

 Nanolayered cobalt–molybdenum sulphide (Co–Mo–S) materials have been established as excellent catalysts for C–S bond construction.
Nanolayered cobalt–molybdenum sulphide (Co–Mo–S) materials have been established as excellent catalysts for C–S bond construction.
Abstract
Nanolayered cobalt–molybdenum sulphide (Co–Mo–S) materials have been established as excellent catalysts for C–S bond construction. These catalysts allow for the preparation of a broad range of thioethers in good to excellent yields from structurally diverse thiols and readily available primary as well as secondary alcohols. Chemoselectivity in the presence of sensitive groups such as double bonds, nitriles, carboxylic esters and halogens has been demonstrated. It is also shown that the reaction takes place through a hydrogen-autotransfer (borrowing hydrogen) mechanism that involves Co–Mo–S-mediated dehydrogenation and hydrogenation reactions. A novel catalytic protocol based on the thioetherification of alcohols with hydrogen sulphide (H2S) to furnish symmetrical thioethers has also been developed using these earth-abundant metal-based sulphide catalysts.
Introduction
Reactions involving carbon–sulphur bond formation are of great interest in organic synthesis and in materials science as well as in the pharmaceutical industry.1 For instance, thioethers exist in natural products and drugs, and they act as key building blocks for the synthesis of various heteroatomic functional groups, such as sulfoxides or sulfones, contained in many biologically and pharmacologically active molecules.2
Traditional synthesis of thioethers involves the condensation of a metal alkyl or aryl thiolate with an alkyl halide under strong basic conditions, resulting in the formation of large amounts of salt and metal waste.1d,3 Consequently, many catalytic methodologies aimed at the preparation of these compounds through more sustainable chemical transformations have been reported to date.1e,f,4 Currently, the most popular routes for their synthesis rely on metal-mediated cross-coupling reactions of organic halides with thiols,5 and on metal-catalysed hydrothiolation of unsaturated carbon–carbon bonds.5h,6 The latter reaction displays high atom efficiency; however, it suffers from low availability of the starting alkenes or alkynes, and presents regioselectivity limitations. The cross-coupling transformations involve substitution reactions, and therefore salts or acids (neutralized with the use of a base) are still produced as by-products.
An alternative methodology consists in the use of readily available alcohols as electrophiles where water is formed as the only stoichiometric residue. However, (primary and secondary) alcohols are usually unreactive because the hydroxyl group displays poor leaving character, their reactivity being slightly increased in the presence of Brønsted or Lewis acid catalysts.7
In this context, the so-called borrowing hydrogen methodology, also known as the transition-metal-catalysed hydrogen autotransfer process, offers compelling benefits.8 In this reaction sequence, alcohol is dehydrogenated to a more reactive aldehyde or ketone intermediate, which is more prone to react with a nucleophile present in the same reaction medium. Subsequent hydrogenation of the resulting unsaturated intermediate with the initially generated hydrogen (or metal hydrides) yields the desired product in a one-pot domino sequence.
The borrowing hydrogen methodology has been widely applied for efficient C–N9 and C–C10 bond formation from alcohols and using amines (including ammonia) or C-nucleophiles as reagents. In contrast, the use of thiols or hydrogen sulphide to accomplish the construction of C–S bonds by means of this catalytic protocol remains largely unexplored. To date, the only example has been reported by our group using a bifunctional solid catalyst based on palladium nanoparticles supported on high-surface area magnesium oxide (MgO). It allows the synthesis of thioethers along with the formation of disulfides.11 The reaction was specifically applied for benzylic alcohols that were dehydrogenated to benzaldehydes and in situ reacted with thiols involving, most probably, the formation of a hemithioacetal (in equilibrium with a hypothetical thionium ion) and its reduction by the previously generated palladium hydride species (Scheme 1). It should be noted that the direct reductive sulfidation of carbonyl compounds with mercaptans in the presence of a reducing agent, such as lithium aluminum hydride/aluminum chloride, triethylsilane, or pyridine-borane in trifluoroacetic acid, has also been described.12 However, the use of more stable and readily available alcohols instead of sensitive carbonyl compounds, and the absence of any additional reducing agent make the borrowing hydrogen methodology a more benign and practical strategy for the synthesis of thioethers.
Scheme 1. Catalytic borrowing hydrogen (BH) synthesis of thioethers from alcohols.

Nowadays, the replacement of precious metals with earth-abundant metal-based catalysts is an exciting goal in catalysis,13 and this substitution would still provide greater benefits to this methodology in terms of sustainability. In general, the activation of alcohols in homogeneous catalysis by the borrowing hydrogen strategy to form C–C and C–N bonds has been commonly performed with Ru or Ir complexes;14 however, recent efforts have led to the design of novel catalysts based on Fe,15 Co,16 Cu,17 Mn18 or Re.19 Although these transformations have been comparatively less investigated in heterogeneous catalysis, they have been accomplished with the use of precious metal-based catalysts to a higher extent,8f while only a handful of Ni- or Cu-based heterogeneous catalysts have been applied.20 Hence, the development of new strategies that make use of non-noble metal-based heterogeneous catalysts for the borrowing hydrogen C–X (X = C, N, S) bond construction from alcohols is an active field of research.
In this context, we herein describe for the first time, borrowing hydrogen synthesis of thioethers catalysed by a non-noble metal-based catalytic system. We show that the use of nanolayered cobalt–molybdenum-sulfides as catalysts allows for efficient C–S bond formation by the reaction of primary as well as secondary alcohols with thiols displaying excellent chemoselectivity even in the presence of sensitive functional groups. In addition, we have also extended this methodology to the use of H2S establishing a novel catalytic protocol for the preparation of symmetrical thioethers by thioetherification of the corresponding alcohols (Scheme 1) for the first time.
Results and discussion
Preparation and catalytic activity of nanolayered cobalt–molybdenum sulphide catalysts
Cobalt–molybdenum sulphide based materials are the most commonly used catalysts for the hydrodesulfurization (HDS) of crude feedstocks in petroleum refineries, a process that involves the C–S bond excision of sulphur-containing molecules.21 Consequently, enormous efforts have been made worldwide to improve the catalytic activity of these hydroprocessing catalysts. In this respect, the most significant breakthrough lay in the development of the so-called Nebula catalyst, an unsupported material with a large population of active sites per unit volume that is currently in use in several industrial units.22 Since its discovery, unsupported materials have emerged as a convenient alternative to the use of conventional supported ones,23 and therefore several methodologies have been reported for their preparation.24
Very recently, we described the hydrothermal synthesis of a series of nanolayered cobalt–molybdenum sulphide materials (Co–Mo–S–X; X = Co/(Mo + Co) mole ratio) and disclosed their useful applications as catalysts for chemo- and regioselective hydrogenation of nitroarenes and N-heteroarenes.25 More specifically, we reacted an aqueous solution containing ammonium molybdate, sulphur and different amounts of cobalt(ii) acetate in an autoclave at 180 °C in the presence of hydrazine as a reductant. Depending on the amount of cobalt(ii) acetate used in their preparation, it was possible to synthesize nanolayered materials with different chemical phase compositions, as they are predominantly constituted by MoS2 (along with the presence of Co–Mo–S-like structures) and cobalt sulphides with diverse stoichiometries, such as CoS2, Co3S4 and/or Co9S8 (Table S1 in the ESI†). The catalytic activity of these cobalt–molybdenum sulphide materials was ascribed to the presence of transient Co–Mo–S-like structures,26 as well as to the nature of the mixed phase of cobalt sulphides, with superior activity when the latter is mainly composed of Co3S4 and lower activity when increasing the relative content of Co9S8, while CoS2 resulted in an inactive phase.25
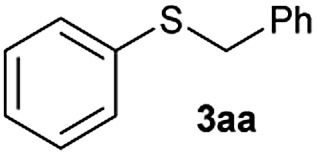
Based on this background and with the aim to extend the toolbox of useful synthetic transformations promoted by these nanolayered cobalt–molybdenum sulphide materials, we decided to investigate their application in hydrogen autotransfer processes. For that, the alkylation of benzenethiol (1a) with benzyl alcohol (2a) to afford benzyl phenyl sulphide (3aa) was selected as a benchmark reaction. Initial catalytic experiments were performed under a nitrogen atmosphere (3.5 bar) at 180 °C and using toluene as a solvent. Reaction profiles, depicted in Fig. 1, reveal diphenyl disulphide (4a) as the primary product, and its relative amount increased parallel to benzenethiol (1a) conversion. After reaching a maximum, the concentration of 4a starts to decrease or is maintained almost constant, likely depending on the efficiency of the catalyst to hydrogenate the disulphide bond. The formation of this product is not surprising since its metal-catalysed formation usually involves lower activation energy than the formation of thioethers.11
Fig. 1. Yield–time diagram of benzyl phenyl sulphide (3aa) and diphenyl disulphide (4a) produced during alkylation of benzenethiol (1a) with benzyl alcohol (2a) in the presence of the catalysts (a) Co–Mo–S-0.39, (b) Co–Mo–S-0.58, (c) Co–Mo–S-0.83 and (d) Co–Mo–S-0.91. Reaction conditions: 1a (0.25 mmol), 2a (0.5 mmol), catalyst (13.1 mg), toluene (1.6 mL), 3.5 bar N2, 180 °C.

