

Abstract
X-linked hypophosphatemia (XLH) is a skeletal disease caused by inactivating mutations in the PHEX gene. Mutated or absent PHEX protein/enzyme leads to a decreased serum phosphate level, which cause mineralization defects in the skeleton and teeth (osteomalacia/odontomalacia). It is not yet altogether clear whether these manifestations are caused solely by insufficient circulating phosphate availability for mineralization or also by a direct, local intrinsic effect caused by impaired PHEX activity. Here, we evaluated the local role of PHEX in a 3-dimensional model of extracellular matrix (ECM) mineralization. Dense collagen hydrogels were seeded either with human dental pulp cells from patients with characterized PHEX mutations or with sex- and age-matched healthy controls and cultured up to 24 d using osteogenic medium with standard phosphate concentration. Calcium quantification, micro–computed tomography, and histology with von Kossa staining for mineral showed significantly lower mineralization in XLH cell-seeded scaffolds, using nonparametric statistical tests. While apatitic mineralization was observed along collagen fibrils by electron microscopy in both groups, Raman microspectrometry indicated that XLH cells harboring the PHEX mutation produced less mineralized scaffolds having impaired mineral quality with less carbonate substitution and lower crystallinity. In the XLH cultures, immunoblotting revealed more abundant osteopontin (OPN), dentin matrix protein 1 (DMP1), and matrix extracellular phosphoglycoprotein (MEPE) than controls, as well as the presence of fragments of these proteins not found in controls, suggesting a role for PHEX in SIBLING protein degradation. Immunohistochemistry revealed altered OPN and DMP1 associated with an increased alkaline phosphatase staining in the XLH cultures. These results are consistent with impaired PHEX activity having local ECM effects in XLH. Future treatments for XLH should target both systemic and local manifestations.
Keywords: tooth, PHEX, extracellular matrix, proteins, osteopontin, collagen scaffold
Introduction
X-linked hypophosphatemia (XLH) is a genetic dominant disorder characterized by hypophosphatemia and growth retardation with bone and tooth deformities (osteomalacia and odontomalacia) (Carpenter 2012). The genetic defect underlying XLH is loss-of-function mutations in the PHEX gene (phosphate-regulating gene with homologies to endopeptidases on the X chromosome) (the HYP consortium). Mineralization defects (hypomineralization) observed by this impairment of PHEX are primarily caused by renal phosphate wasting following an increase in circulating fibroblast growth factor 23 (FGF23), a phosphaturic hormone expressed by osteocytes, osteoblasts, and odontoblasts (Quarles 2003; Yoshiko et al. 2007; Bonewald and Wacker 2013; Murali et al. 2016). Current and in-trial treatment strategies for XLH consist of phosphate and vitamin D supplementation, as well as use of a neutralizing monoclonal antibody against FGF23, respectively. In both cases, treatments for XLH aim at increasing the level of circulating serum phosphate (Carpenter et al. 2011; Linglart et al. 2014), although it is best achieved with the antibody treatment. However, accumulating evidence also points to a direct, extracellular matrix (ECM) role for PHEX in regulating mineralization in bones and teeth. Recently, in vitro and in vivo studies, both in humans and in the Hyp mouse (a murine homolog of XLH), have shown the ability of PHEX to proteolytically degrade proteins and peptides known to influence mineralization (Barros et al. 2013; Boukpessi et al. 2016). More specifically, osteopontin (OPN) of the mineralization-regulating SIBLING protein family (small integrin-binding-ligand-N-linked glycoproteins) was shown to be a substrate for PHEX enzymatic activity; PHEX deficiency resulted in the accumulation of mineralization-inhibiting OPN and OPN fragments in bone and dentin of Hyp mice and XLH patients (Barros et al. 2013; McKee et al. 2013; Boukpessi et al. 2016). Despite these findings, there remains some uncertainty regarding the relative contributions to the mineralization defect of systemic versus local matrix inhibitory effects, since both result in hypomineralization. In the field of tissue engineering, disease-modeling approaches (in this case, so-called disease-in-a-dish culture models) have been used extensively to investigate pathologic mechanisms (Grskovic et al. 2011). In the present study, we have used such an in vitro model of human cell-mediated biomineralization, here consisting of seeding dental pulp cells harvested from the teeth of patients with XLH (with corresponding control cultures) into plastically compressed, collagen hydrogels (Coyac et al. 2013). Here, we tested the hypothesis that local, ECM function of PHEX is physiologically critical and independent of circulating serum phosphate levels during the formation of mineralized tissues. By having the ability to control phosphate supply (in the culture media) in this in vitro model, we explored aspects of the intrinsic, cell-autonomous mineralization defect in human cells having a deficiency in PHEX.
Materials and Methods
Patient Information and Human Teeth
XLH was diagnosed at the collaborating institutions based on the disorder’s characteristic findings and on a pattern of X-linked dominant disease transmission and positive PHEX mutation analysis (Appendix Table). Teeth were obtained from the Dental Department of the HNPVS, France. Deciduous (patients 1′/1) and permanent (patients 2′/2 and 3′/3) teeth were extracted for orthodontic reasons from 3 XLH patients and from 3 sex- and age-matched healthy young individuals (ranging from 11 to 15 y of age) with informed written and oral consent from the patients and the parents according to ethical guidelines set by French law (agreement IRB 00006477 no. DC-2009-927, Cellule Bioéthique DGRI/A5).
Cell Culture
Normal (control) and XLH pulp stem cells from human exfoliated deciduous teeth (SHED cells, patients 1/1′) and dental pulp stem cells (DPSC cells, patients 2/2′ and 3/3′) were collected from 3 control individuals and 3 XLH patients ranging between 11 and 15 y of age with informed consent of the patients and their parents, as well as approval from the institutional review board (6477, subject no. 16-024) of HUPNVS, AP-HP, France. Cells were isolated and expanded following an established protocol (Miura et al. 2003; Gronthos et al. 2011). The absence of mycoplasma contamination of cell cultures was checked by polymerase chain reaction (PCR). For all experiments, pulp cells were used at passage 3 (Huang et al. 2010).
