

Abstract
Temporomandibular joint (TMJ) osteoarthritis (OA) is a degenerative disease of the joint that can produce persistent orofacial pain as well as functional and structural changes to its bone, cartilage, and ligaments. Despite advances in the clinical utility and reliability of the Diagnostic Criteria for Temporomandibular Disorders, clinical tools inadequately predict which patients will develop chronic TMJ pain and degeneration, limiting clinical management. The challenges of managing and treating TMJ OA are due, in part, to a limited understanding of the mechanisms contributing to the development and maintenance of TMJ pain. OA is initiated by multiple factors, including injury, aging, abnormal joint mechanics, and atypical joint shape, which can produce microtrauma, remodeling of joint tissues, and synovial inflammation. TMJ microtrauma and remodeling can increase expression of cytokines, chemokines, and catabolic factors that damage synovial tissues and can activate free nerve endings in the joint. Although studies have separately investigated inflammation-driven orofacial pain, acute activity of the trigeminal nerve, or TMJ tissue degeneration and/or damage, the temporal mechanistic factors leading to chronic TMJ pain are undefined. Limited understanding of the interaction between degeneration, intra-articular chemical factors, and pain has further restricted the development of targeted, disease-modifying drugs to help patients avoid long-term pain and invasive procedures, like TMJ replacement. A range of animal models captures features of intra-articular inflammation, joint overloading, and tissue damage. Although those models traditionally measure peripheral sensitivity as a surrogate for pain, recent studies recognize the brain’s role in integrating, modulating, and interpreting nociceptive inputs in the TMJ, particularly in light of psychosocial influences on TMJ pain. The articular and neural contributors to TMJ pain, imaging modalities with clinical potential to identify TMJ OA early, and future directions for clinical management of TMJ OA are reviewed in the context of evidence in the field.
Keywords: pain, inflammation, central nervous system, animal models, joint diseases, cartilage
Introduction
Temporomandibular joint (TMJ) disorders are very common, with over 70% of the population reporting signs or symptoms (Scrivani et al. 2008). Fifteen percent of TMJ disorder (TMD) cases present as aggressive disease that is recalcitrant to therapies and lead to the development of chronic centralized pain, making TMDs the second most common musculoskeletal pain condition (National Institute of Dental and Craniofacial Research [NIDCR] 2014). Since nomenclature and terminology of TMJ disorders overlap and are used interchangeably, it is helpful to establish common terms. Internal TMJ derangement is defined by articular disc displacement (Fig. 1), pain, and joint dysfunction. Degenerative joint disease of the TMJ can occur secondary to internal derangement of the disc (Tanaka et al. 2008). Using the Diagnostic Criteria for Temporomandibular Disorders (DC/TMD) for clinical and research taxonomy, degenerative joint disease is “a degenerative disorder involving the joint characterized by deterioration of articular tissue with concomitant osseous changes in the condyle and/or articular eminence” (Schiffman et al. 2014).
Figure 1.

MRI and arthroscopic imaging are clinical tools that can identify structural changes in the TMJ as well as synovial inflammation. T1-weighted magnetic resonance imaging (MRI) of a normal temporomandibular joint (TMJ) in the (A) closed position with the yellow arrow marking the transition between the disc and retrodiscal tissues and (B) open position in which the disc is over the condyle. (C) A T2 MRI of a TMJ with condylar degeneration, showing an anteriorly displaced disc and joint effusion. Arthroscopic images of (D) normal-appearing TMJ anatomy in the anterior recess of the joint and (E) with local synovial inflammation/synovitis (arrow). Images D and E courtesy of Dr. Helen Giannakopoulos.
Osteoarthritis (OA) describes joint degeneration with synovitis, cartilage degeneration, and subchondral bone remodeling (Fig. 1) along with joint pain (Wang et al. 2015). For TMJ disease, findings of condylar degeneration and disc displacement often occur without pain, and most patients experience brief pain and dysfunction (Scrivani et al. 2008). For example, anteriorly displaced discs are found in 20% of healthy, asymptomatic individuals (Haiter-Neto et al. 2002). In mild and transient pain, conservative medical management has favorable outcomes (NIDCR 2014). However, for some patients with a recalcitrant clinical course, adaptive mechanisms fail without any pathophysiological reason (Tanaka et al. 2008; Harper et al. 2016), complicating effective management of TMJ disorders. Because the pathophysiology of progressive TMJ OA, joint degeneration, and pain is not well understood, and since imaging modalities do not correlate well with symptoms (Koh et al. 2009), their clinical management is limited and complicated. Also, psychosocial (axis II) conditions affect TMJ pain (Schiffman et al. 2014; Harper et al. 2016), complicating the development and utility of animal models to study these conditions. This review summarizes current clinical approaches for diagnosis and treatment of TMJ OA, along with OA and pain pathophysiology. Based on clinical challenges in understanding this disease, the review discusses new evidence from animal models of TMJ OA to inform emerging clinical approaches in diagnostic imaging and intervention.
Clinical Perspective and Research
Although there are advances in the clinical utility and reliability of the DC/TMD, concerns remain about its clinical use (Steenks et al. 2018) (Appendix Table I). Challenges remain in the examination and accurate diagnosis of patients with temporomandibular disorders, including differentiating myofascial and intra-articular etiologies of pain and dysfunction (Okeson 2018). Despite difficulty distinguishing different TMJ pain etiologies, initial treatment for myofascial TMJ disorders or OA is similar and involves exhausting reversible, noninvasive interventions. Second-line therapies for patients with refractory symptoms require confirming a primary muscular or articular etiology of pain or dysfunction (Liu and Steinkeler 2013).
The American Society of Temporomandibular Joint Surgeons advocates using the Wilkes classification in clinical studies of surgical interventions in TMJ degeneration. That system encompasses patient pain reports, disc position, and condylar degeneration to grade internal joint derangement from early stage anterior disc displacement without pain to late-stage disc perforation and bone changes with episodic or continuous pain (Liu and Steinkeler 2013). Although staging internal TMJ derangement is useful to ensure surgical studies compare similar anatomic degeneration, disc position as the primary driver of disease progression and pain has been questioned (Koh et al. 2009). Increasing evidence supports changes in the TMJ microenvironment as associated with progressive osteoarthritis, dysfunction, and pain regardless of disc position (Wang et al. 2015; Kellesarian et al. 2016).
Nonsteroidal anti-inflammatory drugs (NSAIDs) have the strongest clinical evidence for pain relief with TMJ degeneration. A prospective blinded placebo-controlled study showed naproxen (500 mg twice daily) on a standing schedule for 6 wk improved pain and function compared to celecoxib or placebo (Ta and Dionne 2004). Physical therapy and occlusal splints are also suggested before surgery or other irreversible therapies (Liu and Steinkeler 2013). Although outside this scope, it is worth noting that their low side effect profile is attractive. Minimally invasive procedures like arthrocentesis or arthroscopic lysis and lavage should be used before considering open surgery and demonstrate improved pain and function. Although effective, it is unclear whether there is additional benefit with intra-articular steroid or hyaluronic acid injections over lavage (Bouloux et al. 2017). Considerable controversy remains about arthroplasty for TMJ degeneration, including disc salvage procedures compared to disc removal procedures with or without autogenous grafting. Only a small percentage of patients progresses to open procedures, which should be reserved for end-stage disease after all nonreversible options are exhausted. For patients with severe condylar degeneration, dysfunction, and pain, alloplastic joint replacement may be effective (Wolford et al. 2015).
