

Abstract
Background
Long non-coding RNAs (lncRNAs) play an important role in regulating gene expression and are thus important for determining phenotypes. Most attempts to measure selection in lncRNAs have focused on the primary sequence. The majority of small RNAs and at least some parts of lncRNAs must fold into specific structures to perform their biological function. Comprehensive assessments of selection acting on RNAs therefore must also encompass structure. Selection pressures acting on the structure of non-coding genes can be detected within multiple sequence alignments. Approaches of this type, however, have so far focused on negative selection. Thus, a computational method for identifying ncRNAs under positive selection is needed.
Results
We introduce the SSS-test (test for Selection on Secondary Structure) to identify positive selection and thus adaptive evolution. Benchmarks with biological as well as synthetic controls yield coherent signals for both negative and positive selection, demonstrating the functionality of the test. A survey of a lncRNA collection comprising 15,443 families resulted in 110 candidates that appear to be under positive selection in human. In 26 lncRNAs that have been associated with psychiatric disorders we identified local structures that have signs of positive selection in the human lineage.
Conclusions
It is feasible to assay positive selection acting on RNA secondary structures on a genome-wide scale. The detection of human-specific positive selection in lncRNAs associated with cognitive disorder provides a set of candidate genes for further experimental testing and may provide insights into the evolution of cognitive abilities in humans.
Availability
The SSS-test and related software is available at: https://github.com/waltercostamb/SSS-test. The databases used in this work are available at: http://www.bioinf.uni-leipzig.de/Software/SSS-test/.
Electronic supplementary material
The online version of this article (10.1186/s12859-019-2711-y) contains supplementary material, which is available to authorized users.
Keywords: Long non-coding RNA, Positive selection, RNA secondary structure, Psychiatric disorders, Primate genomes
Background
More than a decade of high-throughput transcriptomics has established wide-spread, pervasive transcription of mammalian genomes as an indisputable fact [1–5]. However, less than a quarter of the total RNA (excluding ribosomal RNAs) accounts for the about 19 000 protein coding genes and their isoforms [6]. The majority of the human transcriptome, in terms of diversity of the products, is composed of other, non-protein-coding, RNAs. These include small non-coding RNAs (ncRNAs) accounting for nearly the same genomic coverage as ORFs [7, 8], mRNA-like long non-coding RNAs (lncRNAs), as well as giant macroRNAs [5, 8, 9]. The current rather conservative estimate predicts 40 000 to 50 000 human lncRNA genes [10].
Although lncRNAs comprise a substantial fraction of the transcriptome, so far only a small minority of them has been assigned a functional annotation. The question thus remains, what fraction of the detectable lncRNAs actually convey biological functions, as opposed to being coherently transcribed and processed byproducts without biological relevance (“junk RNA”). Without experimental testing this question is currently difficult to answer because, in contrast to their protein-coding counterparts, most lncRNAs exhibit only low levels of sequence conservation. From a population genetics point of view, this relatively low level of sequence conservation is interpreted as lack of functional constraints or negative selection [11, 12]. However, as a group, lncRNAs do show signs of negative selection: for example, the cumulative distributions of substitution and transversion rates shows significantly suppressed values relative to neutrally evolving DNA [13], see also [2, 12, 14–17]. Furthermore, while the overall sequence conservation is low, gene structure and splice sites often seem to be highly conserved [18, 19], strongly suggesting that many lncRNAs are evolutionarily old [20–23]. Recent studies also found that lncRNAs are often located in syntenic positions and display similar expression patterns across species [24–26].
In this contribution we are concerned with the identification and quantification of selective pressures on RNA secondary structures. This is by no means a novel topic. From a population genetics point of view, two locus models have been used to study compensatory mutations, i.e., negative and stabilizing selection on RNA structures [27–29]. This line of studies showed that tRNAs are among the molecules with strongest selective pressures [30] and confirmed the influence of the effective population size as a cause of differences in selective constraints on tRNAs across species [31]. Altogether, thousands of well-studied small ncRNAs, mostly compiled in the Rfam [32] and miRBase [33] database, exhibit well-conserved, often nearly immutable, RNA secondary structures that are crucial for the function of the RNA molecule.
It is important to distinguish between the presence of conserved RNA secondary structure and signatures of selection on secondary structure. A conserved structure implies that only small deviations around a well-defined consensus structure are tolerated. In this case one expects sequence variation to have generated a sufficient number of compensatory mutations to test directly for the preservation of the consensus structure. R-scape [34] implements such a method. LncRNAs rarely, if ever, exhibit evidence for this level of structural conservation [34]. This seems to be limited to the “classical” families of small, structured ncRNAs and structured regulatory elements, i.e., the content of Rfam – and even these RNAs at times may show extensive structural variation, see e.g. [35, 36] and the references therein.
Negative selection on secondary structure, on the other hand, is a much less stringent property, and it suffices to show that structural variation is more constrained than what would be expected from the observed, underlying sequence variation. This idea has been used in a series of tools including qrna [37], AlifoldZ [38], EvoFold [39], CMfinder [40], RNAz [41], and SISSIz [42]. Extensive surveys [41, 43–47] of mammalian genomes already compiled evidence that a sizeable fraction of the human genome, possibly as much as 10% of the non-repetitive sequence, is under negative selection on RNA secondary structures. Intriguingly, these studies show that the genomic sequence from which a ncRNA is transcribed often evolves very rapidly, while still showing clear signs of selective constraints on local RNA secondary structures.
While the majority of the human lncRNA sequences evolve at average rates close to the unconstrained background, they show strong evidence for conservation of their gene structure, i.e., the preservation of splice sites, across the eutheria or even deeper phylogenetic groups [22]. It is not surprising therefore, that constrained structural modules have also been reported for some of the best studied examples [48–52]. The selective constraints are not strong enough, however, to enforce large, well-conserved consensus structures [34]. Further evidence for the importance of secondary structure features for the function of lncRNAs comes from disease-related SNPs [53, 54].
In contrast to negative selection on RNA secondary structure, very little is known about positive selection in this context beyond a single, well-studied example that might have a positively selected structure: Rapid, lineage specific changes of the sequence have been reported for the Human Accelerated Region 1 (HAR1). 18 human specific single-nucleotide substitutions in an element that is extremely conserved across non-human mammals make HAR1 the fastest evolving region in the human genome [55]. Interestingly, the human HAR1 forms a stable structure, which differs significantly from the chimpanzee structure of HAR1 [56–58]. Expression patterns of HAR1 suggest that it is involved in the development of the cortex [55]. However, real functional data for HAR1 is still missing. In particular, there is still no direct experimental proof that the function of HAR1 depends on its secondary structure. It is conceivable, therefore, that HAR1 acts based on its sequence, and that this function was human specifically lost. Nevertheless, examples such as HAR1 suggest that lncRNAs can potentially play an important role in species evolution.
To-date, no method is available to detect positive selection on RNA secondary structure in a systematic manner. It is an open question therefore, whether this is a rare phenomenon or whether positive selection on RNA structure is an important contributor to lineage-specific adaptation. In principle, positive selection can be identified by comparing the observed divergence with the expectation for neutral evolution. The Ka/Ks test for coding sequences may serve as the paradigmatic example. The ratio of Ka, the number of non-synonymous substitutions per non-synonymous sites, and Ks, the number of synonymous substitutions per synonymous sites is expected to be larger than 1 when positive selection is acting (see e.g. [59]). This idea, however, does not generalize to non-coding RNAs since there is no analogous distinction between synonymous and non-synonymous substitutions.
An interesting alternative approach is to contrast more generic parameters of divergence and diversity between a functional element and a reference locus in its genomic vicinity. Plausible parameters are e.g. ρ, the fraction of sites under selection, the polymorphism rate λ, and the divergence rate η – in each case normalized by the corresponding parameter in the neutral control [60, 61]. These measures have been applied mostly at the level of groups of loci, which showed strong evidence that regulatory elements are influenced by selective pressures [62, 63].
As for any type of test for selection, an estimate of an effect on the phenotype is desired. As reviewed in [64], many tools have been developed in the past years contributing to unravelling the molecular mechanisms underlying complex phenotypes. Still, the effect of, say indels (insertions or deletions) and structural variation, remain elusive. If secondary structure is important for the function of a ncRNA, a predicted structural change can be taken as proxy for a phenotypic impact. The accumulation of substitutions that change the structure can be interpreted as signs of positive selection, or adaptive evolution.
Several methods have been proposed to quantify the effect of SNPs on RNA structures [65, 66]. Leveraging these methods, we propose here to use an excess of structure changing substitutions as a means of identifying positive selection. Conversely, an excess of substitutions that change the structure less than expected supports negative selection. We use this simple idea, while also accounting for the structural impact of insertions and deletions, to develop a statistical test for lineage-specific positive selection, the SSS-test (“Selection on the Secondary Structure test”). We then use this approach to identify candidate lncRNAs that might have been positively selected on the human lineage relative to their primate background. Among them are genes linked to psychiatric disorders (PDs) to provide further candidates that might have been involved in the evolution of the human brain.
Theory
SSS-test
The basic idea of the SSS-test is to determine whether selection pressures have changed in a particular lineage. The starting point for that is a multiple sequence alignment of orthologous sequences of the RNA gene or element taken from a set of species under consideration. The SSS-test singles out one species, and hence one focal sequence , and checks whether there is evidence of a change in the selection pressures acting on x compared to rest of the alignment . In order to assess how x is different, we consider the input alignment with the focal sequence removed. This alignment , serves as the background. Since the effect of variations can only be computed for individual sequences, we will also need the consensus sequences z of and of . Note that , like , depends on the focal sequence x. We do not indicate this dependence explicitly in the notation since it is clear from the context throughout. The idea of the SSS-test is to determine whether the effect of the individual changes leading from the background consensus to the focal sequence change the secondary structure of more than expected. To this end, we need to identify those sequence changes that set the focal sequence x apart from the background and its consensus .
Since we are interested in testing for lineage-specific positive selection, we consider only sites (alignment columns) i that are well-conserved in the background . In other words, we need to exclude highly variable sites, because these convey no accessible information on the differences between background and focal sequence x. To be considered a well-conserved site, we require that a majority of the sequences in conform to the consensus sequence . As a default, we apply the majority rule and require 60% of the sequences to agree with the consensus. This threshold can be changed by the user.
Given both x and , we determine the set of sites with differences between the consensus and the focal sequence and denote this set of sites by . For this purpose we consider gap characters like regular characters, i.e., also contains insertions and deletions in x relative to the consensus . We denote by the sequence that is equal to except at the single variable site i, where it matches x. Substitutions and indels are scored separately.
An insertion or deletion is treated as a single event independently of its length ℓ. The decision to handle gaps as a single unit was first based on the assumption that a unique evolutionary event is more likely to have caused the indel than two or more events acting on the exact same region. We tested this assumption, by measuring the structural impact of deletions of different lengths in biological RNAs. We found that the length of the deletion did not matter for the impact, but rather its location, specifically if it overlaps a paired region or not (more information in the Additional file 1). This is a consequence of the Turner model [67]: the energy penalty for the different loop types (hairpin, bulge, and interior loops) on slowly changes with the loop length, amounting to only about 1–3 kcal/mol between loop sizes of 3–30 nt.
Compensatory substitutions are those that leave the secondary structure unchanged by replacing one type of base pair (GC, AU, or GU) by another one that differs in one or both paired sites (e.g. AU →GC, or AU →GU). The SSS-test by construction considers single sites separately. Therefore we remove all sites that form compensatory substitutions. In order to identify these, we first compute the consensus structure of using RNAalifold [68] and the structure of sequence x with RNAfold (both tools from the ViennaRNA package [69]). We then compare both structures and consider a substitution or pair of substitutions as compensatory if they form a base pair both in the focal MFE structure of x and the consensus MFE structure of the background alignment . All compensatory sites are removed from .