In the presence of the catalyst Co–Mo–S-0.83 the starting thiol 1a is fully converted affording almost a quantitative yield of the desired product 3aa with traces of the disulphide compound 4a (2% yield) in 8 h, the latter being totally consumed after two additional hours (Fig. 1c and Table 1, entries 1 and 2). This catalyst, which is constituted by a mixture of MoS2, transient Co–Mo–S-like structures (determined through an electrochemical study applying the voltammetry of immobilized particles (VIMP) methodology),27 CoS2 and Co3S4 phases, also proved to be the most active catalytic system in previously reported hydrogenation reactions.25b The use of the catalyst Co–Mo–S-0.91, mainly formed by Co9S8 and MoS2, leads to lower conversion of 1a affording the thioether product 3aa in 70% yield, which co-exists with the non-fully converted disulphide 4a (Fig. 1d). Similar reactivity is achieved in the presence of the catalyst Co–Mo–S-0.39, a cobalt-promoted MoS2-based material with cobalt species homogeneously distributed on MoS2 and containing some separated CoS2 (Fig. 1a).25a It should be noted that the high dispersion of cobalt species on MoS2 benefits the adsorption of cobalt atoms at the edges of the layered structure of MoS2, thus leading to the formation of Co–Mo–S-like active structures to a larger extent.26 In fact, the catalyst Co–Mo–S-0.58, being constituted by the same phases but with more separated and agglomerated non-active CoS2,25b displays considerably lower activity. As revealed in its reaction profile, besides a modest yield of 3aa, once diphenyl disulphide (4a) is formed it remains almost unreactive (Fig. 1b).
Table 1. Alkylation of benzenethiol (1a) with benzyl alcohol (2a) a .

| |||||
| Entry | Catalyst | Solvent | Conversion b [%] | Yield
b
[%] |
|
| 3aa | 4a | ||||
| 1 | Co–Mo–S-0.83 | Toluene | >99 | 96 | 2 |
| 2 c | Co–Mo–S-0.83 | Toluene | >99 | 98 | — |
| 3 | MoS2 | Toluene | 76 | 53 | 18 |
| 4 | Mo-free CoxSy | Toluene | 45 | 15 | 25 |
| 5 d , e | Co–Mo–S-0.83 | Toluene | 94 | 89 | 8 |
| 6 e , f | Co–Mo–S-0.83 | Toluene | 80 | 50 | 10 |
| 7 e | Co–Mo–S-0.83 | 1,4-Dioxane | 98 | 84 | 5 |
| 8 e | Co–Mo–S-0.83 | CH3CN | 82 | 48 | 15 |
| 9 e | Co–Mo–S-0.83 | THF | 64 | 1 | — |
| 10 e | Co–Mo–S-0.83 | Ph-CF3 | >99 | 94 | — |
aReaction conditions: 1a (0.25 mmol), 2a (0.5 mmol), catalyst (13.1 mg), solvent (1.6 mL), 3.5 bar N2, 180 °C, 8 h.
bDetermined by GC with respect to 1a using n-hexadecane as the internal standard.
c10 h.
d 2a (1.5 equiv.).
e18 h.
f150 °C.
In addition, we have also tested MoS2 and a molybdenum-free CoxSy catalyst based on a mixture of CoS2, Co3S4 and Co9S8 (see the ESI† for details of their preparation). MoS2 displays moderate catalytic activity affording 3aa in 53% yield. In contrast, in the presence of the molybdenum-free material as the catalyst, 3aa is achieved in only 15% yield while benzyl alcohol remains largely unreactive (Table 1, entries 3 and 4; see also Fig. S1 in the ESI†).
It is worth noting that all synthesized Co–Mo–S materials are active in the borrowing hydrogen synthesis of benzyl phenyl sulphide (3aa), which means that these catalysts are able to dehydrogenate benzyl alcohol (2a) to benzaldehyde (2a′) as well as hydrogenate the intermediate product formed by the reaction of 2a′ with the initial thiol 1a (see Scheme 2). Typically, in a borrowing hydrogen sequence the hydrogenation stage is thermodynamically favoured, thus driving forward the initial dehydrogenation step. This is in agreement with the catalytic activity tendency followed by these Co–Mo–S catalysts for the synthesis of 3aa, being higher with the increase of their hydrogenation activity (Table S1†). However, the higher efficiency shown by MoS2 in comparison with the molybdenum-free CoxSy catalyst is in contrast since, as previously reported,25b the hydrogenation activity of the latter is higher than that of MoS2. These results clearly show that in the borrowing hydrogen strategy, apart from the hydrogenation activity of the catalyst, its dehydrogenation ability is also important. In this sense, the presence of MoS2 in the investigated Co–Mo–S catalysts seems to be crucial to efficiently accomplish the dehydrogenation of the alcohol,28 since it remains largely unreactive when the molybdenum-free CoxSy material is used as the catalyst.
Scheme 2. Reaction steps for the catalytic borrowing hydrogen (BH) synthesis of benzyl phenyl sulphide (3aa).

Interestingly, when the alkylation of benzenethiol (1a) with benzyl alcohol (2a) was carried out in the presence of the most active catalyst Co–Mo–S-0.83 with an extra supply of hydrogen gas pressure (2 bar N2 and 1.5 bar H2), higher reaction rate for the formation of 3aa was achieved (Fig. S2†), thus suggesting that the rate-controlling step of this reaction sequence is not the dehydrogenation of the alcohol but the direct reductive thiolation. This result is in line with our previous finding that correlates the higher efficiency for the synthesis of 3aa with the increase of the hydrogenation activity of the catalysts when they display similar dehydrogenation activity.
Next, we explored the recycling of the catalyst Co–Mo–S-0.83 in the investigated model reaction for the formation of the thioether 3aa. As shown in Fig. 2a, a decrease in the yield of 3aa is observed in the second run, while it remains almost constant in the third one. This catalytic behaviour suggests that different active species are responsible for the catalytic activity and that the fresh catalyst contains highly active but poorly stable catalytic species, which vanish during the first catalytic run. As we previously reported,25b these active structures correspond to Co–Mo–S-like structures, which is well-established to consist of MoS2 decorated at the edges with cobalt atoms.26 Due to their metastable character, they vanish by the desorption of cobalt atoms from the MoS2 backbone during the first catalytic run as revealed ICP-MS analysis of the filtrate (Table S2 in the ESI;† see also Fig. S3† for hot filtration experiments). Nevertheless, excellent conversions of 1a towards the formation of the desired product 3aa were obtained even after the sixth run by increasing the reaction time, thus suggesting that in addition to the Co–Mo–S like structures other active species are also responsible for the catalytic activity of the catalyst Co–Mo–S-0.83. It is noticeable that no more leaching occurred to the reaction medium in the next runs. X-ray diffraction (XRD) characterization (Fig. 2b; see also Fig. S4–S6† for high-resolution transmission electron microscopy (HRTEM) studies) of the fresh and recycled catalysts shows that they are slowly desulfurized during the reaction cycles producing a subtle change in the composition of the mixed phase of cobalt sulphides. More specifically, CoS2 is continuously transformed to the active phase of Co3S4, and therefore after losing the Co–Mo–S-like active structures in the first run, the catalytic activity is maintained or even slightly increases along the reaction cycles.
Fig. 2. (a) Catalyst recycling for the alkylation of benzenethiol (1a) with benzyl alcohol (2a). Reaction conditions: 1a (0.25 mmol), 2a (0.5 mmol), Co–Mo–S-0.83 (13.1 mg), toluene (1.6 mL), 3.5 bar N2, 180 °C, 10 h (runs 1–3) or 18 h (runs 4–6). (b) XRD patterns of the recycled catalyst.

