2009) Histone deacetylases 1 and 2 form a developmental switch that controls excitatory synapse maturation and function. J. Neurosci. 29, 8288–8297. 10.1523/JNEUROSCI.0097-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev A. G.; Thompson L. M. (2008) Therapeutic application of histone deacetylases for central nervous system disorders. Nat. Rev. Drug Discovery 7, 854–868. 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- Subramanian S.; Bates S. E.; Wright J. J.; Espinoza-Delgado I.; Piekarz R. L. (2010) Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals 3, 2751–2767. 10.3390/ph3092751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziano M. J.; Pilcher G. D.; Walsh K. M.; Kasalil O. B.; Radulovic L. (1997) Preclinical toxicity of a new oral anticancer drug, CI-994 (Acetyldinaline), in rats and dogs. Invest. New Drugs 15, 295–310. 10.1023/A:1005937502511. [DOI] [PubMed] [Google Scholar]
- Gupta P.; Reid R. C.; Iyer A.; Sweet M. J.; Fairlie D. P. (2012) Towards Isozyme-Selective HDAC Inhibitors For Interrogating Disease. Curr. Top. Med. Chem. 12, 1479–1499. 10.2174/156802612802652420. [DOI] [PubMed] [Google Scholar]
- Wagner F. F.; Zhang Y.-L.; Fass D. M.; Joseph N.; Gale J. P.; Weïwer M.; McCarren P.; Fisher S. L.; Kaya T.; Zhao W.-N.; Reis S. A.; Hennig K. M.; Thomas M.; Lemercier B. C.; Lewis M. C.; Guan J. S.; Moyer M. P.; Scolnick E.; Haggarty S. J.; Tsai L.-H.; Holson E. B. (2015) Kinetically selective inhibitors of histone deacetylase 2 (HDAC2) as cognition enhancers. Chem. Sci. 6, 804–815. 10.1039/C4SC02130D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradei O. M.; Mallais T. C.; Frechette S.; Paquin I.; Tessier P. E.; Leit S. M.; Fournel M.; Bonfils C.; Trachy-Bourget M.-C.; Liu J.; Yan T. P.; Lu A.-H.; Rahil J.; Wang J.; Lefebvre S.; Li Z.; Vaisburg A. F.; Besterman J. M. (2007) Novel aminophenyl benzamide-type histone deacetylase inhibitors with enhanced potency and selectivity. J. Med. Chem. 50, 5543–5546. 10.1021/jm701079h. [DOI] [PubMed] [Google Scholar]
- Bantscheff M.; Hopf C.; Savitski M. M.; Dittmann A.; Grandi P.; Michon A.-M.; Schlegl J.; Abraham Y.; Becher I.; Bergamini G.; Boesche M.; Delling M.; Dümpelfeld B.; Eberhard D.; Huthmacher C.; Mathieson T.; Poeckel D.; Reader V.; Strunk K.; Sweetman G.; Kruse U.; Neubauer G.; Ramsden N. G.; Drewes G. (2011) Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 29, 255–265. 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- Gräff J.; Joseph N. F.; Horn M. E.; Samiei A.; Meng J.; Seo J.; Rei D.; Bero A. W.; Phan T. X.; Wagner F.; Holson E.; Xu J.; Sun J.; Neve R. L.; Mach R. H.; Haggarty S. J.; Tsai L.-H. (2014) Epigenetic priming of memory updating during reconsolidation to attenuate fear memories. Cell 156, 261–276. 10.1016/j.cell.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas N.; Grunseich C.; Lu D. D.; Speh J. C.; Mandel G. (2005) REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 121, 645–657. 10.1016/j.cell.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Abrajano J. J.; Qureshi I. A.; Gokhan S.; Zheng D.; Bergman A.; Mehler M. F. (2009) REST and CoREST modulate neuronal subtype specification, maturation, and maintenance. PLoS One 4, e7936 10.1371/journal.pone.0007936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot J. L.; Chakravarty P. K.; Chenard M.; Close J.; Cruz J. C.; Dahlberg W. K.; Fleming J.; Hamblett C. L.; Hamill J. E.; Harrington P.; Harsch A.; Heidebrecht R.; Hughes B.; Jung J.; Kenific C. M.; Kral A. M.; Meinke P. T.; Middleton R. E.; Ozerova N.; Sloman D. L.; Stanton M. G.; Szewczak A. A.; Tyagarajan S.; Witter D. J.; Secrist J. P.; Miller T. A. (2008) Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 18, 973–978. 10.1016/j.bmcl.2007.12.031. [DOI] [PubMed] [Google Scholar]
- Bressi J. C.; Jennings A. J.; Skene R.; Wu Y.; Melkus R.; Jong R. D.; O’Connell S.; Grimshaw C. E.; Navre M.; Gangloff A. R. (2010) Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg. Med. Chem. Lett. 20, 3142–3145. 10.1016/j.bmcl.2010.03.091. [DOI] [PubMed] [Google Scholar]