All single nucleotide substitutions remaining in are scored using RNAsnp [65]. In a nutshell, RNAsnp quantifies the magnitude of structural change in response to a substitution relative to the expected change of secondary structure. The expectation is computed from the same base exchange in random sequences with the same length and GC content. For a given SNP, RNAsnp then returns a p-values for the hypothesis that the structural change caused by the SNP is larger than expected. Small RNAsnpp-values therefore indicate unexpectedly large structural changes in the structure of interest. RNAsnp is conceptually similar to several other tools to evaluate variation of RNA secondary structure, e.g. corRna [70], RNAmute [71], RDMAS [72], or SNPfold [73]. We employed RNAsnp both for its computational efficiency and several features that make its underlying model more realistic. The tool evaluates the Boltzmann ensemble of secondary structures rather than only the minimum energy structure, which provides more accurate information on the structural changes [65]. Instead of using arbitrary sequence windows or simply the global fold of the entire RNA, RNAsnp identifies the region of maximal structural discrepancy and evaluates the changes for this region. This at least approximates the fact that the structural impact of SNPs is expected to be localized e.g. due to proteins bound to a lncRNA.
Since each variation is scored independently, p-values are corrected for multiple testing using the Benjamini-Hochberg [74] procedure (with the more conservative Bonferroni method [75] available as well). The Benjamini-Hochberg procedure performs well with a larger number of p-values, which individually are ≥0.05, as happens quite often in our case with RNAsnp-based p-values. For the correction let p=p1≥p2≥⋯≥pn be the collection of p-values. We then update the corrected set of p-values using:
We then use the to produce the substitution score
| 1 |
measuring the impact of the observed substitution in the focal sequence x relative to the expected changes of the secondary structure.
The RNAsnp tool cannot be used for insertions and deletions since its internal model for evaluating p-values is not designed for this type of variations. We therefore developed a separate model to score indels: for an indel of length ℓ we construct all sequences zj that carry the indel after position j of the consensus. Since zj and differ in length, they cannot have the same structure. We therefore compute a modified reference structure ψj by constraining zj to contain all base pairs of the consensus sequence that are not affected by the indel. To this end we use the option of the ViennaRNA package to fold RNA sequences with user-defined constraints [76]. For comparison we compute the fold ϕj of zj without constraints. To determine the structural impact of the indel we compute the structural difference δ(ϕj,ψj) of ϕj and ψj using RNAforester [77].
We then use a combination of rank statistics and relative structural impact to determine a p-value for the structural impact of indel j: let r(j) be the rank of indel j w.r.t. the size of its structural impact in decreasing order. Then prank=r(j)/n, where n is the number of possible indel ranks. In addition we score the relative structural impact by pstruc=(4l−δ(ϕj,ψj))/4l, with l the length of the sequence and pstruc clamped to 1/4l for extreme δ(·,·) contributions. The complete indel p-values (p=prank+pstruc) are aggregated as described above and yield a corresponding indel score contribution s′(x).
Finally, substitution (s(x)) and indel (s′(x)) scores are added to yield the final SSS-score score for the focal sequence x, using:
| 2 |
As discussed in the previous section, both the assessment of nucleotide changes, and the evaluation of insertions and deletions has heuristic elements. Nevertheless, the nucleotide change model is grounded in a well-established model. Both s(x) and s′(x) are scores that convey information on how unexpectedly large the effect of the observed variations is on the secondary structures. They do not lend themselves to a direct interpretation e.g. as probabilities w.r.t. to a particular probabilistic model. Future versions of the SSS-test thus may well use an improved scoring model for either contribution. Similarly, the weighting factor of 2 in Eq. 2 was empirically determined to improve over equal weights. The scores SSS-score(x) thus serve as test statistics for which relevant cutoffs have to be determined empirically, since a concise statistical model for them is not available.
Manual analysis of families with different scores ranging from 0.0 to 30.0 showed that SSS-score≥10.0 is a suitable threshold determining that an element is under positive selection. For additional details we refer to the Additional file 1. The choice of the weighting factor in Eq. 2 as well as the threshold for SSS-score and the threshold of the family divergence, may vary for different applications. The values observed in this work are valid for primates, i.e., a set of phylogenetically very closely related taxa. For different projects, with more distant or more closely related species, the threshold may need to be adapted to best fit the data. In addition, the candidates should be subjected to functional testing for confirmation of the predictions.
Implementation
The computation of selection scores is implemented in an automated pipeline using Perl and bash scripts (see pseudocode). In our implementation, the test statistic SSS-score is computed for all focal sequences . If the input sequences are not aligned, muscle [78] is used to generate the necessary alignment. Additionally, species distance scores, ds, are computed for each sequence of the alignment to indicate the structural distance of the species to the consensus. The median species distance score is the family divergence score, d, which indicates the family’s structural uniformity (more details in the Additional file 1). However, as for the threshold on the selection score, the user should decide on a meaningful cutoff for the investigated data set.

Alternative approaches
We have also considered alternative ways to score the structural variations. The simplest model classified the substitutions into disruptive and non-disruptive sites based on their classification by RNAsnp. Based on this classification, an equivalent of the Ka/Ks test becomes applicable (more information on the Additional file 1). Usually the number of sites is small so that the power of the test is low, however. In addition, false positive results were often seen in manually checked families, for structures that were extremely similar to their orthologs.
This inconsistency most likely came from categorizing sites only into two categories, either disruptive or non-disruptive, which is very difficult to do for ncRNA structures due to their biochemical properties. As an improvement we also built a statistical model based on a Poisson distribution of the counts of synonymous and non-synonymous sites instead of directly comparing the substitution counts. Although more robust, inconsistencies remained, most likely due to the same problem of using only a binary categorization of sites into either synonymous or non-synonymous. This led to the conclusion that an equivalent of the Ka/Ks test is not appropriate for ncRNAs.
Family divergence
We were most interested in identifying RNA structures that were conserved over a long period of time but showed lineage specific changes. Considering only structures with such conservation ensures to a large degree that the structure is biologically relevant. The SSS-test thus measures the structural divergence within a family of othologs, denoted as d.
Given the alignment of a set of species, we denote by As the basepair probability matrix for the aligned sequence , and by B the basepair probability matrix of the alignment itself. Furthermore, Ps is the set of base pairs in s, while Q is the set of base pairs in the consensus. Then we can calculate the derived sets Ws=Ps∩Q of shared base pairs, Xs=Ps∖Q of unique base pairs, and Ys=Q∖Ps of absent base pairs for each sequence s.
Using these, we can now calculate the divergence of each sequence s compared to its family:
| 3 |
We then calculate the family divergence as the median over the individual divergence scores d=medians ds. Manual analysis of families with different d scores (ranging from 0.0 to 65.0) revealed that (d≤10.0) is a suitable threshold indicating low family divergence (additional details are provided in the Additional file 1).
Results
Benchmarking
Biological controls: SSS-scores indicating negative selection for small ncRNAs and positive selection for human HAR1
As a plausibility check for the SSS-test we used collections of small ncRNAs, which are known to be structurally conserved [14, 61]: miRNAs, snoRNAs, and tRNAs (family conservation overview can be seen in Additional file 1: Figures S8 and S9). These collections of ncRNAs were expected to receive low selection scores, indicating negative selection. As expected, all three groups showed strong evidence for negative selection, while pseudo-tRNAs exhibit the least constraint (Additional file 1: Figure S10).
Despite the lack of direct experimental evidence for HAR1 functioning based on its structure, we also applied our test to HAR1 as the only, at least putative example of a positively selected structure. We detected a signal for positive selection that is exclusive to the human HAR1 structure (s=12.8), while all other seven primate species in the input set displayed strong negative selection signals (s=0.0). This is in agreement with [55].
Synthetic data sets: SSS-test can distinguish between negative and positive selection models
In addition, we also produced synthetic data sets in order to evaluate whether our SSS-test test can distinguish between different degrees of divergence and between positive and negative selection.
One optimization function simulated negative selection, in which changes were kept if they maintained the ancestral structure. Another simulated loss of selection pressure (random evolution), in which every change was kept. The third one simulated positive selection, in which the origin was a Y-shape and the changes were kept if they caused a change towards a cloverleaf structure.
We found that the divergence of the ortholog families under negative selection was distinctly lower than the divergence of families in the random evolution set (Fig. 1). This shows that the SSS-test can correctly distinguish between constrained and highly diverged families.
Fig. 1.
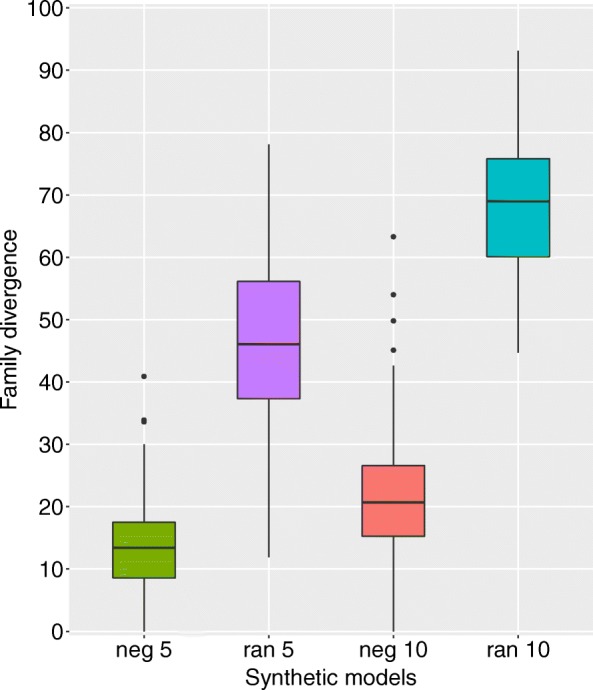
Structural divergence d of synthetic data sets for families evolved under simulated negative selection pressure (neg) compared to unconstrained (ran) evolution. Each data set is composed of 100 families, evolved from one ancestral sequence to five extant sequences, differing by 5 (left) or 10 (right) accepted substitutions from the ancestor
We can also detect a clear difference between the set of families that evolved under negative selection pressure in comparison with the ones that evolved under positive selection pressure, with the families under positive selection having higher scores (Fig 2).
Fig. 2.
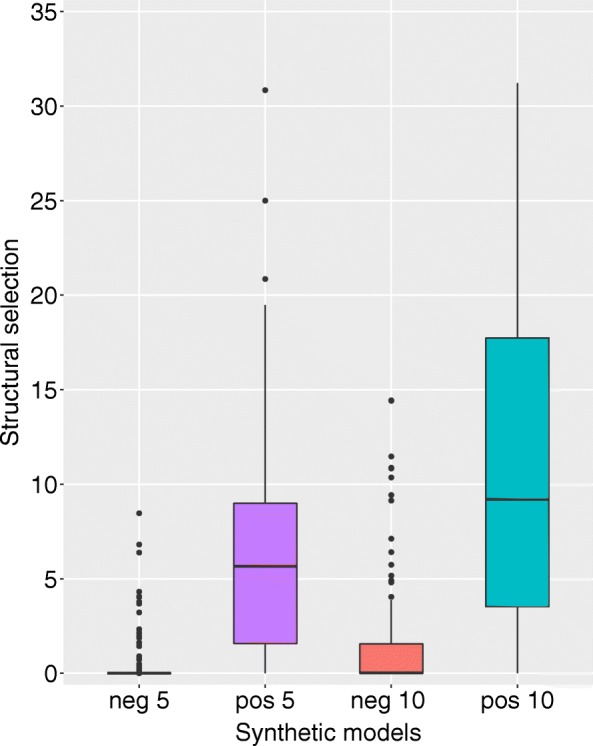
SSS-score s of synthetic data sets with simulated negative evolutionary constraints compared to simulated positive selection. Each data set is composed of 100 sequences, evolved from one ancestral sequence to one extant sequence, differing by 5 (left) or 10 (right) accepted substitutions from the ancestor
Local structures of lncRNAs are mainly negatively selected
To showcase our SSS-test, we conducted a survey of positive selection on structural elements of human lncRNAs. We first computed local structural blocks (conserved local structures shared between at least three species) and then applied the SSS-test on them. For the 15 443 primate lncRNA ortholog families reported in [20], a total of 10 396 blocks were calculated, with orthologs in at least three species and with d≤10.0 (Additional file 1: Table S3).