Preparation of Dense Collagen Hydrogels and Mineralization Assay
Sterile, rat-tail tendon-derived type I collagen (2.10 mg/mL of protein in 0.1% acetic acid) was used as a starting material and processed into a hydrogel as originally described by Brown et al. (2005) and then adapted for use with dental pulp cells (Coyac et al. 2013) (Appendix Figure 1).
Micro–Computed Tomography
Scaffolds (n = 3 per group) were scanned by using high-resolution X-ray micro–computed tomography (CT) (Quantum FX Caliper; Life Sciences, PerkinElmer). Three-dimensional acquisitions were performed using an isotropic voxel size of 40 × 40 × 40 µm3 (90 kV, 160 microA, 180 s). Morphometric measurements were performed using Skyscan software (v1.13.5.1). Measurements were performed at day 24 on 12 scaffolds in each group (Ctrl +BGP, –BGP and XLH +BGP, –BGP) and are expressed as percentage ratio mineralized volume over tissue volume (Mv/Tv%).
Raman Microspectrometry
Scaffolds (n = 3 per group) were fixed in 70% ethanol solution and air-dried at room temperature. Raman analyses were done using a Labram HR800 microspectrometer (Horiba Gr; Jobin Yvon). The spectrometer was equipped with a diode laser (λ = 785 nm), an air-cooled CDD (1024 × 256 pixels), and a ×100 objective (NA = 0.90; Olympus). The illumination spot size was 1 µm. Spectral acquisitions were done in the 300- to 1,700-cm−1 range. The total acquisition time for each spectrum was 1 min with an integration time of 30 s and 2 accumulations. Each spectrum was treated with smoothing filtering (filter width: 3; polynomial order: 2) using Labspec software (Horiba GR; Jobin Yvon). Calibration of the spectrometer was done daily to prevent instrument drift and consisted of checking the band position of a silicon wafer at 520.70 cm–1. A total of 30 spectra were obtained per scaffold. Physicochemical variables were recorded as previously described (Mandair and Morris 2015; Colard et al. 2016). Briefly, mineralization was evaluated as the mineral-to-organic ratio (υ1 (PO4)/δ(CH2)-wag), carbonation was evaluated as type B carbonate substitution (υ1 (CO3)/υ1 (PO4) bands), and crystallinity was considered as the inverse of the full-width-at-half-maximum intensity (FWHM) of the ν1PO4 band (1/FWHM de υ1 (PO4)). All data analyses were performed using Matlab R2010a (Mathworks).
Microscopy of Scaffolds and Cells
After 24 d of culture, scaffolds (n = 3 per group) were rinsed in phosphate-buffered saline (PBS) (–Ca, –Mg) and fixed in 70% ethanol at 4°C and dehydrated in a graded ethanol series. Undecalcified samples were embedded in methyl methacrylate (Merck). Serial sections, 4 µm thick, were cut on a microtome (Polycut E microtome; Leica). Consecutive sections were stained respectively with toluidine blue and with von Kossa reagent (5% silver nitrate solution; Sigma-Aldrich) for mineral staining. Light microscopy (LEITZ DM-RBE microscope [Leica], equipped with a Sony DXC-950 CCD camera) was used to examine control and XLH dental pulp cell-seeded scaffolds.
For scanning electron microscopy (SEM, FEG-SEM Model S-4700; Hitachi High Technologies America), scaffolds were rinsed in PBS and fixed in 1% glutaraldehyde (Electron Microscopy Sciences) and 2% paraformaldehyde in PBS for 1 h at room temperature and then overnight at 4°C, followed by dehydration through a graded ethanol series and critical point drying. Energy-dispersive X-ray spectroscopy (EDS) was performed for elemental analysis of mineral, and the crystalline structure of the mineral was assessed by Raman microspectrometry (Labram HR800 microspectrometer, Horiba Gr; Jobin Yvon).
Alkaline Phosphatase Staining
Alkaline phosphatase (ALP) activity was used as a marker of osteogenic/odontogenic cell differentiation. Histology sections (n = 3 per group) were incubated with naphthol ASTR phosphate (Sigma-Aldrich) and diazonium fast blue RR salt (Sigma-Aldrich) for 30 min at 37°C (pH 9) in the presence of MgCl2.
Immunohistochemistry
Sections (n = 3 per group) were deplastified in a series of methyl glycol acetate baths. After rehydration, sections were blocked using 5% normal horse serum/PBS-T. Sections were incubated for 12 h at 4°C with goat anti-human OPN antibody (AF 1433; R&D Systems) or rabbit anti-human dentin matrix protein 1 (DMP1) antibody (LF148, kindly provided by Larry W. Fisher) or rabbit anti-human matrix extracellular phosphoglycoprotein (MEPE) mid-region antibody (Martin et al. 2008) diluted 1:20, 1:200, and 1:200, respectively, in 2.5% normal horse serum/PBS-T. Sections were washed and then incubated with peroxidase-conjugated anti-IgG diluted 1:1,000 in the same buffer. Peroxidase activity was detected using a diaminobenzadine substrate kit from Abcam following the manufacturer’s instructions. Control incubations to assess nonspecific staining consisted of applying the same procedure except that the primary antibody was substituted by nonimmune serum (control incubations were negative; Appendix Figure 2).