All these results suggest that different active species are responsible of the catalytic activity of the catalyst Co–Mo–S-0.83. More specifically, it depends on the composition of the mixed phase of cobalt sulphides, with superior activity when the latter is mainly composed by Co3S4, and it is also associated with the presence of transient Co–Mo–S-like structures,26 which vanish after the first catalytic run. In addition, MoS2 plays a key role not only in the hydrogenation step, but also in the dehydrogenation of the alcohol.28
Next, the borrowing hydrogen synthesis of 3aa was investigated in more detail. Although benzaldehyde (2a′) appears to be a detectable reaction intermediate when 2a is used as an alkylating reagent, a control experiment was run to rule out a possible mechanism through a direct nucleophilic substitution of alcohols with the formation of carbocation intermediates.7 Under the previously used conditions, benzenethiol (1a) was reacted with 2-phenyl-2-propanol (2b), a tertiary benzylic alcohol that easily generates the stabilized carbocation. Together with the formation of the disulphide product 4a (70% yield), the corresponding thioether was obtained in negligible yield (8%) after 18 h, thus ruling out the direct nucleophilic substitution mechanism (Scheme S1 and eqn (S1)†).
Other evidence that confirms the occurrence of the alcohol dehydrogenation step of the borrowing hydrogen sequence relies on the detection of molecular H2 in the gas-phase of the reaction between 1a and 2a. Moreover, to get more clues on the complete autotransfer mechanism, an additional two step-control experiment was carried out by first reacting the catalyst Co–Mo–S-0.83 with benzyl alcohol (2a) under neat conditions, and then using this pre-treated catalyst in the reaction of 1a with benzaldehyde (2a′) in toluene at 180 °C (for more details see Scheme S1 and eqn (S2a) and (S2b)†). Interestingly, the desired thioether 3aa was obtained in 17% yield, which reveals the presence of hydrogen-derived species on the pre-treated catalyst generated by benzyl alcohol dehydrogenation and their proper transfer to the hemithioacetal intermediate. All of the above results confirm that the alkylation of thiols with alcohols in the presence of the Co–Mo–S-0.83 catalyst is actually taking place through a borrowing hydrogen sequence.
Since diphenyl disulphide (4a) was detected as a transient product during the course of the reaction, we attempted to use it as a reactant in the synthesis of benzyl phenyl sulphide (3aa) by reaction with benzyl alcohol (2a) (Scheme S1 and eqn (S3)†). Interestingly, 4a was almost fully converted (89%) after 18 h at 180 °C affording the thioether 3aa and the thiol 1a in 83 and 4% yields, respectively, which shows the feasibility of synthesizing thioethers from dithiols and alcohols in the presence of the catalyst Co–Mo–S-0.83. In this reaction sequence, longer reaction times are required because the dithiol compound needs to be reduced to the corresponding thiol first.
Another detected by-product, although at the trace level, was the thioacetal α,α-bis(phenylthio)toluene (5a), presumably formed by the over-thiolation of benzyl alcohol. Taking into account the extended used of MoS2-based catalysts in HDS processes,21 an alternative reaction pathway through desulfurization of this by-product to form the thioether 3aa should not be ruled out. Indeed, the reaction of 5a with benzyl alcohol (2a) afforded 3aa and 4a in 74 and 8% yields, respectively, after 10 h under the same reaction conditions used for the previous catalytic reactions (Scheme S1 and eqn (S4)†).
Based on all the above results a mechanism for the synthesis of the thioether 3aa by alkylation of 1a with the alcohol 2a in the presence of the catalyst Co–Mo–S-0.83 is proposed in Scheme 3. Benzyl alcohol (2a) is initially dehydrogenated to benzaldehyde (2a′) generating molecular hydrogen and activated hydrogen species on the Co–Mo–S catalyst. Then, the nucleophile benzenethiol (1a) attacks the carbonyl carbon of 2a′ to give the hemithioacetal intermediate I,11,12c which is Co–Mo–S-mediated reduced to afford the thioether product 3aa. An alternative pathway involving the formation and subsequent catalytic hydrogenolysis of α,α-bis(phenylthio)toluene (5a) can also take place. It should be mentioned that the formation of 5a through a direct dehydration of the hemithioacetal intermediate (I) with benzenethiol is unlikely as revealed by our unsuccessful experiment with a more reactive tertiary alcohol (Scheme S2 and eqn (S1)†). This suggests that the thioacetal 5a is most probably formed through an additional borrowing hydrogen sequence following analogous steps to those previously described.
Scheme 3. Proposed reaction pathway for the Co–Mo–S-catalysed alkylation of benzenethiol (1a) with benzyl alcohol (2a).

In addition, according to the reaction profiles depicted in Fig. 1, the synthesis of 3aa under the present protocol also involves the reversible formation of diphenyl disulphide (4a), which is catalytically reduced to form the starting thiol 1a again. That means reducing species should be formed in more than stoichiometric amounts to get a quantitative yield of 3aa, therefore giving a reasonable explanation for the need of using an excess of the alcohol (2 equiv.) with respect to 1a (Table 1, entry 5). Temperature is also a critical parameter since only 50% yield of the thioether 3aa is obtained when the reaction is conducted at a lower temperature (150 °C) even after a longer reaction time (Table 1, entry 6). The use of other solvents, with the exception of benzotrifluoride, also led to detrimental results (Table 1, entries 7–10).
Reaction scope of the borrowing hydrogen S-alkylation of thiols with alcohols in the presence of nanolayered Co–Mo–S catalysts
To investigate the scope of this catalytic protocol for the general preparation of thioethers, we first tested the alkylation of structurally diverse thiols (1b–1t) with benzyl alcohol (2a) under a nitrogen atmosphere (3.5 bar) at 180 °C using toluene as the solvent. As shown in Table 2, aryl thiols functionalized with alkyl or methoxy groups as well as naphthalene derivatives were converted into the desired thioethers 3ba–3ga in good to excellent yields (Table 2, entries 1–7). Halogen-substituted benzenethiols 1h–1j also reacted efficiently and no dehalogenation products were detected (Table 2, entries 8–10). Interestingly, other reducible functionalities such as ketones, nitriles and carboxylic ester groups remained untouched with the corresponding thioethers 3ka–3ma isolated in nearly 90% yield (Table 2, entries 11–13). The heterocyclic thiol 1n containing the benzothiazole moiety was transformed to the target product 3na, which could be isolated in 82% yield (Table 2, entry 14).
Table 2. Co–Mo–S-catalysed alkylation of thiols with benzyl alcohol (2a) a .

| |||||||||
| Entry | Product | Conv. b [%] | Yield [%] |
Entry | Product | Conv. b [%] | Yield [%] |
||
| 3 c | 4 b | 3 c | 4 b | ||||||
| 1 d |
|
>99 | 91 | — | 11 |

|
>99 | 92 | — |
| 2 |

|
>99 | 85 | — | 12 |

|
>99 | 90 | — |
| 3 |

|
>99 | 90 | 4 | 13 |

|
>99 | 89 | — |
| 4 |

|
>99 | 78 | 6 | 14 e |

|
>99 | 82 | — |
| 5 |

|
>99 | 73 | 6 | 15 f |

|
>99 | 65 | — |
| 6 |

|
>99 | 93 | — | 16 g |

|
>99 | 45 | — |
| 7 |

|
95 | 76 | — | 17 |

|
>99 | 89 | 2 |
| 8 |

|
>99 | 96 | — | 18 |

|
97 | 78 | — |
| 9 |

|
>99 | 93 | — | 19 |

|
95 | 70 | 10 |
| 10 |

|
>99 | 94 | — | 20 h |

|
>99 | 83 | — |
aReaction by-products:conditions: thiol 1a–1t (0.25 mmol), 2a (0.5 mmol), Co–Mo–S-0.83 (13.1 mg), toluene (1.6 mL), 3.5 bar N2, 180 °C, 18 h.
bDetermined by GC with respect to the thiol using n-hexadecane as an internal standard.
cYield of isolated products.
dYield of the isolated product starting from 10 mmol of thiol 1a. GC-yield of the by-products:
eBenzo[d]thiazole (5%).
fBenzyl benzoate (27%).
gBenzyl acetate (47%).
hBenzyl 3-(benzylthio)propanoate (10%).
The alkylation reaction could also be carried out with thiobenzoic and thioacetic acid (1o and 1p, respectively) as starting reactants affording benzyl benzothioate (3oa) and benzyl thioacetate (3pa) in moderate yields along with the formation of the corresponding ethers (Table 2, entries 15 and 16). 2-Phenylethanethiol (1q) and cyclic, linear and even ester functionalized aliphatic thiols (1r–1t) also displayed good reactivity toward the formation of the desired thioethers 3ra–3ta (Table 2, entries 17–20).
After having demonstrated the excellent activity of the catalyst Co–Mo–S-0.83 in the reaction of different thiols with benzyl alcohol (2a), we proposed to extend this methodology to other alcohols (Table 3). Under the same previously used conditions, reaction of benzenethiol (1a) with 2-naphthalenemethanol (2c) and 4-biphenylmethanol (2d) afforded the thioethers 3ac and 3ad in 88 and 89% yields, respectively (Table 3, entries 1 and 2). Different alkyl-substituted benzyl alcohols (2e–2g) and even the sterically hindered trimethyl-substituted 2h also displayed high reactivity towards the formation of the desired products 3ae–3ah (Table 3, entries 3–6).
Table 3. Co–Mo–S-catalysed alkylation of benzenethiol (1a) with alcohols a .

| |||||||||
| Entry | Product | Conv. b [%] | Yield [%] |
Entry | Product | Conv. b [%] | Yield [%] |
||
| 3 c | 4a b | 3 c | 4a b | ||||||
| 1 |