For this collection of lncRNAs 87 613 local blocks were calculated initially (Additional file 1: Table S3). On average, this amounts to 5.7 blocks per lncRNA family. 77 217 of these blocks have orthologs in only one or two species and/or have a d>10.0 and thus are excluded from the analysis. After this filtering step, we retained 0.7 blocks per lncRNA family with at least three species and d≤10.0. Since we do not expect local secondary structures to be functionally important for all RNAs, the relatively small size of the remaining set is not at all unexpected. We note, furthermore, that the data set relies on coordinates and orthology assignments from ref. [20], so that a non-negligible part of the excluded blocks is due to the quality of the primate genome assemblies and alignments available at the time. For the retained blocks, we observed a substantial level of conservation among these local structures, comparable to the small RNA databases (Additional file 1: Figures S8, S9 and S10). This confirms previous reports that lncRNAs as a group are under negative selection [10, 15, 21, 22] and that conserved RNA structures are common throughout the non-protein-coding parts of the genome [43–45, 79].
The results described in this section were obtained by applying the SSS-test with the default threshold of at least 60% of the sequences agreeing with the consensus for a site to be considered well-conserved (for details we refer to the Theory section). For this primate dataset, 98.6% of the sites are well-conserved. By changing the threshold from 0 to 100%, the proportion of well-conserved sites changes only slightly: from 100% to 87.7% of well-conserved sites (Additional file 1: Figure S16). This high value is expected due to the close phylogenetic distances among the primates.
Using the default parameters of the SSS-test, we observed that 0.77% of the sites changed human specifically. 0.74%, 0.98%, 1.57%, and 3.80% of the sites changed compared to the consensus sequence specifically in pan, gorilla, orangutan, and rhesus macaque, respectively (Additional file 1: Table S5). The number of species-specific substitutions varies slightly if the threshold for well-conserved site is changed, (see Additional file 1: Figure S17 for details), however, the overall pattern of more species specific sites with higher evolutionary distance from humans stays the same. In addition, compensatory sites account for 7-10% of the variation among the five primates (Additional file 1: Table S6).
Positively selected RNA structures in human lncRNAs
In order to identify candidates for lineage-specific selection we considered only the subset of 10 396 local blocks with an overall low structural divergence, i.e., a likely well-conserved ancestral structure. Using a stringent selection score cutoff of s≥10.0 (see Methods for details) we detected 1390 local structures that show signs of positive selection. More than half of these structures (738) show significant differences between the rhesus macaque and the Great Ape lineage (in 716 distinct lncRNAs) (Table 1). Among the Great Apes, we identified in the orangutan lineage 315 local structures with high SSS-score in 312 distinct lncRNAs. In the gorilla lineage we found 136 structures with signs of positive selection in 135 distinct lncRNAs. In Pan (chimpazees and bonobos) 90 structures were found as potential positively selected candidated in 89 lncRNAs. High selection scores were detected in 111 local structures of 110 human lncRNAs. The one human lncRNA with two distinct structures under potential positive selection is ENSG00000246548 (LINC02288). The numbers of candidates under positive selection seems roughly proportional to the evolutionary distance between species, which is not unexpected. (Table 1).
Table 1.
Characterization of local structural selection of lncRNAs
| Species | Local structures | Conserved (s≤2) | Positive (s≥10) | |
|---|---|---|---|---|
| Human | 8934 | 8179 (91.6%) | 111 | (1.2%) |
| Pan | 8736 | 7997 (91.5%) | 90 | (1.0%) |
| Gorilla | 8080 | 7199 (89.1%) | 136 | (1.7%) |
| Orangutan | 6435 | 4802 (74.6%) | 315 | (4.9%) |
| Macaque | 5113 | 2659 (52.0%) | 738 | (14.4%) |
Only the low diverged set was considered in this analysis. Percentages of conserved and positive structures are relative to each species’ number of representatives
In order to estimate the FDR of the SSS-test survery, we used the 8934 alignments of local structures containing a human sequence and shuffled them with SISSIz [42] as described in the Methods section. Using the same cutoffs for ds and SSS−score as for the real data, we obtained 50 predictions, amounting to an FDR of 45%. A closer inspection shows that the shuffling does not completely destroy the signal for positive selection in the real data: repeated shuffling of the positive predictions in the real data shows that about 18.5% of these shuffled alignments still yield a positive result with the SSS-test. Hence about 20 of the 50 predictions in the shuffled sets correspond to the predictions on the real data, reducing the estimated FDR to less then 30%. This is comparable to the FDR of most of the surveys for negative selection: For instance RNAz 2.0 reported 54% for a human survey [80], a FOLDALIGN-bases survey on the ENCODE regions obtained about 50% [43]. A hybrid of SISSIz and RNAz achieves 5–22% [44], and a recent cmfinder-based screen with score cutoffs depending on GC content estimates a FDR of 14±5% [45].
We separately investigated SRA, Xist, and HOTAIR for signs of positive selection in humans within the primate group. No signal of positive selection was detected for these three well-studied lncRNAs.
Profile of positively selected structures of human lncRNAs
We next investigated how the local structures with signs of positive selection have been altered. Interestingly, we detected changes in the form (exemplified in Fig. 3) as well as changes in the stability (Fig. 4) of the structures. For instance, local structure 11 of SIX3-AS1 shows little difference in the minimum free energy structure, but has considerably gained in stability in humans, as shown by the increase of the base pair stability in all three inner stems (Fig. 4). Increase in stability could for instance fine-tune interactions, having an important impact in function, as is the case of the human HAR1, which has acquired higher stability in the human lineage [58].
Fig. 3.
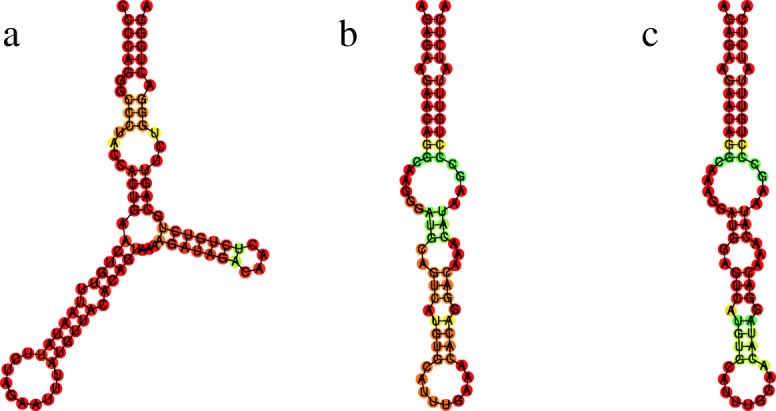
Local lncRNA structure LINC02217sub5: a human, b pan and c gorilla; Only the human structure obtained an SSS-score indicating positive selection with s=16.2, while the data indicates strong negative selection for the other species (s=0.0). Structures are represented by their minimum free energy. Base colors are assigned according to their pairing frequency in the structure’s ensemble. Shades of red occur in ≥90% of the ensemble, shades of green/yellow denote increasing probabilities from ≥50%. For unpaired bases, shades of red denote increasing unpairedness
Fig. 4.

Local lncRNA structure SIX3-AS1sub11: a human, b pan, c orangutan, d gorilla and e macaque. Only the human structure obtained an SSS-score indicating positive selection with s=12.2 while the other species have strong negative selection scores (s=0.0). Structures are represented by their minimum free energy. Base colors are assigned according to their pairing frequency in the structure’s ensemble. Shades of red occur in ≥90% of the ensemble, shades of green/yellow denote increasing probabilities from ≥50%. For unpaired bases, shades of red denote increasing unpairedness
In our initial analysis we had orthologs of SIX3-AS1 only in human, pan, and orangutan. To identify the orthologs in gorilla and rhesus macaque (Additional file 1: Figure S11), we performed genome-wide scans using Infernal v1.1.1 [32]. First, a covariance model was built and calibrated for the sequences of human, pan and orangutan and the consensus structure yielded by RNAalifold [69]. After building the covariance model, we searched for homologous structures in gorilla and macaque, obtaining very likely hits (score of 155.1 and an e-value of 1.5×10−31 for gorilla and score of 150.7 and an e-value of 1.7×10−30 for macaque).
The macaque structure is located in one of the exons of the lncRNA and the gorilla structure is located close but not inside the annotated locus (Additional file 1: Figure S11). The two recovered structures show a similar structural pattern as pan and orangutan, being also less stable than the human structure (Fig. 4). When including these two sequences into the family and recalculating the SSS-scores, the signals are maintained (human SSS-score s=12.2 and the other four species s=0.0). This corroborates our predictions that the human structure is under positive selection when compared to closely related species, and has acquired higher stability during evolution.
In contrast to SIX3-AS1, there is still little or no functional annotation for most candidate lncRNAs. Only 49 of the 110 lncRNAs have an ENSEMBL Gene ID and only 20 of them have been associated an HGNC gene symbol yet (Table 2). To gain more information about the 110 lncRNAs that have positively selected structures in the human lineage, we analyzed in which tissues they are expressed. Based on the expression data reported in [20] we found that six of the lncRNAs are expressed in all nine reported tissues (brain, including developing brain, cerebellum, liver, heart, kidney, placenta, ovary, testis and stem cells), eight have no detected expression in humans and 16 are expressed only in one tissue in humans.
Table 2.
Human lncRNA candidates with signs of positive selection in local structures
| Gene name | Transcription age | Sequence age | Nb species transcribed | Nb species with sequence | ENSEMBL gene ID |
|---|---|---|---|---|---|
| RRS1-AS1 | African apes | Great apes | 3 | 4 | ENSG00000246145 |
| LINC01939 | African apes | Great apes | 3 | 4 | ENSG00000228799 |
| LINC01839 | Primates | Primates | 4 | 4 | ENSG00000227509 |
| LINC01802 | Primates | Primates | 5 | 5 | ENSG00000225064 |
| LINC01724 | Primates | Primates | 5 | 5 | ENSG00000227421 |
| LINC01693 | Primates | Primates | 5 | 5 | ENSG00000227764 |
| MACC1-AS1 | Primates | Primates | 5 | 5 | ENSG00000228598 |
| TRPM2-AS | Primates | Primates | 5 | 5 | ENSG00000230061 |
| LINC01258 | Primates | Primates | 5 | 5 | ENSG00000249534 |
| PLUT1 | Primates | Primates | 5 | 5 | ENSG00000247381 |
| LINC01345 | Primates | Primates | 5 | 5 | ENSG00000226374 |
| MDC1-AS1 | Primates | Eutherians | 2 | 6 | ENSG00000224328 |
| LINC01790 | Therians | Therians | 3 | 7 | ENSG00000230173 |
| LINC02042 | Eutherians | Eutherians | 5 | 5 | ENSG00000240893 |
| LINC02092 | Eutherians | Eutherians | 5 | 6 | ENSG00000234721 |
| LINC01738 | Eutherians | Eutherians | 6 | 6 | ENSG00000227947 |
| LINC02288 | Eutherians | Eutherians | 6 | 6 | ENSG00000246548 |
| LINC02217 | Eutherians | Eutherians | 6 | 6 | ENSG00000248455 |
| DNMBP-AS1 | Mammals | Amniotes | 6 | 9 | ENSG00000227695 |
| SIX3-AS1 | Tetrapods | Tetrapods | 9 | 9 | ENSG00000236502 |
The evolutionary age and expression information was taken from [20]. Gene names were retrieved from the ENSEMBL database. Only transcripts that have been assigned an HGNC ID are shown
Interestingly, the positively selected lncRNAs tend to be expressed in more tissues than lncRNAs in general (Spearman’s rank correlation: ρ=0.78, p =0.0081, Additional file 1: Figure S12). While 2% of the lncRNAs expressed in 8 tissues are under positive selection, this is the case for less than 0.5% of those expressed in a single tissue or not expressed at all in the data set from [20]. Just as a characterization of this data set, we plotted the distribution of number of exons, and saw that most lncRNAs have only one or two exons (Additional file 1: Figure S13).