Immunoblotting
Flash-frozen, cell-seeded scaffolds rapidly scraped from the wells were crushed in liquid nitrogen and solubilized in 15 volumes of Laemmli Sample buffer (Bio-Rad). Lysates were centrifuged at 20,000 × g for 30 min at 4°C, and the supernatants were collected and stored at −80°C until use. Then, 10 µL of total protein extracts of each sample (n = 3 per group) was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on a gradient gel (Mini-Protean TGX Stain-Free gels, any kD; Bio-Rad) and transferred to a nitrocellulose membrane. Before immunoblotting, membranes were scanned with a stain-free gel imaging system (Gel Doc EZ system; Bio-Rad). iBind Flex Western Device (Invitrogen) was used for Western blot workflow. The membranes were probed with goat anti-human OPN antibody (AF 1433; R&D Systems), rabbit anti-human DMP1 antibody (LF148), or rabbit anti-human MEPE mid-region or ASARM (acidic serine aspartate-rich MEPE-associated motif) antibodies (Martin et al. 2008) diluted 1:1,000. Membranes were then incubated with horseradish peroxidase–conjugated anti-goat or anti-rabbit secondary antibody and visualized with ECL Select Prime Western Blotting Detection Reagent (GE Healthcare). Quantitation of digitized images of immunoblots was done using ImageJ software (National Institutes of Health). The intensity of immunoreactive bands was normalized to total proteins for each sample using stain-free imaging technology (Bio-Rad).
Statistical Analysis
Three biological replicates were performed in triplicates per group and for each time point. Data were compared by nonparametric tests (Mann-Whitney U test for group comparisons). Differences were considered significant at P < 0.05. Data are expressed as mean ± standard deviation (SD).
Results
Impaired Mineralization in XLH Cell-Seeded Scaffolds
After 24 d of culture in osteogenic medium (+BGP: 4% fetal bovine serum [FBS], 10 mM β-glycerophosphate), XLH dental pulp cell-seeded scaffolds showed a significant reduction of mineral formation as visualized and measured by micro-CT and the von Kossa staining reaction (Fig. 1A–D) compared with control cell-seeded scaffolds. No differences were observed between SHED and DPSC populations. These observations were further supported by biochemical calcium quantification (Fig. 1E) and Raman microspectrometry (Fig. 1F). When cultured under low-phosphate concentration (osteogenic medium –BGP: 4% FBS), both control (Ctrl) and XLH cell-seeded scaffolds had reduced mineralization compared with the regular osteogenic medium, but the quantity of mineral remained significantly lower in the XLH scaffolds (Appendix Figure 3). In both cell populations, the mineral phase was associated with the collagen fibrils, giving compositional signals for calcium and phosphate by energy-dispersive X-ray microanalysis (Fig. 2A, B) and Raman spectrometry, both of which were consistent with the presence of an apatitic mineral phase (Fig. 2A–C). However, Raman spectra from XLH cell-seeded scaffolds showed an additional immature mineral phase with a recurrent absence of peaks for carbonate type B substitution (: 1,070 cm–1) and a lower level of crystallinity (Fig. 2D, E).
Figure 1.
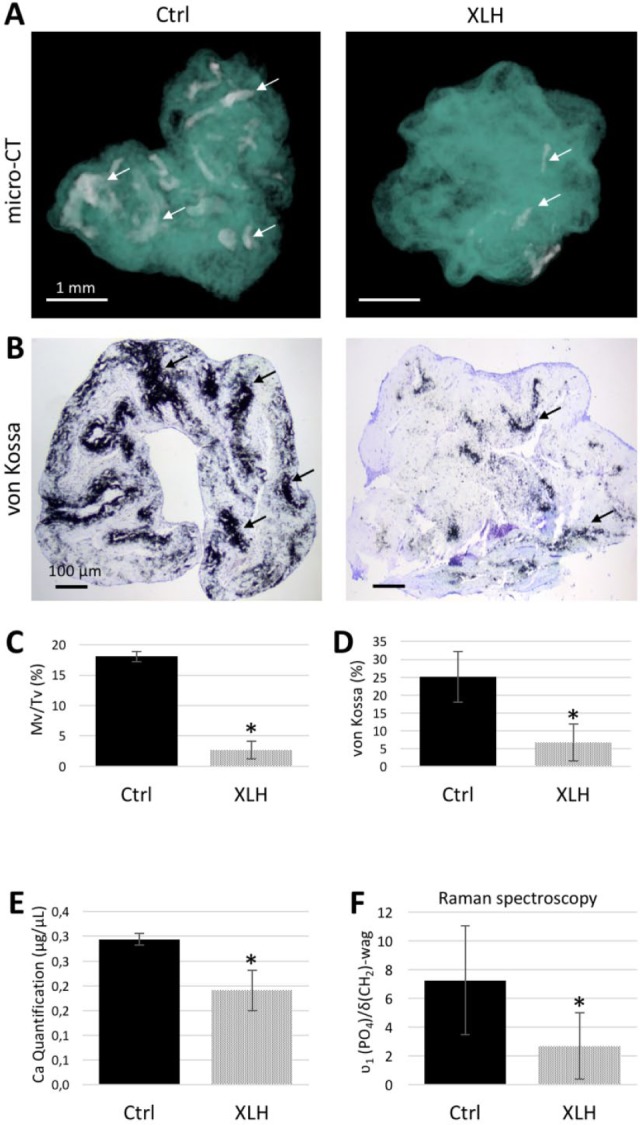
Decreased mineralization in X-linked hypophosphatemia (XLH) cell scaffolds. (A) Representative micro–computed tomography (CT) 3D reconstructions of control (Ctrl, left) and XLH (right) pulp cell-seeded scaffolds at day 24 where both were treated with the same osteogenic conditions: 300 µM L-ascorbic acid sodium salt, 10 nM dexamethasone, 4% fetal bovine serum, and 1% penicillin/streptomycin supplemented with 10 mM β-glycerophosphate. Regions of mineralization (arrows) are found throughout the collagen scaffolds (turquoise) but much less so in the XLH cell-seeded samples. Scale bars = 1 mm. (B) Representative light micrographs of sections of control (left) and XLH (right) cell-seeded scaffolds stained with von Kossa for mineral (black, arrows). Scale bars = 100 µm. (C, D) Mineral phase assessed by quantification of micro-CT and the von Kossa reaction. (E, F) Calcium quantification and mineral-to-organic ratio of the mineral phase. Data are means ± SD, n = 3 per group, *P < 0.05. This figure is available in color online.
