

Abstract
Nucleotide excision repair is a versatile mechanism to repair a variety of bulky DNA adducts. We developed excision repair sequencing (XR-seq) to study nucleotide excision repair of DNA adducts in humans, mice, Arabidopsis thaliana, yeast and Escherichia coli. In this protocol, the excised oligomers, generated in the nucleotide excision repair reaction, are isolated by cell lysis and fractionation, followed by immunoprecipitation with damage- or repair factor-specific antibodies from the non-chromatin fraction. The single-stranded excised oligomers are ligated to adapters and re-immunoprecipitated with damage-specific antibodies. The DNA damage in the excised oligomers is then reversed by enzymatic or chemical reactions before being converted into a sequencing library by PCR amplification. Alternatively, the excised oligomers containing DNA damage, especially those containing irreversible DNA damage such as benzo[a] pyrene-induced DNA adducts, can be converted to a double-stranded DNA (dsDNA) form by using appropriate translesion DNA synthesis (TLS) polymerases and then can be amplified by PCR. The current genome-wide approaches for studying repair measure the loss of damage signal with time, which limits their resolution. By contrast, an advantage of XR-seq is that the repair signal is directly detected above a background of zero. An XR-seq library using the protocol described here can be obtained in 7–9 d.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary.
Introduction
Nucleotide excision repair is widespread in the three domains of life. It is an important, if not the sole, mechanism for removing bulky DNA adducts from the genome, including the major UV photo-products (cyclobutane pyrimidine dimers (CPDs) and (6–4) pyrimidine-pyrimidone photoproducts ((6–4)PPs)), lesions caused by other environmental mutagens such as benzo[a]pyrene and aflatoxins, and anticancer drugs such as cisplatin and oxaliplatin1–3. A defect in nucleotide excision repair greatly exacerbates the lethality and mutagenicity of these damaging agents, and nucleotide excision repair deficiency in humans leads to xeroderma pigmentosum, which is characterized by severe solar sensitivity and early onset of skin cancer1.
Nucleotide excision repair is influenced by numerous factors in vivo such as transcription, chromatin states, DNA replication, epigenetic modifications of DNA and histones, binding of regulatory proteins to DNA and other factors4–6. To investigate the effects of these factors on repair, an approach that directly detects repair events with high resolution at a genome-wide level is required. Although multiple high-resolution methods that map DNA damage sites in the entire genome have been developed7–12, these methods indirectly measure nucleotide excision repair by determining the disappearance of damage at the interval of two time points, which limits their sensitivity and specificity. Here, we describe in detail a robust and broadly applicable genome-wide approach, termed XR-seq, to study nucleotide excision repair of bulky DNA adducts in vivo at single-nucleotide resolution. Since the original development of XR-seq in cultured human cells for UV-induced DNA damage13, we have successfully adapted the XR-seq method to study repair of different DNA-damaging agents in human cells10,14–16, mice17, A. thaliana18, yeast19 and E. coli20,21.
Thus, XR-seq may be applied to diverse species, tissues and biological systems damaged by various substances to investigate nucleotide excision repair directly and other biological processes—most notably transcription and circadian rhythms—indirectly. These applications may help us to understand basic biological functions in a variety of organisms ranging from E. coli to humans and may help improve the efficacy of therapeutic interventions and create crops with more efficient DNA repair mechanisms to better cope with DNA-damaging environmental stresses.
Development and overview of XR-seq
Early mechanistic studies of nucleotide excision repair showed that dual incisions are made in the damaged DNA strand on both sides of the damage. The product is a damage-containing single-stranded oligomer of defined length (mammals and plants, 26–27 nucleotides (nt); budding yeast, 23–24 nt; E. coli, 12–13 nt)1,3,19,22–24. Following the characterization of excision repair proteins and reaction mechanisms1, attention focused on the fate of these excised, damage-containing oligomers generated during the repair reaction25–28. Experimentally, a gentle cell lysis approach was used to isolate excision products from mammalian cells and then directly co-immunoprecipitate them with antibodies against basal repair factors26. Excision products were found to precipitate with TFIIH and XPG, consistent with the known reaction mechanism26. TFIIH, a protein complex with helicase activity, separates the duplex to form a ‘repair bubble’, and XPG cuts the damaged strand on the 3′ side of the damage3. The ability to isolate bona fide excision products generated in vivo constitutes the basis for the XR-seq procedure.
In XR-seq, the excised oligomers are isolated, ligated to adapters and sequenced; then the valid sequencing reads are identified and aligned to the genome sequence to create maps of where repair has occurred (Fig. 1). As originally developed for cultured mammalian cells13, gentle lysis is followed by isolation of excision products by TFIIH or XPG immunoprecipitation or immunoprecipitation with damage-specific antibodies, and then by ligation of excision products to 5′ and 3′ adapters that are compatible with the Illumina TruSeq small RNA protocol. Ligation products are then immunoprecipitated with DNA damage–specific antibodies (available for CPD-, (6–4)PP-, cisplatin-, and benzo[a]pyrene diol epoxide (BPDE)-damaged DNA). Before the next step, PCR amplification of ligation products, the damage must be reversed or removed. This is accomplished for UV photo-products by photoreactivation with the appropriate CPD- or (6–4)PP-photolyase. Photolyases bind specifically to either a CPD or a (6–4)PP adduct in DNA and photo-catalytically repair the photo-product by direct reversal to the original dipyrimidine1. The recombinant photolyases used in XR-seq can be prepared by conventional biochemical methods. Platinum damage is reversed by treatment with sodium cyanide (NaCN)10. Samples of the repaired, ligated products and control unrepaired products are subjected to pilot PCR. The purpose is threefold: to demonstrate successful PCR of an appropriate-sized product, to demonstrate that PCR is repair dependent (i.e., photoreactivation or cyanide treatment worked) and to determine the fewest number of PCR cycles needed to generate a library for sequencing in the subsequent preparative-scale PCR reaction.
Fig. 1 |. Schematic overview of the XR-seq protocol.
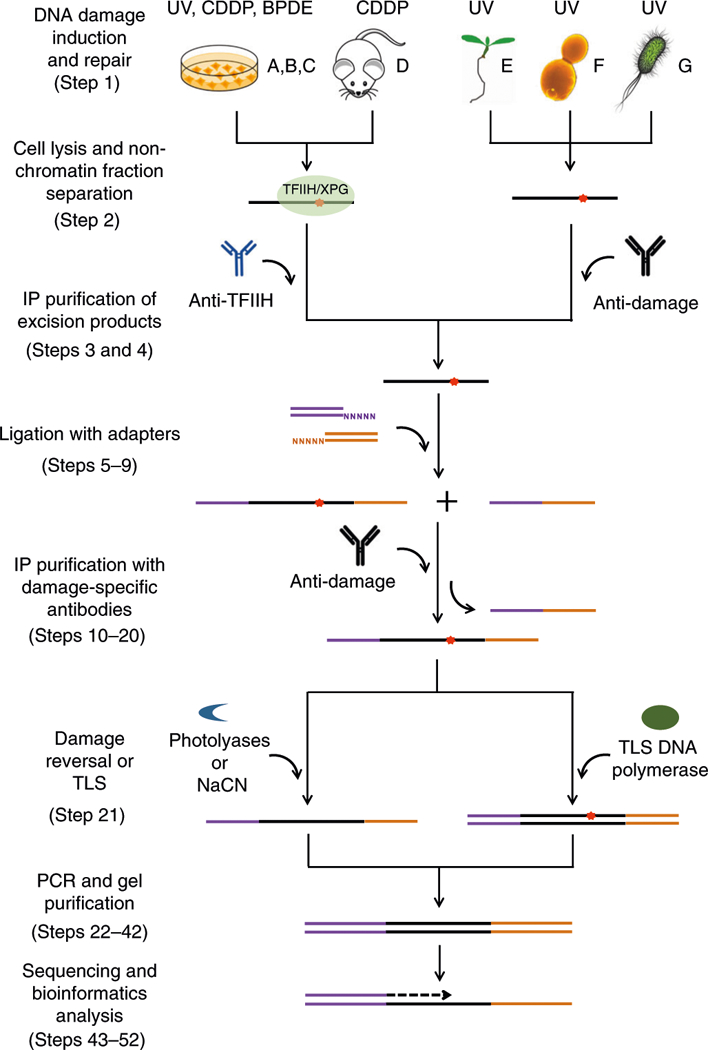
The red star denotes the DNA damage in the excised oligomer released during nucleotide excision repair. The purple and orange lines represent the 5′ and 3′ adapters, respectively. BPDE, benzo[a]pyrene diol epoxide; CDDP, cisplatin; NaCN, sodium cyanide; UV, ultraviolet.
For some lesions that cannot be reversed in vitro such as BPDE adducts, we have developed translesion XR-seq (tXR-seq) by using an appropriate TLS polymerase to bypass the damage and enable subsequent high-fidelity PCR amplification29. In the tXR-seq method29, the BPDE adduct–containing excision products, immunoprecipitated with anti-BPDE antibody, are subjected to a single round of primer extension by DNA polymerase K, which is able to bypass the damage in an error-free manner. Primer extension by TLS polymerase has also been used to replace the photo-reactivation step required in CPD XR-seq. For CPD-containing excision products, DNA polymerase η is used to accurately bypass the CPD in the primer extension reaction29. The extension products are amplified to generate a library for next-generation sequencing (NGS).
Modifications to the procedure were also made to apply XR-seq to species for which immuno-precipitation with anti-human repair protein antibodies is not applicable. For XR-seq in E. coli20, cell lysis is followed by anti-DNA damage–specific immunoprecipitation. The remaining procedure, including a second damage-specific immunoprecipitation following ligation of excision products to adapters, is the same as in the original procedure. Experiments with yeast19 and A. thaliana18 follow this same strategy, with the exception that very harsh methods for cell wall lysis are applied. By contrast, excision products in mice are complexed with TFIIH, and only modest procedural modifications of the original XR-seq protocol are needed to isolate the excised oligomers from various mouse tissues in a form amenable to immunoprecipitation with anti-mammalian TFIIH antibodies.
Advantages and applications of XR-seq
XR-seq is a powerful tool that enables genome-wide analysis of nucleotide excision repair of different DNA-damaging agents in a wide variety of organisms and formats, including cultured cells and entire organisms or tissues. The two-step immunoprecipitation procedure with different antibodies excludes most nonspecific genomic DNA fragments, which reduces the background of XR-seq to nearly zero. During excision repair, the excision products are continually degraded by nucleases, thus the repair map generated at each time point following damage illustrates a snapshot of a dynamic process of ongoing repair. The method indirectly but robustly detects transcription as well.
XR-seq has been applied to explore species-specific and damage-specific characteristics of repair, as well as regulation of repair by chromatin structure and states. The ‘chromatin state’ of a genomic region can refer to the set of chromatin-associated proteins and histone modifications in that region30. The most striking finding is the extent of the effect of transcription on repair. A transcription-coupled repair pathway exists in which RNA polymerase II (RNAP II) blocked by a template strand lesion targets the lesion for rapid repair by the basal repair factors5. For lesions such as CPDs and cisplatin-DNA adducts that are poorly repaired by the basal repair factors, this pathway greatly accelerates repair of template strand lesions, whereas the readily recognized (6–4)PP is rapidly repaired in both strands. Strong strand-specific repair has been observed so far in mouse17, A. thaliana18, cultured human cells13, budding yeast19 and E. coli20,21 genomes, although in the bacterium it is somewhat obscured due to the prevalence of overlapping genes and antisense transcription31–33.