- Lauffer B. E. L.; Mintzer R.; Fong R.; Mukund S.; Tam C.; Zilberleyb I.; Flicke B.; Ritscher A.; Fedorowicz G.; Vallero R.; Ortwine D. F.; Gunzner J.; Modrusan Z.; Neumann L.; Koth C. M.; Lupardus P. J.; Kaminker J. S.; Heise C. E.; Steiner P. (2013) Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 288, 26926–26943. 10.1074/jbc.M113.490706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner F. F.; Weïwer M.; Steinbacher S.; Schomburg A.; Reinemer P.; Gale J. P.; Campbell A. J.; Fisher S. L.; Zhao W.-N.; Reis S. A.; Hennig K. M.; Thomas M.; Müller P.; Jefson M. R.; Fass D. M.; Haggarty S. J.; Zhang Y.-L.; Holson E. B. (2016) Kinetic and structural insights into the binding of histone deacetylase 1 and 2 (HDAC1,2) inhibitors. Bioorg. Med. Chem. 24, 4008–4015. 10.1016/j.bmc.2016.06.040. [DOI] [PubMed] [Google Scholar]
- Berger M. R.; Richter H.; Seelig M. H.; Eibl J.; Schmähl D. (1990) New cytostatics – more activity and less toxicity. Cancer Treat. Rev. 17, 143–154. 10.1016/0305-7372(90)90039-I. [DOI] [PubMed] [Google Scholar]
- Becher I.; Dittmann A.; Savitski M. M.; Hopf C.; Drewes G.; Bantscheff M. (2014) Chemoproteomics reveals time-dependent binding of histone deacetylase inhibitors to endogenous repressor complexes. ACS Chem. Biol. 9, 1736–1746. 10.1021/cb500235n. [DOI] [PubMed] [Google Scholar]
- Millard C. J.; Watson P. J.; Fairall L.; Schwabe J. W. R. (2017) Targeting Class I Histone Deacetylases in a “Complex” Environment. Trends Pharmacol. Sci. 38, 363–377. 10.1016/j.tips.2016.12.006. [DOI] [PubMed] [Google Scholar]
- Robertson J. C.; Hurley N. C.; Tortorici M.; Ciossani G.; Borrello M. T.; Vellore N. A.; Ganesan A.; Mattevi A.; Baron R. (2013) Expanding the druggable space of the LSD1/CoREST epigenetic target: new potential binding regions for drug-like molecules, peptides, protein partners, and chromatin. PLoS Comput. Biol. 9, e1003158 10.1371/journal.pcbi.1003158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalin J. H.; Wu M.; Gomez A. V.; Song Y.; Das J.; Hayward D.; Adejola N.; Wu M.; Panova I.; Chung H. J.; Kim E.; Roberts H. J.; Roberts J. M.; Prusevich P.; Jeliazkov J. R.; Burman S. S. R.; Fairall L.; Milano C.; Eroglu A.; Proby C. M.; Dinkova-Kostova A. T.; Hancock W. W.; Gray J. J.; Bradner J. E.; Valente S.; Mai A.; Anders N. M.; Rudek M. A.; Hu Y.; Ryu B.; Schwabe J. W. R.; Mattevi A.; Alani R. M.; Cole P. A. (2018) Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat. Commun. 9, 53. 10.1038/s41467-017-02242-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milelli A.; Marchetti C.; Turrini E.; Catanzaro E.; Mazzone R.; Tomaselli D.; Fimognari C.; Tumiatti V.; Minarini A. (2018) Novel polyamine-based histone deacetylases-Lysine demethylase 1 dual binding inhibitors. Bioorg. Med. Chem. Lett. 28, 1001–1004. 10.1016/j.bmcl.2018.02.034. [DOI] [PubMed] [Google Scholar]
- Pessina A.; Albella B.; Bayo M.; Bueren J.; Brantom P.; Casati S.; Croera C.; Gagliardi G.; Foti P.; Parchment R.; Parent-Massin D.; Schoeters G.; Sibiril Y.; Van Den Heuvel R.; Gribaldo L. (2003) Application of the CFU-GM assay to predict acute drug-induced neutropenia: an international blind trial to validate a prediction model for the maximum tolerated dose (MTD) of myelosuppressive xenobiotics. Toxicol. Sci. 75, 355–367. 10.1093/toxsci/kfg188. [DOI] [PubMed] [Google Scholar]
- Karmodiya K.; Krebs A. R.; Oulad-Abdelghani M.; Kimura H.; Tora L. (2012) H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genomics 13, 424. 10.1186/1471-2164-13-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W.; Gillard M.; Sands Z. A.; Kaminski R. M.; Klitgaard H. (2016) Synaptic vesicle glycoprotein 2A ligands in the treatment of epilepsy and beyond. CNS Drugs 30, 1055–1077. 10.1007/s40263-016-0384-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt C. A.; Schenker L. J.; Kennedy M. B. (1996) PSD-95 is associated with the postsynaptic density and not with the presynaptic membrane at forebrain synapses. J. Neurosci. 16, 1380–1388. 10.1523/JNEUROSCI.16-04-01380.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenberg T.; Mohammed A. K.; Henriksson B.; Persson H.; Winblad B.; Lindefors N. (1992) Increased expression of brain-derived neurotrophic factor mRNA in rat hippocampus is associated with improved spatial memory and enriched environment. Neurosci. Lett. 138, 153–156. 10.1016/0304-3940(92)90494-R. [DOI] [PubMed] [Google Scholar]