Figure 2.

Impaired mineral quality of X-linked hypophosphatemia (XLH) cell-seeded scaffolds. (A, B) Representative micrographs at the ultrastructural level: scanning electron microscopy observations show mineral-encrusted collagen fibrils (arrows). Scale bars = 20 µm. Insets display energy-dispersive X-ray (EDS) microanalysis of mineralized areas in control (Ctrl) and XLH scaffolds with major spectral peaks for calcium (Ca) and phosphorus (P), consistent with the mineral phase being hydroxyapatite. (C) Raman spectra of control and XLH pulp cell-seeded scaffolds at day 24 were treated with the same osteogenic conditions showing characteristic peaks for phosphate. (D, E) Compared with control scaffolds, no carbonate band (arrow, highlighting the type B CO3–2 peak at 1,070 cm–1) and a reduction of the relative value of the crystallinity ratio are found in XLH cell scaffolds. Data are means ± SD, n = 3 per group, *P < 0.05.
Defective PHEX Impacts ALP and SIBLING Proteins
We then sought to analyze the expression of matrix proteins involved in the mineralization process and frequently investigated in the pathobiology of XLH (Martin et al. 2008; Gaucher et al. 2009; Boukpessi et al. 2016). Increased ALP activity was observed in XLH cell-seeded scaffolds compared with control scaffolds, as evidenced by the increased number of ALP-positive cells (Fig. 3A–C). Although ALP (a mineralization promoter) was strongly expressed by XLH cells, OPN (a mineralization inhibitor) accumulated in the ECM, more specifically associating with the mineralized regions within the XLH scaffolds (Fig. 3D, E). Immunoblotting of total protein extract showed an accumulation of SIBLING proteins and peptide fragments in XLH cell scaffolds compared to controls cultured under identical +β-glycerophosphate conditions. More specifically, full-length OPN (~50 kDa) was significantly increased in the XLH cell scaffolds compared with controls. Two additional fragments were observed in XLH samples, including a fragment detected at ~60 kDa that was not found in the controls (Fig. 3F) and a lower molecular weight fragment (~40 kDa) that was faintly detected in the XLH DPSC cultures but not in either control cell cultures or XLH SHED cultures (patient 1′). Unlike OPN, DMP1 accumulated with the mineral phase in the control scaffolds and in XLH cells (Fig. 4A, B). The DMP1 57-kDa fragment, which corresponds to the active C-terminal fragment of the protein (Padovano et al. 2015), was significantly increased in XLH cell scaffolds, and the DMP1 N-terminal 37-kDa fragment also increased in XLH cell scaffolds, although its detection in controls was not measurable (Fig. 4B). MEPE was localized within cells in both control and XLH groups, with an increased localization in mineralization nodules of control and XLH scaffolds (Fig. 4C). MEPE 55 kDa and some lower bands appeared with the same intensity in control and XLH scaffolds after immunodetection with antibodies raised against the mid-region (Fig. 4D) and the ASARM region of the protein (Fig. 4E). However, the MEPE-ASARM antibody further revealed a 27-kDa fragment (including the ASARM domain) that was increased in the XLH cell scaffolds (Fig. 4E). No differences were observed in terms of DMP1 and MEPE processing between SHED (patient 1′) and DPSC (patients 2′ and 3′) populations.
Figure 3.
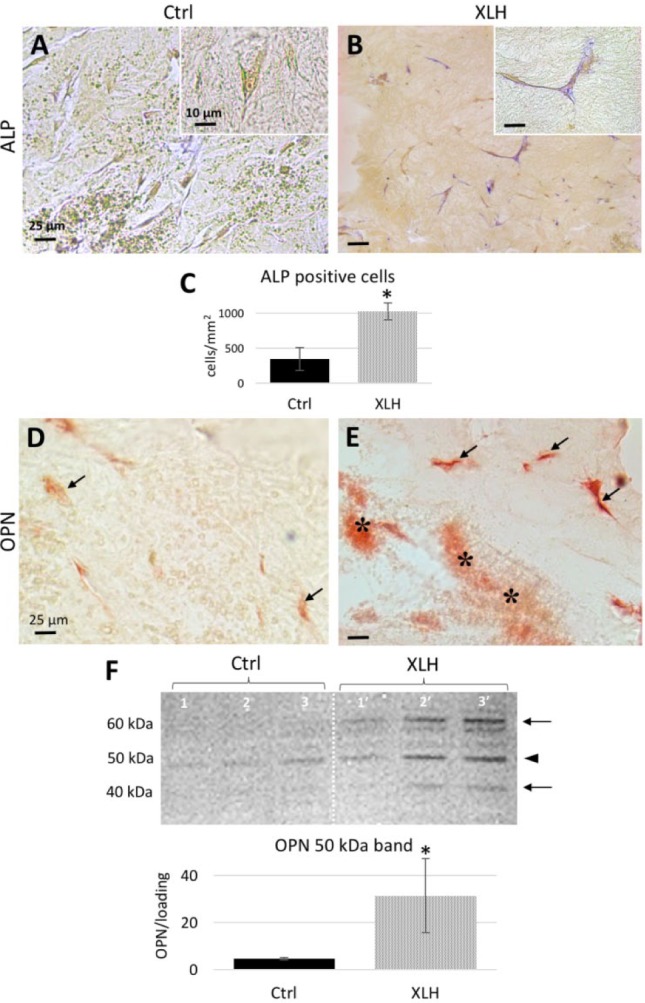
Increased alkaline phosphatase (ALP) enzyme activity and osteopontin (OPN) accumulation in X-linked hypophosphatemia (XLH) cell scaffolds. (A, B) ALP activity (magenta staining) is strong in the XLH cultures compared to control cultures. (C) The number of ALP-positive cells is higher in XLH cell scaffolds than in control scaffolds. Scale bars = 25 µm; insets = 10 µm. Data are means ± SD, n = 15 per group, *P < 0.05. (D, E) Representative images of immunohistochemistry for OPN. Whereas a faint immunostaining for OPN (red) in control cultures occurs in pulp cells (arrows), in XLH cell cultures, OPN prominently localizes to the mineral phase in the collagen scaffold (right, asterisks) and to the cells. Scale bars = 25 µm. (F) Western blotting for OPN with a normalized quantitative analysis showing an increased abundance of the 50-kDa form (arrowhead) in XLH cell scaffolds; additional bands (arrows) are visible in XLH patients 1′, 2′, and 3′. Data are means ± SD, n = 3 per group, *P < 0.05. This figure is available in color online.