XR-seq has been applied to reveal numerous features of transcription-coupled repair. Analyses of genes from higher organisms have revealed associations between RNAP II density and repair and between transcription level and repair, and have also detected divergent transcription at promoters13. These analyses have also shown a strong correspondence between sites of RNAP II pausing (by native elongating transcript sequencing (NET-seq)) and repair, and shown progression of repair, with time, from the transcribed strand to the nontranscribed strand19. XR-seq maps have also shown repair to be prioritized by chromatin accessibility. Time course studies showed that, in mammalian cells, there is a bias toward repair of more accessible regions, which is most pronounced at early time points14,15.
Interestingly, studies with A. thaliana plants18 and mice17 have demonstrated that repair exhibits circadian rhythmicity owing to the rhythmic control of transcription (and thus transcription-coupled repair) of many genes. In mice, the basal repair also oscillates34,35, with a peak activity just before dusk. This leads to complex patterns of repair in mice, and the startling observation that, in some genes, the transcribed strand is preferentially repaired in the morning whereas the nontranscribed strand is repaired in the evening.
At the molecular level, DNA metabolic processes such as replication and processing of interstrand crosslinks can be studied to examine their effects on the distribution of repair. Also, XR-seq data have been used to examine how repair heterogeneity determines the distribution of mutation frequency in some cancers12,36–40. These comparisons are possible in well-characterized systems in which genome-wide chromatin structure and mutagenesis data are available. In these and in less-well-defined systems, the procedure will be valuable toward defining the basic repair and repair-associated proteins for a given species by the use of appropriate mutant or transgenic cells or organisms. XR-seq can be applied to study a variety of mutagens individually, or to study the consequences of exposure to multiple drugs or toxins, such as in complex environmental mixtures or anticancer therapy regimens, and possible interactions and non-additive effects, and thereby guide the development of drugs and assessments of their toxicity.
Cells exist in various stages of growth and differentiation. In light of the utility of XR-seq as a sensitive probe of gene expression, both gene activity and regions of open chromatin can be probed by XR-seq. The effects of disruption of growth by nutrient deprivation or growth stimulation and different types of stress (altered oxygen availability, pH and temperature) on DNA repair and transcription can be studied by XR-seq. These and other factors, including pathogenic responses and cell-cell interactions, can be investigated using XR-seq to obtain repair and transcription data relevant to normal physiological conditions.
At the organismal level, XR-seq studies allow detailed illustrations of circadian rhythmicity in transcription and repair in plants and mice. Embryonic and mature tissues under various conditions are also amenable to the procedure. Practical applications may be found in assessing transcription and repair in normal and diseased tissues during cancer therapy, in addressing issues such as drug resistance and evaluating treatments such as chronotherapy (therapy applied at a specific time of day to maximize the therapeutic index). Other applications may be found in improving the performance of biological systems such as crops.
Limitations
An XR-seq experiment typically yields on the order of 10 million excision product reads per sample. For human and mouse genomes, the coverage that this provides is less than ideal; that is, each individual excision product is unlikely to be represented more than once. There is less constraint with organisms having smaller genomes; several dozen reads or more for each excision site are commonly obtained for E. coli20,21.
XR-seq studies have revealed variations in repair in different regions of the genome. These variations have been attributed to factors that are known to influence repair, such as transcription. Analyses of these data have assumed nearly uniform distribution of damage, and experimental evidence for uniformity is available15. However, measurements of damage are perhaps even more impacted by constraints of coverage than are measurements of repair. The relatively low coverage in measurement of both damage and repair has hindered the ability to compare, genome-wide, repair as a function of damage level, and to explore factors relevant to repair, such as sequence context.
Perhaps the most serious limitation of XR-seq is the inability to measure true rates of repair. XR-seq can be used to determine relative rates, but because the excised oligomers are concurrently formed and degraded3, and rates of degradation in different species are unknown, actual repair rates cannot be calculated.
Degradation of the excision product is also an important factor in determining the amount of specimen needed for XR-seq. The level of degradation varies among species, cell lines and tissues, and thus specimen requirements are determined empirically. At early time points following DNA damage, the repair rate exceeds degradation, and full-length product is most readily available (Fig. 2a). However, as repair slows, excision products are available in diminishing amounts26. In isolated cases, it has been possible to ‘boost’ the repair signal. In one set of experiments, E. coli triple exonuclease mutant cells (exoI−, exoVII−, recJ−) were used to reduce digestion of the repair product. In a separate set of experiments, interestingly, uvrD− E. coli cells gave an exceptionally high yield of full-length product20. In these cells, the excision product is protected from nucleases by remaining annealed to the chromosome; the UvrD helicase assists in the release of the excision products from the genome. Unfortunately, eukaryotes have an excision reaction mechanism that is different from the bacterial mechanism. In mammalian repair, the damaged region is melted (in the reaction intermediate, the damage exists in a single-stranded region of a ‘repair bubble’) before incisions are made, and a comparable protective mechanism does not exist1.
Fig. 2 |. Representative gels showing the excision assay and isolation of PCR products during XR-seq library preparation.
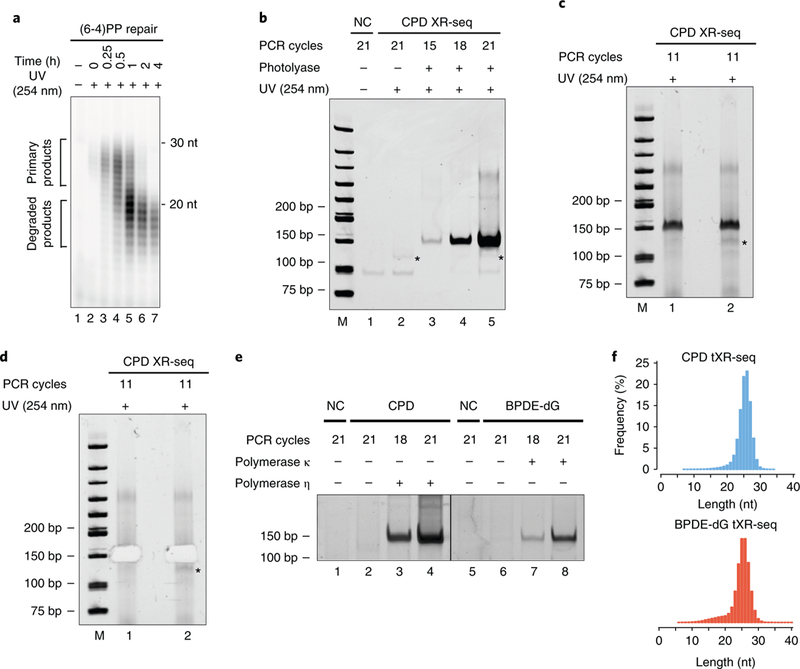
a, Image showing the excised oligomers containing (6–4)PPs at different time points after 10 J/m2 UV irradiation of A375 cells. For each lane, the excised oligomers were isolated by Hirt extraction from one 150-mm tissue culture dish of cells and then immunoprecipitated with anti-(6–4)PP antibody. Purified excised oligomers were 3′-end-labeled and resolved on a sequencing gel. Adapted from Hu et al.26 under a Creative Commons Attribution 4.0 license (https://creativecommons.org/licenses/by/4.0/legalcode). b, Image showing the pilot PCR products from Step 29. Lanes 3, 4 and 5 show the pilot PCR products after damage reversal. The PCR products in Lane 4, as shown by the band intensity, represent the optimal number of PCR cycles, which are sufficient for NGS. On the basis of this image, the optimal number of PCR cycles for the library preparation is 12 (18 – 6 = 12). c,d, Images showing a gel used to isolate the final PCR products of two biological replicates before (c, Step 35) and after (d, Step 37) excision from the gel. The black asterisk denotes the PCR products of adapter dimers. e, Image showing dsDNA libraries for tXR-seq29 in a pilot PCR reaction. CPD and BPDE-dG damage were bypassed by DNA polymerases η and κ, respectively. Lanes 3 and 8 represent the optimum pilot PCR results, and based on this result, 12 (18 – 6 = 12) and 15 (21 – 6 = 15) PCR cycles were chosen for CPD and BPDE-dG tXR-seq, respectively. f, Length distribution of excised oligomers from CPD tXR-seq and CPD XR-seq29. e,f adapted from Li et al., Human genome-wide repair map of DNA damage caused by the cigarette smoke carcinogen benzo[a]pyrene. Proc. Natl. Acad. Sci. USA 114, 6752–6757 (2017). M, DNA size marker; NC, non-template control.
The diminishing availability of excision repair product at late time points, when overall repair nears completion, may limit the opportunity to perform XR-seq in some cases. Opportunities may also be limited by the availability of desired biological samples, including clinical specimens.
Chemical mutagens introduce an additional timing issue, although this is not specific to the XR-seq method. UV light is an ideal mutagen in that it is readily delivered in controlled doses, with irradiation taking only seconds to minutes. By contrast, chemicals such as cisplatin and oxaliplatin can take considerably longer between initial dosing and DNA damage, even in cultured cells. Thus, repair begins as damage continues to accumulate. In animals, time is also needed for distribution throughout the body, and drugs such as benzo[a]pyrene require additional time for biotransformation to the ultimate mutagenic form41.
Comparison with other methods
The repair field has continually developed novel methods to successfully investigate interesting problems. To date, in vivo methods have commonly measured repair as loss of DNA damage with time following exposure to a mutagen. These in vivo methods have also commonly measured repair at the gene- and/or genome-wide levels, and adduct detection methods have been based on nuclease or chemical sensitivity, or immunoreactivity7–12,24,34,42–46.
A representative and illustrative example of these in vivo methods is the slot blot assay, which measures repair as loss of DNA damage from the entire genome with time34,35. At a given time point, slot blot data reveal what percentage of the total genome has been repaired, whereas XR-seq data reveal where in the genome repair occurred. The same anti-DNA damage antibodies can be used in both methods. Thus, slot blot is valuable because it complements XR-seq. In slot blot, genomic DNA is isolated from control untreated cells and mutagen-treated cells either unrepaired or repaired for various times. An apparatus is used to apply equivalent amounts of each DNA sample to a membrane in uniform-sized ‘slots’. The membrane is probed with anti-DNA damage–specific antibodies, and repair is inferred from the loss of the damaged DNA signal with time. This method is applicable to diverse species. For example, it was used to demonstrate overall repair of UV damage in plant cells24 and to confirm in vitro studies47 by demonstrating circadian rhythmicity in overall excision repair in mice35.
Other related methods use different strategies to measure damage and repair (as loss of damage) in specific genomic fragments or genome-wide. One such strategy uses a UV damage-specific nuclease to digest DNA at damage sites, and then different detection methods are used to measure the damage level. Among these methods, Southern blot can detect the total damage level in a specific region of the genome at low resolution42. Three high-resolution methods, ligation-mediated PCR43,48,49, radiolabeled primer extension of damaged templates50,51 and direct end labeling of DNA fragments cleaved at lesions44,52, are able to map DNA damage sites at nucleotide resolution in a specific region. Recently, Excision-seq8 and CPD-seq6,11 have been developed to map damage sites at single-nucleotide resolution in the whole genome. Another strategy uses damage-specific antibodies to enrich damaged fragments, followed by microarray chip detection7,9,45 or high-throughput sequencing12,46 to map damage sites genome-wide but at low resolution (approximately several hundred base pairs). To overcome this limitation, Damage-seq10 utilizes a high-fidelity DNA polymerase, which is blocked by damage, to detect the accurate positions of damage in immunoprecipitation-enriched damaged fragments, in order to measure damage at single-nucleotide resolution in the whole genome.