|
>99 | 88 | 3 | 11 |

|
>99 | 93 | 2 |
| 2 |

|
>99 | 89 | — | 12 |

|
93 | 76 | 7 |
| 3 |

|
>99 | 96 | — | 13 d |

|
>99 | 73 | 9 |
| 4 |

|
>99 | 95 | — | 14 |

|
>99 | 79 | 13 |
| 5 |

|
>99 | 91 | — | 15 |

|
97 | 86 | 6 |
| 6 |

|
>99 | 82 | 11 | 16 |

|
>99 | 86 | 3 |
| 7 |

|
>99 | 84 | — | 17 |

|
94 | 81 | 3 |
| 8 |

|
>99 | 81 | — | 18 |

|
>99 | 87 | 9 |
| 9 |

|
82 | 51 | 15 | 19 |

|
92 | 81 | — |
| 10 |

|
95 | 80 | 8 | 20 |

|
>99 | 82 | 13 |
aReaction conditions: 1a (0.25 mmol), alcohol 2c–2v (0.5 mmol), Co–Mo–S-0.83 (13.1 mg), toluene (1.6 mL), 3.5 bar N2, 180 °C, 18 h.
bDetermined by GC with respect to 1a using n-hexadecane as an internal standard.
cYield of isolated products. GC-yield of by-products:
d 3aa (4%).
In the presence of other electron-rich groups, such as methoxy and thiomethyl, the alkylation reaction of benzenethiol (1a) proceeds in a similar manner (Table 3, entries 7 and 8), whereas in general the introduction of electron-withdrawing substituents leads to a slight decrease in the efficiency (Table 3, entries 9 and 13). A drastic decline was observed with benzyl alcohol functionalized with two fluorine groups at the ortho position, likely associated with electronic more than steric effects (Scheme S2 in the ESI†). From the viewpoint of selectivity, the sensitive unsaturated double bond and ester group of benzyl alcohols 2p and 2q remain untouched in the final thioethers 3ap and 3aq, which were isolated in 79 and 86% yields, respectively (Table 3, entries 14 and 15). Furthermore, heteroaromatic alcohols also underwent the alkylation reaction of 1a in high yields (Table 3, entries 16 and 17). Interestingly, this catalytic methodology could also be extended to the allylic alcohol 2t affording 3at in 87% isolated yield (Table 3, entry 18). However, the use of aliphatic alcohols failed with this catalytic system (Scheme S3 in the ESI†).
Next, we were interested in the use of more challenging secondary alcohols (Table 3, entries 19 and 20). Gratifyingly, the alkylation of 1a with alcohols 2u and 2v proceeded efficiently, allowing the preparation of thioethers 3au and 3av in high yields. It is worth noting that the formation of the dehydrogenated ketones was detected during the reaction and, in accordance with our previous unsuccessful experiment with the tertiary alcohol 2b (see Scheme S1, eqn. (S1)†), this suggests that a borrowing hydrogen mechanism instead of a direct nucleophilic substitution of the alcohol functionality takes place.7
Thioetherification of alcohols with H2S
Hydrogen sulphide (H2S) is a major by-product produced in petroleum refineries during hydrotreating processes carried out for upgrading heavy crude feed-stocks. Due to its toxicity, the release of H2S to the atmosphere is environmentally restricted. Nowadays, the emission of the H2S produced in most of the refineries is commonly mitigated through the conventional Claus process, which converts this harmful gas into elemental sulphur.29 However, the consumption of sulphur relative to its current production is too high, and therefore the development of new methodologies to transform H2S into valuable and more in-demand products is highly desirable.30
In this regard, after the general development of the alkylation of structurally diverse thiols with a wide range of alcohols, we proposed to extend this methodology to the reaction of alcohols with H2S. We therefore reacted benzyl alcohol (2a) at 180 °C in the presence of the catalyst Co–Mo–S-0.83 under a N2/H2S (10% v/v in H2S) atmosphere (4 bar) using toluene as the solvent. Initially, as shown in Fig. 3, benzyl mercaptan (6a) and dibenzyl sulphide (7a) are formed as reaction products, but the concentration of 6a reaches a maximum and then starts to decrease concomitant with the progressive formation of 7a, which is afforded in 70% yield within 10 h. Benzaldehyde (2a′) and dibenzyl disulphide (8a) are also formed during the reaction. These results suggest that the formation of the symmetrical thioether 7a in this thioetherification reaction takes place through two consecutive borrowing hydrogen sequences with 6a as the reaction intermediate (see Scheme 1; R3 = H).
Fig. 3. Yield–time diagram for the thioetherification of benzyl alcohol (2a) with H2S. Reaction conditions: 2a (0.25 mmol), Co–Mo–S-0.83 (13.1 mg), toluene (1.6 mL), 4 bar N2/H2S (10% v/v in H2S), 180 °C. Traces of dibenzyl ether (<5%) were also detected.

Notably, as shown in Fig. 4a the catalyst was conveniently recycled affording the symmetrical thioether 7a in moderate yields even after the sixth run. As in the prior use of benzenethiol (vide supra), a significant decrease of the catalytic activity is achieved in the second run likely as result of the consumption of the Co–Mo–S-like active structures. In contrast, in the present case the catalyst continues to be slightly deactivated with subsequent reaction cycles affording 7a in similar to lower yields even after longer reaction times. As revealed by the XRD patterns of the recycled catalyst after different catalytic runs (Fig. 4b; see also Fig. S4, S7 and S8† for HRTEM characterization), the catalyst is progressively sulfurized with H2S transforming the active phase Co3S4 to CoS2, thus resulting in the progressive decrease of the catalytic activity.
Fig. 4. (a) Catalyst recycling for the thioetherification of benzyl alcohol (2a) with H2S. Reaction conditions: 2a (0.25 mmol), Co–Mo–S-0.83 (13.1 mg), toluene (1.6 mL), 4 bar N2/H2S (10% v/v in H2S), 180 °C, 10 h (runs 1–3) or 18 h (runs 4–6). (b) XRD patterns of the recycled catalyst.

We next explored the synthesis of other symmetrical thioethers from the corresponding functionalized alcohols (Table 4). In addition to 2-naphthalenemethanol (2c) other benzyl alcohols substituted with electron-donating groups, like alkyl, methoxy and thiomethyl moieties, reacted efficiently producing the desired derivatives in good yields (Table 4, entries 2–5). A slight decrease in efficiency is observed in the presence of electron-withdrawing substituents, such as halogen groups, which were well-retained in the final symmetrical thioethers (Table 4, entries 6–8). Gratifyingly, the carboxylic ester functionality of alcohol 2q remained untouched and the product 7q could be isolated in 56% yield (Table 4, entry 9). In addition, the heterocyclic alcohol 2r also successfully accomplished this thioetherification reaction affording the corresponding symmetrical product 7r in 70% isolated yield (Table 4, entry 10).
Table 4. Co–Mo–S-catalysed thioetherification of alcohols with H2S a .

| |||||||||||
| Entry | Substrate | Conv. b [%] | Yield [%] |
Entry | Substrate | Conv. b [%] | Yield [%] |
||||
| 6 b | 7 c | 8 b | 6 b | 7 c | 8 b | ||||||
| 1 d , e |