Figure 4.
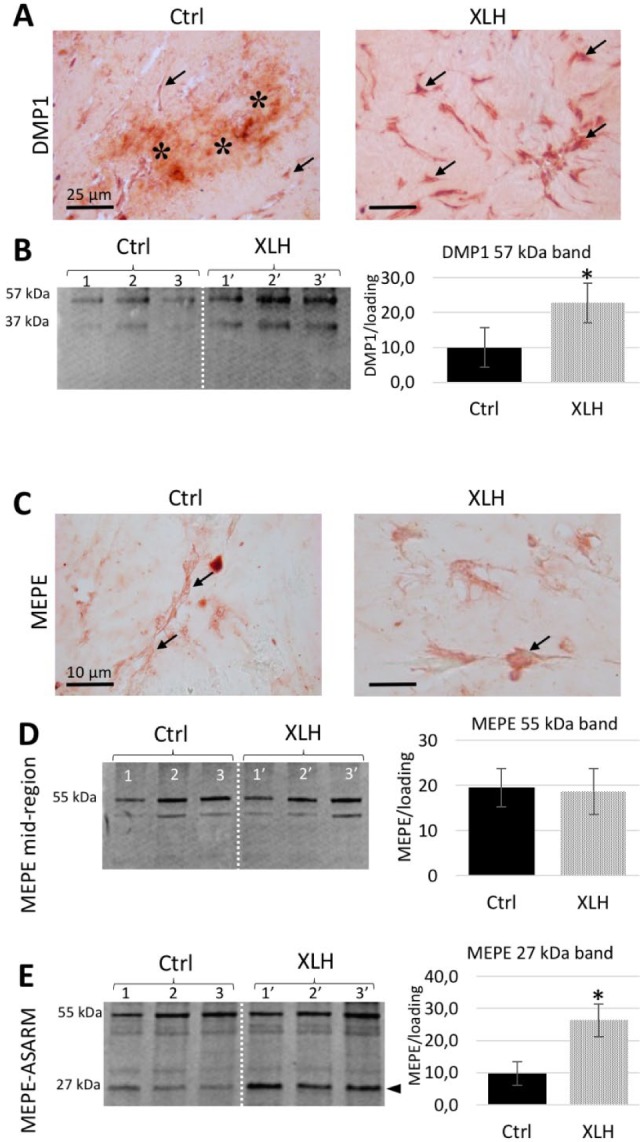
Dentin matrix protein 1 (DMP1) and matrix extracellular phosphoglycoprotein (MEPE) in cell-seeded cultures. (A) Representative images of immunohistochemistry for DMP1 (red) show strong staining in control cultures in both pulp cells (arrows) and with the mineral phase in the collagen scaffold (asterisks). In X-linked hypophosphatemia (XLH) cell scaffolds, DMP1 localizes prominently to the cells (arrows). Scale bars = 25 µm. (B) Western blotting for DMP1 with quantitative analysis of the C-terminal 57-kDa fragment. (C) Representative images of MEPE immunohistochemistry (red) showing localization in pulp cells (arrows) of both control and XLH cell scaffolds. Scale bars = 10 µm. (D) Western blotting using MEPE mid-region antibody with a normalized quantitative analysis showing no differences in terms of the 55-kDa fragment accumulation between groups. (E) Western blotting for the MEPE-ASARM region with a normalized quantitative analysis showing the accumulation of a 27-kDa fragment in the XLH cell scaffolds (arrowhead). Data are means ± SD, n = 3 per group, *P < 0.05. Patient samples are numbered 1′, 2′, and 3′. This figure is available in color online.
Discussion
In this study, using an in vitro osteogenic 3-dimensional model with human dental pulp cells from patients with XLH and sex- and aged-matched control individuals (Coyac et al. 2013), we showed a local cell-autonomous abnormal mineralization in XLH cultures, independent of hypophosphatemia (Fig. 5). These findings are consistent with previous studies in the context of murine bone cell biology suggesting abnormal intrinsic mineralization by Hyp osteoblasts (Ecarot-Charrier et al. 1988; Xiao et al. 1998).
Figure 5.