- Elmariah S. B.; Crumling M. A.; Parsons T. D.; Balice-Gordon R. J. (2004) Postsynaptic TrkB-mediated signaling modulates excitatory and inhibitory neurotransmitter receptor clustering at hippocampal synapses. J. Neurosci. 24, 2380–2393. 10.1523/JNEUROSCI.4112-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassoé-Pognetto M.; Utvik J. K.; Camoletto P.; Watanabe M.; Stephenson F. A.; Bredt D. S.; Ottersen O. P. (2003) Organization of postsynaptic density proteins and glutamate receptors in axodendritic and dendrodendritic synapses of the rat olfactory bulb. J. Comp. Neurol. 463, 237–248. 10.1002/cne.10745. [DOI] [PubMed] [Google Scholar]
- Cooke S. F.; Bliss T. V. P. (2006) Plasticity in the human central nervous system. Brain 129, 1659–1673. 10.1093/brain/awl082. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M.; Honkura N.; Ellis-Davies G. C. R.; Kasai H. (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766. 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H.; Cole S. L.; Logan S.; Maus E.; Shao P.; Craft J.; Guillozet-Bongaarts A.; Ohno M.; Disterhoft J.; Van Eldik L.; Berry R.; Vassar R. (2006) Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder F. A.; Lewis M. C.; Fass D. M.; Wagner F. F.; Zhang Y.-L.; Hennig K. M.; Gale J.; Zhao W.-N.; Reis S.; Barker D. D.; Berry-Scott E.; Kim S. W.; Clore E. L.; Hooker J. M.; Holson E. B.; Haggarty S. J.; Petryshen T. L. (2013) A selective HDAC1/2 inhibitor modulates chromatin and gene expression in brain and alters mouse behavior in two mood-related tests. PLoS One 8 (8), e71323 10.1371/journal.pone.0071323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson P. J.; Fairall L.; Santos G. M.; Schwabe J. W. R. (2012) Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 481, 335–340. 10.1038/nature10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard C. J.; Watson P. J.; Celardo I.; Gordiyenko Y.; Cowley S. M.; Robinson C. V.; Fairall L.; Schwabe J. W. R. (2013) Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol. Cell 51, 57–67. 10.1016/j.molcel.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. U.S. Department of Health and Human Services, https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM078932.pdf.
- Hahne H.; Pachl F.; Ruprecht B.; Maier S. K.; Klaeger S.; Helm D.; Médard G.; Wilm M.; Lemeer S.; Kuster B. (2013) DMSO enhances electrospray response, boosting sensitivity of proteomic experiments. Nat. Methods 10, 989–991. 10.1038/nmeth.2610. [DOI] [PubMed] [Google Scholar]
- Shevchenko A.; Wilm M.; Vorm O.; Mann M. (1996) Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal. Chem. 68, 850–858. 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Tyanova S.; Temu T.; Cox J. (2016) The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319. 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- Baumgartel K.; Green A.; Hornberger D.; Lapira J.; Rex C.; Wheeler D. G.; Peters M. (2018) PDE4D regulates spine plasticity and memory in the retrosplenial cortex. Sci. Rep. 8, 3895. 10.1038/s41598-018-22193-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. Y.; Rex C. S.; Casale M. S.; Gall C. M.; Lynch G. (2007) Changes in synaptic morphology accompany actin signaling during LTP. J. Neurosci. 27 (20), 5363–5372. 10.1523/JNEUROSCI.0164-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex C. S.; Chen L. Y.; Sharma A.; Liu J.; Babayan A. H.; Gall C. M.; Lynch G. (2009) Different Rho GTPase–dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J. Cell Biol. 186, 85–97. 10.1083/jcb.200901084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.