Osteoarthritis Pain
OA is the most common joint pathology affecting the TMJ and is characterized by cartilage deterioration, inflammation, dysfunctional joint remodeling, and pain (Tanaka et al. 2008) (Fig. 1). Since these key features are shared across different synovial joints with OA (Tonge et al. 2014; Sperry, Ita, et al. 2017), the broader literature provides mechanistic insights into joint degeneration and disability. OA is initiated by multiple factors, including injury, aging, abnormal joint mechanics, and atypical joint shape (Loeser et al. 2012). Their combination can produce microtrauma, functional tissue remodeling, and variable degrees of synovial inflammation (Tanaka et al. 2008; Loeser et al. 2012).
Synovial, chondrocytic, and inflammatory cells in the joint express and respond to inflammatory cytokines (Miller et al. 2014). Tumor necrosis factor α (TNFα) is one of the earliest and most ubiquitously expressed cytokines in OA (Miller et al. 2014). TNFα both activates osteoclasts (Wu et al. 2012) and can directly stimulate nociceptors (Durham et al. 2017), making it a key mediator of both joint damage and pain. Cytokines also influence the cartilage microenvironment by dysregulating hypoxia inducible factors (HIFs) (Yang et al. 2010), which maintain normal chondrocyte function (Mino-Oka et al. 2017) but activate destructive pathways when absent or highly upregulated. Increased HIF2α leads to cartilage destruction through catabolic pathways (Yang et al. 2010). Vascular endothelial growth factor (VEGF) and matrix metalloproteinases (MMPs) are also upregulated in OA through HIF1α-mediated pathways (Loeser et al. 2012), leading to atypical vasculature growth and extracellular matrix dissolution in the cartilage (Shen et al. 2015; Hamilton et al. 2016). Tissue catabolism and extracellular matrix breakdown can produce further inflammation by recruiting macrophages (Tanaka et al. 2008). Similar to TNFα, VEGF, MMPs, and downstream effectors like nerve growth factor (NGF) are associated with sensory neuron hyperexcitability (Kras et al. 2015; Hamilton et al. 2016). Because peripheral hyperexcitability is modulated at multiple anatomic sites (Sessle 2011), effects of peripheral sensitizers are described in the following section.
Unlike OA in other joints, premenopausal females are disproportionately affected by TMJ dysfunction, reporting jaw pain at twice the rate of males (Lipton et al. 1993) and seeking care at 3 to 9 times that of males (Scrivani et al. 2008). In contrast, symptomatic knee OA is only slightly higher in females (Lawrence et al. 2008) than males (10%) (Dillon et al. 2006). Estrogen receptors, particularly on the TMJ disc and articular cartilage, are hypothesized to be associated with TMJ pain and destruction. Although TMJ dysfunction is associated with increased estrogen receptors in human TMJs with painful degeneration (Abubaker et al. 1993), cell signaling differences in the central nervous system (CNS) also have been proposed as an explanation of more pain (Rosen et al. 2017).
Neural Processing of TMJ Pain
Peripheral Innervation and Trigeminal Ganglia
The TMJ contains numerous free nerve endings, many of which are nociceptors connected to small-diameter A-delta or C-fibers activated by noxious inflammatory or injurious inputs (Sessle 1999). The joint capsule, articular disc, and periosteal bone are most densely innervated and contain nerve fibers immunoreactive for the neuropeptides substance P and calcitonin gene-related peptide (CGRP) (Kido et al. 1993). Although TMJ condyle fibrocartilage is not innervated, cartilage damage can activate nociceptors when macrophages and mast cells release chemical mediators (i.e., bradykinin, TNFα, prostaglandin E2, NGF) (Sessle 2011; Loeser et al. 2012; Kellesarian et al. 2016). In some cases, repeated activation of nerve endings by damage-induced cytokines, chemokines, and neurotrophic factors causes peripheral sensitization (Okamoto et al. 2003; Durham et al. 2017). Peripheral sensitization decreases the activation threshold of nociceptors and can induce pain, even for typically nonnoxious joint motions (Scrivani et al. 2008; Sekiguchi et al. 2016).
Nerve fibers from TMJ tissues coalesce to form the auriculotemporal nerve, a branch of the mandibular nerve (V3; third branch of trigeminal nerve). The mandibular nerve relays to the trigeminal ganglion (Fig. 2), where the first-order, unipolar neurons are, and is a site of nociceptive processing and modulation (Okeson 2004). Stimulation by inflammation can activate the satellite glial cells and release inflammatory mediators and neuropeptides that further excite trigeminal ganglion neurons (Takeda et al. 2007; Villa et al. 2010). Substance P and NGF are commonly identified in trigeminal neurons and can sensitize nociceptive-specific and wide dynamic range neurons (Fig. 2) (Sessle 2011). The central branch of the unipolar neuron synapses with secondary neurons in the spinal trigeminal nucleus (Fig. 2), which extends through the brainstem from the upper cervical spinal cord to the midbrain.
Figure 2.

Nociceptive signals are detected by nerve endings in the temporomandibular joint (TMJ) and sent to neuron cell bodies in the trigeminal ganglion, which express neuropeptides like nerve growth factor (NGF) (green) and substance P (magenta) when activated. Those first-order neurons synapse with second-order neurons (purple) in the spinal trigeminal nucleus caudalis (Sp5C) of the brainstem. From there, nociceptive signals are transmitted to third-order neurons (green) and other higher-order brain centers, like the ventral medioposterior (VPM) of thalamus, where they are integrated and interpreted for pain sensation. BS, brainstem; CN5, cranial nerve 5; TG, trigeminal ganglion.
Trigeminal Nucleus
Nociceptive signals from the TMJ are further modulated at the trigeminal nucleus. Nociceptive neurons are primarily located in the superficial layers of the dorsal trigeminal nucleus caudalis (Sp5C) (Takeshita et al. 2001), which spans from the caudal medulla to the C2 spinal cord (Fig. 2). Inflammatory and neuropathic sensitization in the TMJ activates microglia in the trigeminal nucleus, which release proinflammatory cytokines that regulate synaptic pain transmission (Villa et al. 2010) both acutely by direct interaction with ion channels and persistently by altering gene transcription and protein expression (Miller et al. 2014). In acute pain conditions that resolve, excitation is temporary and signaling returns to baseline after stimulus removal (Takeshita et al. 2001; Okamoto et al. 2003). In TMJ pain, a loss of GABAergic inhibitory neurons increases pain and neuronal activity in the Sp5C (Puri et al. 2012). TMJ pain patients exhibit reduced gray matter at the trigeminal nucleus and altered diffusivity properties in the trigeminal tract (Wilcox et al. 2015); structural alterations along the trigeminal axons and at synapses in the Sp5C nucleus may contribute to aberrant signaling by trigeminal nociceptive neurons. Altered signaling at the trigeminal nucleus can also be propagated to higher processing sites and modify brain activity (Youssef et al. 2014).
Brain
The brain integrates, modulates, and interprets neuronal inputs. Sensory signals travel from the trigeminal nucleus to the thalamus, with most connections in the ventral medioposterior (VPM) region (Fig. 2) (Upadhyay et al. 2008), which integrates and propagates information to cortical and subcortical brain regions (Malfait and Schnitzer 2013). Glia in the VPM can also become activated and modulate brain signaling via neuroexcitatory signals (Milligan and Watkins 2009; Sessle 2011). Thalamic neurons project to the prefrontal cortex, somatosensory cortex, and hypothalamus (Farmer et al. 2012; Kucyi and Davis 2017).