Dual putative local/systemic function of PHEX in normal mineralization relevant to X-linked hypophosphatemia. PHEX promotes intrinsic mineralization at the level of the extracellular matrix by directly degrading specific SIBLING proteins and their peptides, particularly osteopontin, and also (either directly or indirectly) MEPE-ASARM peptide. Green indicates a mineralization promoter; red indicates a mineralization inhibitor. DMP1, dentin matrix acidic phosphoprotein 1; FGF23, fibroblast growth factor 23; MEPE, matrix extracellular phosphoglycoprotein; OPN, osteopontin; Pi, phosphate; PHEX, phosphate-regulating gene with homologies to endopeptidases on the X chromosome; SIBLING, small integrin-binding-ligand-N-linked glycoprotein. This figure is available in color online.
In humans, similar phenotypes can result from endocrine conditions characterized by mishandling of phosphate metabolism, such as XLH (defective PHEX) and autosomal recessive hypophosphatemic rickets (ARHR, DMP1 deficiency) (Li et al. 2016; Tahir et al. 2016). This observation, among others, suggested that these pathologic phenotypes may have a stronger association with the documented high levels of FGF23 (Ichikawa et al. 2017) that lead to renal phosphate wasting and may not result from direct local effects of the loss of PHEX or DMP1. Accordingly, current and novel treatment strategies for XLH and other examples of genetic hypophosphatemia were designed to target only the systemic hypophosphatemia component, either by supplementation with phosphate and vitamin D or by neutralizing with monoclonal antibodies the phosphatonin effects of FGF23 hormone.
However, intrinsic, cell-autonomous mineralization defects have long been proposed for XLH tissues. Conventional (2-dimensional) mineralization assays for osteoblast cultures derived from the Hyp mouse, the murine model of XLH, showed impaired mineralization, with diminished calcium accumulation compared with wild-type (WT) mice (Xiao et al. 1998). In vivo, Hyp osteoblasts transplanted into WT mice were also associated with mineralization defects (Ecarot et al. 1992; Ecarot-Charrier et al. 1988). In addition, Hyp;Fgf23–/– compound mice had resolution of hypophosphatemia (Sitara et al. 2004) but not osteomalacia (Liu et al. 2006), revealing that although much of the phenotype in the Hyp mouse is attributable to the high levels of FGF23, local defects unrelated to FGF23 or hypophosphatemia still lead to osteomalacia. Indeed, Hyp mice having osteoblast-targeted Phex expression had partial rescue of the Hyp phenotype, showing improved bone and dentin mineralization, without improving systemic hypophosphatemia (Bai et al. 2002). The differential role played by PHEX and FGF23 in the Hyp mouse pathogenesis was further investigated by actin-driven transgenic expression of Phex in the Hyp mouse, which corrected much of the mutant bone phenotype without improving phosphate metabolism deficiencies (Erben et al. 2005). In humans, it has been observed that XLH-related hypophosphatemia and osteomalacia improved with phosphate and vitamin D supplementation treatments, but XLH-specific periosteocytic lesions were only partially resolved, suggesting an intrinsic pathologic mechanism in XLH, resulting in impaired mineralization and perilacunar lesions (Marie and Glorieux 1983). Thus, our data suggest that the mineralization induced by human cells is disturbed independently of FGF23-mediated hypophosphatemia, supporting a local role for PHEX in matrix mineralization. However, an excessive osteocytic FGF23 secretion was shown to contribute to pyrophosphate accumulation and mineralization defect (Murali et al. 2016). Associated with the recent discovery showing that KLOTHO is expressed in bone (Komaba et al. 2017), together with the fact that KLOTHO also plays a role in dentinogenesis (Suzuki et al. 2008), these findings, taken together, suggest that besides PHEX deficiency and hypophosphatemia, the intrinsic mineralization defects observed in XLH dentin or bone ECM might also be a consequence of disturbed FGF23/KLOTHO signaling in these tissues.
Here, despite an identical +β-glycero-phosphate supply used in our in vitro conditions, XLH tooth cells produced defective mineralization (Coyac et al. 2013). Like in human appendicular bone tissues, where hypophosphatemia is believed to cause osteomalacia and impaired quality of the mineral phase (Appendix Figure 4), we observed the same defects in the mineral phase obtained using XLH tooth cells cultured under standard phosphate concentrations. It has been shown previously that PHEX completely degrades full-length inhibitory OPN to promote mineralization, but in the absence of PHEX activity, an OPN protein fragment accumulates in Hyp mouse bone (Barros et al. 2013), and OPN/OPN fragments also accumulate in human XLH bone and tooth (Boukpessi et al. 2016) (Appendix Figure 4F, G). Our data show that XLH human dental pulp cell cultures also exhibit the OPN fragment accumulation associated with decreased mineralization, consistent with our previous findings (Boukpessi et al. 2016) and underscoring the importance of a local ECM effect for PHEX function. In addition, we show that 2 other SIBLING proteins—DMP1 and MEPE—are affected by PHEX deficiency, where there is an increase in specific fragments, including the bioactive C-terminal DMP1 peptide (Padovano et al. 2015) and a fragment encompassing the ASARM domain of MEPE, which has been shown to be a potent inhibitory motif in the mineralization process (Addison et al. 2008; Salmon et al. 2013). Thus, in the absence of PHEX, the processing (by both cleavage and degradation) of these additional SIBLING proteins is impaired, resulting in the accumulation of both a promoter (C-terminal DMP1 peptide) and an inhibitor (MEPE-ASARM peptide) of mineralization, despite the identical +β-glycerophosphate supply in the culture media. Of note, ALP activity was also found to be increased in XLH scaffolds despite the “normalized” phosphate supply, suggesting a link between PHEX function and ALP activity. It is not clear under these conditions why mineralization remained lower in XLH scaffolds despite the increased ALP and C-terminal DMP1. It is likely that the increased levels of these mineralization promoters cannot overcome the inhibitory effects of increased OPN and MEPE (and their peptides) on mineralization.