|
>99 | 2 | 66 | 2 | 6 b , j |

|
97 | — | 67 | 3 |
| 2 b , f |

|
>99 | — | 72 | 6 | 7 b , k |

|
95 | 9 | 63 | 5 |
| 3 g |

|
>99 | 3 | 75 | 1 | 8 l |

|
99 | — | 54 | — |
| 4 h |

|
98 | 3 | 70 | — | 9 m |

|
93 | — | 56 | 5 |
| 5 i |

|
>99 | — | 64 | 4 | 10 n |

|
>99 | — | 70 | 4 |
aReaction conditions: alcohol 2 (0.25 mmol), Co–Mo–S-0.83 (13.1 mg), toluene (1.6 mL), 4 bar N2/H2S (10% v/v in H2S), 180 °C, 18 h.
bDetermined by GC with respect to the alcohol using n-hexadecane as an internal standard.
cYield of isolated products.
dYield of the isolated product on a 10 mmol scale. GC-yield of by-products:
e 2a′ (3%).
f 2c′ (3%) and 2-methylnaphthalene (11%).
g 2e′ (4%).
h 2i′ (3%) and 4-methylanisole (5%).
i4-(Methylthio)toluene (5%).
j4-Chlorotoluene (4%).
k3-Bromotoluene (10%).
l 2o′ (1%) and 2-iodotoluene (9%).
m 2q′ (6%) and methyl p-toluate (15%).
n3,4-(Methylenedioxy)toluene (3%). Variable amounts (3–5%) of the corresponding symmetrical ethers were also detected in all tested reactions.
Conclusions
In summary, we have disclosed for the first time that, apart from its well established use as hydrotreating catalysts, cobalt–molybdenum sulphide (Co–Mo–S) unsupported materials are also excellent catalysts for the inverse reaction, that is, carbon–sulfur bond formation. Indeed, different Co–Mo–S catalysts have been shown to be active in the reaction of benzenethiol (1a) with benzyl alcohol (2a) to form benzyl phenyl sulphide (3a). The reaction proceeds through a borrowing hydrogen sequence involving Co–Mo–S-mediated dehydrogenation and hydrogenation reactions. The catalytic activity of the most active catalyst (Co–Mo–S-0.83) for the aforementioned reaction is in line with its previously reported hydrogenation activity, which was associated with highly active but unstable Co–Mo–S-like structures and with the composition of the mixed phase of cobalt sulphides, the activity being increased in the presence of Co3S4 to a higher extent. In addition, the presence of MoS2 in the investigated Co–Mo–S catalysts seems to be crucial to efficiently accomplish the dehydrogenation step of this borrowing hydrogen sequence.
Application of the catalyst Co–Mo–S-0.83 allowed us the preparation of a broad range of thioethers in good to excellent yields from structurally diverse thiols and primary as well as secondary alcohols. In general, this catalyst displays excellent chemoselectivity towards carbon–sulfur bond formation even in the presence of sensitive functionalities, such as halogens, double bonds, ketones, nitriles and carboxylic esters, which were well-retained in the final thioethers. Furthermore, we have also developed a novel methodology for the synthesis of symmetrical thioethers by reaction of alcohols with H2S in the presence of the catalyst Co–Mo–S-0.83.
Experimental details
General procedure for the alkylation of thiols with alcohols
Reactions were performed in a 7 mL reinforced glass reactor equipped with a pressure controller. The glass pressure tube containing a stirring bar was sequentially charged with Co–Mo–S catalyst (13.1 mg), thiol (0.25 mmol), alcohol (0.5 mmol), n-hexadecane (30 μL) and toluene (1.6 mL). The pressure tube was then closed and the reactor was repeatedly purged with 5 bar of N2, pressurized to 3.5 bar and stirred and heated at 180 °C in an aluminium block previously preheated to this temperature. After the desired reaction time, the reactor was cooled down to room temperature and carefully depressurized. The reaction mixture was diluted with ethyl acetate and an aliquot was taken to be analysed by gas chromatography. To determine the isolated yields, the diluted reaction mixture was filtered over Celite to separate off the catalyst, and then purified by silica gel chromatography affording the corresponding thioethers in the reported yields.
General procedure for the synthesis of symmetrical thioethers from H2S and alcohols
The general procedure described above for the alkylation of thiols with alcohols was applied with minor modifications. The glass pressure tube containing a stirring bar was sequentially charged with Co–Mo–S catalyst (13.1 mg), alcohol (0.25 mmol), n-hexadecane (30 μL) and toluene (1.6 mL). After the reactor was sealed and purged with N2, it was pressurized to 4 bar with N2/H2S (10% v/v in H2S).
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
Financial support by the Spanish Government-MINECO through the program ‘‘Severo Ochoa” (SEV-2016-0683) is gratefully acknowledged. I. S. also acknowledges the Vice-Rectorate for Research, Innovation and Transfer of the Universitat Politècnica de València (UPV) for a postdoctoral fellowship and the Spanish Government-MINECO for a “Juan de la Cierva-Incorporación” fellowship. The authors also acknowledge the Microscopy Service of the UPV and Dr José María Moreno for kind help with TEM and STEM measurements.
Footnotes
†Electronic supplementary information (ESI) available: General information, additional tables, schemes, figures, characterization data and NMR spectra of isolated products. See DOI: 10.1039/c8sc05782f
References
- (a) Block E., Reactions of Organosulfur Compounds, Academic Press, New York, 1978. [Google Scholar]; (b) Peach M. E., in The Chemistry of the Thiol Group, ed. S. Patai, John Wiley & Sons, London, 1979, pp. 721–723. [Google Scholar]; (c) Bernardi F., Csizmadia I. G. and Mangini A., Organic Sulfur Chemistry. Theoretical and Experimental Advances, Elsevier, Amsterdam, 1985. [Google Scholar]; (d) Cremlyn R. J., An Introduction to Organosulfur Chemistry, John Wiley & Sons, New York, 1996. [Google Scholar]; (e) Kondo T., Mitsudo T.-a. Chem. Rev. 2000;100:3205–3220. doi: 10.1021/cr9902749. [DOI] [PubMed] [Google Scholar]; (f) Liu H., Jiang X. Chem.–Asian J. 2013;8:2546–2563. doi: 10.1002/asia.201300636. [DOI] [PubMed] [Google Scholar]
- (a) Artico M., Silvestri R., Pagnozzi E., Bruno B., Novellino E., Greco G., Massa S., Ettorre A., Loi A. G., Scintu F., La Colla P. J. Med. Chem. 2000;43:1886–1891. doi: 10.1021/jm9901125. [DOI] [PubMed] [Google Scholar]; (b) Sun Z.-Y., Botros E., Su A.-D., Kim Y., Wang E., Baturay N. Z., Kwon C.-H. J. Med. Chem. 2000;43:4160–4168. doi: 10.1021/jm9904957. [DOI] [PubMed] [Google Scholar]; (c) Wang Y., Chackalamannil S., Chang W., Greenlee W., Ruperto V., Duffy R. A., McQuade R., Lachowicz J. E. Bioorg. Med. Chem. Lett. 2001;11:891–894. doi: 10.1016/s0960-894x(01)00100-7. [DOI] [PubMed] [Google Scholar]; (d) Faucher A.-M., White P. W., Brochu C., Grand-Maître C., Rancourt J., Fazal G. J. Med. Chem. 2004;47:18–21. doi: 10.1021/jm034206x. [DOI] [PubMed] [Google Scholar]; (e) Clader J. W., Billard W., Binch H., Chen L.-Y., Crosby G., Duffy R. A., Ford J., Kozlowski J. A., Lachowicz J. E., Li S., Liu C., McCombie S. W., Vice S., Zhou G., Greenlee W. J. Biorg. Med. Chem. 2004;12:319–326. doi: 10.1016/j.bmc.2003.11.005. [DOI] [PubMed] [Google Scholar]; (f) Sciabola S., Carosati E., Baroni M., Mannhold R. J. Med. Chem. 2005;48:3756–3767. doi: 10.1021/jm049162m. [DOI] [PubMed] [Google Scholar]; (g) Gangjee A., Zeng Y., Talreja T., McGuire J. J., Kisliuk R. L., Queener S. F. J. Med. Chem. 2007;50:3046–3053. doi: 10.1021/jm070165j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Minghao F., Bingqing T., Steven H. L. a. X. J. Curr. Top. Med. Chem. 2016;16:1200–1216. [Google Scholar]