Evolution of lncRNAs associated with psychiatric disorders
Given that HAR1’s expression is dysregulated in Huntington’s disease [81, 82] and that two of the lncRNAs with potentially positively selected structures on the human lineage (SIX3-AS1 and TRPM2-AS) are antisense to genes involved in brain disorders [83, 84], we decided to have a closer look at lncRNAs that have been reported to be related to psychiatric disorders (PDs). Positive selection in such lncRNAs might indicate functional changes related to human brain evolution.
We collected 26 lncRNAs from public databases and the literature that have been reported to be involved in PDs (Suppl. Tab. 4). These 26 lncRNAs contain 362 local blocks consisting of 1331 local structures. 942 of those had a low family divergence (d≤10.0). From those, 32 have a positive selection score (s≥10.0), with three of them in the human lineage (Table 3). Considering the small number of analysed families, secondary structures could be analysed by manual inspection. We saw that if the threshold for considering positive selection is relaxed in this data set from s≥10.0 to s≥4.5, another 11 local structures in humans can also be considered as candidates for having evolved under positive selection (Table 3).
Table 3.
ENSEMBL IDs and SSS-scores of local structures with signs of a positive selection/weak positive selection in humans
| lncRNA family (block ID) | Selection score |
|---|---|
| MIATsub92 | 21.2 |
| NEAT1sub2 | 14.5 |
| LINC00689sub32 | 11.0 |
| LINC00689sub40 | 8.9 |
| LINC00689sub38 | 7.4 |
| MEG3sub15 | 7.0 |
| H19sub7 | 6.8 |
| SOX2-OTsub27 | 5.9 |
| MEG3sub1 | 5.4 |
| BDNF-ASsub18 | 5.0 |
| LINC02151sub5 | 4.9 |
| MIATsub86 | 4.7 |
| MIATsub31 | 4.6 |
| NEAT1sub120 | 4.6 |
Marked in bold are local structures with SSS-score above 10.0
Among the positively selected human structures, MIATsub92 shows the highest selection score (SSS-score = 21.2). The human structure and its ortholog in chimpanzees contains a tandem duplication of the sequence TTTGAACTTGGCTAACACAGG (Fig. 5), with a substition of a G to A in one of the duplicates (TTTGAACTTG(G/A)CTAACACAGG). Unlike the chimpanzee ortholog, the human structure contains another duplication of the TTTGAACTTGGCTAACACAGG sequence. It seems that this duplication has had an essential impact on the human MIATsub92 structure and might have contributed also to an increase in stability of the human structure compared to its counterparts. It is worth noting that TTTGAACTTGGCTAACACAGG also has a deletion variant in humans with highest population frequency of 0.36. This means that some human individuals still possess the ancestral version of MIATsub92, indicating that this human specific change likely occurred very recently and is not yet fixed. Although this deletion variant is not reported to have a phenotypic effect, it could lead to destabilization of structure, as denoted by decreasing base pair probabilities and shortening of the lower stem, as seen in the chimpanzee ortholog (Fig. 5).
Fig. 5.
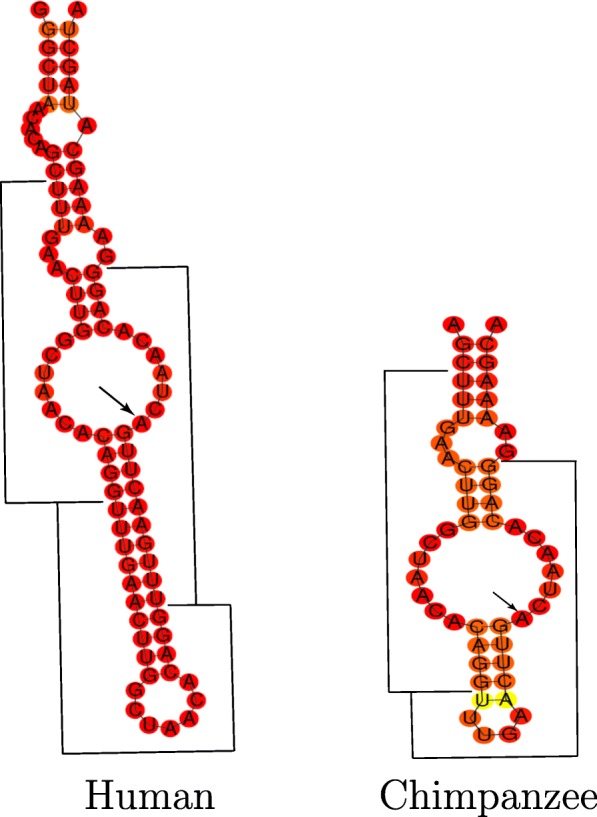
Minimum Free Energy (MFE) structures of MIATsub92 local structures of Human and Pan. The duplication of a TTTGAACTTGGCTAACACAGG sequence in the human lineage might have driven the evolution of the structure towards a more stable structure. Prevailing red regions exhibit well-defined structures with probabilities close to 1 for paired and unpaired bases. Duplicated regions are labeled with horizontal and vertical lines, and G/A nucleotide substitution is marked with an arrow. Bonobo has the same sequence as the chimpanzee
Apart from MIATsub92, two other local structures in MIAT (MIATsub31 and MIATsub86) also show high structural differences in the human lineage in comparison to non-human primates. Greater structural stability of MIATsub31 in humans is noticeable when comparing the centroid structures (Additional file 1: Figures S14 and S15). MIATsub31 contains UACUAAC repeats, which are known to be important for binding of splicing factors [85, 86]. Importantly, in both, humans and non-human primates, these repeats are always found within unpaired regions, and the red coloring of the nucleotides in these internal loops indicates high probability of unpairedness. It seems as if selection was driving the evolution of MIATsub31 towards higher stability in humans, while at the same time it was important that UACUAAC remained unpaired. This suggests that the binding specificity of splicing factors is dependent on internal loops, and not simply a sequence of repeats.
Discussion
We have introduced here a statistical test for positive selection on RNA secondary structure. It focuses on orthologous structures that are otherwise conserved across taxa, thereby identifying candidates with species-specific functional changes in ncRNAs. The SSS-test is implemented as an easy to use command line tool, that we expect to be relevant for a number of future genomic and evolutionary studies. It requires as input a simple FASTA file, and can be used for any type of ncRNA, including the yet largely uncharacterised group of lncRNAs.
While the SSS-test was designed primarily to detect positive selection, it can also be used to identify ncRNAs under negative selection. Since genes with very small scores are candidates for evolving under negative selection, the SSS-test may be employed to complement other available methods for assessing structural conservation. In addition, orthologous groups of local structures with high divergence scores within the family might indicate genes that evolve under relaxed selective constraints. For instance, lncRNAs that contain only local structures with high family divergence are probably transcripts for which the secondary structure is not functionally relevant.
In the study reported here we required at least three orthologs for the analysis. Depending on the system under consideration, a sufficient number of closely related species may not always be available, however. In this case, one could consider a pairwise version of the SSS-test. While this could easily be implemented, the interpretation of the results will necessarily be quite different: In a pairwise setting, it is unknown which sequence represents the ancestral state, hence one can only test whether the structures are unexpectedly different. While positive selection in one of the lineages is possible, divergent evolution should also be considered.
The current version of the SSS-test uses the relatively simple SSS−score as decision variable. Although it is based on established measures of structural variability and it is empirically capable of distinguishing modes of selection, it does not derive from a stochastic model of the evolution of RNA secondary atructure. In particular, the way in which in/dels are evaluated is rather ad hoc. It will be of interest for future work to find a parameter with a better theoretical foundation as a replacement for the SSS−score. This will likely imply that the covariation of paired nucleotides will have to be taken into consideration as well.
Gene duplications are often – but not necessarily – accompanied by positive selection, leading to functional changes in one or both duplicates [87, 88], see e.g. [89] for a case study. In this context, it is imperative to accurately distinguish between (co)orthologs and paralogs. As any statistical test for positive selection, also our test could report false positives when inadvertently including paralogs. This is a general concern in the analysis of protein-coding genes and also pertains to many ncRNAs including miRNAs, snoRNAs, and tRNAs that are frequently duplicated [90–92]. On the other hand, the pairwise version of our SSS test mentioned above could also be applied to a pair of duplicated ncRNAs to assess whether they might evolve under positive selection. In our case study of local structures of lncRNAs in primates, we found an extra duplication in the human structure of MIATsub92, which increases the stability of the local structure and elongates its lower stem, confering it a signal for positive selection (Fig. 5). This example suggests that tandem duplications could greatly impact the evolutionary trajectories of lncRNAs, and should therefore be considered in further studies of ncRNA selection. The contribution of segmental duplications to lncRNAs appears to be much smaller than for the families of small RNAs [93], despite some exceptional cases such as FAM230 [94]. Short local duplications may cause alignment problems that may translate into erroneous results of the SSS-test. Thus we strongly recommend to manually inspect sequence alignments passed to the SSS-test.
We applied our test to an evolutionary analysis of primate lncRNAs, identifying 111 local structures with signs of positive selection on the human lineage. These comprise 110 lncRNA genes, with one of those containing two local structures with signs of positive selection. Most of the candidates are unknown, and those with some description are: PDX1 associated lncRNA (PLUT1) and another six candidates that overlap proteins that are antisense to them (RRS1, MACC1, TRPM2, SIX3, DNMBP and MDC1). The power of the test is inherently limited by the amount of sequence variation among the lineages considered. Most likely, therefore, we experience a large false negative rate due to small divergence between primate genomes. Given that the power of the test is limited by the moderate sequence divergence between primates, the lncRNAs identified in this study are probably only the proverbial tip of the iceberg.
Of the proteins mentioned above, two have known important functions in the brain: TRPM2 (Transient receptor potential melastatin 2) is an ion channel expressed in the brain. It is essential for cell survival by modulating mitochondrial responses and has also been associated with neuroblastoma [83]; The SIX3 protein is a transcriptional regulator that plays a role in eye development and is associated with cephalic disorder [84]. The SIX3-AS1 lncRNA (also known as SIX3OS) modulates the SIX3 protein by acting in trans to regulate retinal development. SIX3-AS1 has an essential role in regulating retinal cell specification. Although it is directly antisense to SIX3, it does not regulate its expression, but is rather acting as a molecular scaffold directly binding to histone modification enzymes directing them to SIX3 target genes [95]. The specifics on the SIX3-AS1 interaction with its partner proteins still remain unknown, but it seems worthwhile to study the role of this positively selected structure in such interactions, and whether the increased stability in humans changes or fine-tunes interactions, when compared to other primates.
The proteins antisense to the other candidates have varying functions outside of the brain. MACC1 is an immunogene [96]. PDX1 is a regulator of pancreas development and β cell differentiation and its antisense lncRNA, PLUT1, is potentially associated with diabetes and affects chromatic structure and the transcription of PDX1 [97]. RRS1 is involved in ribosome biogenesis [98]. MDC1 has a role in cell cycle and cancer control [99, 100]. And the function of C5orf66 has not been characterised yet.
While functional annotation of most other candidate lncRNAs is still pending, it is interesting to note that there is a tendency for lncRNA that are expressed in many tissues to show more frequently signs of positive selection than lncRNAs that are expressed in only a few tissues. The broader tissue expression suggests that structural changes might often have a ubiquitous effect instead of a very localized one restricted to a few tissues.
LncRNAs have been strongly associated with brain development, synaptic plasticity, neural functioning as well as neurodegenerative and psychiatric disorders [101–104]. Human evolution has been characterized by an incease in brain size and complexity, followed by an improvement of cognitive abilities. Notably, dysfunctions of cognitive skills are observed in psychiatric patients [105]. There might be a causal link between human brain evolution and increased susceptability to PDs, as those are mostly human specific disorders. Candidates such as HAR1 and SIX-AS1 prompted us to investigate if also other lncRNAs associated with PDs have been positively selected in humans.