Rodent studies of trigeminal inflammatory pain suggest that central sensitization—enhanced function of nociceptive neurons and circuits due to increased neuronal membrane excitability and heightened synaptic efficiency—produces widespread changes in the somatosensory network (Latremoliere and Woolf 2009; Spisák et al. 2016). The anterior cingulate cortex, an affective brain region responsible for emotion, decision making, and impulse control, exhibits increased activity and connectivity with the somatosensory cortex after TMJ inflammation (Spisák et al. 2016). Similarly, TMJ disorder patients exhibit increased anterior cingulate and somatosensory cortices activity (Youssef et al. 2014; Harper et al. 2016). The involvement of sensory and affective brain regions in chronic pain aligns with clinical observations that sensory and psychosocial factors contribute to TMJ OA pain (Scrivani et al. 2008; Schiffman et al. 2014). Psychological traits, like anxiety and depression, overlap with centralized pain in TMJ disorders, which may be due to similar neurotransmitter dysfunction (Harper et al. 2016).
Measuring the affective component of pain is challenging and relies primarily on patient reports but is being combined with emerging neuroimaging techniques to predict chronic pain development (Vachon-Presseau et al. 2016). Studies in rodents frequently measure withdrawal thresholds to stimuli as quantitative pain measurements (Nicoll et al. 2010; Villa et al. 2010; Wang et al. 2012; Kartha et al. 2016); yet, that does not capture spontaneous pain. Increasingly, facial grimace scales are employed to estimate affective pain (Langford et al. 2010; Sotocinal et al. 2011; Philips et al. 2016). The Rat Grimace Scale (RGS) detects orofacial pain within 3 h after an initial jaw-loading exposure, lasting for up to 24 h after loading is stopped (Fig. 3), suggesting that sustained, affective orofacial pain is evident. RGS is a useful new tool for translational research of the affective components of chronic pain, particularly in animal models (Sperry et al. 2018). Although animal models of TMJ and OA pain are limited, they provide platforms to investigate biological mechanisms of joint degeneration and pain.
Figure 3.
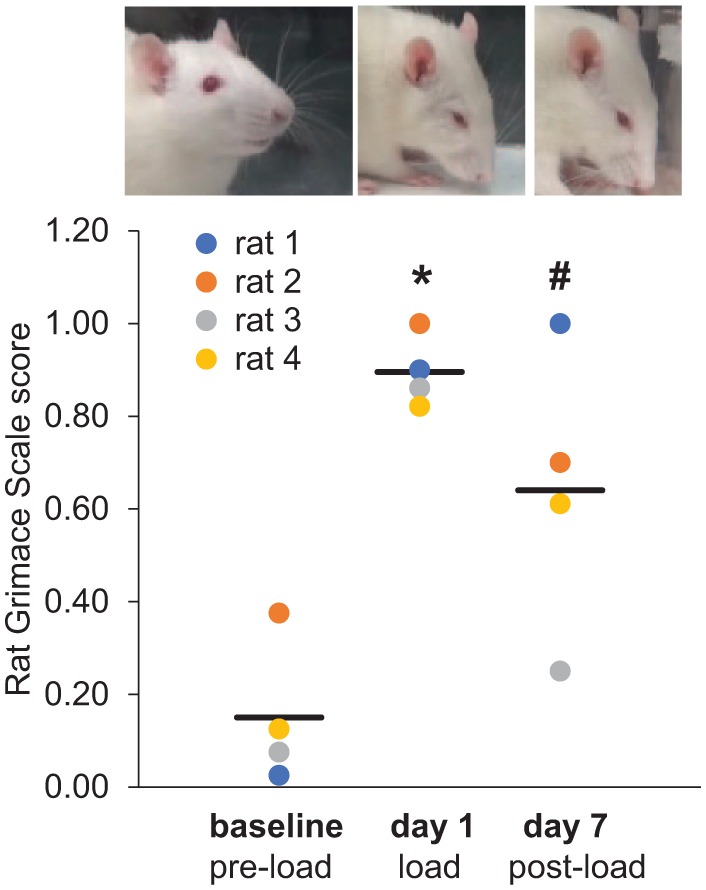
The Rat Grimace Scale (RGS) is increased over baseline levels at day 1, which is 3 h after temporomandibular joint (TMJ) loading (*P = 0.003), and at day 7, which is 24 h after the cessation of daily TMJ loading (#P = 0.026) (comparison between baseline and each day by a 1-way analysis of variance). The examples of typical rat faces reflecting the corresponding RGS scoring are above each time point.
Animal Models of TMJ Disorders
Experimental models of TMJ pain define causative factors in OA progression, joint pathology, and pain symptoms and attempt to replicate any/all of the mechanical loading, inflammatory conditions, and tissue damage observed in degenerative TMJs (Scrivani et al. 2008; Tanaka et al. 2008). Models simulate either the symptoms or signs of TMJ pain and degeneration using chemical and inflammatory agents, surgical manipulation, or mechanical TMJ loading.
Despite having heterogeneous etiologies, since TMJ disorders share the common traits of inflammation and pain, inducing inflammation enables studying its effects on the TMJ tissues and cells (Shen et al. 2015) and on pain signaling (Iwata et al. 1999; Bereiter et al. 2005; Tashiro et al. 2009). Intra-articular injection of inflammatory or catabolic agents, like proinflammatory cytokines or collagenase, promotes OA and chemically simulates microdamage (Kawai et al. 2000; Wang et al. 2012). Within 1 to 6 wk, inflammation upregulates expression of inflammatory and catabolic proteins (HIFs, TNFα, VEGF, MMPs) in chondrocytes, alters the normally organized cartilage structure, and eventually causes resorption of the subchondral bone (Kawai et al. 2000; Shen et al. 2015; reviewed by Wang et al. 2015). TMJ inflammation also enhances excitability of trigeminal neurons in the C2 junction of the brainstem and spinal cord (Bereiter et al. 2005) and alters brain connectivity in the somatosensory and anterior cingulate regions (Spisák et al. 2016), similar to changes in humans with chronic TMJ disorder (Youssef et al. 2014; Wilcox et al. 2015).
Inflammatory models also enable studying direct effects of immune cells and cytokines on the TMJ (Kuroki et al. 2011; Wang et al. 2012), as well as the role of glia pain (Villa et al. 2010). For example, VEGF activates osteoclasts, which initiates OA through destruction of the subchondral bone matrix in the mouse (Shen et al. 2015). However, the artificial inflammatory condition does not accurately model the early stages of TMJ pathology that typically includes articular surface microdamage, limiting understanding of TMJ disorder mechanisms. Clinical analyses hypothesize that tissue microtrauma from parafunctional loading can damage the collagen networks and alter the synovial microenvironment (Okeson 2004). In fact, several animal models simulate the mechanical loading leading to microtrauma from disc derangement or parafunctional habits like bruxism or clenching (Nicoll et al. 2010; Embree et al. 2015; Kartha et al. 2016).
Surgical methods altering TMJ kinematics and stability model the derangement and displacement of the condyle/disc complex. For example, severing the discal attachments with anterior disc displacement has been used in rabbits (Ali and Sharawy 1994) and disc perforation and scraping of the condylar surface in sheep (Kim et al. 2001). Similar to human disc displacement, surgical models produce neovascularization of the TMJ disc, osteoarthritic condylar changes (Ali and Sharawy 1994), erosion, and osteophyte formation (Kim et al. 2001). However, those interventions produce severe trauma that is unlikely in clinical TMJ disorder. A less extreme surgical procedure, the small punch disc biopsy (Embree et al. 2015), induces pathology more closely replicating that in humans. Both condylar cartilage degeneration and disc heterotopic bone formation occur in a rabbit disc perforation model of TMJ disease (Embree et al. 2016). A small punch biopsy, restricted to the inferior layers of the disc in minipigs, similarly induces cartilage degeneration, which can be repaired using a tissue-engineered disc implant (Vapniarsky et al. 2018). Although disc punch biopsies and perforation induce OA paralleling the human condition, surgical models artificially damage the joint.
