

A 62-year-old Caucasian man was hospitalized after he presented to an ophthalmologist with 5 months of painless vision loss in his left eye and was found to have a 2+ afferent pupillary defect oculus sinister (OS, left eye), 20/50 visual acuity OS, and severe pallor of the temporal retina OS. Magnetic resonance imaging of the brain showed diffuse hypertrophic pachymeningitis (HP) with enhancing, expansile soft tissue infiltration of the medial wall of middle cranial fossa bilaterally (Figure 1A, B). Routine labs showed an elevated creatinine of 1.9 mg/dL, with computed tomography of the abdomen showing retroperitoneal fibrosis (RPF) encasing the right ureter, causing hydronephrosis. IgG4-related disease (IgG4-RD) was suspected and serum IgG4 was found elevated at 108 mg/dL. A meningeal biopsy was performed demonstrating diffuse lymphoplasmacytic infiltrate with >30 IgG4+ cells/hpf and a IgG4+/IgG ratio of >50% (Figure 1C, D). While diffuse fibrosis was present, its organization into the characteristic irregularly whorled (storiform) pattern was not seen (often subject to sampling errors, especially with needle biopsies).1 Obliterative phlebitis is uncommon in neuro-ophthalmologic disease but almost always present with pancreatic/submandibular gland disease.1 Clearly defined diagnostic criteria remain lacking, with consensus guidelines recommending that clinical (subacute progression), radiographic (HP, hypophysitis), serological (elevated serum IgG4), and pathological evidence be taken together, as no finding is individually pathognomonic.1 He was started on the recommended oral prednisone regimen (induction at 0.6 mg/kg/d for 2-4 weeks, followed by a 3 to 6 month maintenance period and a slow taper over 3 years).2 Follow-up examination after 2 months showed improvement in both visual acuity (20/25 OS) and the HP (Figure 1E, F); however, optical coherence tomography not only showed the expected thinning of the left temporal nerve fiber layer but also showed neuronal dropout in the right temporal retina (Figure 2I, J). This prompted the initiation of rituximab. IgG4-RD has rapidly become a common entity in a neurohospitalist’s differential diagnosis for middle-aged men (male:female ratio ∼ 3:1) in their fifth to seventh decade of life presenting with subacute symptoms of a mass lesion or atopy.3 Constitutional symptoms (fevers, malaise, weight loss and night sweats) can be present.3 IgG4-related disease has now been described in every organ except for the spinal cord parenchyma, dorsal root ganglia, brachial–lumbosacral plexi, and skeletal muscles outside the head.3 It has become the commonest cause of nonmalignant HP and RPF.3 The incidence of neurologic IgG4-RD in order of frequency is HP > hypophysitis > peripheral neuropathy > brain parenchymal disease.3 IgG4-related disease is a multisystem disorder; however, meningeal and hypophyseal involvement often occur as a single-organ disease.3 Positron emission tomography scan can identify the extra central nervous system disease burden to guide an optimal biopsy site and thus avoid a meningeal/brain parenchymal biopsy.3 The first line of treatment is oral prednisone; however, given the concern for subclinical neuronal injury, current expert recommendations are to initiate rituximab right from the start (2 doses of 1 g intravenously, separated by 2 weeks and repeated at 6-month intervals) with follow-up neuroimaging at 3 months.3
Figure 1.
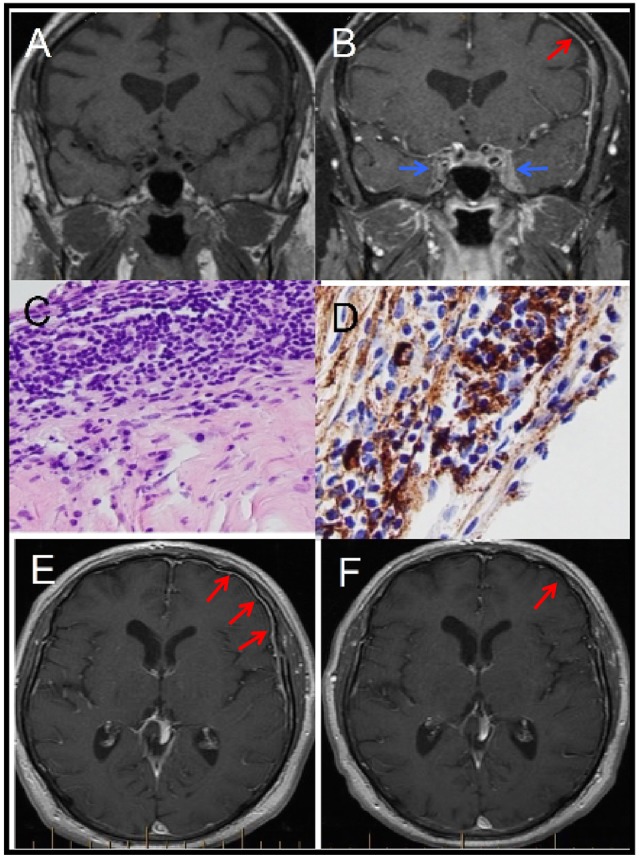
Magnetic resonance imaging of the brain pre (A) and post (B) gadolinium showing diffuse hypertrophic pachymeningitis (HP; red arrow) with expansile soft tissue infiltration of the cavernous sinuses (blue arrow) bilaterally. Meningeal biopsy showing diffuse lymphoplasmacytic infiltrate (C) and >30 IgG4+ cells/hpf with an IgG4+/IgG ratio of >50% (D). Postcontrast axial T1 sequences taken before (E) and 2 months after (F) steroid treatment showing interval reduction of HP.
Figure 2.

Magnetic resonance imaging orbits axial (A) and coronal (B) sections showing enhancement of the left optic nerve with Humphrey visual fields (C) at baseline (January) and on presentation (August). Optical coherence tomography (D) performed 2 months after steroid initiation showing thinning of the left temporal nerve fiber layer OS (39 um) specifically involving the papillomacular bundle with neuronal dropout occurring even in the right temporal retina (asterisk).
Footnotes
Authors’ Note: Karan Topiwala contributed to draft of the manuscript, acquisition of data, and primary clinical care of the patient. Christopher Hampton contributed to draft of the manuscript and primary clinical care of the patient. Carl Boland contributed to review of the manuscript and primary clinical care of the patient. David Waitzman contributed to review of the manuscript and primary clinical care of the patient.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25(9):1181–1192. [DOI] [PubMed] [Google Scholar]
- 2. Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366(6):539–551. [DOI] [PubMed] [Google Scholar]
- 3. AbdelRazek MA, Venna N, Stone JH. IgG4-related disease of the central and peripheral nervous systems. Lancet Neurol. 2018;17(2):183–92. [DOI] [PubMed] [Google Scholar]