Taken together, our data show that hypophosphatemia alone does not fully explain the pathologic phenotype observed in XLH. Loss of PHEX independently and locally affects ECM mineral formation and quality, likely through the accumulation and impaired degradation of OPN and possibly through changes in DMP1 and MEPE cleavage. In vivo, the local dysfunction of PHEX—although seemingly less physiologically critical than circulating serum levels of FGF23 causing renal phosphate wasting—likely leads to defects in mineral quantity and quality during formation of mineralized tissues, a function that also needs to be addressed for therapy.
Author Contributions
B.R. Coyac, contributed to conception, design, data acquisition, analysis, and interpretation, drafted the manuscript; B. Hoac, M.D. McKee, contributed to data analysis and interpretation, critically revised the manuscript; P. Chafey, G. Falgayrac, contributed to data acquisition and analysis, critically revised the manuscript; L. Slimani, G. Penel, A. Linglart, contributed to data acquisition, critically revised the manuscript; P.S. Rowe, contributed to data interpretation, critically revised the manuscript; C. Chaussain, contributed to conception, design, data analysis, and interpretation, drafted the manuscript; C. Bardet, contributed to conception, design, data analysis, and interpretation, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplementary Material
Acknowledgments
The authors thank Dimitra Athanasiadou, Lydia Malynowsky, Brigitte Baroukh, and Annie Llorens for their help with the microscopy methods and the Showan N. Nazhat laboratory for teaching us how to prepare the dense collagen hydrogel scaffolds.
Footnotes
A supplemental appendix to this article is available online.
This work was supported by grants from Paris Descartes University, Fondation pour la Recherche Médicale for Plateforme d’imagerie du Vivant Paris Descartes (FRM DGE20111 123012), and the Agence Nationale de la Recherche (ANR PulpCell 2014-2017). BRC was supported by the Fondation pour la Recherche Médicale (PhD scholarship FDM20140731354) (France).
M.D.M. is a member of the FRQ-S Network for Oral and Bone Health Research, as well as the McGill Centre for Bone and Periodontal Research. A.L. receives research grant and/or consulting funds from Ultragenyx Pharmaceutical, Novato, CA. The other authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Addison WN, Nakano Y, Loisel T, Crine P, McKee MD. 2008. MEPE-ASARM peptides control extracellular matrix mineralization by binding to hydroxyapatite: an inhibition regulated by PHEX cleavage of ASARM. J Bone Miner Res. 23(10):1638–1649. [DOI] [PubMed] [Google Scholar]
- Bai X, Miao D, Panda D, Grady S, McKee MD, Goltzman D, Karaplis AC. 2002. Partial rescue of the Hyp phenotype by osteoblast-targeted PHEX (phosphate-regulating gene with homologies to endopeptidases on the X chromosome) expression. Mol Endocrinol (Baltimore, Md). 16(12):2913–2925. [DOI] [PubMed] [Google Scholar]
- Barros NM, Hoac B, Neves RL, Addison WN, Assis DM, Murshed M, Carmona AK, McKee MD. 2013. Proteolytic processing of osteopontin by PHEX and accumulation of osteopontin fragments in Hyp mouse bone, the murine model of X-linked hypophosphatemia. J Bone Miner Res. 28(3):688–699. [DOI] [PubMed] [Google Scholar]
- Bonewald LF, Wacker MJ. 2013. FGF23 production by osteocytes. Pediatr Nephrol (Berlin, Germany). 28(4):563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukpessi T, Hoac B, Coyac BR, Leger T, Garcia C, Wicart P, Whyte MP, Glorieux FH, Linglart A, Chaussain C, et al. 2016. Osteopontin and the dento-osseous pathobiology of X-linked hypophosphatemia. Bone. 95:151–161. [DOI] [PubMed] [Google Scholar]
- Brown RA, Wiseman M, Chuo CB, Cheema U, Nazhat SN. 2005. Ultrarapid engineering of biomimetic materials and tissues: fabrication of nano- and microstructures by plastic compression. Adv Funct Mater. 15(11):1762–1770. [Google Scholar]
- Carpenter TO. 2012. The expanding family of hypophosphatemic syndromes. J Bone Miner Metab. 30(1):1–9. [DOI] [PubMed] [Google Scholar]
- Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL. 2011. A clinician’s guide to X-linked hypophosphatemia. J Bone Miner Res. 26(7):1381–1388. Erratum in J Bone Miner Res. 2015;30(2): 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colard T, Falgayrac G, Bertrand B, Naji S, Devos O, Balsack C, Delannoy Y, Penel G. 2016. New insights on the composition and the structure of the acellular extrinsic fiber cementum by Raman analysis. PLoS One. 11(12):e0167316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyac BR, Chicatun F, Hoac B, Nelea V, Chaussain C, Nazhat SN, McKee MD. 2013. Mineralization of dense collagen hydrogel scaffolds by human pulp cells. J Dent Res. 92(7):648–654. [DOI] [PubMed] [Google Scholar]
- Ecarot B, Glorieux FH, Desbarats M, Travers R, Labelle L. 1992. Defective bone formation by Hyp mouse bone cells transplanted into normal mice: evidence in favor of an intrinsic osteoblast defect. J Bone Miner Res. 7(2):215–220. [DOI] [PubMed] [Google Scholar]
- Ecarot-Charrier B, Glorieux FH, Travers R, Desbarats M, Bouchard F, Hinek A. 1988. Defective bone formation by transplanted Hyp mouse bone cells into normal mice. Endocrinology. 123(2):768–773. [DOI] [PubMed] [Google Scholar]
- Erben RG, Mayer D, Weber K, Jonsson K, Jüppner H, Lanske B. 2005. Overexpression of human PHEX under the human β-actin promoter does not fully rescue the Hyp mouse phenotype. J Bone Miner Res. 20(7):1149–1160. [DOI] [PubMed] [Google Scholar]