- (a) Glass H. B., Reid E. E. J. Am. Chem. Soc. 1929;51:3428–3430. [Google Scholar]; (b) Dougherty G., Hammond P. D. J. Am. Chem. Soc. 1935;57:117–118. [Google Scholar]; (c) Kharasch N., Potempa S. J., Wehrmeister H. L. Chem. Rev. 1946;39:269–332. doi: 10.1021/cr60123a004. [DOI] [PubMed] [Google Scholar]; (d) Parham W. E., Wynberg H. Org. Synth. 1955;35:51. [Google Scholar]; (e) Patai S., The Chemistry of the Functional Groups – The Chemistry of the Thiol Group, Wiley, London, 1974. [Google Scholar]; (f) Herriott A. W., Picker D. J. Am. Chem. Soc. 1975;97:2345–2349. [Google Scholar]; (g) Landini D., Rolla F. Org. Synth. 1978;58:143. [Google Scholar]; (h) Boscato J. F., Catala J. M., Franta E., Brossas J. Tetrahedron Lett. 1980;21:1519–1520. [Google Scholar]; (i) Kosugi M., Ogata T., Terada M., Sano H., Migita T. Bull. Chem. Soc. Jpn. 1985;58:3657–3658. [Google Scholar]; (j) Hundscheid F. J. A., Tandon V. K., Rouwette P. H. F. M., van Leusen A. M. Tetrahedron. 1987;43:5073–5088. [Google Scholar]; (k) Harpp D. N., Gingras M. J. Am. Chem. Soc. 1988;110:7737–7745. [Google Scholar]; (l) Gingras M., Chan T. H., Harpp D. N. J. Org. Chem. 1990;55:2078–2090. [Google Scholar]; (m) Li C. J., Harpp D. N. Tetrahedron Lett. 1992;33:7293–7294. [Google Scholar]; (n) Yin J., Pidgeon C. Tetrahedron Lett. 1997;38:5953–5954. [Google Scholar]; (o) Malmström J., Gupta V., Engman L. J. Org. Chem. 1998;63:3318–3323. [Google Scholar]; (p) Blanchard P., Jousselme B., Frère P., Roncali J. J. Org. Chem. 2002;67:3961–3964. doi: 10.1021/jo025627+. [DOI] [PubMed] [Google Scholar]; (q) Ichiishi N., Malapit C. A., Woźniak Ł., Sanford M. S. Org. Lett. 2018;20:44–47. doi: 10.1021/acs.orglett.7b03305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Shen C., Zhang P., Sun Q., Bai S., Hor T. S. A., Liu X. Chem. Soc. Rev. 2015;44:291–314. doi: 10.1039/c4cs00239c. [DOI] [PubMed] [Google Scholar]; (b) Qiao Z., Jiang X. Org. Biomol. Chem. 2017;15:1942–1946. doi: 10.1039/c6ob02833k. [DOI] [PubMed] [Google Scholar]
- (a) Page P. C. B., Klair S. S., Brown M. P., Harding M. M., Smith C. S., Maginn S. J., Mulley S. Tetrahedron Lett. 1988;29:4477–4480. [Google Scholar]; (b) Beletskaya I. P., Cheprakov A. V. Coord. Chem. Rev. 2004;248:2337–2364. [Google Scholar]; (c) Fernández-Rodríguez M. A., Shen Q., Hartwig J. F. J. Am. Chem. Soc. 2006;128:2180–2181. doi: 10.1021/ja0580340. [DOI] [PubMed] [Google Scholar]; (d) Arisawa M., Suzuki T., Ishikawa T., Yamaguchi M. J. Am. Chem. Soc. 2008;130:12214–12215. doi: 10.1021/ja8049996. [DOI] [PubMed] [Google Scholar]; (e) Correa A., Carril M., Bolm C. Angew. Chem., Int. Ed. 2008;47:2880–2883. doi: 10.1002/anie.200705668. [DOI] [PubMed] [Google Scholar]; (f) Wu J.-R., Lin C.-H., Lee C.-F. Chem. Commun. 2009:4450–4452. doi: 10.1039/b907362k. [DOI] [PubMed] [Google Scholar]; (g) Fernández-Rodríguez M. A., Hartwig J. F. Chem.–Eur. J. 2010;16:2355–2359. doi: 10.1002/chem.200902313. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Beletskaya I. P., Ananikov V. P. Chem. Rev. 2011;111:1596–1636. doi: 10.1021/cr100347k. [DOI] [PubMed] [Google Scholar]; (i) Sayah M., Organ M. G. Chem.–Eur. J. 2011;17:11719–11722. doi: 10.1002/chem.201102158. [DOI] [PubMed] [Google Scholar]; (j) Lan M.-T., Wu W.-Y., Huang S.-H., Luo K.-L., Tsai F.-Y. RSC Adv. 2011;1:1751–1755. [Google Scholar]; (k) Cabrero-Antonino J. R., García T., Rubio-Marqués P., Vidal-Moya J. A., Leyva-Pérez A., Al-Deyab S. S., Al-Resayes S. I., Díaz U., Corma A. ACS Catal. 2011;1:147–158. [Google Scholar]; (l) Baig R. B. N., Varma R. S. Chem. Commun. 2012;48:2582–2584. doi: 10.1039/c2cc17283f. [DOI] [PubMed] [Google Scholar]; (m) Liao Y., Jiang P., Chen S., Qi H., Deng G.-J. Green Chem. 2013;15:3302–3306. [Google Scholar]; (n) Liu T.-J., Yi C.-L., Chan C.-C., Lee C.-F. Chem.–Asian J. 2013;8:1029–1034. doi: 10.1002/asia.201300045. [DOI] [PubMed] [Google Scholar]; (o) Kamal A., Srinivasulu V., Murty J. N. S. R. C., Shankaraiah N., Nagesh N., Srinivasa Reddy T., Subba Rao A. V. Adv. Synth. Catal. 2013;355:2297–2307. [Google Scholar]; (p) Timpa S. D., Pell C. J., Ozerov O. V. J. Am. Chem. Soc. 2014;136:14772–14779. doi: 10.1021/ja505576g. [DOI] [PubMed] [Google Scholar]; (q) Lee C.-F., Liu Y.-C., Badsara S. S. Chem.–Asian J. 2014;9:706–722. doi: 10.1002/asia.201301500. [DOI] [PubMed] [Google Scholar]; (r) Thomas A. M., Asha S., Sindhu K. S., Anilkumar G. Tetrahedron Lett. 2015;56:6560–6564. [Google Scholar]; (s) Oderinde M. S., Frenette M., Robbins D. W., Aquila B., Johannes J. W. J. Am. Chem. Soc. 2016;138:1760–1763. doi: 10.1021/jacs.5b11244. [DOI] [PubMed] [Google Scholar]; (t) Kanemoto K., Sugimura Y., Shimizu S., Yoshida S., Hosoya T. Chem. Commun. 2017;53:10640–10643. doi: 10.1039/c7cc05868c. [DOI] [PubMed] [Google Scholar]; (u) Chen C.-W., Chen Y.-L., Reddy D. M., Du K., Li C.-E., Shih B.-H., Xue Y.-J., Lee C.-F. Chem.–Eur. J. 2017;23:10087–10091. doi: 10.1002/chem.201701671. [DOI] [PubMed] [Google Scholar]; (v) Lian Z., Bhawal B. N., Yu P., Morandi B. Science. 2017;356:1059–1063. doi: 10.1126/science.aam9041. [DOI] [PubMed] [Google Scholar]; (w) Fang Y., Rogge T., Ackermann L., Wang S.-Y., Ji S.-J. Nat. Commun. 2018;9:2240. doi: 10.1038/s41467-018-04646-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; (x) Jones K. D., Power D. J., Bierer D., Gericke K. M., Stewart S. G. Org. Lett. 2018;20:208–211. doi: 10.1021/acs.orglett.7b03560. [DOI] [PubMed] [Google Scholar]
- (a) Kumar P., Pandey R. K., Hegde V. R. Synlett. 1999:1921–1922. [Google Scholar]; (b) Kanagasabapathy S., Sudalai A., Benicewicz B. C. Tetrahedron Lett. 2001;42:3791–3794. [Google Scholar]; (c) Morita N., Krause N. Angew. Chem., Int. Ed. 2006;45:1897–1899. doi: 10.1002/anie.200503846. [DOI] [PubMed] [Google Scholar]; (d) Kawatsura M., Komatsu Y., Yamamoto M., Hayase S., Itoh T. Tetrahedron Lett. 2007;48:6480–6482. [Google Scholar]; (e) Banerjee S., Das J., Alvarez R. P., Santra S. New J. Chem. 2010;34:302–306. [Google Scholar]; (f) Cabrero-Antonino J. R., Leyva-Pérez A., Corma A. Adv. Synth. Catal. 2012;354:678–687. [Google Scholar]; (g) Cabrero-Antonino J. R., Leyva-Pérez A., Corma A. Chem.–Eur. J. 2013;19:8627–8633. doi: 10.1002/chem.201300386. [DOI] [PubMed] [Google Scholar]; (h) Castarlenas R., Di Giuseppe A., Pérez-Torrente J. J., Oro L. A. Angew. Chem., Int. Ed. 2013;52:211–222. doi: 10.1002/anie.201205468. [DOI] [PubMed] [Google Scholar]; (i) Zeng X. Chem. Rev. 2013;113:6864–6900. doi: 10.1021/cr400082n. [DOI] [PubMed] [Google Scholar]; (j) Kuciński K., Pawluć P., Hreczycho G. Adv. Synth. Catal. 2015;357:3936–3942. [Google Scholar]; (k) Kumar R., Saima, Shard A., Andhare N. H., Richa, Sinha A. K. Angew. Chem., Int. Ed. 2015;54:828–832. doi: 10.1002/anie.201408721. [DOI] [PubMed] [Google Scholar]; (l) Pérez M., Mahdi T., Hounjet L. J., Stephan D. W. Chem. Commun. 2015;51:11301–11304. doi: 10.1039/c5cc03572d. [DOI] [PubMed] [Google Scholar]; (m) Palacios L., Di Giuseppe A., Artigas M. J., Polo V., Lahoz F. J., Castarlenas R., Pérez-Torrente J. J., Oro L. A. Catal. Sci. Technol. 2016;6:8548–8561. [Google Scholar]; (n) Kennemur J. L., Kortman G. D., Hull K. L. J. Am. Chem. Soc. 2016;138:11914–11919. doi: 10.1021/jacs.6b07142. [DOI] [PubMed] [Google Scholar]; (o) Palacios L., Meheut Y., Galiana-Cameo M., Artigas M. J., Di Giuseppe A., Lahoz F. J., Polo V., Castarlenas R., Pérez-Torrente J. J., Oro L. A. Organometallics. 2017;36:2198–2207. [Google Scholar]; (p) Cabrero-Antonino J. R., Tejeda-Serrano M., Quesada M., Vidal-Moya J. A., Leyva-Pérez A., Corma A. Chem. Sci. 2017;8:689–696. doi: 10.1039/c6sc03335k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Martin M. T., Thomas A. M., York D. G. Tetrahedron Lett. 2002;43:2145–2147. [Google Scholar]; (b) Saxena A., Kumar A., Mozumdar S. J. Mol. Catal. A: Chem. 2007;269:35–40. [Google Scholar]; (c) Bandgar B. P., Gawande S. S., Muley D. B. Green Chem. Lett. Rev. 2010;3:49–54. [Google Scholar]; (d) Bahrami K., Khodaei M. M., Khodadoustan N. Synlett. 2011:2206–2210. [Google Scholar]; (e) Basu B., Kundu S., Sengupta D. RSC Adv. 2013;3:22130–22134. [Google Scholar]; (f) Hikawa H., Toyomoto M., Kikkawa S., Azumaya I. Org. Biomol. Chem. 2015;13:11459–11465. doi: 10.1039/c5ob01717c. [DOI] [PubMed] [Google Scholar]; (g) Hikawa H., Machino Y., Toyomoto M., Kikkawa S., Azumaya I. Org. Biomol. Chem. 2016;14:7038–7045. doi: 10.1039/c6ob01140c. [DOI] [PubMed] [Google Scholar]; (h) Santoro F., Mariani M., Zaccheria F., Psaro R., Ravasio N. Beilstein J. Org. Chem. 2016;12:2627–2635. doi: 10.3762/bjoc.12.259. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Kucinski K., Hreczycho G. Eur. J. Org. Chem. 2017:5572–5581. [Google Scholar]