We detected at least three lncRNAs with strong signs of positive selection on the human lineage, MIAT, NEAT, and LINC00689. Several lines of evidence demonstrate an important role of MIAT RNA in the development of schizophrenia [85, 106, 107] and in substance dependence, as its expression is upregulated in the nucleus accumbens of cocaine and heroin abusers [108, 109]. In addition, aberrant expression of MIAT is observed in neurovascular dysfunction contributing to the pathogenesis of Alzheimer’s disease [110]. The strongest signal of positive selection in our data was in the local structure MIATsub92, which contains a human specific duplication that seems to be very recent and not yet fixed in the human population. This duplication has an effect on the shape and stability of the structure. MIAT RNA was shown to co-localize within a nuclear compartment that is enriched in splicing factors [111]. Its expression is down-regulated upon activation of neurons, which allows disassociation of splicing factors that further mediate splicing of targeted genes [85]. In post-mortem brains of schizophrenic patients, expression of MIAT is also down-regulated, and changes in expression level of MIAT result in dysregulation of alternative splicing [85]. MIATsub31, with weak signs of positive selection in humans, contains repeats that are important for the interaction of MIAT with the splicing machinery. These repeats have always been observed with high probability to be located within unpaired parts of the structure, implying importance of internal loops in recognition and binding of splicing factors. It is tempting to speculate that differences in splicing patterns between human and non-human primate brains are in part caused by evolutionary changes in MIAT.
Conclusions
The SSS-test provides an efficient statistical approach to assess whether small ncRNAs or local structures of lncRNAs might evolve under positive selection. Our work thus complements previous studies on detecting negative selection of ncRNAs. The SSS-test evaluates whether ncRNA genes harbour an excess of evolutionary events, such as substitutions and/or indels, that lead to a rather large structural change. Therefore, it can also provide information on whether secondary structure is under negative selection, and whether relaxed constraints make a functional role of the RNA structure unlikely. An advantage of our test is that we consider the structure directly as the phenotype, instead of analyzing the sequence conservation to detect positive selection. One has to keep in mind, however, that RNA secondary structure prediction is not perfect and hence false positive predictions are unavoidable; additional experimental verification thus is strongly advised.
We demonstrated that the SSS-test is capable of detecting lineage-specific positive selection on secondary structures in genome-wide surveys. Given the limited power of the method, we suspect that the approximately one hundred candidates in the human lineage are a lower bound. In addition, the detection of lineage-specific positive selection in genes associated with cognitive disorders in humans lends further credibility to our method.
Data and methods
Evaluation of the SSS-test on small ncRNA databases
We applied the SSS-test to known examples of positive and negative selection. To the best of our knowledge the 118 nucleotide region of the Human accelerated region 1 [55] is the only available control for positive selection on non-coding structures to date [58]. It is well established that HAR1 is stable in humans and differs from its orthologous structure in chimpanzee and other non-human primates [56, 57]. It is extremely conserved across vertebrates but has 18 fixed human changes [55], which stabilized its structure, likely caused by positive selection [58].
As negative controls for our test, we used three databases of structurally conserved small ncRNAs: (i) miRNAs (miRBase [33], release 21), (ii) CD and HACA box snoRNAs [90], and (iii) tRNAs (personal communication). We only selected sequences of the following primates from each database: human, chimpanzee, gorilla, orangutan, and rhesus macaque (with the exception of the snoRNA database which does not contain orangutan sequences). We analysed 167 microRNA families, 176 snoRNA families (containing CD and HACA box snoRNAs) and 511 tRNA families (containing functional tRNAs as well as pseudo tRNAs). Each family of these databases contained only one sequence per primate species, to avoid species bias. Only families with low-divergence (d≤10.0) were retained for further analysis, resulting in 142 microRNA families, 78 snoRNAs families and 141 tRNA families for our analysis of selection.
Evaluation of the SSS-test on synthetic data sets
After evaluating the SSS-test using biological RNAs, we evaluated it using in silico designed sequences. In this way, it is possible to simulate evolution and keep a tighter control on the selective pressures and how the families are constructed. We designed two experiments. The first is designed to test if the SSS-test can differentiate between low and high divergence of individual families. The second experiment tests if the SSS-test can differentiate between negative and positive selection within low-diverged families.
To answer the first question, we simulated evolution from one origin or ancestral sequence to five extant branches. This provides us with five evolved sequences that compose one family, similar to the real biological data we worked with.
To answer the second question, we simulated evolution from one origin to one extant branch but kept the other four branches unchanged compared to the ancestral origin. This simulates a case in which the family is composed of four species that have kept the ancestral sequence (due to extreme negative selection) and one species that changed its sequence due to a different evolutionary pressure.
The synthetic data sets were created with RNAdesign [112], with each family starting from a randomly created RNA sequence of 150 nt. For each database, 100 families were generated and subjected to the SSS-test. To simulate evolutionary pressures, the starting sequence is randomly mutated, whereby a mutation is accepted or rejected according to the different optimization functions which we detail below. The simulation evolves the origin until n changes are accepted. We performed two simulations for each set, with n=5 and n=10.
We simulate the following cases:
negative selection (fneg), as a pressure to maintain the original structure, where deviation from the ancestral secondary structure is penalized;
random evolution (frand), with no pressure towards any goal, with any mutation being accepted; and
positive selection (fpos), where an ancestral Y-shaped structure experiences mutations and the optimization function provides pressure towards a cloverleaf structure.
Denote by a be the ancestral sequence and let m be the current sequence being designed by RNAdesign. Consider ε as a stabilizing parameter that keeps the energy of the evolved sequence at least at half of the original sequence to prevent degenerate structures from forming:
| 4 |
As long as the sequence m has a minimum-free energy at least half of the ancestral sequence, ε(a,m) accepts the proposal m. Otherwise, the large penalty will make acceptance extremely improbable.
Similarly, we can constrain both the basepair and shape distance [113] of a and m. For the basepair distance, the penalty function reads as in Eq. 5, while the shape distance is based on a simplified alignment cost of the two shapes.
| 5 |
Shapes [113] are coarse-grained representations of secondary-structures. Each shape represents a wide range of sequences and their secondary structures that fold into the same “rough” structure. There are different levels of shape representations, ranging from 1 to 5, which indicate the abstraction level. We used the most abstract representation (level 5), which encompass a wide set of possible sequences that fold into the same abstract structure.
For case (i) of negative selection, we constrain the basepair distance of the centroids of the origin and extant sequence. Using a very large penalty for a basepair distance >0, we prevent structural divergence of the centroid. This penalty is given in addition to the energy penalty ε discussed above. This results in the following optimization function
| 6 |
In contrast, case (ii), random evolution, has no penalties at all, here the optimization function is constant 0, independent of the extant sequence:
| 7 |
Finally, for case (iii), positive selection, we compute the RNA shapes (level 5) of the centroid of the mutating extant sequence. We penalize distance to a cloverleaf-shaped target (level 5). This simulates the pressure on the new structure, which is constrained to move from a Y-shaped origin towards a cloverleaf ([[][][]]) target:
| 8 |
It is important to notice that these experiments are intended to provide a control for the SSS-test and its ability to differentiate between differently constructed families. The intention is not to provide a full model of simulated evolution in a biological sense. The latter is a very difficult problem, and out of scope for this contribution.
Structural selection of lncRNA local structures
To illustrate an interesting application of the test, we searched for lncRNA structures that are positively selected in human using a primate group which includes human, pan (including both chimpanzee and bonobo), gorilla, orangutan, and rhesus macaque. The data of [20] provides coordinates in BED format for 15 443 lncRNA families, including orthologs of these five primates. We used an in-house C-program to retrieve the sequence information from the genomic DNA data based on the coordinates provided. We used muscle to compute alignments of orthologous lncRNAs.
It has been observed that most base-pairing interactions in longer RNAs occur within a short span of 150-200 bp [114]. Taking this into consideration, it is also expected that evolution acts on these smaller modules of lncRNAs (local folds), rather than on the entire structure. Therefore, it is more reasonable to search for positive selection locally than globally in lncRNAs. Local structural elements were identified separately for each species using RNALfold, a component of the ViennaRNA package that computes minimum energy structures with restricted base pair span [115].
The most energetically stable local structures were chosen for each species in a way that all chosen structures can co-exist with each other (they do not overlap). Local structures from different species were considered orthologous if they overlap at the starting position with regard to the alignment. To allow for a little bit of freedom, the starting positions could diverge by at most 30% of the length of the sequences. Only regions containing orthologous structures from at least three species were considered, and these are defined as conserved blocks. In total we identified 19 408 blocks with at least three ortholog species. Of these 10 396 have low family divergence (d≤10.0) and were kept for complete selection analysis with the SSS-test.
Information on the evolutionary age and tissue-specific expression patterns were extracted from the supplemental files provided in [20]. This data also includes the number of species with orthologous sequences in the lncRNAs and detectable expression.
Estimation of the false discovery rate
The false discovery rate (FDR) is defined as the expected fraction of false discoveries among all discoveries. It can be estimated for a given “foreground” data set by comparing the number F of positive test results in the “foreground” with the number R of positive test results a “background” data set of the same size. The latter is conveniently obtained by shuffling each of the “foreground” alignments using SISSIz -s [42]. Since this shuffling method destroys the correlation of alignment columns, and hence the secondary structure, we may consider all positive test results on the shuffled alignments as false positives. If this assumption is violated, and the shuffled set retains some of the foreground signal, we only obtain an upper bound, i.e., FDR=R/F.
Empirically we found that our shuffling procedure indeed does not completely remove the “foreground” signal. Using SISSIz -s to produce 20 independent randomizations of the “foreground” predictions, we estimate the fraction of tests f at which the signal is retained. Under these circumstances, we can refine the estimate of the FDR and use FDR=(1−f)R/F.
LncRNAs involved in psychiatric disorders
Candidates of human lncRNAs associated with PDs were obtained from the lncRNA Disease database [116], a publicly available database of disease-associated lncRNAs. In addition, we performed a literature survey to identify further lncRNAs with PD association that are not listed in the lncRNA Disease database. In total, 26 human candidate lncRNAs were obtained (Additional file 1: Table S4 for IDs).
To annotate the orthologous lncRNAs in the other primate species, orthologous splice sites were first calculated in bonobo, chimpanzee, orangutan and rhesus macaque using the SpliceMap tool [22]. In addition to the splice sites, orthologous start and end sites were also calculated using both SpliceMap and BLASTN. A greedy approach was applied to retrieve the full set of orthologous, transcripts in BED12 format, based on the positions of starts, ends and splice sites (unpublished). To obtain the FASTA sequences from the BED12 coordinates, an in-house C program was used. Subsequently, local structure blocks were calculated, and the blocks with a d value below the threshold were submitted to the SSS-test. The same approach was used to identify the primate orthologs of Xist and HOTAIR. For SRA, we retrieved the orthologs from ENSEMBL’s BioMart.
Additional file
The Supplemental Material contains additional information on Methods and Data as well as additional Results. (PDF 1517 kb)
Acknowledgements
The authors would like to thank Irma Lozada for the very fruitful discussions regarding the indel structural impact and for providing the tRNA database, Guilherme Telles for helping with retrieve-fasta, Jana Schor and Stephanie Kehr for providing the microRNA and snoRNA databases. PFS and CHzS thank Maria EMT Walter for her hospitality in Brasilía, where the work was completed.
Funding
This work was supported by CNPq Brasil/scholarship of Science without Borders (246039/2012-4) (MBWC), the Volkswagen Foundation within the initiative “Evolutionary Biology” (KN), the Deutsche Forschungsgemeinschaft as part of the SPP 1738 (MBWC & KN & PFS), the Center for International Cooperation of the Freie Universitaet Berlin (MD) and the German Academic Exchange Service (DAAD grant no. 57390771). We acknowledge support from the German Research Foundation (DFG) and Universität Leipzig within the program of Open Access Publishing.