These methods have yielded information about the influences of chromatin structure and transcription on damage formation and repair. However, these methods have inherent limitations in either the fraction of the genome characterized (in the case of studying specific genomic fragments) or the resolution (in the case of genomic DNA fragmentation and microarray detection). The most serious drawback common to all these approaches is that repair is measured indirectly by subtraction, that is, as loss of the damage signal with time. By contrast, with XR-seq, the repair signal is directly detected above a background of zero. This is an important difference, especially at early time points when only a small percentage of the damage has been repaired. Subtraction of two large percentages of damage has very low sensitivity and low resolution, whereas the direct-detection approach of XR-seq provides high resolution and sensitivity. Early time points are especially informative because they directly show where and when repair is initially prioritized.
The XR-seq technology is within the range of time (7–9 d) and cost (~$2,000) of other genomics methods used in any modern molecular biology laboratory.
Experimental design
The step-by-step protocol described in the Procedure has been optimized for one XR-seq experiment applicable to different species. We strongly recommend using a control (as explained further below) to monitor the success of Step 20 in the Procedure. For CPD damage, damage reversal and TLS in Step 21 are two alternative options in the protocol. At least two biological replicates are needed to obtain meaningful data in each of the options described in this protocol.
DNA damage induction and repair incubation
The quantity of excised oligomer isolated in this protocol is critical to the success of XR-seq. As the induction of DNA damage and incubation to allow repair are the most critical steps of XR-seq, we recommend performing an excision assay26 (Box 1) to determine the ideal amount of DNA-damaging agent, the number of cells and the repair incubation time before starting the XR-seq experiment. For example, excision assay of (6–4)PP, as shown in Fig. 2a, indicates that XR-seq should be done following relatively short repair incubation times (in minutes), whereas comparable assays of CPD repair (not shown) indicate that XR-seq of these lesions should be assayed after ≥1 h of repair. For A. thaliana, yeast and E. coli, which have photolyases, until proteins are denatured and separated from excision products in Step 2D(iv), 2E(x) and 2F(viii), respectively, it is very important to use yellow light for illumination and avoid exposure to white and blue light, which can initiate reversal of the UV damage.
Box 1 |. Excision assay  Timing 3 d.
Timing 3 d.
This procedure describes the excision assay used to determine the ideal amount of DNA-damaging agent, the number of cells, and the repair incubation time.
Procedure
-
1
Perform the DNA-damage induction and repair incubation as described in Step 1.
-
2Perform the cell lysis and low-molecular-weight DNA isolation by following one of the options (A-F) according to the organism and DNA-damaging agent used in the experiment.
-
(A)Human cells treated with UV
-
(i)Collect the cells as described in Step 2A(i-iv).
-
(ii)Resuspend the cells in 360 μl of 1× TE, add 40 μl of 10% (wt/vol) SDS, gently invert the tube ten times and incubate it at room temperature for 10 min.
-
(iii)Add 100 μl of 5 M NaCl, gently invert the tube ten times and incubate it at 4 °C overnight.
-
(iv)Centrifuge the tube at 16,800g for 1 h at 4 °C.
-
(v)Transfer the supernatant to a new 1.5-ml tube.
-
(vi)Add 5 μl of RNase A to the tube, mix well by pipetting and incubate the tube at 37 °C for 1 h.
-
(vii)Add 5 μl of proteinase K to the tube, mix it well by pipetting and incubate the tube in a water bath at 55 °C for 1 h.
-
(viii)Isolate the low-molecular-weight DNA as described in Box 2, and dissolve the pellet in 80 μl of distilled water.
-
(i)
-
(B)Human cells treated with cisplatin or BPDE
-
(i)Collect the cells as described in Step 2B(i-iii).
-
(ii)Lyse the cells and isolate the low-molecular-weight DNA as described in step 2A(ii-viii) above.
-
(i)
-
(C)Mice treated with cisplatin
-
(i)Harvest and homogenize the organ as described in Step 2C(i-ix) of the main Procedure.
-
(i)
-
(D)A. thaliana treated with UV
-
(i)Lyse the cells and isolate the low-molecular-weight DNA as described in Step 2D(i-vii) of the main Procedure.
-
(i)
-
(E)S. cerevisiae treated with UV
-
(i)Lyse the cells and isolate the low-molecular-weight DNA as described in Step 2E(i-xiv) of the main Procedure.
-
(i)
-
(F)E. coli treated with UV
-
(i)Lyse the cells and isolate the low-molecular-weight DNA as described in Step 2F(i-xvii) of the main Procedure.
-
(i)
-
(A)
-
3Perform the immunoprecipitation with anti-TFIIH/XPG or anti-DNA damage-specific antibodies by following one of the options (A-D).
-
(A)Human cells treated with UV, BPDE or cisplatin
-
(i)Perform the immunoprecipitation as described in Steps 13–18 of the main Procedure.
-
(ii)Extract the excised oligomers by following the protocol described in Box 2 and dissolve the pellet with 35 μl of distilled water.
-
(i)
-
(B)Mice treated with cisplatin
-
(i)Perform the immunoprecipitation with anti-TFIIH antibody and elution of the excised oligomers as described in Steps 3B(i) and (ii) and 4A(i-vii) of the main Procedure.
-
(ii)Isolate the excised oligomers as described in steps 3–7 of Box 2 and dissolve the pellet in 80 μl of distilled water.
-
(iii)Perform the immunoprecipitation with anti-DNA damage-specific antibody as described in Steps 13–18 of the main Procedure.
-
(iv)Extract the excised oligomers by following the protocol described in Box 2 and dissolve the pellet with 35 μl of distilled water.
-
(i)
-
(C)A. thaliana and S. cerevisiae treated with UV
-
(i)Perform the immunoprecipitation as described in Step 3C(i-vi) and Step 4 B(i-iii) of the main Procedure.
-
(ii)Extract the excised oligomers by following the protocol described in Box 2 and dissolve the pellet with 35 μl of distilled water.
-
(i)
-
(D)E. coli treated with UV
-
(i)Perform the immunoprecipitation and elution as described in Step 3D(i-iii) and Step 4B(i-iii) of the main Procedure.
-
(ii)Extract the excised oligomers by following the protocol described in Box 2 and dissolve the pellet with 35 μl of distilled water.
-
(i)
-
(A)
-
4.
Add 5 μl of 5× TdT buffer, 5 μl of CoCl2,1 μCi of [α−32P]-3′ dATP (Cordycepin) and 4 μl of RP1 oligo (1 μM) to the tube containing the excised oligomers from step 3 above, and mix well by pipetting at least ten times.
-
5.
Add 1 μl of TdT to the mixture from step 4 above, mix well by pipetting at least ten times and incubate the reaction in a water bath at 37 °C for 1 h.
-
6.
Extract the radiolabeled excised oligomers by following the protocol described in Box 2 and dissolve the pellet in 6 μl of loading buffer.
-
7.
Load 6 μl of the samples into a 10% denaturing acrylamide gel.
-
8.
Run the gel at 25–35 W (2,000 V, 35 mA) until the blue dye is 5–8 cm from the bottom. Then run at 60 W until the blue dye is 1–3 cm from the bottom.
-
9.
Stop running and transfer the gel to a filter paper, and cover it with a layer of plastic wrap.
-
10.
Put the gel on a gel dryer and dry it at 80 °C for 1 h.
-
11.
Expose the gel to a phosphor screen for 12 h in a radiography phosphor cassette.
-
12.
Scan the screen using a Typhoon phosphor imager.
Cell lysis and non-chromatin fraction separation
For mammalian tissues and cells, we use lysis buffer supplemented with 0.5% (vol/vol) NP40 and an ice-cold Dounce homogenizer to gently break the cells and nuclear membranes, and, importantly, to keep the repair complex intact during the manipulation. We have observed that storing pelleted cells, mouse organs or the non-chromatin fraction at −80 °C can decrease the efficiency of immunoprecipitation. We recommend performing the cell lysis and the separation of the non-chromatin fraction consecutively.
For yeast cells, we use bead beating to disrupt the cell walls and a Hirt lysis53 procedure to separate the non-chromatin fraction. To avoid degradation of the excised oligomers by nucleases, we resuspend the cells in TE buffer with proteinase K before bead beating. For E. coli cells, we directly use the Hirt lysis procedure with 1% (wt/vol) SDS lysis buffer19. For A. thaliana, we first freeze the seedlings in liquid nitrogen, grind them into a powder and then resuspend them in STES buffer with phenol/ chloroform/isoamyl alcohol before vortexing with glass beads. For other species, we recommend optimizing the cell lysis approach to avoid the degradation of the excised oligomers (such as seen in Fig. 2a) during their isolation.
Immunoprecipitation and isolation of excised oligomers
In human cells, the full-length excision products form a tight complex with TFIIH and XPG. These full-length excision products can be isolated by co-immunoprecipitation with TFIIH or XPG. For XR-seq of mouse samples, precipitation with anti-TFIIH antibodies alone has been found to work best. For E. coli, A. thaliana and yeast, the co-immunoprecipitation with TFIIH or XPG is replaced with immunoprecipitation with DNA damage-specific antibodies. Precipitation of yeast or plant excision products with anti-mammalian repair proteins has not been attempted, and the E. coli excision repair proteins and reaction mechanism are completely different from those of eukaryotes. When using DNA damage-specific antibodies other than the ones we describe, we recommend analyzing the efficiency of those antibodies by radiolabeling of excised, immunoprecipitated oligomers and running them on an 11% sequencing gel as described in Box 1 (Fig. 2a).
Controls
We recommend including a no-UV treatment group in Step 1. In Step 20, we strongly recommend including a negative control in which the enzyme for DNA damage reversal or TLS is omitted. After the pilot PCR, this will result in the absence of PCR product in Step 29 (Fig. 2b). Success of the DNA damage reversal or TLS is indicated by the presence of a PCR product of the expected size (Fig. 2b).
Materials
Biological materials
Human lymphocytes (Coriell Institute, ID no. GM12878) cultured in RPMI 1640 medium containing 15% (vol/vol) FBS and 2 mM glutamine at 37 °C in a 5% CO2 humidified chamber
 CAUTION The cell lines used in your research should be regularly checked to ensure they are authentic and are not infected with mycoplasma.
CAUTION The cell lines used in your research should be regularly checked to ensure they are authentic and are not infected with mycoplasma.NHF1 cells, telomerase-immortalized normal human fibroblasts derived from the foreskin of a normal newborn (a gift from the W. K. Kaufmann lab, University of North Carolina School of Medicine54), cultured in DMEM containing 10% (vol/vol) FBS and 2 mM glutamine at 37 °C in a 5% CO2 humidified chamber
CAUTION The cell lines used in your research should be regularly checked to ensure they are authentic and are not infected with mycoplasma.  CRITICAL The BJ-5ta cell line (ATCC, cat. no. CRL-4001) is a commercially available hTERT-immortalized normal human foreskin fibroblast cell line and can be used instead of NHF1.