Repeated mechanical TMJ loading recapitulates clinically observed parafunctional habits or disc dislocations that alter normal loading (Okeson 2004). Light loads (<0.49 N) to the rat TMJ produce cartilage thickening (Zhang et al. 2015; Utreja et al. 2016), suggesting adaptive remodeling. Conversely, forces above that magnitude applied over multiple days induce cartilage damage, decreased proteoglycan expression, and chondrocyte apoptosis (Zhang et al. 2015; Zhang et al. 2017). Sustained loading to the rat TMJ by a sliding plate at the incisors induces condylar cartilage degeneration, increases HIF1α, and activates osteoclasts (Shirakura et al. 2010). Although these models share characteristics with clinical TMJ OA, the relationship between loading and pain is not addressed.
A noninvasive OA pain model was developed by applying mouth opening to the rat TMJ (Nicoll et al. 2010). In that model, the mandible is stationary and the maxilla is held open by 2-N and 3.5-N force separately, applied for 1 h/d for 7 d (Nicoll et al. 2010; Kartha et al. 2016). Both loads produce behavioral sensitivity during the loading phase; however, sensitivity only persists for 3.5 N (Kartha et al. 2016) (Fig. 4). Interestingly, the 3.5-N load induces similar sensitivity as does intra-articular injection of the inflammatory stimulus, complete Freund’s adjuvant (CFA), remaining sensitive for at least 7 d after loading cessation (Fig. 4). In fact, TMJ sensitivity is detectable until day 27 (20 d after loading stops) (Fig. 4B), demonstrating that joint overloading produces sustained peripheral sensitivity, possibly due to TMJ changes and/or altered signaling in the trigeminal nerve. The similar profiles between loading and inflammatory models suggest they may induce similar molecular mediators and/or structural changes.
Figure 4.
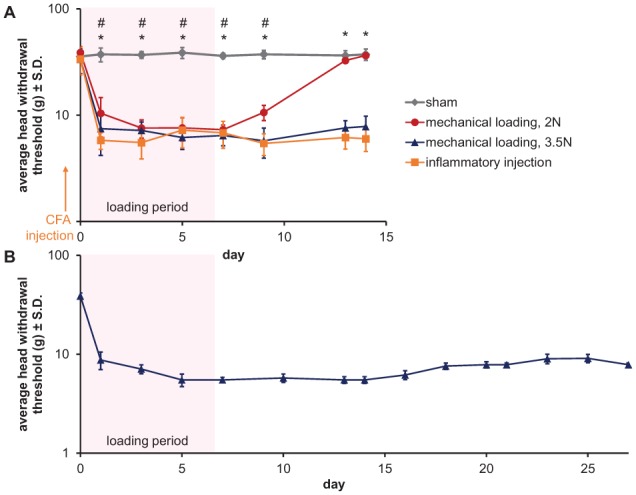
Orofacial sensitivity in rats exposed to repeated TMJ loading or intraarticular inflammatory injection. (A) The average head withdrawal threshold for sham, 2-N loading, 3.5-N loading, and complete Freund’s adjuvant (CFA)–injected rats (25 µg CFA in 50 µL saline). Except sham controls, all groups have decreased thresholds after the first day of loading or inflammatory injection. Reduced thresholds are maintained through day 14, except in the 2-N loading group, for which the withdrawal thresholds returned to sham levels by day 13 (2-way analysis of variance [ANOVA] with day and group as factors; #sham versus 2 N, P < 0.001; *sham versus 3.5-N loading or CFA injected, P < 0.001). (B) After 3.5-N loading, the head withdrawal threshold decreases from baseline through the latest day tested—day 27 (days compared by a 1-way ANOVA; all time points compared to baseline, P < 0.001).
Signs of hypoxic damage (increased HIF1α/HIF2α) and inflammation (increased TNFα) are evident with the persistently painful 3.5-N, but not the transiently painful 2-N, loading (Kartha et al. 2016; Sperry, Moody, et al. 2017), suggesting that OA-like changes are induced early and only when persistent pain develops. Histology at day 15 reveals moderate osteoarthritic pathology by Mankin scoring but little change from normal in the transiently painful (2-N) group (Sperry et al. 2018). Functional overloading also produces signs of subchondral bone erosion at day 15, particularly in the posterior region of the condyle (Fig. 5). Similar flattening, osteophytes, and cortical thickening are also evident in a patient undergoing joint replacement surgery (Fig. 5). Despite these shared OA characteristics, the human TMJ has a much finer trabecular structure that extends further down the condyle, whereas the rat’s TMJ trabeculae are denser and restricted to the mandibular head (Fig. 5). Because the articular eminence is more prominent in the human, the anterior condyle also experiences greater tissue-on-tissue contact during jaw opening, which may produce different patterns of osteophyte outgrowth (Fig. 5) (Scrivani et al. 2008). Although loading models replicate the repeated microtrauma that is observed clinically, the ambiguity of applied loads and required repeated exposures restricts their translational ability. Furthermore, the role of muscles is not well defined, and jaw mechanics vary between species (Liu and Herring 2000; Herring 2003).
Figure 5.

Comparison of the human temporomandibular joint (TMJ) with diagnosed osteoarthritis (OA) and the rat TMJ after exposure to 3.5-N jaw opening reveals similar condylar flattening and formation of osteophytes. Prominent osteophytes are visible on the anterior head of the human condyle, whereas the beginning of an osteophyte is found on the posterior side of the rat condyle. The condyle shape and trabecular structure are also markedly different, with thicker trabeculae in the head of rat condyle compared to the human.
Imaging in Painful TMJ Osteoarthritis
The most useful and widely used clinical screening tool for the TMJ is the panoramic radiograph or panorex (Scrivani et al. 2008). However, it is rarely used to evaluate complex pathology due to its poor reliability and sensitivity for detecting osseous changes (Ahmad et al. 2009). Computed tomography (CT) is the preferred diagnostic modality, providing excellent resolution of joint OA (Ahmad et al. 2009) (Fig. 6A). Cone-beam CT (CBCT) provides more detailed images at lower radiation doses, with similar levels of detail to that achieved in the rat using the MILabs ultra-focus CT protocol (Fig. 6B), and has high diagnostic accuracy for TMJ bone changes (Ma et al. 2016). Although CT provides anatomical detail, it can only detect calcified tissues and does not provide information about soft tissues—the cartilage, disc, and ligaments—that are important in OA.
Figure 6.

Multiple imaging modalities are useful for studying and diagnosing temporomandibular joint (TMJ) OA. Peripherally, (A) computed tomography (CT) imaging is widely used. This example is a frontal, bilateral view of the rat TMJ. (B) Ultra-focus CT imaging provides improved views of the trabecular details of the condyle. 18F-FDG positron emission tomography imaging of the central nervous system is able to capture (C) sagittal and (D) coronal views of brain metabolism. (E) The white matter structure of the brain can be visualized by diffusion tensor imaging.
Magnetic resonance imaging (MRI) can visualize soft tissues and identifies altered disc position and effusion. MRI provides a useful platform for evaluating the morphology and location of the articular disc in both open- and closed-mouth positions. Disc location is crucial since a displaced disc is a sign of disc derangement, which can progress to microtrauma and OA if not treated. Although MRI has become the imaging modality of choice for identifying TMJ soft tissue pathology, it is not without incidental findings; 20% of asymptomatic volunteers have positive findings of anterior disc displacement (Haiter-Neto et al. 2002). Advances in MRI may distinguish precise anatomical features and detect pathological processes earlier.