Availability of data and material
The SSS-test and the local.sh script to calculate local conserved blocks for lncRNAs are available at: https://github.com/waltercostamb/SSS-test/
All databases used in this work are available at:
http://www.bioinf.uni-leipzig.de/Software/SSS-test/
The retrieve-fasta program for retrieving FASTA sequences from BED12 files and the greedy approach to reconstruct lncRNA orthologs from ortholog splice sites can be found at:
https://github.com/waltercostamb/lncRNA-ortholog-reconstruction/
Abbreviations
- ENSG
Ensembl gene
- FDR
False discovery rate
- HAR1
Human accelerated region 1
- HGNC
HUGO gene numenclature committee
- HOTAIR
Hox antisense intergenic RNA
- indel
Insertion or deletion
- LINC
Long intergenic non-protein coding
- lncRNA
Long non-coding RNA
- MFE
Minimum free energy
- MIAT
Myocardial infarction associated transcript
- miRNA
microRNA
- ncRNA
(small) non-coding RNA
- ORF
Open reading frame
- PD
Psychiatric disorder
- SIX3-AS1
SIX3 antisense RNA 1
- snoRNA
Small nucleolar RNA
- SNP
Single nucleotide polymorphism
- SRA
Steroid receptor RNA activator
- SSS
Selection on the secondary structure
- tRNA
Transfer RNA
- TRPM2-AS
TRPM2 antisense RNA
- Xist
X inactive-specific transcript
Authors’ contributions
KN and PFS designed the study, MBWC, CHzS, and PFS developed the theory, MBWC implemented the test and conducted the computational analysis, MD analysed the lncRNAs associated with psychiatric diseases. All authors contributed to writing the manuscript and approved of the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Maria Beatriz Walter Costa, Email: bia.walter@gmail.com.
Christian Höner zu Siederdissen, Email: choener@bioinf.uni-leipzig.de.
Marko Dunjić, Email: marko.dunjic94@gmail.com.
Peter F. Stadler, Email: studla@bioinf.uni-leipzig.de
Katja Nowick, Email: katja.nowick@fu-berlin.de.
References
- 1.Clark MB, Amaral PP, Schlesinger FJ, Dinger ME, Taft RJ, Rinn JL, Ponting CP, Stadler PF, Morris KJ, Morillon A, Rozowsky JS, Gerstein M, Wahlestedt C, Hayashizaki Y, Carninci P, Gingeras TR, Mattick JS. The reality of pervasive transcription. PLoS Biol. 2011; 9:1000625. 10.1371/journal.pbio.1000625. [DOI] [PMC free article] [PubMed]
- 2.Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, Ruan Y, Lassmann T, Carninci P, Brown JB, Lipovich L, Gonzalez JM, Thomas M, Davis CA, Shiekhattar R, Gingeras TR, Hubbard TJ, Notredame C, Harrow J, Guigò R. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012; 22:1775–89. 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed]
- 3.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed]
- 4.FANTOM Consortium and the RIKEN PMI and CLST (DGT). A promoter-level mammalian expression atlas. Nature. 2014; 507:462–70. 10.1038/nature13182. [DOI] [PMC free article] [PubMed]
- 5.St. Laurent G, Wahlestedt C, Kapranov P. The landscape of long noncoding RNA classification. Trends Genet. 2015; 31:239–51. 10.1016/j.tig.2015.03.007. [DOI] [PMC free article] [PubMed]
- 6.St. Laurent G, Shtokalo D, Tackett MR, Yang Z, Eremina T, Wahlestedt C, Urcuqui-Inchima S, Seilheimer B, McCaffrey TA, Kapranov P. Intronic RNAs constitute the major fraction of the non-coding RNA in mammalian cells. BMC Genomics. 2012; 13:504. 10.1186/1471-2164-13-504. [DOI] [PMC free article] [PubMed]
- 7.Kapranov P, Cheng J, Dike S, Nix D, Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermüller J, Hofacker IL, Bell I, Cheung E, Drenkow J, Dumais E, Patel S, Helt G, Madhavan G, Piccolboni A, Sementchenko V, Tammana H, Gingeras TR. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007; 316:1484–8. 10.1126/science.1138341. [DOI] [PubMed]
- 8.Kapranov P, Ozsolak F, Milos PM. Profiling of short RNAs using Helicos single-molecule sequencing. Methods Mol Biol. 2012; 822:219–32. 10.1007/978-1-61779-427-8_15. [DOI] [PMC free article] [PubMed]
- 9.Binder S, Hösler N, Riedel D, Zipfel I, Buschmann T, Kämpf C, Reiche K, Burger R, Gramatzki M, Hackermüller J, Stadler PF, Horn F. STAT3-induced long noncoding RNAs in multiple myeloma cellsi display different properties in cancer. Sci Rep. 2017; 7:7976. 10.1038/s41598-017-08348-5. [DOI] [PMC free article] [PubMed]
- 10.Managadze D, Lobkovsky AE, Wolf YI, Shabalina SA, Rogozin IB, Koonin EV. The vast, conserved mammalian lincRNome. PLoS Comput Biol. 2013;9(2):1002917. doi: 10.1371/journal.pcbi.1002917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haerty W, Ponting CP. Mutations within lncRNAs are effectively selected against in fruitfly but not in human. Genome Biol. 2013; 14:49. 10.1186/gb-2013-14-5-r49. [DOI] [PMC free article] [PubMed]
- 12.Wiberg RAW, Halligan DL, Ness RW, Necsulea A, Kaessmann H, Keightley PD. Assessing recent selection and functionality at long noncoding rna loci in the mouse genome. Genome Biol Evol. 2015; 7:2432–44. 10.1093/gbe/evv155. [DOI] [PMC free article] [PubMed]
- 13.Ponjavic J, Ponting CP, Lunter G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007; 17:556–65. 10.1101/gr.6036807. [DOI] [PMC free article] [PubMed]
- 14.Pang KC, Frith MC, Mattick JS. Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet. 2006; 22:1–5. 10.1016/j.tig.2005.10.003. [DOI] [PubMed]
- 15.Marques AC, Ponting CP. Catalogues of mammalian long noncoding RNAs: modest conservation and incompleteness. Genome Biol. 2009; 10:124. 10.1186/gb-2009-10-11-r124. [DOI] [PMC free article] [PubMed]
- 16.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009; 458:223–7. 10.1038/nature07672. [DOI] [PMC free article] [PubMed]
- 17.Pegueroles C, Gabaldón T. Secondary structure impacts patterns of selection in human lncRNAs. BMC Biology. 2016; 14(1):60. 10.1186/s12915-016-0283-0. [DOI] [PMC free article] [PubMed]
- 18.Schüler A, Ghanbarian AT, Hurst LD. Purifying selection on splice-related motifs, not expression level nor RNA folding, explains nearly all constraint on human lincRNAs. Mol Biol Evol. 2014; 31:3164–83. 10.1093/molbev/msu249. [DOI] [PMC free article] [PubMed]
- 19.Chodroff RA, Goodstadt L, Sirey TM, Oliver PL, Davies KE, Green ED, Molnár Z, Ponting CP. Long noncoding RNA genes: conservation of sequence and brain expression among diverse amniotes. Genome Biology. 2010; 11(7):1. 10.1186/gb-2010-11-7-r72. [DOI] [PMC free article] [PubMed]
- 20.Necsulea A, Soumillon M, Warnefors M, Liechti A, Daish T, Zeller U, Baker JC, Grützner F, Kaessmann H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature. 2014; 505:635–40. 10.1038/nature12943. [DOI] [PubMed]
- 21.Washietl S, Kellis M, Garber M. Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mammals. Genome Res. 2014; 24:616–28. 10.1101/gr.165035.113. [DOI] [PMC free article] [PubMed]
- 22.Nitsche A, Rose D, Fasold M, Reiche K, Stadler PF. Comparison of splice sites reveals that long non-coding RNAs are evolutionarily well conserved. RNA. 2015; 21:801–12. 10.1261/rna.046342.114. [DOI] [PMC free article] [PubMed]
- 23.Chen J, Shishkin AA, Zhu X, Kadri S, Maza I, Guttman M, Hanna JH, Regev A, Garber M. Evolutionary analysis across mammals reveals distinct classes of long non-coding RNAs. Genome Biol. 2016; 17:19. 10.1186/s13059-016-0880-9. [DOI] [PMC free article] [PubMed]
- 24.Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011; 147:1537–50. 10.1016/j.cell.2011.11.055. [DOI] [PMC free article] [PubMed]
- 25.Young RS, Marques AC, Tibbit C, Haerty W, Bassett AR, Liu JL, Ponting CP. Identification and properties of 1,119 candidate lincRNA loci in the Drosophila melanogaster genome. Genome Biol Evol. 2012; 4:427–42. 10.1093/gbe/evs020. [DOI] [PMC free article] [PubMed]
- 26.Hezroni H, Koppstein D, Schwartz MG, Avrutin A, Bartel DP, Ulitsky I. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell. 2015; 11:1110–22. 10.1016/j.celrep.2015.04.023. [DOI] [PMC free article] [PubMed]
- 27.Stephan W. The rate of compensatory evolution. Genetics. 1996;144:419–26. doi: 10.1093/genetics/144.1.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Innan H, Stephan W. Selection intensity against deleterious mutations in RNA secondary structures and rate of compensatory nucleotide substitutions. Genetics. 2001;159:389–99. doi: 10.1093/genetics/159.1.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kusumi J, Ichinose M, Takefu M, Piskol R, Stephan W, Iizuka M. A model of compensatory molecular evolution involving multiple sites in RNA molecules. J Theor Biol. 2016; 388:96–107. 10.1016/j.jtbi.2015.10.008. [DOI] [PubMed]
- 30.Piskol R, Stephan W. The role of the effective population size in compensatory evolution. Genome Biol Evol. 2011; 3:528–38. 10.1093/gbe/evr057. [DOI] [PMC free article] [PubMed]
- 31.Piskol R, Stephan W. Selective constraints in conserved folded RNAs of drosophilid and hominid genomes. Mol Biol Evol. 2011; 28:1519–29. 10.1093/molbev/msq343. [DOI] [PubMed]
- 32.Nawrocki EP, Burge SW, Bateman A, Daub J, Eberhardt RY, Eddy SR, Floden EW, Gardner PP, Jones TA, Tate J, Finn RD. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 2014; 43(D1):130–7. 10.1093/nar/gku1063. [DOI] [PMC free article] [PubMed]
- 33.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence micrornas using deep sequencing data. Nucleic Acids Res. 2013; 42(D1):68–73. 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed]