CRITICAL The BJ-5ta cell line (ATCC, cat. no. CRL-4001) is a commercially available hTERT-immortalized normal human foreskin fibroblast cell line and can be used instead of NHF1.C57BL/6J mice (Jackson Laboratory, stock no. 000664)
CAUTION Any experiments involving live mice must conform to relevant institutional and national regulations. Our mice were handled in accordance with the guidelines of the NIH and the University of North Carolina School of Medicine Institutional Animal Care and Use Committee.A. thaliana strain Col-0 (a gift from the J. Dangl lab, University of North Carolina School of Medicine), grown on Murashige and Skoog (MS) agar medium at room temperature (18–27 °C)
CRITICAL Any other repair-proficient strain could also be used in this protocol.Saccharomyces cerevisiae strain Y452 (MATα, ura3–52, his3–1, leu2–3, leu2–112, cir°; a gift from the S. Li lab, Louisiana State University), cultured in YPD medium at 30 °C in a shaking incubator at 250 r.p.m.
CRITICAL Any other repair-proficient strain could also be used in this protocol.E. coli strain STL4150 (a gift from V. Burdett (the P. Modrich lab, Duke University)20), cultured in LB medium at 37 °C in a shaking incubator at 160 r.p.m.
CRITICAL Any other repair-proficient strain could also be used in this protocol.
Reagents
High-glucose DMEM (Thermo Fisher, cat. no. 11995065)
RPMI 1640 medium (Thermo Fisher, cat. no. 11875093)
FBS (Sigma-Aldrich, cat. no. TMS-013-B)
Trypsin-EDTA (Thermo Fisher, cat. no. 25300054)
L-Glutamine (Thermo Fisher, cat. no. 25030081)
DPBS (Thermo Fisher, cat. no. 14190144)
LB base (Thermo Fisher, cat. no. 12780029)
Murashige and Skoog (MS) basal medium (Sigma-Aldrich, cat. no. M5519)
Sucrose (Fisher Scientific, cat. no. S5–3)
Agar (for plant; Sigma-Aldrich, cat. no. A7921)
Yeast extract (Fisher Scientific, cat. no. BP1422–500)
Peptone (Fisher Scientific, cat. no. BP1420–500)
Dextrose (glucose; Sigma-Aldrich, cat. no. D9434)
Glass beads (acid-washed; Sigma-Aldrich, cat. no. G8772)
CO2 (Airgas, cat. no. CD USP50)
Liquid nitrogen (N2; Airgas, cat. no. NI NF180LT350)
CAUTION N2 is extremely cold. Handle it with care and wear protective clothing, thick gloves and safety goggles.Dimethyl sulfoxide (DMSO; VWR, cat. no. 97063–136)
CAUTION DMSO is an irritant and is flammable. Wear protective clothing, gloves and safety goggles when handling.Cis-diammineplatinum(II) dichloride (cisplatin; in vitro use, Sigma-Aldrich, cat. no. P4394; in vivo use, Fresenius Kabi Pharmaceutical, cat. no. 63323–103-51)
CAUTION Cisplatin is toxic and carcinogenic. Wear protective clothing, gloves and safety goggles when handling.Benzo[a]pyrene-r-7,t-8-dihydrodiol-t-9,10-epoxide(±)(anti) (BPDE; MRIGlobal, cat. no. 477)
CAUTION BPDE is mutagenic and carcinogenic. Wear protective clothing, gloves and safety goggles when handling.Nuclease-free H2O (Corning, cat. no. 46–000-CM)
Tris base (Fisher Scientific, cat. no. BP152–1)
Hydrochloric acid (HCl; Avantor, cat. no. 9535–02)
CAUTION HCl is corrosive and harmful. Handle it in a fume hood and wear protective clothing, gloves and safety goggles.Potassium hydroxide (KOH; Avantor, cat. no. 6984–04)
CAUTION KOH is corrosive and harmful. Wear protective clothing, gloves and safety goggles when handling.EDTA (Fisher Scientific, cat. no. 02793–500)
SDS (Fisher Scientific, cat. no. BP8200–500)
CAUTION SDS powder is harmful. Handle it in a fume hood and wear protective clothing, gloves and safety goggles.Sodium chloride (NaCl; Fisher Scientific, cat. no. BP358–212)
PBS (10×; Corning, cat. no. 46013CM)
HEPES (Fisher Scientific, cat. no. BP310–1)
Potassium phosphate, dibasic (K2HPO4; Avantor, cat. no. 3252–01)
Potassium phosphate, monobasic (KH2PO4; Avantor, cat. no. 7100–12)
Potassium chloride (KCl; Avantor, cat. no. 6858–06)
Magnesium chloride hexahydrate (MgCl2–6H2O; Fisher Scientific, cat. no. M35–500)
DTT (Fisher Scientific, cat. no. BP172–25)
CAUTION DTT is toxic. Handle it in a fume hood and wear protective clothing, gloves and safety goggles.Glycerol (Fisher Scientific, cat. no. G33–20)
Igepal CA-630 (NP-40 substitute; US Biological, cat. no. N3500)
CAUTION Igepal CA-630 is an irritant and is harmful. Wear protective clothing, gloves and safety goggles when handling.RNase A (Sigma-Aldrich, cat. no. R4642)
Anti-p89 (Santa Cruz Biotechnology, cat. no. sc-271500)
CRITICAL Antibodies from other suppliers may not be as efficient.Anti-p62 (Santa Cruz Biotechnology, cat. no. sc-25329)
CRITICAL Antibodies from other suppliers may not be as efficient.Anti-XPG (Santa Cruz Biotechnology, cat. no. sc-13563)
CRITICAL Antibodies from other suppliers may not be as efficient.Protein A/G-PLUS agarose (Santa Cruz Biotechnology, cat. no. sc-2003)
RNase A/T1 (Thermo Fisher, cat. no. EN0551)
Proteinase K (NEB, cat. no. P8107S)
Phenol/chloroform/isoamyl alcohol (25:24:1 (vol/vol/vol); Thermo Fisher, cat. no. 15593031)
CAUTION This item is toxic and corrosive. Handle it in a fume hood and wear protective clothing, gloves and safety goggles.Ethanol (Decon Labs, cat. no. 2716)
CAUTION Ethanol is flammable. Wear protective clothing, gloves and safety goggles when handling.Glycogen (Roche, cat. no. 10901393001)
Sodium acetate (NaAc; Amresco, cat. no. 0602–500G)
Acetic acid (HAc; VWR, cat. no. BDH3094)
CAUTION HAc is an irritant and is flammable. Handle it in a fume hood and wear protective clothing, gloves and safety goggles.T4 DNA ligase (5 U/μl; Thermo Fisher, cat no. 15224041)
PEG 8000 (50% (wt/vol), from T4 RNA ligase reaction buffer; NEB, cat. no. B0216L)
Anti-(6–4)PP (Cosmo Bio, cat. no. CAC-NM-DND-002)
CRITICAL Antibodies from other suppliers may not be as efficient.Anti-CPD (Cosmo Bio, cat. no. CAC-NM-DND-001)
CRITICAL Antibodies from other suppliers may not be as efficient.Rabbit-anti-mouse IgG (Abcam, cat. no. ab46540)
Anti-BPDE-DNA (clone 8E11; Trevigen, cat. no. 4360-MC-100)
CRITICAL Antibodies from other suppliers may not be as efficient.Anti-cisplatin-DNA (Abcam, cat. no. ab103261)
CRITICAL Antibodies from other suppliers may not be as efficient.Dynabeads Protein G (Thermo Fisher, cat. no. 10004D)
Dynabeads sheep-anti-rabbit IgG (Thermo Fisher, cat. no. 11203D)
Dynabeads sheep-anti-rat IgG (Thermo Fisher, cat. no. 11035)
Triton X-100 (Millipore, cat. no. 9400)
CAUTION Triton X-100 is an irritant and is harmful. Wear protective clothing, gloves and safety goggles when handling.Sodium deoxycholate (Sigma-Aldrich, cat. no. D6750–100G)
CAUTION Sodium deoxycholate is harmful. Handle it in a fume hood and wear protective clothing, gloves and safety goggles.Lithium chloride (LiCl; Sigma-Aldrich, cat. no. L9650–100G)
CAUTION LiCl is harmful. Handle it in a fume hood and wear protective clothing, gloves and safety goggles.Salmon sperm DNA (Thermo Fisher, cat. no. 15632011)
BSA (NEB, cat. no. B9000S)
Sodium cyanide (NaCN; Sigma-Aldrich, cat. no. 205222–100G)
CAUTION NaCN is highly toxic. Handle it in a fume hood with extreme care and wear protective clothing, gloves and safety goggles.Human DNA polymerase η (Enzymax, cat. no. 19)
CRITICAL Enzymes from other suppliers may not be as efficient.Human DNA polymerase k (Enzymax, cat. no. 27)
CRITICAL Enzymes from other suppliers may not be as efficient.dNTP mix (2.5 mM; Thermo Fisher, cat. no. R72501)
Terminal transferase (NEB, cat. no. M0315L)
[α−32P]-3′ dATP (Cordycepin; PerkinElmer, cat. no. NEG026250UC)
CAUTION This is a radioactive product. Handle it behind proper shielding and wear protective clothing, gloves and safety goggles. A license may be needed for using radioactive materials.Terminal transferase (TdT) (NEB, cat. no. M0315S)
Kapa HiFi PCR HotStart ReadyMix, (Kapa Biosystems, cat. no. KK2602)
Gel-loading dye (purple, with SDS; NEB, cat. no. B7024S)
Gel-loading dye (purple, no SDS; NEB, cat. no. B7025S)
Acrylamide (Thermo Fisher, cat. no. 15512–023)
CAUTION Acrylamide is an irritant and is toxic and carcinogenic. Handle it in a fume hood and wear protective clothing, gloves and safety goggles.Bisacrylamide (Millipore, cat. no. 2620–100GM)
CAUTION Bisacrylamide is an irritant and is toxic. Handle it in a fume hood and wear protective clothing, gloves and safety goggles.Urea (Fisher Scientific, cat. no. BP169–212)
Boric acid (Fisher Scientific, cat. no. BP168–1)
Ammonium persulfate (APS; Fisher Scientific, cat. no. BP179–25)
CAUTION APS is an irritant and is harmful. Handle it in a fume hood and wear protective clothing, gloves and safety goggles.TEMED (Thermo Fisher, cat. no. 15524010)
CAUTION TEMED is an irritant and is toxic and flammable. Handle it in a fume hood and wear protective clothing, gloves and safety goggles.Buffer EB (Qiagen, cat. no. 19086)
Qubit dsDNA HS Kit (Thermo Fisher, cat. no. Q32854)
Low-molecular-weight DNA ladder (NEB, cat. no. N3233S)
SYBR Gold (Thermo Fisher, cat. no. S33102)
CAUTION SYBR Gold is toxic and mutagenic. Handle it in a fume hood and wear protective clothing, gloves and safety goggles.DNA oligonucleotides (IDT, custom orders; see Table 1 for sequences)