Nuclear medicine techniques that image active metabolic processes show promise for providing early stage clinical assessment of active disease. Positron emission tomography (PET) with the radioisotope 18F-FDG is taken up by metabolically active inflammatory cells (Basu et al. 2009). Combined with MRI, 18F-FDG PET identified increased uptake in periarticular lesions of patients with active symptoms (Nakamura et al. 2007), demonstrating it may be sensitive enough to measure low-grade inflammatory conditions. The tracer fluoride-18 PET is also an option, which is incorporated into the matrix of remodeling bone. Fluoride-18 can detect OA in the TMJ (Lee et al. 2013) and may be a more sensitive alternative to bone scans in detecting arthritic changes in the condyle. TMJ OA is also characterized by hypoxia that disrupts cellular homeostasis in the condyle. Using hypoxia-detecting radioactive tracers like 18F-EF5 and 18F-FMISO, it may be possible to detect active hypoxia in the early stages of TMJ overloading. A pilot study in rats with 18F-EF5 detected increases in uptake, accompanied by increased HIF1α and HIF2α in the chondrocytes of the TMJ after painful loading (Sperry, Moody, et al. 2017), suggesting TMJ hypoxia may be an early indicator of pain and degeneration. Last, single photon emission computed tomography (SPECT) with 99mTc-MDP can detect experimental TMJ OA and is a potentially useful clinical tool for its improved accuracy and resolution over traditional bone scans (Piscaer et al. 2013). Biologic imaging shows active disease before anatomic damage is evident and serves as a novel modality to image inflammation and predict the development of chronic pain associated with degeneration of the TMJ.
Imaging techniques to visualize the CNS have also emerged for understanding mechanisms of pain development and potentially predicting chronic pain (Vachon-Presseau et al. 2016). CNS imaging combined with validated algorithms to identify predictive features of brain activity and connectivity could estimate susceptibility to develop chronic pain based on neuronal hyperexcitability, functional brain networks, or structural information.
Blood oxygen level–dependent functional MRI (BOLD fMRI) is used to investigate changes in brain activity, and it is sensitive enough to identify brain network and subcircuit alterations in chronic pain (Farmer et al. 2012). In TMJ disease, BOLD fMRI measurements suggest that the insular and cingulate cortices are both involved in chronic pain experiences (Ichesco et al. 2012). Changes in gray matter content of the brain and brainstem in TMJ disorder patients also suggest that alterations to the trigeminal and limbic systems occur (Younger et al. 2010; Wilcox et al. 2015). Quantitative arterial spin labeling (qASL) measures blood flow and identifies increases in blood flow to the anterior cingulate cortex and the trigeminal nucleus (Youssef et al. 2014). PET imaging using 18F-FDG can provide complementary information (Riedl et al. 2014) by measuring cellular metabolism concentrated at synapses (Fig. 6) (Stoessl 2017). 18F-FDG PET has identified plastic changes in brain networks in neuropathic pain models (Kim et al. 2014) and between chronic and acute TMJ pain (Sperry et al. 2016). Detailed structural imaging of the brain by diffusion tensor imaging (DTI) (Fig. 6E) could aid in predicting patient outcomes; when combined with functional imaging, it can predict chronic pain development with 80% to 100% accuracy based on preexisting functional and morphologic risk factors (Vachon-Presseau et al. 2016). DTI scans have also identified microstructural differences in the trigeminal nerve, thalamus, and internal capsule in patients with chronic TMJ disorder (Moayedi et al. 2012).
Future Directions for Clinical Management
Managing temporomandibular disorders includes improved prognostic indicators of disease progression, biologic solutions for tissue reconstruction, and disease-modifying medications. Profound clinical success has been obtained using disease-modifying medications in rheumatologic conditions, which highlights potential for disease-modifying OA medications (DMOADs) to treat recalcitrant painful TMJ OA disease (Tonge et al. 2014). A major obstacle with DMOADs is the small but real associated side effects with many cytokine specific medications—in particular, systemic etanercept presents increased risk of infection and/or hematologic cancers, limiting its general use in the TMJ OA population (Bongartz et al. 2006). Intra-articular DMOADs are particularly promising since they focus drug delivery while minimizing systemic exposure. Mesenchymal stem cell therapies show promise for the treatment and reconstruction of the TMJ (Salash et al. 2016).
Summary and Conclusions
TMJ disorders are complex with multiple etiologies, including the most common joint pathology, OA. Best clinical practice for patients with TMJ OA is pain reduction with NSAIDs and/or replacement of the degenerative TMJ. The development and clinical use of directed DMOADs in TMJ OA is extremely limited because multiple molecular pathways are disrupted on different timescales, requiring customized therapies. Animal models that better replicate both the tissue degeneration and pain associated with TMJ OA can help more accurately define the mechanisms and timeline of this disease. In addition, combining techniques to measure affective pain and investigate brain circuits (e.g., optogenetics, electrophysiology, brain network analysis) in animal models of TMJ OA could provide insights into chronic pain maintenance.
Disease-modifying treatments can be challenging to integrate into clinical practice because there are few tools accurately predicting who develops persistent pain and degeneration. CT and MRI, the most commonly used imaging modalities in patients with TMJ pain, primarily detect late-stage disease, and there is frequently poor correspondence between anatomical findings and disease progression. New imaging approaches for the TMJ focus on function over anatomy, with the goal of detecting key features early in disease progression, before permanent structural damage and central sensitization occurs.
Author Contributions
M.M. Sperry, contributed to conception, design, data acquisition, analysis, and interpretation, drafted the manuscript; S. Kartha, contributed to data acquisition and analysis, critically revised the manuscript; B.A. Winkelstein, contributed to conception, design, and data interpretation, critically revised the manuscript; E.J. Granquist, contributed to conception and data interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
Supplemental material, DS_10.1177_0022034519828731 for Experimental Methods to Inform Diagnostic Approaches for Painful TMJ Osteoarthritis by M.M. Sperry, S. Kartha, B.A. Winkelstein and E.J. Granquist in Journal of Dental Research
Acknowledgments
We thank Dr. Ya-Hsin Yu for assistance with collecting RGS data, Rachel Welch for scoring RGS data, and Meagan Ita for the human TMJ specimen. Dr. Granquist is a consultant for Zimmer Biomet.
Footnotes
A supplemental appendix to this article is available online.
This project was funded by the Catherine D. Sharpe Foundation, Oral and Maxillofacial Surgery Foundation, Oral and Maxillofacial Surgery Schoenleber Research Fund from the University of Pennsylvania School of Dental Medicine, and a training grant from NIH/NIAMS (T32-AR007132).