- 34.Rivas E, Clements J, Eddy SR. A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat Methods. 2017; 14:45–8. 10.1038/nmeth.4066. [DOI] [PMC free article] [PubMed]
- 35.Yazbeck A, Tout KR, Stadler PF. Detailed secondary structure models of invertebrate 7SK RNAs. RNA Biol. 2018; 15:158–64. 10.1080/15476286.2017.1412913. [DOI] [PMC free article] [PubMed]
- 36.Waldl M, Thiel B, Ochsenreiter R, Holzenleiter A, de Araujo Oliveira JV, Walter MEMT, Wolfinger MT, Stadler PF. TERribly difficult: Searching for telomerase RNAs in Saccharomycetes. Genes. 2018; 9:372. 10.3390/genes9080372. [DOI] [PMC free article] [PubMed]
- 37.Rivas E, Eddy SR. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatics. 2001; 2:8. 10.1186/1471-2105-2-8. [DOI] [PMC free article] [PubMed]
- 38.Washietl S, Hofacker IL. Consensus folding of aligned sequences as a new measure for the detection of functional RNAs by comparative genomics. J Mol Biol. 2004; 342:19–30. 10.1016/j.jmb.2004.07.018. [DOI] [PubMed]
- 39.Pedersen JS, Bejerano G, Siepel A, Rosenbloom K, Lindblad-Toh K, Lander ES, Kent J, Miller W, Haussler D. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput Biol. 2006; 2:33. 10.1371/journal.pcbi.0020033. [DOI] [PMC free article] [PubMed]
- 40.Yao Z, Weinberg Z, Ruzzo WL. CMfinder – a covariance model based RNA motif finding algorithm. Bioinformatics. 2006; 22:445–52. 10.1093/bioinformatics/btk008. [DOI] [PubMed]
- 41.Washietl S, Hofacker IL, Stadler PF. Fast and reliable prediction of noncoding RNAs. Proc Natl Acad Sci USA. 2005; 102:2454–9. 10.1073/pnas.0409169102. [DOI] [PMC free article] [PubMed]
- 42.Gesell T, Washietl S. Dinucleotide controlled null models for comparative RNA gene prediction. BMC Bioinformatics. 2008; 9:248. 10.1186/1471-2105-9-248. [DOI] [PMC free article] [PubMed]
- 43.Torarinsson E, Yao Z, Wiklund ED, Bramsen JB, Hansen C, Kjems J, Tommerup N, Ruzzo WL, Gorodkin J. Comparative genomics beyond sequence-based alignments: RNA structures in the ENCODE regions. Genome Res. 2008; 18:242–51. 10.1101/gr.6887408. [DOI] [PMC free article] [PubMed]
- 44.Smith MA, Gesell T, Stadler PF, Mattick JS. Widespread purifying selection on RNA structure in mammals. Nucleic Acids Res. 2013; 41:8220–36. 10.1093/nar/gkt596. [DOI] [PMC free article] [PubMed]
- 45.Seemann SE, Mirza AH, Hansen C, Bang-Berthelsen CH, Garde C, Christensen-Dalsgaard M, Torarinsson E, Yao Z, Workman CT, Pociot F, Nielsen H, Tommerup N, Ruzzo WL, Gorodkin J. The identification and functional annotation of RNA structures conserved in vertebrates. Genome Res. 2017; 27:1371–83. 10.1101/gr.208652.116. [DOI] [PMC free article] [PubMed]
- 46.Thiel BC, Ochsenreiter R, Gadekar VP, Tanzer A, Hofacker IL. RNA structure elements conserved between mouse and 59 other vertebrates. Genes. 2018; 9:392. 10.3390/genes9080392. [DOI] [PMC free article] [PubMed]
- 47.Kirsch R, Seemann SE, Ruzzo WL, Cohen SM, Stadler PF, Gorodkin J. Identification and characterization of novel conserved RNA structures in Drosophila. BMC Genomics. 2018; 19:899. 10.1186/s12864-018-5234-4. [DOI] [PMC free article] [PubMed]
- 48.Duszczyk MM, Sattler M. 1H, 13C, 15N and 31P chemical shift assignments of a human Xist RNA A-repeat tetraloop hairpin essential for X-chromosome inactivation. Biomolecular NMR Assignments. 2012; 6(1):75–7. 10.1007/s12104-011-9328-z. [DOI] [PubMed]
- 49.Johnsson P, Lipovich L, Grandér D, Morris KV. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim Biophys Acta. 2014; 1840:1063–71. 10.1016/j.bbagen.2013.10.035. [DOI] [PMC free article] [PubMed]
- 50.Somarowthu S, Legiewicz M, Chillón I, Marcia M, Liu F, Pyle AM. HOTAIR forms an intricate and modular secondary structure. Mol Cell. 2015; 58(2):353–61. 10.1016/j.molcel.2015.03.006. [DOI] [PMC free article] [PubMed]
- 51.Blythe AJ, Fox AH, Bond CS. The ins and outs of lncRNA structure: How, why and what comes next?. Biochim Biophys Acta (BBA) - Gene Regul Mech. 2016; 1859(1):46–58. 10.1016/j.bbagrm.2015.08.009. [DOI] [PubMed]
- 52.Lin Y, Schmidt BF, Bruchez MP, McManus CJ. Structural analyses of NEAT1 lncRNAs suggest long-range RNA interactions that may contribute to paraspeckle architecture. Nucleic Acids Res. 2018; 46:3742–52. 10.1093/nar/gky046. [DOI] [PMC free article] [PubMed]
- 53.Castellanos-Rubio A, Fernandez-Jimenez N, Kratchmarov R, Luo X, Bhagat G, Green PH, Schneider R, Kiledjian M, Bilbao JR, Ghosh S. A long noncoding RNA associated with susceptibility to celiac disease. Science. 2016; 352(6281):91–5. 10.1126/science.aad0467. [DOI] [PMC free article] [PubMed]
- 54.Gong J, Liu W, Zhang J, Miao X, Guo A-Y. lncRNASNP: a database of SNPs in lncRNAs and their potential functions in human and mouse. Nucleic Acids Res. 2015; 43(D1):181–6. 10.1093/nar/gku1000. [DOI] [PMC free article] [PubMed]
- 55.Pollard KS, Salama SR, Lambert N, Lambot M-A, Coppens S, Pedersen JS, Katzman S, King B, Onodera C, Siepel A, Kern AD, Dehay C, Igel H, Ares Jr M, Vanderhaeghen P, Haussler D. An RNA gene expressed during cortical development evolved rapidly in humans. Nature. 2006; 443(7108):167–72. 10.1038/nature05113. [DOI] [PubMed]
- 56.Ziegeler M, Cevec M, Richter C, Schwalbe H. NMR studies of HAR1 RNA secondary structures reveal conformational dynamics in the human RNA. ChemBioChem. 2012; 13(14):2100–12. 10.1002/cbic.201200401. [DOI] [PubMed]
- 57.Beniaminov A, Westhof E, Krol A. Distinctive structures between chimpanzee and human in a brain noncoding RNA. RNA. 2008; 14(7):1270–5. 10.1261/rna.1054608. [DOI] [PMC free article] [PubMed]
- 58.Walter Costa MB, Höner zu Siederdissen C, Tulpan D, Stadler PF, Nowick K. Temporal ordering of substitutions in RNA evolution: Uncovering the structural evolution of the human accelerated region 1. J Theor Biol. 2018; 438:143–50. 10.1016/j.jtbi.2017.11.015. [DOI] [PubMed]
- 59.Hurst L. The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 2002; 18:486–9. 10.1016/S0168-9525(02)02722-1. [DOI] [PubMed]
- 60.Huang Y-F, Gulko B, Siepel A. Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat Genet. 2017; 49(4):618. 10.1038/ng.3810. [DOI] [PMC free article] [PubMed]
- 61.Gronau I, Arbiza L, Mohammed J, Siepel A. Inference of natural selection from interspersed genomic elements based on polymorphism and divergence. Mol Biol Evol. 2013; 30:1159–71. 10.1093/molbev/mst019. [DOI] [PMC free article] [PubMed]
- 62.Arbiza L, Gronau I, Aksoy BA, Hubisz MJ, Gulko B, Keinan A, Siepel A. Genome-wide inference of natural selection on human transcription factor binding sites. Nat Genet. 2013; 45:723–9. 10.1038/ng.2658. [DOI] [PMC free article] [PubMed]
- 63.Gulko B, Hubisz MJ, Gronau I, Siepel A. A method for calculating probabilities of fitness consequences for point mutations across the human genome. Nat Genet. 2015; 47:276–83. 10.1038/ng.3196. [DOI] [PMC free article] [PubMed]
- 64.Lee PH, Lee C, Li X, Wee B, Dwivedi T, Daly M. Principles and methods of in-silico prioritization of non-coding regulatory variants. Hum Genet. 2017; 137:15–30. 10.1007/s00439-017-1861-0. [DOI] [PMC free article] [PubMed]
- 65.Sabarinathan R, Tafer H, Seemann SE, Hofacker IL, Stadler PF, Gorodkin J. RNAsnp: Efficient detection of local RNA secondary structure changes induced by SNPs. Hum Mut. 2013; 34:546–56. 10.1002/humu.22273. [DOI] [PMC free article] [PubMed]
- 66.Salari R, Kimchi-Sarfaty C, Gottesman MM, Przytycka TM. Sensitive measurement of single-nucleotide polymorphism-induced changes of RNA conformation: application to disease studies. Nucleic Acids Res. 2012; 41:44–53. 10.1093/nar/gks1009. [DOI] [PMC free article] [PubMed]
- 67.Turner DH, Mathews DH. NNDB: the nearest neighbor parameter database for predicting stability of nucleic acid secondary structure. Nucleic Acids Res. 2010; 38:280–2. 10.1093/nar/gkp892. [DOI] [PMC free article] [PubMed]
- 68.Bernhart SH, Hofacker IL, Will S, Gruber AR, Stadler PF. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics. 2008;9:474. doi: 10.1186/1471-2105-9-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lorenz R, Bernhart SH, Höner zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. ViennaRNA package 2.0. Algoritm Mol Biol. 2011; 6(1):1. 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed]
- 70.Lam E, Kam A, Waldispühl J. corRna: a web server for predicting multiple-point deleterious mutations in structural RNAs. Nucleic Acids Res. 2011; 39:160–6. 10.1093/nar/gkr358. [DOI] [PMC free article] [PubMed]
- 71.Churkin A, Gabdank I, Barash D. The RNAmute web server for the mutational analysis of RNA secondary structures. Nucleic Acids Res. 2011; 39:92–9. 10.1093/nar/gkr207. [DOI] [PMC free article] [PubMed]
- 72.Shu W, Bo X, Liu R, Zhao D, Zheng Z, Wang S. RDMAS: a web server for RNA deleterious mutation analysis. BMC Bioinformatics. 2006; 7:404. 10.1186/1471-2105-7-404. [DOI] [PMC free article] [PubMed]
- 73.Halvorsen M, Martin JS, Broadaway S, Laederach A. Disease-associated mutations that alter the RNA structural ensemble. PLoS Genet. 2010; 6:1001074. 10.1371/journal.pgen.1001074. [DOI] [PMC free article] [PubMed]
- 74.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Statist Soc Ser B. 1995; 57:289–300. 10.2307/2346101.