Formamide (Sigma, cat. no. F9037)
Bromophenol blue (Sigma, cat. no. B0126)
Xylene cyanol FF (Sigma, cat. no. X4126)
Bleach (Fisher, cat. no. 50371478)
High Sensitivity DNA Analysis Kit (Agilent, cat. no. 5067–4626)
Equipment
Cell culture dishes (Corning, cat. no. 639160)
T175 flasks (Thermo Fisher, cat. no. 159910)
Cell scrapers (Corning, cat. no. 3011)
Syringes (BD, cat. no. 309659)
Needles (BD, cat. no. 305122)
Forceps (Fine Science Tools, cat. no. 11051–10)
Scissors (Fine Science Tools, cat. no. 14058–11)
Petri dishes (for plant) (VWR, cat. no. 25384–342)
Porcelain mortar (CoorsTek, cat. no. 60310)
Porcelain pestle (CoorsTek, cat. no. 60311)
Micropore surgical tape (3M, cat. no.1530–1)
Centrifuge tubes (15 ml; Corning, cat. no. 430790)
Centrifuge tubes (50 ml; Corning, cat. no. 430828)
Centrifuge tubes (40 ml; Sorvall, cat. no. 03718)
Oak Ridge tubes (polypropylene copolymer; Thermo Fisher, cat. no. 3119–0028)
Oak Ridge tubes (polycarbonate; Thermo Fisher, cat. no. 3118–0028)
DNA LoBind 1.5-ml tubes (Eppendorf, cat. no. 022431021)
Small PCR tubes (VWR, cat. no. 20170–010)
Countess II FL cell counter (Thermo Fisher, cat. no. AMQAF1000)
Countess Cell Counting Chamber Slides (Thermo Fisher, cat. no. C10228)
MicroSpin G50 columns (GE Healthcare, cat. no. 27–5330-01)
Gel Breaker Tubes (IST Engineering, cat. no. 3388100)
Spin-X centrifuge tube filter (Corning, cat. no. CLS8161–100EA)
Blade (Integra Miltex, cat. no. 4–122)
Thermal cycler (C1000 Touch model; Bio-Rad, cat. no. 1851148)
Centrifuge (1.5 ml; model no. 5418; Eppendorf, cat. no. 022620304)
Centrifuge (50 ml; model no. CL2; Thermo Fisher, cat. no. 004260F)
Centrifuge (for E. coli; Sorvall RC3BP Plus; Thermo Fisher; cat. no. 75007530)
Incubator shaker (for E. coli and yeast; model no. Innova 4330; Eppendorf, cat. no. M1193)
CO2 incubator (37 °C and 5% CO2; Fisher Scientific, cat. no. 20–235-32)
Digital heating shaking dry bath (Thermo Fisher, cat. no. 88880027)
Electrophoresis equipment (for native gel; Mini-PROTEAN Tetra model; Bio-Rad, cat. no. 1658001)
Power supply for electrophoresis (for native gel; PowerPac HC model; Bio-Rad, cat. no. 1645052)
Imager (ChemiDoc XRS+ model; Bio-Rad, cat. no. 1708265)
Gibco S2 Sequencing Gel Electrophoresis Apparatus System (Life Technologies, cat. no. 21105–010)
PowerPac 3000 electrophoresis power supply (Bio-Rad Laboratories, cat. no. 165–5056)
GD2000 vacuum gel-drying system (Hoefer, cat. no. GD2000–115V)
Phosphor screen (model no. BAS-IP SR 2040 E; GE Healthcare, cat. no. 28956477)
Phosphor screen scanner (Typhoon TRIO+ model; GE Healthcare, cat. no. 28998418)
Qubit 3.0 Fluorometer (Thermo Fisher, cat. no. Q33216)
Agilent Bioanalyzer 2100 (Agilent, cat. no. G2940CA)
Mixer (Vortex Genie 2 model; Scientific Industries, cat. no. SI-0236)
Tube revolver/rotator (Thermo Fisher, cat. no. 88881001)
Magnetic stand (DynaMag-2 Magnet; Thermo Scientific, cat. no. 12321D)
Electronic timer (GraLab, model no. 451)
Germicidal lamp (254 nm; model no. G8T5; GE Lighting, cat. no. 11077)
CAUTION UV irradiation is harmful and carcinogenic. Handle it in a hood and wear protective clothing, gloves and safety goggles.Black light bulb (model no. F15T8/BLB; GE Lighting, cat. no. 35885)
CAUTION UV-A generated by this bulb is harmful and carcinogenic. Handle it behind a glass plate and wear protective clothing, gloves and safety goggles.Mini-beadbeater-16 (Mini-Beadbeater-16 model; Bio Spec Products, cat. no. 607)
XXTuff reinforced vial (2 ml, non-sterile; BioSpec Products, cat. no. 330TX).
2.0-ml Vial adapter for Mini-BeadBeater-16 (BioSpec Products, cat. no. 607TC16)
Dounce homogenizers (Wheaton, cat. nos. 357538 (1 ml), 357542 (7 ml), 357544 (15 ml))
Sequencing system (Illumina, model no. HiSeq 2500)
Reagent setup
Liquid LB medium
To prepare liquid LB medium, dissolve 20 g of LB base (powder) per liter of distilled water. Autoclave the medium and store at 4 °C for up to 12 months.
MS agar medium
To prepare MS agar medium, dissolve 4.33 g of MS basal medium, 20 g of sucrose and 8 g of agar per liter of distilled water, and adjust the pH to 5.7 with KOH. Autoclave and store at 4 °C for up to 12 months.
YPD medium
To prepare YPD medium, dissolve 10 g of yeast extract, 20 g of peptone and 20 g of dextrose per liter of distilled water. Autoclave and store at 4 °C for up to 12 months.
1 M Tris-HCl buffer
To prepare 1 M, pH 8.0, Tris-HCl buffer, dissolve 121.2 g of Tris base in 800 ml of distilled water, adjust the pH to 8.0 with HCl, and then add distilled water to bring the volume to 1 liter. Autoclave and store at room temperature for up to 12 months.
0.5 M EDTA stock solution
To prepare 0.5 M, pH 8.0, EDTA stock solution, add 186.12 g of EDTA to 700 ml of distilled water, adjust the pH to 8.0 with NaOH (EDTA will dissolve when the pH is adjusted to 8.0), and then add distilled water to bring the volume to 1 liter. Autoclave and store at room temperature for up to 12 months.
TE buffer
TE buffer is 10 mM Tris and 1 mM EDTA, pH 8.0. To prepare the buffer, mix 10 ml of 1 M Tris-HCl (pH 8.0), and 2 ml of 0.5 M EDTA, and add sterilized distilled water to bring the volume to 1 liter. Store at room temperature for up to 12 months.
10% (wt/vol) SDS
To prepare 10% (wt/vol) SDS solution, dissolve 1 g of SDS powder in a total volume of 10 ml of distilled water. Store at room temperature for up to 12 months.
5 M NaCl
To prepare 5 M NaCl solution, dissolve 292 g of NaCl in a total volume of 1 liter of distilled water. Autoclave and store at room temperature for up to 12 months.
2% (wt/vol) Glucose (dextrose)
To prepare 2% (wt/vol) glucose solution, dissolve 20 g of dextrose in a total volume of 1 liter of distilled water. Filter-sterilize and store at 4 °C for up to 12 months.
3 M NaAc
To prepare 3 M, pH 5.2, NaAc solution, dissolve 40.8 g of NaAc in 40 ml of distilled water, adjust the pH to 5.2 with HAc, and then add distilled water to bring the volume to 100 ml. Autoclave and store at room temperature for up to 12 months.
20 mM Cisplatin solution (in vitro)
To prepare 20 mM cisplatin solution, dissolve 6 mg of cisplatin in 1 ml of DMSO. Prepare this solution fresh before use.
4 mM BPDE solution
To prepare 4 mM BPDE solution, dissolve 1.2 mg of BPDE in 1 ml of DMSO. Prepare fresh solution before use.
4 M NaCN
To prepare 4 M NaCN solution, dissolve 196 mg of NaCN in 1 ml of buffer EB. Prepare fresh solution before use.
1 M HEPES buffer
To prepare 1 M, pH 7.9, HEPES buffer, dissolve 238.3 g of HEPES in 800 ml of distilled water, adjust the pH to 7.9 with NaOH, and then add distilled water to bring the volume to 1 liter. Filter-sterilize and store at 4 °C for up to 12 months.
2 M KCl stock solution
To prepare 2 M KCl stock solution, dissolve 74.55 g of KCl in a total volume of 500 ml of distilled water. Autoclave and store at room temperature for up to 12 months.
1 M MgCl2 stock solution
To prepare 1 M MgCl2 stock solution, dissolve 20.33 g of MgCl2-6H2O in a total volume of 100 ml of distilled water. Autoclave and store at room temperature for up to 12 months.
1 M DTT stock solution
To prepare 1 M DTT stock solution, dissolve 1.54 g of DTT in a total volume of 10 ml of distilled water. Filter-sterilize and store at −20 °C for up to 12 months.
Buffer A (for TFIIH immunoprecipitation)
Buffer A is 25 mM HEPES, 100 mM KCl, 12 mM MgCl2, 0.5 mM EDTA, 2 mM DTT, 12.5% (vol/vol) glycerol and 0.5% (vol/vol) Igepal CA-630 (pH 7.9). To prepare the buffer, mix 25 ml of 1 M HEPES (pH 7.9), 50 ml of 2 M KCl, 12 ml of 1 M MgCl2, 1 ml of 0.5 M EDTA, 2 ml of 1 M DTT, 125 ml of glycerol and 5 ml of Igepal CA-630, and then add sterilized distilled water to bring the volume to 1 liter. Store at 4 °C for up to 6 months.
Buffer B (for TFIIH immunoprecipitation)
Buffer B is 25 mM HEPES, 100 mM KCl, 12 mM MgCl2, 0.5 mM EDTA, 2 mM DTT, 12.5% (vol/vol) glycerol and 1% (vol/vol) Igepal CA-630 (pH 7.9). To prepare the buffer, mix 25 ml of 1 M HEPES (pH 7.9), 50 ml of 2 M KCl, 12 ml of 1 M MgCl2, 1 ml of 0.5 M EDTA, 2 ml of 1 M DTT, 125 ml of glycerol and 10 ml of Igepal CA-630, and then add sterilized distilled water to bring the volume to 1 liter. Store at 4 °C for up to 6 months.
STES buffer
STES buffer is 200 mM Tris-HCl buffer, 500 mM NaCl, 0.1% (wt/vol) SDS and 10 mM EDTA, pH 8.0. To prepare the buffer, mix 20 ml of 1 M Tris-HCl buffer (pH 8.0), 10 ml of 5 M NaCl, 1 ml of 10% (wt/vol) SDS and 2 ml of 0.5 M EDTA, and then add sterilized distilled water to bring the volume to 100 ml. Store at room temperature for up to 12 months.
5× TBE stock solution
To prepare 5× TBE stock solution, dissolve 54 g of Tris base, 27.5 g of boric acid and 20 ml of 0.5 M EDTA (pH 8.0) in a total volume of 1 liter of distilled water. Pass the solution through a 0.22-μm filter and store at room temperature for up to 12 months.