The authors declare no other potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Abubaker A, Raslan W, Sotereanos G. 1993. Estrogen and progesterone receptors in temporomandibular joint discs of symptomatic and asymptomatic persons: a preliminary study. J Oral Maxillofac Surg. 51(10):1096–1100. [DOI] [PubMed] [Google Scholar]
- Ahmad M, Hollender L, Anderson Q, Kartha K, Ohrbach R, Truelove EL, John MT, Schiffman EL. 2009. Research diagnostic criteria for temporomandibular disorders (RDC/TMD): development of image analysis criteria and examiner reliability for image analysis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 107(6):844–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A, Sharawy M. 1994. Histopathological changes in rabbit TMJ associated with experimentally induced anterior disk displacement (ADD). J Oral Pathol Med. 23(8):364–374. [DOI] [PubMed] [Google Scholar]
- Basu S, Chryssikos T, Moghadam-Kia S, Zhuang H, Torigian DA, Alavi A. 2009. Positron emission tomography as a diagnostic tool in infection: present role and future possibilities. Semin Nucl Med. 39(1):36–51. [DOI] [PubMed] [Google Scholar]
- Bereiter DA, Okamoto K, Bereiter DF. 2005. Effect of persistent monoarthritis of the temporomandibular joint region on acute mustard oil-induced excitation of trigeminal subnucleus caudalis neurons in male and female rats. Pain. 117(1–2):58–67. [DOI] [PubMed] [Google Scholar]
- Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. 2006. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA. 295(19):2275–2285. [DOI] [PubMed] [Google Scholar]
- Bouloux GF, Chou J, Krishnan D, Aghaloo T, Kahenasa N, Smith JA, Giannakopoulos H. 2017. Is hyaluronic acid or corticosteroid superior to lactated ringer solution in the short-term reduction of temporomandibular joint pain after arthrocentesis? Part 1. J Oral Maxillofac Surg. 75(1):52–62. [DOI] [PubMed] [Google Scholar]
- Dillon CF, Rasch EK, Gu Q, Hirsch R. 2006. Prevalence of knee osteoarthritis in the United States: arthritis data from the Third National Health and Nutrition Examination Survey 1991–94. J Rheumatol. 33(11):2271–2279. [PubMed] [Google Scholar]
- Durham ZL, Hawkins JL, Durham PL. 2017. Tumor necrosis factor–alpha stimulates cytokine expression and transient sensitization of trigeminal nociceptive neurons. Arch Oral Biol. 75:100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embree MC, Chen M, Pylawka S, Kong D, Iwaoka GM, Kalajzic I, Yao H, Shi C, Sun D, Sheu T, et al. 2016. Exploiting endogenous fibrocartilage stem cells to regenerate cartilage and repair joint injury. Nat Commun. 7(13073):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embree MC, Iwaoka GM, Kong D, Martin BN, Patel RK, Lee AH, Nathan JM, Eisig SB, Safarov A, Koslovsky DA, et al. 2015. Soft tissue ossification and condylar cartilage degeneration following TMJ disc perforation in a rabbit pilot study. Osteoarthritis Cartilage. 23(4):629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer MA, Baliki MN, Apkarian AV. 2012. A dynamic network perspective of chronic pain. Neurosci Lett. 520(2):197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haiter-Neto F, Hollender L, Barclay P, Maravilla KR. 2002. Disk position and the bilaminar zone of the temporomandibular joint in asymptomatic young individuals by magnetic resonance imaging. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 94(3):372–378. [DOI] [PubMed] [Google Scholar]
- Hamilton JL, Nagao M, Levine BR, Chen D, Olsen BR, Im HJ. 2016. Targeting VEGF and its receptors for the treatment of osteoarthritis and associated pain. J Bone Miner Res. 31(5):911–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper DE, Schrepf A, Clauw DJ. 2016. Pain mechanisms and centralized pain in temporomandibular disorders. J Dent Res. 95(10):1102–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring SW. 2003. TMJ anatomy and animal models. J Musculoskelet Neuronal Interact. 3(4):391–394. [PMC free article] [PubMed] [Google Scholar]
- Ichesco E, Quintero A, Clauw DJ, Peltier S, Sundgren PM, Gerstner GE, Schmidt-Wilcke T. 2012. Altered functional connectivity between the insula and the cingulate cortex in patients with temporomandibular disorder: a pilot study. Headache. 52(3):441–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata K, Tashiro A, Tsuboi Y, Imai T, Sumino R, Morimoto T, Dubner R, Ren K. 1999. Medullary dorsal horn neuronal activity in rats with persistent temporomandibular joint and perioral inflammation. J Neurophysiol. 82(3):1244–1253. [DOI] [PubMed] [Google Scholar]
- Kartha S, Zhou T, Granquist EJ, Winkelstein BA. 2016. Development of a rat model of mechanically induced tunable pain and associated temporomandibular joint responses. J Oral Maxillofac Surg. 74(1):54.e1–54e10. [DOI] [PubMed] [Google Scholar]
- Kawai Y, Kubota E, Okabe E. 2000. Reactive oxygen species participation in experimentally induced arthritis of the temporomandibular joint in rats. J Dent Res. 79(7):1489–1495. [DOI] [PubMed] [Google Scholar]
- Kellesarian SV, Al-Kheraif AA, Vohra F, Ghanem A, Malmstrom H, Romanos GE, Javed F. 2016. Cytokine profile in the synovial fluid of patients with temporomandibular joint disorders: a systematic review. Cytokine. 77:98–106. [DOI] [PubMed] [Google Scholar]
- Kido M, Kiyoshima T, Kondo T, Ayasaka N, Moroi R, Terada Y, Tanaka T. 1993. Distribution of Substance P and calcitonin gene-related peptide-like immunoreactive nerve fibers in the rat temporomandibular joint. J Dent Res. 72(3):592–598. [DOI] [PubMed] [Google Scholar]
- Kim CE, Kim YK, Chung G, Jeong JM, Lee DS, Kim J, Kim SJ. 2014. Large-scale plastic changes of the brain network in an animal model of neuropathic pain. Neuroimage. 98:203–215. [DOI] [PubMed] [Google Scholar]
- Kim CH, Lee BJ, Yoon J, Seo KM, Park JH, Lee JW, Cho ES, Hong JJ, Lee YS, Park JH. 2001. Therapeutic effect of hyaluronic acid on experimental osteoarthrosis of ovine temporomandibular joint. J Vet Med Sci. 63(10):1083–1089. [DOI] [PubMed] [Google Scholar]
- Koh KJ, List T, Petersson A, Rohlin M. 2009. Relationship between clinical and magnetic resonance imaging diagnoses and findings in degenerative and inflammatory temporomandibular joint diseases: a systematic literature review. J Orofac Pain. 23(2):123–139. [PubMed] [Google Scholar]
- Kras JV, Kartha S, Winkelstein BA. 2015. Intra-articular nerve growth factor regulates development, but not maintenance, of injury-induced facet joint pain & spinal neuronal hypersensitivity. Osteoarthritis Cartilage. 23(11):1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucyi A, Davis KD. 2017. The neural code for pain: from single-cell electrophysiology to the dynamic pain connectome. Neuroscientist. 23(4):397–414. [DOI] [PubMed] [Google Scholar]
- Kuroki Y, Honda K, Kijima N, Wada T, Arai Y, Matsumoto N, Iwata K, Shirakawa T. 2011. In vivo morphometric analysis of inflammatory condylar changes in rat temporomandibular joint. Oral Dis. 17(5):499–507. [DOI] [PubMed] [Google Scholar]