- (a) Hamid M. H. S. A., Slatford P. A., Williams J. M. J. Adv. Synth. Catal. 2007;349:1555–1575. [Google Scholar]; (b) Nixon T. D., Whittlesey M. K., Williams J. M. J. Dalton Trans. 2009:753–762. doi: 10.1039/b813383b. [DOI] [PubMed] [Google Scholar]; (c) Hollmann D. ChemSusChem. 2014;7:2411–2413. doi: 10.1002/cssc.201402320. [DOI] [PubMed] [Google Scholar]; (d) Muzart J. Eur. J. Org. Chem. 2015:5693–5707. [Google Scholar]; (e) Shimizu K.-i. Catal. Sci. Technol. 2015;5:1412–1427. [Google Scholar]; (f) Corma A., Navas J., Sabater M. J. Chem. Rev. 2018;118:1410–1459. doi: 10.1021/acs.chemrev.7b00340. [DOI] [PubMed] [Google Scholar]
- (a) Guillena G., Ramon D. J., Yus M. Chem. Rev. 2010;110:1611–1641. doi: 10.1021/cr9002159. [DOI] [PubMed] [Google Scholar]; (b) Baehn S., Imm S., Neubert L., Zhang M., Neumann H., Beller M. ChemCatChem. 2011;3:1853–1864. [Google Scholar]; (c) Yang Q., Wang Q., Yu Z. Chem. Soc. Rev. 2015;44:2305–2329. doi: 10.1039/c4cs00496e. [DOI] [PubMed] [Google Scholar]; (d) Ma X., Su C., Xu Q. Top. Curr. Chem. 2016;374:27. doi: 10.1007/s41061-016-0027-1. [DOI] [PubMed] [Google Scholar]
- (a) Guillena G., Ramon D. J., Yus M. Angew. Chem., Int. Ed. 2007;46:2358–2364. doi: 10.1002/anie.200603794. [DOI] [PubMed] [Google Scholar]; (b) Huang F., Liu Z., Yu Z. Angew. Chem., Int. Ed. 2016;55:862–875. doi: 10.1002/anie.201507521. [DOI] [PubMed] [Google Scholar]; (c) Obora Y. Top. Curr. Chem. 2016;374:11. doi: 10.1007/s41061-016-0012-8. [DOI] [PubMed] [Google Scholar]
- Corma A., Navas J., Ródenas T., Sabater M. J. Chem.–Eur. J. 2013;19:17464–17471. doi: 10.1002/chem.201302226. [DOI] [PubMed] [Google Scholar]
- (a) Glass R. S. Synth. Commun. 1976;6:47–51. [Google Scholar]; (b) Kikugawa Y. Chem. Lett. 1981:1157–1158. [Google Scholar]; (c) Olah G. A., Wang Q., Trivedi N. J., Surya Prakash G. K. Synthesis. 1992:465–466. [Google Scholar]; (d) Olah G. A., Wang Q., Li X.-y., Surya Prakash G. K. Synlett. 1993:32–34. [Google Scholar]
- (a) Liu L., Concepción P., Corma A. J. Catal. 2016;340:1–9. [Google Scholar]; (b) Liu L., Gao F., Concepción P., Corma A. J. Catal. 2017;350:218–225. [Google Scholar]; (c) Milian R., Liu L., Boronat M., Corma A. J. Catal. 2018;364:19–30. [Google Scholar]; (d) Filonenko G. A., van Putten R., Hensen E. J. M., Pidko E. A. Chem. Soc. Rev. 2018;47:1459–1483. doi: 10.1039/c7cs00334j. [DOI] [PubMed] [Google Scholar]
- (a) Taguchi K., Nakagawa H., Hirabayashi T., Sakaguchi S., Ishii Y. J. Am. Chem. Soc. 2004;126:72–73. doi: 10.1021/ja037552c. [DOI] [PubMed] [Google Scholar]; (b) Burling S., Paine B. M., Nama D., Brown V. S., Mahon M. F., Prior T. J., Pregosin P. S., Whittlesey M. K., Williams J. M. J. J. Am. Chem. Soc. 2007;129:1987–1995. doi: 10.1021/ja065790c. [DOI] [PubMed] [Google Scholar]; (c) Iuchi Y., Obora Y., Ishii Y. J. Am. Chem. Soc. 2010;132:2536–2537. doi: 10.1021/ja9106989. [DOI] [PubMed] [Google Scholar]; (d) Blank B., Kempe R. J. Am. Chem. Soc. 2010;132:924–925. doi: 10.1021/ja9095413. [DOI] [PubMed] [Google Scholar]; (e) Obora Y., Anno Y., Okamoto R., Matsu-ura T., Ishii Y. Angew. Chem., Int. Ed. 2011;50:8618–8622. doi: 10.1002/anie.201104452. [DOI] [PubMed] [Google Scholar]; (f) Peña-Lopez M., Neumann H., Beller M. Chem. Commun. 2015;51:13082–13085. doi: 10.1039/c5cc01708d. [DOI] [PubMed] [Google Scholar]; (g) Guo L., Ma X., Fang H., Jia X., Huang Z. Angew. Chem., Int. Ed. 2015;54:4023–4027. doi: 10.1002/anie.201410293. [DOI] [PubMed] [Google Scholar]; (h) Shen D., Poole D. L., Shotton C. C., Kornahrens A. F., Healy M. P., Donohoe T. J. Angew. Chem., Int. Ed. 2015;54:1642–1645. doi: 10.1002/anie.201410391. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Zou Q., Wang C., Smith J., Xue D., Xiao J. Chem.–Eur. J. 2015;21:9656–9661. doi: 10.1002/chem.201501109. [DOI] [PubMed] [Google Scholar]; (j) Ramachandran R., Prakash G., Selvamurugan S., Viswanathamurthi P., Malecki J. G., Linert W., Gusev A. RSC Adv. 2015;5:11405–11422. [Google Scholar]; (k) Peña-Lopez M., Neumann H., Beller M. Angew. Chem., Int. Ed. 2016;55:7826–7830. doi: 10.1002/anie.201600698. [DOI] [PubMed] [Google Scholar]; (l) Wang Q., Wu K., Yu Z. Organometallics. 2016;35:1251–1256. [Google Scholar]; (m) Marichev K. O., Takacs J. M. ACS Catal. 2016;6:2205–2210. doi: 10.1021/acscatal.6b00175. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Said Stålsmeden A., Belmonte Vázquez J. L., van Weerdenburg K., Rae R., Norrby P.-O., Kann N. ACS Sustainable Chem. Eng. 2016;4:5730–5736. [Google Scholar]
- (a) Yang J., Liu X., Meng D.-L., Chen H.-Y., Zong Z.-H., Feng T.-T., Sun K. Adv. Synth. Catal. 2012;354:328–334. [Google Scholar]; (b) Bala M., Verma P. K., Sharma U., Kumar N., Singh B. Green Chem. 2013;15:1687–1693. [Google Scholar]; (c) Yan T., Feringa B. L., Barta K. Nat. Commun. 2014;5:5602. doi: 10.1038/ncomms6602. [DOI] [PubMed] [Google Scholar]; (d) Elangovan S., Sortais J.-B., Beller M., Darcel C. Angew. Chem., Int. Ed. 2015;54:14483–14486. doi: 10.1002/anie.201506698. [DOI] [PubMed] [Google Scholar]; (e) Pan H.-J., Ng T. W., Zhao Y. Chem. Commun. 2015;51:11907–11910. doi: 10.1039/c5cc03399c. [DOI] [PubMed] [Google Scholar]; (f) Emayavaramban B., Roy M., Sundararaju B. Chem.–Eur. J. 2016;22:3952–3955. doi: 10.1002/chem.201505214. [DOI] [PubMed] [Google Scholar]; (g) Mastalir M., Stöger B., Pittenauer E., Puchberger M., Allmaier G., Kirchner K. Adv. Synth. Catal. 2016;358:3824–3831. [Google Scholar]; (h) Peña-Lopez M., Neumann H., Beller M. ChemSusChem. 2016;9:2233–2238. doi: 10.1002/cssc.201600587. [DOI] [PubMed] [Google Scholar]; (i) Yan T., Feringa B. L., Barta K. ACS Catal. 2016;6:381–388. [Google Scholar]; (j) Polidano K., Allen B. D. W., Williams J. M. J., Morrill L. C. ACS Catal. 2018;8:6440–6445. [Google Scholar]
- (a) Rösler S., Ertl M., Irrgang T., Kempe R. Angew. Chem., Int. Ed. 2015;54:15046–15050. doi: 10.1002/anie.201507955. [DOI] [PubMed] [Google Scholar]; (b) Deibl N., Kempe R. J. Am. Chem. Soc. 2016;138:10786–10789. doi: 10.1021/jacs.6b06448. [DOI] [PubMed] [Google Scholar]; (c) Yin Z., Zeng H., Wu J., Zheng S., Zhang G. ACS Catal. 2016;6:6546–6550. [Google Scholar]; (d) Freitag F., Irrgang T., Kempe R. Chem.–Eur. J. 2017;23:12110–12113. doi: 10.1002/chem.201701211. [DOI] [PubMed] [Google Scholar]; (e) Zhang G., Wu J., Zeng H., Zhang S., Yin Z., Zheng S. Org. Lett. 2017;19:1080–1083. doi: 10.1021/acs.orglett.7b00106. [DOI] [PubMed] [Google Scholar]
- Liao S., Yu K., Li Q., Tian H., Zhang Z., Yu X., Xu Q. Org. Biomol. Chem. 2012;10:2973–2978. doi: 10.1039/c1ob06739g. [DOI] [PubMed] [Google Scholar]