30% (wt/vol) 29:1 acrylamide/bisacrylamide stock solution
To prepare 30% (wt/vol) 29:1 acrylamide/bisacrylamide stock solution, dissolve 29 g of acrylamide powder and 1 g of bisacrylamide powder in a total volume of 100 ml of distilled water. Pass the solution through a 0.22-μm filter and store at 4 °C for up to 6 months (avoid light).
10% (wt/vol) APS solution
To prepare 10% (wt/vol) APS solution, dissolve 0.1 g of APS powder in a total volume of 1 ml of distilled water. Store at 4 °C for up to 1 week (avoid light).
Native polyacrylamide gel (10%)
Native polyacrylamide gel (10%) is 10% (wt/vol) acrylamide/bisacrylamide 29:1, 1× TBE, 1.67% (wt/vol) APS and 0.83% (vol/vol) TEMED. To prepare two gels, mix 4 ml of 30% (wt/vol) acrylamide/ bisacrylamide solution, 5.6 ml of distilled water, 2.4 ml of 5× TBE, 200 μl of 10% (wt/vol) APS and 10 μl of TEMED. Prepare fresh gels before use.
Denaturing gel (10%) working solution
Denaturing gel (10%) working solution is 10% (wt/vol) acrylamide/bisacrylamide 19:1, 2× TBE and 7 M urea. Dissolve 47.5 g of acrylamide powder, 2.5 g of bisacrylamide powder and 210.2 g of urea in 200 ml of 5× TBE stock, then add distilled water to a total volume of 500 ml. Heat the mixture to dissolve the powders (do not exceed 30 °C during dissolving). Pass the solution through a 0.22-μm filter and store at room temperature for up to 6 months (avoid light). To prepare the gel, add 200 μl of 10% (wt/vol) APS and 10 μl of TEMED to 40 ml of this solution immediately before use.
Gel-loading dye for denaturing gel
Gel-loading dye for denaturing gel is 95% (vol/vol) deionized formamide, 0.025% (wt/vol) bromophenol blue, 0.025% (wt/vol) xylene cyanol FF and 5 mM EDTA. To prepare the gel, mix 9.5 ml of formamide, 2.5 mg of bromophenol blue, 2.5 mg of xylene cyanol FF, 100 μl of 0.5 M EDTA (pH 8.0) and 400 μl of distilled water. Store in aliquots at −20 °C for up to 12 months.
Elution buffer I (for immunoprecipitation)
Elution buffer I is 10 mM Tris-HCl, 1 mM EDTA and 1% (wt/vol) SDS, pH 8.0. To prepare the buffer, mix 1 ml of 1 M Tris-HCl (pH 8.0), 10 ml of 10% (wt/vol) SDS and 0.2 ml of 0.5 M EDTA, and then add sterilized distilled water to bring the volume to 100 ml. Store at room temperature for up to 12 months.
10× Hybridization buffer
10× Hybridization buffer is 100 mM Tris-HCl, 1 M NaCl and 1 mM EDTA, pH 8.0. To prepare the buffer, mix 1 ml of 1 M Tris-HCl (pH 8.0), 2 ml of 5 M NaCl and 20 μl of 0.5 M EDTA, and then add sterilized distilled water to bring the volume to 10 ml. Store at room temperature for up to 12 months.
10% (wt/vol) Sodium deoxycholate
To prepare 10% (wt/vol) sodium deoxycholate solution, dissolve 1 g of sodium deoxycholate powder in a total volume of 10 ml of distilled water. Store at room temperature for up to 12 months.
Reaction buffer
Reaction buffer is 20 mM Tris-HCl, 2 mM EDTA, 150 mM NaCl, 1% (vol/vol) Triton X-100 and 0.5% (wt/vol) sodium deoxycholate, pH 8.0. To prepare the buffer, mix 1 ml of 1 M Tris-HCl (pH 8.0), 1.5 ml of 5 M NaCl, 0.2 ml of 0.5 M EDTA, 0.5 ml of Triton X-100 and 2.5 ml of 10% (wt/vol) sodium deoxycholate, and then add sterilized distilled water to bring the volume to 50 ml. Store at room temperature for up to 12 months.
5× Reaction buffer
5× Reaction buffer is 100 mM Tris-HCl, 10 mM EDTA, 750 mM NaCl, 5% (vol/vol) Triton X-100 and 2.5% (wt/vol) sodium deoxycholate, pH 8.0. To prepare the buffer, mix 1 ml of 1 M Tris-HCl (pH 8.0), 1.5 ml of 5 M NaCl, 0.2 ml of 0.5 M EDTA, 0.5 ml of Triton X-100 and 2.5 ml of 10% (wt/vol) sodium deoxycholate, and then add sterilized distilled water to bring the volume to 10 ml. Store at room temperature for up to 12 months.
Wash buffer I
Wash buffer I is 20 mM Tris-HCl, 2 mM EDTA, 150 mM NaCl, 1% (vol/vol) TritonX-100 and 0.1% (wt/vol) SDS, pH 8.0. To prepare the buffer, mix 1 ml of 1 M Tris-HCl (pH 8.0), 1.5 ml of 5 M NaCl, 0.2 ml of 0.5 M EDTA, 0.5 ml of Triton X-100 and 0.5 ml of 10% (wt/vol) SDS, and then add sterilized distilled water to bring the volume to 50 ml. Store at room temperature for up to 12 months.
Wash buffer II
Wash buffer II is 20 mM Tris-HCl, 2 mM EDTA, 500 mM NaCl, 1% (vol/vol) TritonX-100 and 0.1% (wt/vol) SDS, pH 8.0. To prepare the buffer, mix 1 ml of 1 M Tris-HCl (pH S.G), 5 ml of 5 M NaCl, 0.2 ml of 0.5 M EDTA, 0.5 ml of Triton X-100 and 0.5 ml of 10% (wt/vol) SDS, and then add sterilized distilled water to bring the volume to 50 ml. Store at room temperature for up to 12 months.
5 M LiCl stock solution
To prepare 5 M LiCl stock solution, dissolve 42.39 g of LiCl in a total volume of 200 ml of distilled water. Autoclave and store at room temperature for up to 12 months.
Wash buffer III
Wash buffer III is 10 mM Tris-HCl, 1 mM EDTA, 150 mM LiCl, 1% (vol/vol) Igepal CA-630and 1% (wt/vol) sodium deoxycholate, pH 8.0. To prepare the buffer, mix 0.5 ml of 1 M Tris-HCl (pH 8.0), 1.5 ml of 5 M LiCl, 0.1 ml of 0.5 M EDTA, 0.5 ml of Igepal CA-630 and 5 ml of 10% (wt/vol) sodium deoxycholate, and then add sterilized distilled water to bring the volume to 50 ml. Store at room temperature for up to 12 months.
Wash buffer IV
Wash buffer IV is 10 mM Tris-HCl, 1 mM EDTA, 500 mM LiCl, 1% (vol/vol) Igepal CA-630 and 1% (wt/vol) sodium deoxycholate, pH 8.0. To prepare the buffer, mix 0.5 ml of 1 M Tris-HCl (pH 8.0), 5 ml of 5 M LiCl, 0.1 ml of 0.5 M EDTA, 0.5 ml of Igepal CA-630 and 5 ml of 10% (wt/vol) sodium deoxycholate, and then add sterilized distilled water to bring the volume to 50 ml. Store at room temperature for up to 12 months.
PEX buffer
PEX buffer is 1× PBS, 2 mM EDTA and G.1% (vol/vol) Triton X-100. To prepare the buffer, mix 5 ml of 10× PBS, 0.2 ml of 0.5 M EDTA and 50 μl of Triton X-100, and then add sterilized distilled water to bring the volume to 50 ml. Store at room temperature for up to 12 months.
1× PEXB buffer
1× PEXB buffer is 1× PBS, 2 mM EDTA, G.1% (vol/vol) Triton X-100 and 0.025% (wt/vol) BSA. To prepare the buffer, mix 5 ml of 10× PBS, 0.2 ml of 0.5 M EDTA, 50 μl of Triton X-100 and 0.625 ml of 20 mg/ml BSA, and then add sterilized distilled water to bring the volume to 50 ml. Store at 4 °C for up to 6 months.
5× PEXB buffer
5× PEXB buffer is 5× PBS, 10 mM EDTA, G.S% (vol/vol) Triton X-100 and 0.125% (wt/vol) BSA. To prepare the buffer, mix 5 ml of 10× PBS, 0.2 ml of 0.5 M EDTA, 50 μl of Triton X-100 and 0.620 ml of 20 mg/ml BSA, and then add sterilized distilled water to bring the volume to10 ml. Store at 4 °c for up to 6 months.
10× Photoreactivation buffer
10× Photoreactivation buffer is 500 mM Tris-HCl, 1,000 mM NaCl, 10 mM EDTA and 100 mM DTT, pH 8.0. To prepare the buffer, mix 0.5 ml of 1 M Tris-HCl (pH 8.0), 0.2 ml of S M NaCl, 20 μl of 0.5 M EDTA and 0.1 ml of 1 M DTT, and then add sterilized distilled water to bring the volume to 1 ml. Store at −20 °c for up to 12 months.
1 M Potassium phosphate buffer
To prepare 1 M potassium phosphate buffer (pH 7.0), dissolve 1.07 g of K2HPO4 and 0.524 g of KH2PO4 in a total volume of 10 ml of distilled water. Autoclave and store at room temperature for up to 12 months.
2× TLS polymerase buffer
2× TLS polymerase buffer is 50 mM potassium phosphate, 10 mM MgCl2, 5 mM DTT, 200 μg/ml BSA, 20% (vol/vol) glycerol and 200 μM dNTP mix, pH 7.0. To prepare the buffer, mix 50 μl of 1 M potassium phosphate buffer (pH 7.0), 10 μl of 1 M MgCl2, 5 μl of 1 M DTT, 10 μl of 20 mg/ml BSA, 200 μl of glycerol and 80 μl of 2.5 mM dNTP mix, and then add nuclease-free water to bring the volume to 1 ml. Store at −20 °c for up to 12 months.
5× TBE buffer
5× TBE buffer is 445 mM Tris, 445 mM boric acid and 10 mM EDTA, pH 8.3. To prepare the buffer, dissolve 54 g of Tris base, 27.5 g of boric acid and 20 ml of 0.5 M EDTA (pH 8.0) in a total volume of 1 liter of distilled water. Store at room temperature for up to 12 months.
Elution buffer II (for recovering DNA from polyacrylamide gel)
Elution buffer II is 10 mM Tris, 300 mM NaCl and 1 mM EDTA, pH 8.0. To prepare the buffer, mix 1 ml of 1 M Tris-HCl (pH 8.0), 6 ml of 5 M NaCl and 0.2 ml of 0.5 M EDTA, and then add sterilized distilled water to bring the volume to 100 ml. Store at room temperature for up to 12 months.
A3 and A5 adapters
To prepare the adapters, combine 20 μl of A3F or A5F (250 μM) with 20 μl of A3R or A5R (250 μM), respectively (the oligonucleotides are listed in Table 1); add 5 μl of 10× hybridization buffer and 5 μl of nuclease-free H2O; and mix well by pipetting up and down five times. Boil for 1 min in water, then turn off the heater and let the solution naturally cool to 25 °C to obtain 100 μM A3 or A5 stock solution. Combine 10 μl of A3 or A5 stock solution (100 μM) with 4 μl of 10× hybridization buffer and 36 μl of nuclease-free H2O, and then mix well by pipetting up and down five times to obtain 20 μM A3 or A5 working solutions. Store the stock solutions and working solutions at −20 °C for up to 24 months (stock solutions) or 12 months (working solutions).