- Langford DJ, Bailey AL, Chanda ML, Clarke SE, Drummond TE, Echols S, Glick S, Ingrao J, Klassen-Ross T, Lacroix-Fralish ML, et al. 2010. Coding of facial expressions of pain in the laboratory mouse. Nat Methods. 7(6):447–449. [DOI] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. 2009. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 10(9):895–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence RC, Gabriel S, Hirsch R, Kwoh CK, Liang MH, Kremers HM, Mayes MD, Merkel PA, Pillemer SR, Reveille JD, et al. 2008. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Arthritis Rheum. 58(1):15–25. [DOI] [PubMed] [Google Scholar]
- Lee JW, Lee SM, Kim SJ, Choi JW, Baek KW. 2013. Clinical utility of fluoride-18 positron emission tomography/CT in temporomandibular disorder with osteoarthritis: comparisons with 99mTc-MDP bone scan. Dentomaxillofacial Radiol. 42(2):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton JA, Ship JA, Larach-Robinson D. 1993. Estimated prevalence and distribution of reported orofacial pain in the United States. J Am Dent Assoc. 124(10):115–121. [DOI] [PubMed] [Google Scholar]
- Liu F, Steinkeler A. 2013. Epidemiology, diagnosis, and treatment of temporomandibular disorders. Dent Clin North Am. 57(3):465–479. [DOI] [PubMed] [Google Scholar]
- Liu ZJ, Herring SW. 2000. Masticatory strains on osseous and ligamentous components of the temporomandibular joint in miniature pigs. J Orofac Pain. 14(4):265–278. [PubMed] [Google Scholar]
- Loeser RF, Goldring SR, Scanzello CR, Goldring MB. 2012. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 64(6):1697–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma R, Yin S, Li G. 2016. The detection accuracy of cone beam CT for osseous defects of the temporomandibular joint: a systematic review and meta-analysis. Sci Rep. 6(May):34714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malfait AM, Schnitzer TJ. 2013. Towards a mechanism-based approach to pain management in osteoarthritis. Nat Rev Rheumatol. 9(11):654–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RE, Miller RJ, Malfait AM. 2014. Osteoarthritis joint pain: the cytokine connection. Cytokine. 70(2):185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR. 2009. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 10(1):23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mino-Oka A, Izawa T, Shinohara T, Mori H, Yasue A. 2017. Roles of hypoxia inducible factor-1α in the temporomandibular joint. Arch Oral Biol. 73:274–281. [DOI] [PubMed] [Google Scholar]
- Moayedi M, Weissman-Fogel I, Salomons TV, Crawley AP, Goldberg MB, Freeman BV, Tenenbaum HC, Davis KD. 2012. White matter brain and trigeminal nerve abnormalities in temporomandibular disorder. Pain. 153(7):1467–1477. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Masuko K, Yudoh K, Kato T, Nishioka K, Sugihara T, Beppu M. 2007. Positron emission tomography with 18F-FDG in osteoarthritic knee. Osteoarthritis Cartilage. 15(6):673–681. [DOI] [PubMed] [Google Scholar]
- Nicoll SB, Hee CK, Davis MB, Winkelstein BA. 2010. A rat model of temporomandibular joint pain with histopathologic modifications. J Orofac Pain. 24(3):298–304. [PubMed] [Google Scholar]
- National Institute of Dental and Craniofacial Research. 2014. Facial pain [accessed 2019 Jan 10]. https://www.nidcr.nih.gov/research/data-statistics/facial-pain.
- Okamoto K, Hirata H, Takeshita S, Bereiter DA. 2003. Response properties of TMJ units in superficial laminae at the spinomedullary junction of female rats vary over the estrous cycle. J Neurophysiol. 89(3):1467–1477. [DOI] [PubMed] [Google Scholar]
- Okeson J. 2004. Bell’s orofacial pains: the clinical management of orofacial pain. 6th ed. New Malden, UK: Quintessence Publishing Co Ltd. [Google Scholar]
- Okeson J. 2018. Critical commentary 1: reliability and validity of the DC/TMD axis I. J Oral Facial Pain Headache. 32(1):19–21. [DOI] [PubMed] [Google Scholar]
- Philips B, Weisshaar C, Winkelstein B. 2016. The rat grimace scale can evaluate spontaneous neuropathic pain in a model of cervical radiculopathy. Comp Med. 67(1):34–42. [PMC free article] [PubMed] [Google Scholar]
- Piscaer TM, Sandker M, van der Jagt OP, Verhaar JA, de Jong M, Weinans H. 2013. Real-time assessment of bone metabolism in small animal models for osteoarthritis using multi pinhole-SPECT/CT. Osteoarthritis Cartilage. 21(6):882–888. [DOI] [PubMed] [Google Scholar]
- Puri J, Vinothini P, Reuben J, Bellinger LL, Ailing L, Peng YB, Kramer PR. 2012. Reduced GABA-(A) receptor α6 expression in the trigeminal ganglion alters inflammatory TMJ hypersensitivity. Neuroscience. 213:179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl V, Bienkowska K, Strobel C, Tahmasian M, Grimmer T, Forster S, Friston KJ, Sorg C, Drzezga A. 2014. Local activity determines functional connectivity in the resting human brain: a simultaneous FDG-PET/fMRI study. J Neurosci. 34(18):6260–6266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen S, Ham B, Mogil JS. 2017. Sex differences in neuroimmunity and pain. J Neurosci Res. 95(1–2):500–508. [DOI] [PubMed] [Google Scholar]
- Salash JR, Hossameldin RH, Almarza AJ, Chou JC, McCain JP, Mercuri LG, Wolford LM, Detamore MS. 2016. Potential indications for tissue engineering in temporomandibular joint surgery. J Oral Maxillofac Surg. 74(4):705–711. [DOI] [PubMed] [Google Scholar]
- Schiffman E, Ohrbach R, Truelove E, Look J, Anderson G, Goulet JP, List T, Svensson P, Gonzalez Y, Lobbezoo F, et al. ; International RDC/TMD Consortium Network, International Association for Dental Research; Orofacial Pain Special Interest Group, International Association for the Study of Pain. 2014. Diagnostic Criteria for Temporomandibular Disorders (DC/TMD) for Clinical and Research Applications: recommendations of the International RDC/TMD Consortium Network and Orofacial Pain Special Interest Group. J Oral Facial Pain Headache. 28(1):6–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrivani SJ, Keith DA, Kaban LB. 2008. Temporomandibular disorders. N Engl J Med. 359(25):2693–2705. [DOI] [PubMed] [Google Scholar]
- Sekiguchi K, Takehana S, Shibuya E, Matsuzawa N, Hidaka S, Kanai Y, Inoue M, Kubota Y, Shimazu Y, Takeda M. 2016. Resveratrol attenuates inflammation-induced hyperexcitability of trigeminal spinal nucleus caudalis neurons associated with hyperalgesia in rats. Mol Pain. 12: 1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessle BJ. 1999. The neural basis of temporomandibular joint and masticatory muscle pain. J Orofac Pain. 13(4):238–245. [PubMed] [Google Scholar]
- Sessle BJ. 2011. Peripheral and central mechanisms of orofacial inflammatory pain. Int Rev Neurobiol. 97:179–206. [DOI] [PubMed] [Google Scholar]
- Shen P, Jiao Z, Zheng JS, Xu WF, Zhang SY, Qin A, Yang C. 2015. Injecting vascular endothelial growth factor into the temporomandibular joint induces osteoarthritis in mice. Sci Rep. 5:16244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakura M, Tanimoto K, Eguchi H, Miyauchi M, Nakamura H, Hiyama K, Tanimoto K, Tanaka E, Takata T, Tanne K. 2010. Activation of the hypoxia-inducible factor-1 in overloaded temporomandibular joint, and induction of osteoclastogenesis. Biochem Biophys Res Commun. 393(4):800–805. [DOI] [PubMed] [Google Scholar]