- Gaucher C, Boukpessi T, Septier D, Jehan F, Rowe PS, Garabédian M, Goldberg M, Chaussain-Miller C. 2009. Dentin noncollagenous matrix proteins in familial hypophosphatemic rickets. Cells Tissues Organs. 189(1–4):219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronthos S, Arthur A, Bartold PM, Shi S. 2011. A method to isolate and culture expand human dental pulp stem cells. Methods Mol Biol. 698:107–121. [DOI] [PubMed] [Google Scholar]
- Grskovic M, Javaherian A, Strulovici B, Daley GQ. 2011. Induced pluripotent stem cells—opportunities for disease modelling and drug discovery. Nat Rev Drug Discov. 10(12):915–929. [DOI] [PubMed] [Google Scholar]
- Huang GT, Yamaza T, Shea LD, Djouad F, Kuhn NZ, Tuan RS, Shi S. 2010. Stem/progenitor cell-mediated de novo regeneration of dental pulp with newly deposited continuous layer of dentin in an in vivo model. Tissue Eng Part A. 16(2):605–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa S, Gerard-O’Riley RL, Acton D, McQueen AK, Strobel IE, Witcher PC, Feng JQ, Econs MJ. 2017. A mutation in the DMP1 gene alters phosphate responsiveness in mice. Endocrinology. 158(3):470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komaba H, Kaludjerovic J, Hu DZ, Nagano K, Amano K, Ide N, Sato T, Densmore MJ, Hanai JI, Olauson H, et al. 2017. Klotho expression in osteocytes regulates bone metabolism and controls bone formation. Kidney Int. 92(3):599-611. [DOI] [PubMed] [Google Scholar]
- Li SS, Gu JM, Yu WJ, He JW, Fu WZ, Zhang ZL. 2016. Seven novel and six de novo PHEX gene mutations in patients with hypophosphatemic rickets. Int J Mol Med. 38(6):1703–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linglart A, Biosse-Duplan M, Briot K, Chaussain C, Esterle L, Guillaume-Czitrom S, Kamenicky P, Nevoux J, Prie D, Rothenbuhler A, et al. 2014. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect. 3(1):R13–R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. 2006. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab. 291(1):E38–E49. [DOI] [PubMed] [Google Scholar]
- Mandair GS, Morris MD. 2015. Contributions of Raman spectroscopy to the understanding of bone strength. Bonekey Rep. 4:620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie PJ, Glorieux FH. 1983. Relation between hypomineralized periosteocytic lesions and bone mineralization in vitamin D–resistant rickets. Calcif Tissue Int. 35(4–5):443–448. [DOI] [PubMed] [Google Scholar]
- Martin A, David V, Laurence JS, Schwarz PM, Lafer EM, Hedge AM, Rowe PS. 2008. Degradation of MEPE, DMP1, and release of SIBLING ASARM-peptides (minhibins): ASARM-peptide(s) are directly responsible for defective mineralization in HYP. Endocrinology. 149(4):1757–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee MD, Hoac B, Addison WN, Barros NM, Millan JL, Chaussain C. 2013. Extracellular matrix mineralization in periodontal tissues: noncollagenous matrix proteins, enzymes, and relationship to hypophosphatasia and X-linked hypophosphatemia. Periodontol 2000. 63(1):102–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura M, Gronthos S, Zhao M, Lu B, Fisher LW, Robey PG, Shi S. 2003. SHED: stem cells from human exfoliated deciduous teeth. Proc Natl Acad Sci USA. 100(10):5807–5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murali SK, Andrukhova O, Clinkenbeard EL, White KE, Erben RG. 2016. Excessive osteocytic Fgf23 secretion contributes to pyrophosphate accumulation and mineralization defect in Hyp mice. PLoS Biol. 14(4):e1002427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padovano JD, Ravindran S, Snee PT, Ramachandran A, Bedran-Russo AK, George A. 2015. DMP1-derived peptides promote remineralization of human dentin. J Dent Res. 94(4):608–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarles LD. 2003. FGF23, PHEX, and MEPE regulation of phosphate homeostasis and skeletal mineralization. Am J Physiol Endocrinol Metab. 285(1):E1–E9. [DOI] [PubMed] [Google Scholar]
- Salmon B, Bardet C, Khaddam M, Naji J, Coyac BR, Baroukh B, Letourneur F, Lesieur J, Decup F, Le Denmat D, et al. 2013. MEPE-derived ASARM peptide inhibits odontogenic differentiation of dental pulp stem cells and impairs mineralization in tooth models of X-linked hypophosphatemia. PLoS One. 8(2):e56749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG, Juppner H, Lanske B. 2004. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in PHEX-deficient mice. Matrix Biol. 23(7):421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Amizuka N, Oda K, Noda M, Ohshima H, Maeda T. 2008. Involvement of the Klotho protein in dentin formation and mineralization. Anat Rec (Hoboken). 291(2):183–190. [DOI] [PubMed] [Google Scholar]
- Tahir S, Demirbilek H, Ozbek MN, Baran RT, Tanriverdi S, Hussain K. 2016. Genotype and phenotype characteristics in 22 patients with vitamin D–dependent rickets type I. Horm Res Paediatr. 85(5):309–317. [DOI] [PubMed] [Google Scholar]
- Xiao ZS, Crenshaw M, Guo R, Nesbitt T, Drezner MK, Quarles LD. 1998. Intrinsic mineralization defect in Hyp mouse osteoblasts. Am J Physiol. 275(4 Pt 1):E700–E708. [DOI] [PubMed] [Google Scholar]
- Yoshiko Y, Wang H, Minamizaki T, Ijuin C, Yamamoto R, Suemune S, Kozai K, Tanne K, Aubin JE, Maeda N. 2007. Mineralized tissue cells are a principal source of Fgf23. Bone. 40(6):1565–1573. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.