Equipment setup
Bioinformatics pipeline
The pipeline is run in a Linux environment. We provide a documented script library to help users to set up an environment and install the necessary prerequisites (https://github.com/adebali/NGStoolkit). The sequence processing steps (Steps 47–52) are collected in a single. bash script, which can be found at https://github.com/adebali/NGStoolkit/blob/master/stable/XR-seq-basics.sh. To bypass the dependency installation process (e.g., Cutadapt, Bowtie2, SAMtools, BEDTools), we provide a Docker image (see the repository) that provides a Linux container containing all the necessary programs and scripts needed to perform the basic analysis provided here. See the documentation on the repository home page for setup instructions.
Procedure
DNA damage induction and repair incubation ●Timing 10 min-48 h
-
1Induce DNA damage with UV (254 nm), cisplatin or BPDE and allow nucleotide excision repair to function for appropriate time periods. Perform this step by choosing one of the options (A–G) based on the organism and DNA-damaging agent.
-
(A)Treatment of human cells with UV
- CRITICAL The UV treatment has been optimized for NHF1 cells. Protocol optimization might be needed if other cell lines are used (Experimental design).
-
(i)Grow the NHF1 cells to 80% confluence in 150-mm tissue culture dishes in DMEM with 10% (vol/vol) FBS. Use one dish for (6–4)PP XR-seq and five dishes for CPD XR-seq.
-
(ii)Discard the DMEM, add 15 ml of 1× PBS per dish to wash the cells and then discard the 1× PBS.
-
(iii)With the cover off, place the cells under a germicidal lamp emitting 254-nm UV (1 J/m2/s) for 20 s, add 20 ml of DMEM to the tissue culture dish, and incubate the cells at 37 °C for an appropriate time period in a humidified chamber. Proceed directly to cell lysis and fractionation (Step 2).
-
(i)
- CAUTION UV irradiation is harmful and carcinogenic. Handle the lamp in a hood and wear protective clothing, gloves and safety goggles.
- CRITICAL STEP Count the incubation time from the beginning of UV irradiation. The ideal incubation durations for (6–4)PP XR-seq and CPD XR-seq range from 20 min to 2 h and from 2 h to 24 h, respectively.
-
(B)Treatment of human cells with cisplatin
- CRITICAL The following cisplatin treatment procedure has been optimized for human GM12878 cells. The total cell numbers and treatment procedures should be further optimized if other cell lines are used in the experiment.
-
(i)Grow the GM12878 cells in a T175 flask containing 50 ml of RPMI 1640 medium with 15% (vol/vol) FBS to ~8 × 105 cells/ml.
-
(ii)Disperse clumps of cells by pipetting up and down until no clumps are observed, determine the cell number using a cell counter, and use 1.2 × 108 cells (150 ml of cell culture) for each biological replicate.
-
(i)
- CRITICAL STEP It is important to break up all the clumps of cells before cell counting.
-
(iii)Add 500 μl of fresh cisplatin solution (20 mM) per flask to the cell suspension to a final concentration of 200 μM, mix well by gentle shaking and incubate the cells at 37 °C for 2 h in a humidified incubator. Proceed directly to cell lysis and fractionation (Step 2).
-
(iii)
- CRITICAL STEP The repair incubation duration should be adjusted according to the specific experiment (Experimental design).
-
(C)Treatment of human cells with BPDE
- CRITICAL The following BPDE treatment procedure has been optimized for human GM12878 cells. The total cell numbers and treatment procedures should be further optimized if other cell lines are used in the experiment (Experimental design).
-
(i)Prepare the GM12878 cells as described in Step 1B(i) and (ii).
-
(ii)Add 25 μl of BPDE stock solution (4 mM) per flask to the cell suspension to a final concentration of 2 μM, mix well by gentle shaking and incubate the cells at 37 °C for 1 h in a humidified chamber. Proceed directly to cell lysis and fractionation (Step 2).
-
(i)
- CRITICAL STEP The repair incubation duration should be adjusted according to the specific experiment.
-
(D)Treatment of mice with cisplatin
- CAUTION All steps of the treatment procedure must follow institutional regulatory board guidelines for the care and use of experimental animals.
- CRITICAL The following cisplatin treatment was optimized to study nucleotide excision repair in the liver of 6-month-old female C57BL/6J wild-type mice.
-
(i)Receive the mice, and allow them 7 d to acclimate before starting the experiment.
-
(ii)Pick up one mouse, weigh it, and calculate the cisplatin dose needed to deliver a final concentration of 10 mg/kg.
-
(iii)Inject the appropriate volume of cisplatin (in vivo use) into the mouse intraperitoneally.
-
(i)
- CRITICAL STEP It is important to avoid puncturing the intestine and/or the bladder during the injection.
-
(iv)Place the mouse in the cage after injection, and allow repair of cisplatin-induced DNA damage for 2 h. Proceed directly to cell lysis and fractionation (Step 2).
-
(iv)
- CRITICAL STEP The time period for repair should be optimized according to the specific experimental design and mouse strain.
-
(E)Treatment of A. thaliana with UV
- CRITICAL The following procedure has been optimized for A. thaliana. The growth conditions and stage should be further optimized if other plant species are used in the experiment.
-
(i)Treat 1 mg of A. thaliana seeds in a 15-ml centrifuge tube with 10 ml of 70% (vol/vol) ethanol for 2 min by rotating. After the seeds settle to the bottom, pour off the 70% ethanol. Add 10 ml of 50% (vol/vol) bleach solution and rotate for 8 min. Let the seeds settle to the bottom, then pour off the 50% bleach. Then wash the seeds with 10 ml of sterile H2O by vortexing for ~10 s. Repeat the washing step two more times. Allow the seeds to imbibe H2O at 4 °C under light for 2 d, and then place the cold-imbibed seeds on MS agar medium in a 100-mm Petri dish. Seal the plate with 3M micropore surgical tape.
-
(ii)Grow the seedlings for 10 d in 16-h light/8-h dark conditions at 22 °C.
-
(iii)Open the cover of the Petri dish and place the seedlings under a GE germicidal lamp emitting 254-nm UV (1 J/m2/s) for 2 min.
-
(i)
- CAUTION UV irradiation is harmful and carcinogenic. Handle the lamp in a hood and wear protective clothing, gloves and safety goggles.
- CRITICAL STEP Handle the irradiated A. thaliana under yellow light to avoid the repair of UV damage by photoreactivation.
-
(iv)Cover the plate with aluminum foil immediately, and incubate the irradiated A. thaliana seedlings at room temperature for 30 min. Proceed directly to cell lysis and fractionation (Step 2).
-
(iv)
- CRITICAL STEP The incubation duration can be optimized based on the experimental setup.
- (F) Treatment of yeast S. cerevisiae with UV
- CRITICAL In the following steps, the volume of yeast culture for each time point used in this protocol depends on the yeast strain genotype, growth conditions (growth medium, temperature and phase), UV dose and the duration of repair incubation. The total culture volume should be calculated according to the specific experimental design. Here, we describe the protocol for the yeast Y452 strain cultured in YPD medium at 30 °C with shaking.
-
(i)Culture the yeast Y452 strain in 100 ml of YPD medium at 30 °C with shaking (250 r.p.m.) until the OD600 value reaches 0.6–1.0.
-
(ii)Transfer 15 ml of cell culture for each time point to a 50-ml tube and spin the tube at 5,000g for 5 min at 4 °C.
-
(iii)Discard the supernatant, resuspend the cell pellet in 10 ml of ice-cold ddH2O and spin the tube at 5,000g for 5 min at 4 °C. Repeat the ice-cold ddH2O wash and resuspend the cell pellet in 9 ml of ice-cold 2% (wt/vol) glucose.
-
(iv)Spread the 9 ml of suspension from one time point sample onto an ice-cold 150-mm tissue culture dish and place the uncovered dish under a GE germicidal lamp emitting primarily 254-nm UV light (2 J/m2/s) for 60 s (120 J/m2 in total). Shake the cell suspension gently during the UV irradiation process. A wire attached to a dish beneath the sample allows the cells to be shaken remotely, avoiding exposure of the investigator to UV.
-
(i)
- CAUTION UV irradiation is harmful and carcinogenic. Handle the lamp in a hood and wear protective clothing, gloves and safety goggles.
- CRITICAL STEP It is important that the cells be shaken well while being irradiated by UV. To avoid photoreactivation of the UV damage, it is extremely important to perform the following steps under yellow light until the cells are lysed.
-
(v)Transfer the 9 ml of irradiated cell suspension to a 50-ml tube, add 1 ml of 10× YPD medium, mix the cell suspension by inverting the tube five times, and immediately put the tube into the culture incubator at 30 °C.
-
(vi)Incubate the irradiated cells at 30 °C with shaking (250 r.p.m.) for different time periods according to the experimental design. Proceed directly to cell lysis and fractionation (Step 2).
-
(v)
-
(G)Treatment of E. coli with UV
- CRITICAL In the following procedure, the volume of E. coli culture needed for each time point depends on the E. coli strain genotype, growth conditions (growth medium, temperature and phase), UV dose and the duration of repair incubation. ~300 ml of log-phase wild-type, phr− (photolyase deficient mutant) or other mutant cells is needed for one XR-seq experiment, although for uvrD− cells, 30 ml is sufficient. Here, we describe the protocol for the STL4150 strain (Biological materials) cultured in LB medium at 37 °C with shaking.
-
(i)Inoculate 320 ml of LB medium with a 1:15 to 1:50 dilution of overnight culture of the strain, grow at 37 °C with shaking (160 r.p.m.) until the OD600 value reaches 0.3–0.8 to achieve mid-log phase (1–3 h), or to an OD600 value dictated by experimental needs.
-
(ii)Transfer 15 ml of E. coli cells to a 150-mm tissue culture dish, place the dish with the cover off under a GE germicidal lamp emitting primarily 254-nm UV light (2 J/m2/s) for 60 s. Shake the cells gently during the UV irradiation process. A wire attached to a dish beneath the sample allows the cells to be shaken remotely, avoiding exposure of the investigator to UV.
-
(i)
- CAUTION UV irradiation is harmful and carcinogenic. Handle the lamp in a hood and wear protective clothing, gloves and safety goggles.
- CRITICAL STEP To prevent photoreactivation, either use phr− cells or handle UV-irradiated cells under yellow light.
-
(iii)Incubate the irradiated cells at room temperature or 37 °C for an appropriate repair time period according to the specific experiment. Then place the cells on ice. Repeat irradiation and repair 20 times to accumulate 300 ml of irradiated, repaired cells. Then proceed directly to cell lysis and fractionation (Step 2).
-
(iii)
- CRITICAL STEP Irradiation and repair of 20 plates takes time. The OD600 value of the culture started in Step 1G(i) may change substantially during irradiation and repair. If it is important that the OD600 value at the time of irradiation be fixed, then the irradiation and repair of plates of cells can be done in multiple smaller batches (e.g., five plates per batch or ten plates per batch) at different times so that the cells can be irradiated at a given OD600 value. In this case, each batch will require a separate culture in Step 1G(i). Delayed inoculation of these separate cultures will provide cultures at the appropriate OD600 value at delayed times.