- Sotocinal SG, Sorge RE, Zaloum A, Tuttle AH, Martin LJ, Wieskopf JS, Mapplebeck JC, Wei P, Zhan S, Zhang S, et al. 2011. The Rat Grimace Scale: a partially automated method for quantifying pain in the laboratory rat via facial expressions. Mol Pain. 7:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperry M, Moody V, Frank J, Granquist E, Winkelstein B. 2017. 18F-EF5 PET imaging can detect hypoxia in TMJs that later exhibit cartilage degradation. Abstract presented at: 2017 Biomedical Engineering Society Annual Meeting; 2017 Oct 11-14; Phoenix, AZ. [Google Scholar]
- Sperry MM, Ita ME, Kartha S, Zhang S, Yu Y-H, Winkelstein B. 2017. The interface of mechanics and nociception in joint pathophysiology: insights from the facet and temporomandibular joints. J Biomech Eng. 139(2):021003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperry MM, Kartha S, Granquist EJ, Winkelstein BA. 2016. Meso-scale reorganization of metabolic brain networks is associated with persistent TMJ pain. Abstract presented at: Summer Biomechanics, Bioengineering and Biotransport Conference 2016; 2016 Jun 29-Jul 2 National Harbor, MD. [Google Scholar]
- Sperry MM, Yu YH, Welch RL, Granquist EJ, Winkelstein BA. 2018. Grading facial expression is a sensitive means to detect grimace differences in orofacial pain in a rat model. Sci Rep. 8(1):13894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spisák T, Pozsgay Z, Aranyi C, Dávid S, Kocsis P, Nyitrai G, Gajári D, Emri M, Czurkó A, Kincses ZT. 2016. Central sensitization-related changes of effective and functional connectivity in the rat inflammatory trigeminal pain model. Neuroscience. 344:133–147. [DOI] [PubMed] [Google Scholar]
- Steenks M, Türp J, de Wijer A. 2018. Reliability and validity of the diagnostic criteria for temporomandibular disorders axis I in clinical and research settings: a critical appraisal. J Oral Facial Pain Headache. 32(1):7–18. [DOI] [PubMed] [Google Scholar]
- Stoessl AJ. 2017. Glucose utilization: still in the synapse. Nat Neurosci. 20(3):382–384. [DOI] [PubMed] [Google Scholar]
- Ta LE, Dionne RA. 2004. Treatment of painful temporomandibular joints with a cyclooxygenase-2 inhibitor: a randomized placebo-controlled comparison of celecoxib to naproxen. Pain. 111(1–2):13–21. [DOI] [PubMed] [Google Scholar]
- Takeda M, Tanimoto T, Kadoi J, Nasu M, Takahashi M, Kitagawa J, Matsumoto S. 2007. Enhanced excitability of nociceptive trigeminal ganglion neurons by satellite glial cytokine following peripheral inflammation. Pain. 129(1–2):155–166. [DOI] [PubMed] [Google Scholar]
- Takeshita S, Hirata H, Bereiter DA. 2001. Intensity coding by TMJ-responsive neurons in superficial laminae of caudal medullary dorsal horn of the rat. J Neurophysiol. 86(5):2393–2404. [DOI] [PubMed] [Google Scholar]
- Tanaka E, Detamore MS, Mercuri LG. 2008. Degenerative disorders of the temporomandibular joint: etiology, diagnosis, and treatment. J Dent Res. 87(4):296–307. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Okamoto K, Bereiter DA. 2009. Chronic inflammation and estradiol interact through MAPK activation to affect TMJ nociceptive processing by trigeminal caudalis neurons. Neuroscience. 164(4):1813–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonge DP, Pearson MJ, Jones SW. 2014. The hallmarks of osteoarthritis and the potential to develop personalised disease-modifying pharmacological therapeutics. Osteoarthritis Cartilage. 22(5):609–621. [DOI] [PubMed] [Google Scholar]
- Upadhyay J, Knudsen J, Anderson J, Becerra L, Borsook D. 2008. Noninvasive mapping of human trigeminal brainstem pathways. Magn Reson Med. 60(5):1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utreja A, Dyment NA, Yadav S, Villa MM, Li Y, Jiang X, Nanda R, Rowe DW. 2016. Cell and matrix response of temporomandibular cartilage to mechanical loading. Osteoarthritis Cartilage. 24(2):335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachon-Presseau E, Centeno MV, Ren W, Berger SE, Tetreault P, Ghantous M, Baria A, Farmer M, Baliki MN, Schnitzer TJ, et al. 2016. The emotional brain as a predictor and amplifier of chronic pain. J Dent Res. 95(6):605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vapniarsky N, Huwe LW, Arzi B, Houghton MK, Wong ME, Wilson JW, Hatcher DC, Hu JC, Athanasiou KA. 2018. Tissue engineering toward temporomandibular joint disc regeneration. Sci Transl Med. 10(446):1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa G, Ceruti S, Zanardelli M, Magni G, Jasmin L, Ohara PT, Abbracchio MP. 2010. Temporomandibular joint inflammation activates glial and immune cells in both the trigeminal ganglia and in the spinal trigeminal nucleus. Mol Pain. 6:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XD, Kou XX, Mao JJ, Gan YH, Zhou YH. 2012. Sustained inflammation induces degeneration of the temporomandibular joint. J Dent Res. 91(5):499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XD, Zhang JN, Gan YH, Zhou YH. 2015. Current understanding of pathogenesis and treatment of TMJ osteoarthritis. J Dent Res. 94(5):666–673. [DOI] [PubMed] [Google Scholar]
- Wilcox SL, Gustin SM, Macey PM, Peck CC, Murray GM, Henderson LA. 2015. Anatomical changes within the medullary dorsal horn in chronic temporomandibular disorder pain. Neuroimage. 117:258–266. [DOI] [PubMed] [Google Scholar]
- Wolford LM, Mercuri LG, Schneiderman ED, Movahed R, Allen W. 2015. Twenty-year follow-up study on a patient-fitted temporomandibular joint prosthesis: the techmedica/TMJ concepts device. J Oral Maxillofac Surg. 73(5):952–960. [DOI] [PubMed] [Google Scholar]
- Wu L, Huang X, Li L, Huang H, Xu R, Luyten W. 2012. Insights on biology and pathology of HIF-1α/-2α, TGFβ/BMP, Wnt/β-catenin, and NF-κB pathways in osteoarthritis. Curr Pharm Des. 18(22):3293–3312. [DOI] [PubMed] [Google Scholar]
- Yang S, Kim J, Ryu J-H, Oh H, Chun C-H, Kim BJ, Min BH, Chun J-S JS. 2010. Hypoxia-inducible factor-2α is a catabolic regulator of osteoarthritic cartilage destruction. Nat Med. 16(6):687–693. [DOI] [PubMed] [Google Scholar]
- Younger JW, Shen YF, Goddard G, Mackey SC. 2010. Chronic myofascial temporomandibular pain is associated with neural abnormalities in the trigeminal and limbic systems. Pain. 149(2):222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef AM, Gustin SM, Nash PG, Reeves JM, Petersen ET, Peck CC, Murray GM, Henderson LA. 2014. Differential brain activity in subjects with painful trigeminal neuropathy and painful temporomandibular disorder. Pain. 155(3):467–475. [DOI] [PubMed] [Google Scholar]
- Zhang C, Lin S, Li T, Jiang Y, Huang Z, Wen J, Cheng W, Li H. 2017. Mechanical force-mediated pathological cartilage thinning is regulated by necroptosis and apoptosis. Osteoarthritis Cartilage. 25(8):1324–1334. [DOI] [PubMed] [Google Scholar]
- Zhang C, Xu Y, Cheng Y, Wu T, Li H. 2015. Effect of asymmetric force on the condylar cartilage, subchondral bone and collagens in the temporomandibular joints. Arch Oral Biol. 60(4):650–663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, DS_10.1177_0022034519828731 for Experimental Methods to Inform Diagnostic Approaches for Painful TMJ Osteoarthritis by M.M. Sperry, S. Kartha, B.A. Winkelstein and E.J. Granquist in Journal of Dental Research