- 75.Ranstam J. Multiple p-values and Bonferroni correction. Osteoarthr Cartil. 2016; 5(24):763–4. 10.1016/j.joca.2016.01.008. [DOI] [PubMed]
- 76.Lorenz R, Hofacker IL, Stadler PF. RNA folding with hard and soft constraints. Alg Mol Biol. 2016; 11:8. 10.1186/s13015-016-0070-z. [DOI] [PMC free article] [PubMed]
- 77.Höchsmann M, Voss B, Giegerich R. Pure multiple RNA secondary structure alignments: a progressive profile approach. IEEE/ACM Trans Comput Biol Bioinform. 2004; 53:53–62. 10.1109/TCBB.2004.11. [DOI] [PubMed]
- 78.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004; 32(5):1792–7. 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed]
- 79.Washietl S, Hofacker IL, Lukasser M, Hüttenhofer A, Stadler PF. Mapping of conserved RNA secondary structures predicts thousands of functional non-coding RNAs in the human genome. Nature Biotech. 2005; 23:1383–90. 10.1038/nbt1144. [DOI] [PubMed]
- 80.Gruber AR, Findeiß S, Washietl S, Hofacker IL, Stadler PF. RNAz 2.0: improved noncoding RNA detection. Pac Symp Biocomput. 2010; 15:69–79. 10.1142/9789814295291_0009. [PubMed]
- 81.Riva P, Ratti A, Venturin M. The long non-coding RNAs in neurodegenerative diseases: novel mechanisms of pathogenesis. Curr Alzheimer Res. 2016; 13:1219–31. 10.2174/1567205013666160622112234. [DOI] [PubMed]
- 82.Johnson R, Richter N, Jauch R, Gaughwin PM, Zuccato C, Cattaneo E, Stanton LW. Human accelerated region 1 noncoding RNA is repressed by REST in Huntington’s disease. Physiol Genomics. 2010; 41:269–74. 10.1152/physiolgenomics.00019.2010. [DOI] [PubMed]
- 83.Bao L, Chen S-j, Conrad K, Keefer K, Abraham T, Lee JP, Wang J, Zhang X-Q, Hirschler-Laszkiewicz I, Wang H-G, Dovat S, Gans B, Madesh M, Cheung JY, Miller BA. Depletion of the human ion channel TRPM2 in neuroblastoma demonstrates its key role in cell survival through modulation of mitochondrial reactive oxygen species and bioenergetics. J Biol Chem. 2016; 291(47):24449–64. 10.1074/jbc.M116.747147. [DOI] [PMC free article] [PubMed]
- 84.Domené S, Roessler E, El-Jaick KB, Snir M, Brown JL, Vélez JI, Bale S, Lacbawan F, Muenke M, Feldman B. Mutations in the human SIX3 gene in holoprosencephaly are loss of function. Hum Mol Genet. 2008; 17(24):3919–28. 10.1093/hmg/ddn294. [DOI] [PMC free article] [PubMed]
- 85.Barry G, Briggs JA, Vanichkina DP, Poth EM, Beveridge NJ, Ratnu VS, Nayler SP, Nones K, Hu J, Bredy TW, Nakagawa S, Rigo F, Taft RJ, Cairns MJ, Blackshaw S, Wolvetang EJ, Mattick JS. The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol Psychiatry. 2014; 19:486–94. 10.1038/mp.2013.45. [DOI] [PubMed]
- 86.Tsuiji H, Yoshimoto R, Hasegawa Y, Furuno M, Yoshida M, Nakagawa S. Competition between a noncoding exon and introns: Gomafu contains tandem UACUAAC repeats and associates with splicing factor-1. Genes Cells. 2011; 16(5):479–90. 10.1111/j.1365-2443.2011.01502.x. [DOI] [PMC free article] [PubMed]
- 87.Hahn MW. Distinguishing among evolutionary models for the maintenance of gene duplicates. J Heredity. 2009; 100:605–17. 10.1093/jhered/esp047. [DOI] [PubMed]
- 88.Innan H, Kondrashov F. The evolution of gene duplications: classifying and distinguishing between models. Nature Rev Genet. 2010; 11:97–108. 10.1038/nrg2689. [DOI] [PubMed]
- 89.Cardoso-Moreira M, Arguello JR, Gottipati S, Harshman LG, Grenier JK, Clark AG. Evidence for the fixation of gene duplications by positive selection in Drosophila. Genome Res. 2016; 26:787–98. 10.1101/gr.199323.115. [DOI] [PMC free article] [PubMed]
- 90.Kehr S, Bartschat S, Tafer H, Stadler PF, Hertel J. Matching of soulmates: coevolution of snoRNAs and their targets. Mol Biol Evol. 2014; 31:455–67. 10.1093/molbev/mst209. [DOI] [PubMed]
- 91.Hertel J, Stadler PF. The expansion of animal microRNA families revisited. Life. 2015; 5:905–20. 10.3390/life5010905. [DOI] [PMC free article] [PubMed]
- 92.Velandia-Huerto CA, Berkemer SJ, Hoffmann A, Retzlaff N, Romero Marroquín LC, Hernández Rosales M, Stadler PF, Bermúdez-Santana CI. Orthologs, turn-over, and remolding of tRNAs in primates and fruit flies. BMC Genomics. 2016; 17:617. 10.1186/s12864-016-2927-4. [DOI] [PMC free article] [PubMed]
- 93.Sun J, Zhou M, Mao Z-T, Hao D-P, Wang Z-z, Li C-X. Systematic analysis of genomic organization and structure of long non-coding RNAs in the human genome. FEBS Letters. 2013; 587:976–82. 10.1016/j.febslet.2013.02.036. [DOI] [PubMed]
- 94.Delihas N. Formation of a family of long intergenic noncoding RNA genes with an embedded translocation breakpoint motif in human chromosomal low copy repeats of 22q11.2 — some surprises and questions. Noncoding RNA. 2018; 4:16. 10.3390/ncrna4030016. [DOI] [PMC free article] [PubMed]
- 95.Rapicavoli NA, Poth EM, Zhu H, Blackshaw S. The long noncoding RNA Six3OS acts in trans to regulate retinal development by modulating Six3 activity. Neural Dev. 2011; 6(1):32. 10.1186/1749-8104-6-32. [DOI] [PMC free article] [PubMed]
- 96.Sueta A, Yamamoto Y, Yamamoto-Ibusuki M, Hayashi M, Takeshita T, Yamamoto S, Omoto Y, Iwase H. Differential role of MACC1 expression and its regulation of the HGF/c-Met pathway between breast and colorectal cancer. Int J Oncol. 2015; 46(5):2143–53. 10.3892/ijo.2015.2907. [DOI] [PubMed]
- 97.Akerman I, Tu Z, Beucher A, Rolando DMY, Sauty-Colace C, Benazra M, Nakic N, Yang J, Wang H, Pasquali L, Moran I, Garcia-Hurtado J, Castro N, Gonzalez-Franco R, Stewart AF, Bonner C, Piemonti L, Berney T, Groop L, Kerr-Conte J, Pattou F, Argmann C, Schadt E, Ravassard P, Ferrer J. Human pancreatic β cell lncRNAs control cell-specific regulatory networks. Cell Metab. 2017; 25:400–11. 10.1016/j.cmet.2016.11.016. [DOI] [PMC free article] [PubMed]
- 98.Gambe AE, Matsunaga S, Takata H, Ono-Maniwa R, Baba A, Uchiyama S, Fukui K. A nucleolar protein RRS1 contributes to chromosome congression. FEBS Lett. 2009; 583(12):1951–6. 10.1016/j.febslet.2009.05.033. [DOI] [PubMed]
- 99.Wang S, Zou Z, Luo X, Mi Y, Chang H, Xing D. LRH1 enhances cell resistance to chemotherapy by transcriptionally activating MDC1 expression and attenuating DNA damage in human breast cancer. Oncogene. 2018; 37:3243–59. 10.1038/s41388-018-0193-4. [DOI] [PubMed]
- 100.Qin Y, Zhuang S, Wen J, Zheng K. Long non-coding RNA MDC1-AS inhibits human gastric cancer cell proliferation and metastasis through an MDC1-dependent mechanism. Exp Ther Med. 2018; 15(1):191–7. 10.3892/etm.2017.5370. [DOI] [PMC free article] [PubMed]
- 101.Zuo L, Tan Y, Wang Z, Wang K-S, Zhang X, Chen X, Li C-SR, Wang T, Luo X. Long non-coding RNAs in psychiatric disorders. Psychiatr Genet. 2016; 26(3):109. 10.1097/YPG.0000000000000129. [DOI] [PMC free article] [PubMed]
- 102.Roberts TC, Morris KV, Wood MJ. The role of long non-coding RNAs in neurodevelopment, brain function and neurological disease. Phil Trans R Soc B. 2014; 369(1652):20130507. 10.1098/rstb.2013.0507. [DOI] [PMC free article] [PubMed]
- 103.Wu P, Zuo X, Deng H, Liu X, Liu L, Ji A. Roles of long noncoding RNAs in brain development, functional diversification and neurodegenerative diseases. Brain Res Bull. 2013; 97:69–80. 10.1016/j.brainresbull.2013.06.001. [DOI] [PubMed]
- 104.Qureshi IA, Mattick JS, Mehler MF. Long non-coding RNAs in nervous system function and disease. Brain Res. 2010; 1338:20–35. 10.1016/j.brainres.2010.03.110. [DOI] [PMC free article] [PubMed]
- 105.Millan MJ, Agid Y, Brüne M, Bullmore ET, Carter CS, Clayton NS, Connor R, Davis S, Deakin B, DeRubeis RJ, Dubois B, Geyer MA, Goodwin GM, Gorwood P, Jay TM, Joëls M, Mansuy IM, Meyer-Lindenberg A, Murphy D, Rolls E, Saletu B, Spedding M, Sweeney J, Whittington M, Young LJ. Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapy. Nat Rev Drug Discov. 2012; 11(2):141–68. 10.1038/nrd3628. [DOI] [PubMed]
- 106.Rao S-Q, Hu H-L, Ye N, Shen Y, Xu Q. Genetic variants in long non-coding RNA MIAT contribute to risk of paranoid schizophrenia in a chinese Han population. Schizophr Res. 2015; 166(1):125–30. 10.1016/j.schres.2015.04.032. [DOI] [PubMed]
- 107.Takahashi S, Ohtsuki T, Yu S-Y, Tanabe E-i, Yara K, Kamioka M, Matsushima E, Matsuura M, Ishikawa K, Minowa Y, Noguchi E, Nakayama J, Yamakawa-Kobayashi K, Arinami T, Kojima T. Significant linkage to chromosome 22q for exploratory eye movement dysfunction in schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2003; 123(1):27–32. 10.1002/ajmg.b.10046. [DOI] [PubMed]
- 108.Michelhaugh SK, Lipovich L, Blythe J, Jia H, Kapatos G, Bannon MJ. Mining affymetrix microarray data for long non-coding RNAs: altered expression in the nucleus accumbens of heroin abusers. J Neurochem. 2011; 116(3):459–66. 10.1111/j.1471-4159.2010.07126.x. [DOI] [PMC free article] [PubMed]
- 109.Albertson DN, Schmidt CJ, Kapatos G, Bannon MJ. Distinctive profiles of gene expression in the human nucleus accumbens associated with cocaine and heroin abuse. Neuropsychopharmacology. 2006; 31(10):2304–12. 10.1038/sj.npp.1301089. [DOI] [PMC free article] [PubMed]
- 110.Jiang Q, Shan K, Qun-Wang X, Zhou R-M, Yang H, Liu C, Li Y-J, Yao J, Li X-M, Shen Y, Cheng H, Yuan J, Zhang Y-Y, Yan B. Long non-coding RNA-MIAT promotes neurovascular remodeling in the eye and brain. Oncotarget. 2016; 7:49688. 10.18632/oncotarget.10434. [DOI] [PMC free article] [PubMed]
- 111.Sone M, Hayashi T, Tarui H, Agata K, Takeichi M, Nakagawa S. The mRNA-like noncoding RNA Gomafu constitutes a novel nuclear domain in a subset of neurons. J Cell Sci. 2007; 120(15):2498–506. 10.1242/jcs.009357. [DOI] [PubMed]
- 112.Höner zu Siederdissen C, Hammer S, Abfalter I, Hofacker IL, Flamm C, Stadler PF. Computational design of RNAs with complex energy landscapes. Biopolymers. 2013; 99(12):1124–36. 10.1002/bip.22337. [DOI] [PubMed]
- 113.Steffen P, Voß B, Rehmsmeier M, Reeder J, Giegerich R. RNAshapes: an integrated RNA analysis package based on abstract shapes. Bioinformatics. 2006; 22(4):500–3. 10.1093/bioinformatics/btk010. [DOI] [PubMed]
- 114.Lange SJ, Maticzka D, Möhl M, Gagnon JN, Brown CM, Backofen R. Global or local? predicting secondary structure and accessibility in mRNAs. Nucleic Acids Res. 2012; 40(12):5215–26. 10.1093/nar/gks181. [DOI] [PMC free article] [PubMed]
- 115.Hofacker IL, Priwitzer B, Stadler PF. Prediction of locally stable RNA secondary structures for genome-wide surveys. Bioinformatics. 2004; 20:191–8. 10.1093/bioinformatics/btg388. [DOI] [PubMed]
- 116.Chen G, Wang Z, Wang D, Qiu C, Liu M, Chen X, Zhang Q, Yan G, Cui Q. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013; 41:983–6. 10.1093/nar/gks1099. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Supplemental Material contains additional information on Methods and Data as well as additional Results. (PDF 1517 kb)
Data Availability Statement
The SSS-test and the local.sh script to calculate local conserved blocks for lncRNAs are available at: https://github.com/waltercostamb/SSS-test/
All databases used in this work are available at:
http://www.bioinf.uni-leipzig.de/Software/SSS-test/
The retrieve-fasta program for retrieving FASTA sequences from BED12 files and the greedy approach to reconstruct lncRNA orthologs from ortholog splice sites can be found at:
https://github.com/waltercostamb/lncRNA-ortholog-reconstruction/