- (a) Elangovan S., Neumann J., Sortais J.-B., Junge K., Darcel C., Beller M. Nat. Commun. 2016;7:12641. doi: 10.1038/ncomms12641. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mukherjee A., Nerush A., Leitus G., Shimon L. J. W., Ben David Y., Espinosa Jalapa N. A., Milstein D. J. Am. Chem. Soc. 2016;138:4298–4301. doi: 10.1021/jacs.5b13519. [DOI] [PubMed] [Google Scholar]; (c) Peña-Lopez M., Piehl P., Elangovan S., Neumann H., Beller M. Angew. Chem., Int. Ed. 2016;55:14967–14971. doi: 10.1002/anie.201607072. [DOI] [PubMed] [Google Scholar]; (d) Bruneau-Voisine A., Wang D., Dorcet V., Roisnel T., Darcel C., Sortais J.-B. J. Catal. 2017;347:57–62. doi: 10.1021/acs.orglett.7b01657. [DOI] [PubMed] [Google Scholar]; (e) Deibl N., Kempe R. Angew. Chem., Int. Ed. 2017;56:1663–1666. doi: 10.1002/anie.201611318. [DOI] [PubMed] [Google Scholar]; (f) Fu S., Shao Z., Wang Y., Liu Q. J. Am. Chem. Soc. 2017;139:11941–11948. doi: 10.1021/jacs.7b05939. [DOI] [PubMed] [Google Scholar]; (g) Neumann J., Elangovan S., Spannenberg A., Junge K., Beller M. Chem.–Eur. J. 2017;23:5410–5413. doi: 10.1002/chem.201605218. [DOI] [PubMed] [Google Scholar]; (h) Barman M. K., Waiba S., Maji B. Angew. Chem., Int. Ed. 2018;57:9126–9130. doi: 10.1002/anie.201804729. [DOI] [PubMed] [Google Scholar]; (i) Das U. K., Ben-David Y., Diskin-Posner Y., Milstein D. Angew. Chem., Int. Ed. 2018;57:2179–2182. doi: 10.1002/anie.201712593. [DOI] [PubMed] [Google Scholar]
- Piehl P., Peña-Lopez M., Frey A., Neumann H., Beller M. Chem. Commun. 2017;53:3265–3268. doi: 10.1039/c6cc09977g. [DOI] [PubMed] [Google Scholar]
- (a) Carlini C., Macinai A., Marchionna M., Noviello M., Galletti A. M. R., Sbrana G. J. Mol. Catal. A: Chem. 2003;206:409–418. [Google Scholar]; (b) Alonso F., Riente P., Yus M. Eur. J. Org. Chem. 2008:4908–4914. [Google Scholar]; (c) Shimizu K.-i., Kanno S., Kon K., Hakim Siddiki S. M. A., Tanaka H., Sakata Y. Catal. Today. 2014;232:134–138. [Google Scholar]; (d) Onyestyák G., Novodárszki G., Barthos R., Klébert S., Wellisch Á. F., Pilbáth A. RSC Adv. 2015;5:99502–99509. [Google Scholar]; (e) Xu J., Yue H., Liu S., Wang H., Du Y., Xu C., Dong W., Liu C. RSC Adv. 2016;6:24164–24174. [Google Scholar]
- (a) Topsøe H., Clausen B. S. and Massoth F. E., Hydrotreating Catalysis, Science and Technology, Springer-Verlag, Heidelberg, 1996. [Google Scholar]; (b) Toshiaki K., Atsushi I. and Weihua Q., Hydrodesulfurization and Hydrodenitrogenation: Chemistry and Engineering, Wiley-VCH, Tokyo, 1999. [Google Scholar]; (c) Sánchez-Delgado R. A., Organometallic Modeling of the Hydrodesulfurization and Hydrodenitrogenation Reactions, Springer Netherlands, Kluwer, Dordrecht, 2002. [Google Scholar]
- (a) Exxonmobil Pat., US Pat. 6,299,760, 2001.; (b) Plantenga F. L., Cerfontain R., Eijsbouts S., van Houtert F., Anderson G. H., Miseo S., Soled S., Riley K., Fujita K., Inoue Y. Stud. Surf. Sci. Catal. 2003;145:407–410. [Google Scholar]
- Eijsbouts S., Mayo S. W., Fujita K. Appl. Catal., A. 2007;322:58–66. [Google Scholar]
- (a) Yoosuk B., Song C., Kim J. H., Ngamcharussrivichai C., Prasassarakich P. Catal. Today. 2010;149:52–61. [Google Scholar]; (b) Yoosuk B., Tumnantong D., Prasassarakich P. Fuel. 2012;91:246–252. [Google Scholar]; (c) Liu N., Wang X., Xu W., Guo D., Tang J., Zhang B. Prog. Chem. 2013;25:726–734. [Google Scholar]; (d) Wang W., Zhang K., Li L., Wu K., Liu P., Yang Y. Ind. Eng. Chem. Res. 2014;53:19001–19009. [Google Scholar]; (e) Itthibenchapong V., Ratanatawanate C., Oura M., Faungnawakij K. Catal. Commun. 2015;68:31–35. [Google Scholar]; (f) Wang W., Li L., Wu K., Zhu G., Tan S., Li W., Yang Y. RSC Adv. 2015;5:61799–61807. [Google Scholar]; (g) Wang W., Li L., Wu K., Zhang K., Jie J., Yang Y. Appl. Catal., A. 2015;495:8–16. [Google Scholar]; (h) Wang W., Li L., Wu K., Zhu G., Tan S., Liua Y., Yang Y. RSC Adv. 2016;6:31265–31271. [Google Scholar]; (i) Wang W., Li L., Tan S., Wu K., Zhu G., Liu Y., Xu Y., Yang Y. Fuel. 2016;179:1–9. [Google Scholar]; (j) Wang W., Wu K., Li L., Tan S., Zhu G., Li W., He Z., Yang Y. Catal. Commun. 2016;74:60–64. [Google Scholar]; (k) Wang W., Zhu G., Li L., Tan S., Wu K., Zhang X., Yang Y. Fuel. 2016;174:1–8. [Google Scholar]
- (a) Sorribes I., Liu L., Corma A. ACS Catal. 2017;7:2698–2708. [Google Scholar]; (b) Sorribes I., Liu L., Doménech-Carbó A., Corma A. ACS Catal. 2018;8:4545–4557. [Google Scholar]
- (a) Topsøe H., Clausen B. S., Candia R., Wivel C., Morup S. J. Catal. 1981;68:433–452. [Google Scholar]; (b) Wivel C., Candia R., Clausen B. S., Morup S., Topsøe H. J. Catal. 1981;68:453–463. [Google Scholar]; (c) Clausen B. S., Topsøe H., Candia R., Villadsen J., Lengeler B., Alsnielsen J., Christensen F. J. Phys. Chem. 1981;85:3868–3872. [Google Scholar]; (d) Breysse M., Bennett B. A., Chadwick D., Vrinat M. Bull. Soc. Chim. Belg. 1981;90:1271–1277. [Google Scholar]; (e) Topsøe N. Y., Topsøe H. J. Catal. 1983;84:386–401. [Google Scholar]; (f) Kasztelan S., Toulhoat H., Grimblot J., Bonnelle J. P. Appl. Catal. 1984;13:127–159. [Google Scholar]; (g) Topsøe H., Clausen B. S. Appl. Catal. 1986;25:273–293. [Google Scholar]; (h) Byskov L. S., Nørskov J. K., Clausen B. S., Topsøe H. J. Catal. 1999;187:109–122. [Google Scholar]; (i) Schweiger H., Raybaud P., Toulhoat H. J. Catal. 2002;212:33–38. [Google Scholar]; (j) Lauritsen J. V., Bollinger M. V., Lægsgaard E., Jacobsen K. W., Nørskov J. K., Clausen B. S., Topsøe H., Besenbacher F. J. Catal. 2004;221:510–522. [Google Scholar]; (k) Topsøe H. Appl. Catal., A. 2007;322:3–8. [Google Scholar]; (l) Lauritsen J. V., Kibsgaard J., Olesen G. H., Moses P. G., Hinnemann B., Helveg S., Nørskov J. K., Clausen B. S., Topsøe H., Lægsgaard E., Besenbacher F. J. Catal. 2007;249:220–233. [Google Scholar]; (m) Berhault G., Perez De la Rosa M., Mehta A., Yácaman M. J., Chianelli R. R. Appl. Catal., A. 2008;345:80–88. [Google Scholar]; (n) Besenbacher F., Brorson M., Clausen B. S., Helveg S., Hinnemann B., Kibsgaard J., Lauritsen J. V., Moses P. G., Nørskov J. K., Topsøe H. Catal. Today. 2008;130:86–96. [Google Scholar]; (o) Gandubert A. D., Krebs E., Legens C., Costa D., Guillaume D., Raybaud P. Catal. Today. 2008;130:149–159. [Google Scholar]; (p) Krebs E., Silvi B., Raybaud P. Catal. Today. 2008;130:160–169. [Google Scholar]; (q) Kibsgaard J., Tuxen A., Knudsen K. G., Brorson M., Topsøe H., Lægsgaard E., Lauritsen J. V., Besenbacher F. J. Catal. 2010;272:195–203. [Google Scholar]; (r) Zhu Y., Ramasse Q. M., Brorson M., Moses P. G., Hansen L. P., Kisielowski C. F., Helveg S. Angew. Chem., Int. Ed. 2014;53:10723–10727. doi: 10.1002/anie.201405690. [DOI] [PubMed] [Google Scholar]
- (a) Scholz F., Schröder U., Gulaboski R. and Doménech-Carbó A., Electrochemistry of Immobilized Particles and Droplets, Springer, Berlin-Heidelberg, 2nd edn, 2014. [Google Scholar]; (b) Domenech-Carbo A., Labuda J., Scholz F. Pure Appl. Chem. 2012;85:609–631. [Google Scholar]
- (a) McCullough L. R., Childers D. J., Watson R. A., Kilos B. A., Barton D. G., Weitz E., Kung H. H., Notestein J. M. ACS Sustainable Chem. Eng. 2017;5:4890–4896. [Google Scholar]; (b) McCullough L. R., Cheng E. S., Gosavi A. A., Kilos B. A., Barton D. G., Weitz E., Kung H. H., Notestein J. M. J. Catal. 2018;366:159–166. [Google Scholar]
- (a) Piéplu A., Saur O., Lavalley J.-C., Legendre O., Nédez C. Catal. Rev.: Sci. Eng. 1998;40:409–450. [Google Scholar]; (b) Eow J. S. Environ. Prog. 2002;21:143–162. [Google Scholar]
- (a) Huang H., Yu Y., Chung K. H. Energy Fuels. 2009;23:4420–4425. [Google Scholar]; (b) Singh G., Nakade P. G., Chetia D., Jha P., Mondal U., Kumari S., Sen S. Ind. Eng. Chem. 2016;37:190–197. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.