-
(A)
Cell lysis and non-chromatin fraction separation Timing 1 h plus overnight and ~6 h the next day
-
2Lyse cells treated with UV, cisplatin or BPDE and separate the non-chromatin fraction containing the excised oligomers. Perform this step by choosing one of the options (A–F) based on the organism and DNA-damaging agent that are used.
- CRITICAL Before performing one of the following procedures, we recommend performing an optional excision assay as described in Box 1 to validate the successful isolation of the excision products. By visualizing and estimating the amount of excision products, this excision assay will be useful for determining the optimal parameters in XR-seq, such as the amount of DNA-damaging agent, the total number of cells and the duration of repair incubation.
-
(A)Cell lysis and fractionation of human cells treated with UV
-
(i)Remove the cells from the incubator at the appropriate time point, discard the medium, and put the cells on ice immediately.
-
(ii)Wash the cells with 10 ml of ice-cold 1× PBS per dish and discard the 1× PBS.
-
(iii)Add 10 ml of ice-cold 1× PBS per dish and detach the cells using a cell scraper.
-
(iv)Transfer the cells to a 15-ml tube, pellet them by centrifugation at 5,000g at 4 °C for 5 min, and discard the supernatant.
-
(v)Resuspend the cells in 500 μl of ice-cold buffer A per dish and incubate them on ice for 10 min.
-
(vi)Transfer the cell suspension to an ice-cold Dounce homogenizer and disrupt the cells using 50 strokes of a tight (type B) pestle in ice.
-
(i)
- CRITICAL STEP Be sure to use the tight pestle and keep the homogenizer in the ice while doing the strokes.
-
(vii)Transfer the lysed cells to one 1.5-ml tube (for (6–4)PP XR-seq) or two 1.5-ml tubes(for CPD XR-seq) and pellet the chromatin fraction by centrifugation at 16,800g for 30 min at 4 °C.
-
(viii)Transfer the supernatant to one 1.5-ml tube (for (6–4)PP XR-seq) or two 1.5-ml tubes(for CPD XR-seq). Proceed immediately to the relevant option of Step 3 of the protocol.
-
(vii)
-
(B)Cell lysis and fractionation of human cells treated with cisplatin or BPDE
-
(i)Remove the cells from the incubator at the appropriate time point and immediately put them in ice water for at least 2 min.
-
(ii)Transfer the cells to three 50-ml tubes, pellet them by centrifugation at 5,000g at 4 °C for 5 min, and then discard the supernatant.
-
(iii)Resuspend the cells in 25 ml of ice-cold 1× PBS per tube, pellet them by centrifugation at 5,000g at 4 °C for 5 min, and then discard the 1× PBS. Repeat the washing step once with 25 ml of ice-cold 1× PBS per tube.
-
(iv)Lyse the cells and separate the non-chromatin fraction as described in Step 2A(v–viii). Use 500 μl of ice-cold buffer A per 10 ml of the original cell culture. Proceed immediately to the relevant option of Step 3 of the protocol.
-
(i)
-
(C)Harvesting, lysis and fractionation of organs from mice treated with cisplatin
-
(A)
- CRITICAL The total number of organs should be optimized according to the experimental design. For example, sufficient excised oligomers for XR-seq can be obtained from one liver, whereas it generally requires 4–6 kidneys. In this procedure, we use one mouse to perform XR-seq in liver.
-
(i)Sacrifice the mouse by CO2 exposure for 5 min, harvest the liver, and immediately wash it two times with 30 ml ice-cold 1× PBS in a 50-ml tube.
-
(i)
- CAUTION Euthanasia should be carried out according to your institution’s approved protocol.
-
(ii)Transfer the liver to a 10-ml Dounce homogenizer, add 5 ml of ice-cold 1× PBS, and homogenize using 15 strokes on ice.
-
(ii)
- CRITICAL STEP Be sure to use the loose (type A) pestle and keep the homogenizer in the ice while doing the strokes.
-
(iii)Transfer the homogenized organ to a 50-ml tube, spin at 3,000g for 4 min at 4 °C, and discard the supernatant.
-
(iv)Wash the homogenized organ: resuspend the pellet in 30 ml of ice-cold 1× PBS, spin at 3,000g for 4 min at 4 °C, and discard the supernatant. Repeat the wash two times to remove fat.
-
(v)Resuspend the pellet in 5 ml of ice-cold buffer A and incubate on ice for 10 min.
-
(vi)Transfer the cell suspension to a 15-ml ice-cold Dounce homogenizer and disrupt the cells using 60 strokes of a tight (type B) pestle.
-
(iii)
- CRITICAL STEP Be sure to use the tight pestle and keep the homogenizer in the ice while doing the strokes.
-
(vii)Split the 5 ml of lysed cells into four 1.25-ml aliquots, transfer each aliquot to a 1.5-ml tube and pellet the chromatin fraction by centrifugation at 16,800g for 30 min at 4 °C.
-
(viii)Combine the supernatants from the four aliquots in a new 15-ml tube. Proceed immediately to the relevant option of Step 3 of the protocol.
-
(vii)
- CRITICAL STEP Keep the samples in ice water while handling the other samples.
-
(D)Cell lysis and fractionation of A. thaliana treated with UV
-
(D)
- CRITICAL Perform all the steps under yellow light to avoid the repair of UV damage by photoreactivation.
-
(i)Transfer the UV-irradiated seedlings to a pre-cooled mortar, immediately freeze them by adding liquid nitrogen, and then grind them into powder using a pestle.
-
(ii)Resuspend the powder in 400 μl of STES buffer and 400 μl of phenol/chloroform (20:1).
-
(iii)Transfer the solution to a 1.5-ml tube containing 200 μl of acid-washed glass beads, tape the tube securely to the rubber cover of a Vortex Genie 2 mixer, and then vortex at the highest speed for 30 min at 4 °C.
-
(iv)Spin the tube at 16,800g for 10 min at 4 °C and transfer the supernatant to a new 1.5-ml tube.
-
(v)Add 10 μl of RNase A to the supernatant, mix it thoroughly by pipetting, and incubate at 37 °C for 1 h.
-
(vi)Add 10 μl of proteinase K to the supernatant, mix it thoroughly by pipetting, and incubate at 55 °C for 1 h.
-
(vii)Isolate the excised oligomers as described in Box 2, and dissolve the pellet in 10 μl of TE buffer.
-
(i)
 PAUSE POINT The samples can be stored at −20 °C until further use.
PAUSE POINT The samples can be stored at −20 °C until further use.
-
(E)Lysis and fractionation of S. cerevisiae treated with UV
-
(i)Remove the 50-ml tube containing the irradiated cells from the incubator at the appropriate time and immediately place it in ice water for at least 2 min.
-
(i)
- CRITICAL STEP Keep the sample in ice water while handling the other samples.
-
(ii)Spin the tube at 5,000g for 5 min at 4 °C and discard the supernatant.
-
(iii)Resuspend the cell pellet in 25 ml of ice-cold ddH2O, spin the tube at 5,000g for 5 min at 4 °C, and discard the supernatant.
-
(iv)Repeat the ice-cold ddH2O wash one time, add 590 μl of ice-cold TE buffer and 10 μl of proteinase K, and then resuspend the cell pellet.
-
(v)Transfer the suspension to a 2-ml sterile vial filled with 600 μl of acid-washed glass beads.
-
(vi)Load the vial into a vial adapter, place the adapter into the Mini-Beadbeater-16, and disrupt the cell wall by agitating for 2.5 min at 4 °C.
-
(ii)
- CRITICAL STEP It is important to distribute the vials symmetrically before starting agitation.
-
(vii)Add 66 μl of 10% (wt/vol) SDS to the vial containing the disrupted cells and mix it gently by inverting ten times.
-
(viii)Punch a hole in the bottom side of the vial using a pushpin, insert the punctured vial into a new 1.5-ml tube, and spin at 750g for 1 min at 4 °C.
-
(vii)
- CRITICAL STEP Do not spin with a centrifugal force >2,000g, as higher centrifugation speeds may ruin the sample and damage the centrifuge.
-
(ix)Discard the vial, resuspend the cell lysate by pipetting up and down gently ten times, and keep it at room temperature for 10 min.
-
(ix)
- CRITICAL STEP The duration of incubation at room temperature should be no more than 15 min.
-
(x)Add 165 μl of 5 M NaCl to the cell suspension, invert the tube gently ten times, and incubate it at 4 °C overnight.
-
(xi)Spin the cell suspension at 16,800g for 40 min at 4 °C and transfer the supernatant to a new1.5-ml tube.
-
(xii)Perform the steps described in Step 2D(v–vii).
-
(xiii)Add 40 μl of TE buffer to the tube, and purify the excised oligomers by following the protocol in Box 3.
-
(xiv)Isolate the excised oligomers as described in steps 3–7 of Box 2 and dissolve the pellet in 10 μl of TE buffer.
-
(x)
- PAUSE POINT The samples can be stored at −20 °C until further use.
-
(F)Cell lysis and fractionation of E. coli treated with UV
-
(E)
- CRITICAL Handle the irradiated cells under yellow light at all times until they are lysed to avoid the repair of UV damage by photoreactivation if phr− strains are not used.
-
(i)Place the 150-mm tissue culture dish containing the irradiated cells in ice water to stop repair.
-
(i)
- CRITICAL STEP
E. coli repair their genome very rapidly, more than ten times faster than human cells, and direct contact of the culture dish bottom with ice water rapidly stops repair at a predetermined time.
-
(ii)Transfer the cells to 50-ml tubes, spin the tubes at 5,000g for 10 min at 4 °C, and discard the supernatant.
-
(ii)
- CRITICAL STEP Each 50-ml tube can be filled with cells from multiple dishes.
-
(iii)Resuspend the pellet in 1 ml of ice-cold TE buffer per 15 ml of cell culture. Spin the tube at 5,000g for 10 min at 4 °C, and discard the supernatant.
-
(iii)
- CRITICAL STEP Pellets distributed among several 50-ml tubes can be collected into one 50-ml tube.
- PAUSE POINT Pellets can be frozen on dry ice and stored at −80 °C for up to 12 months.
-
(iv)Add 340 μl of ice-cold TE buffer per 15 ml of the original culture to the pellet. Vortex to resuspend. Transfer suspensions from 50-ml conical tubes to 25-ml Oak Ridge polypropylene copolymer tubes.
-
(v)Add 40 μl of room-temperature SDS (10% (wt/vol)) per 15 ml of the original culture. Mix by gently inverting eight times.
-
(vi)Incubate at room temperature for 25 min.
-
(vii)Add 100 μl of room-temperature NaCl (5 M) per 15 ml of original culture. Mix by gently inverting eight times.
-
(viii)Incubate at 4 °C overnight.
-
(ix)Pellet in an ultracentrifuge at 30,000g for 1 h at 4 °C.
-
(x)Transfer the supernatants to 15-ml conical tubes.
-
(xi)Add 12 μl of RNase A per 15 ml of the original culture, mix well and incubate for 1 h at 37 °C in a water bath or equivalent.
-
(xii)Add 12 μl of proteinase K per 15 ml of the original culture, mix well and incubate for 1.5 h at 60 °C in a water bath or equivalent.
-
(xiii)Extract DNA twice with an equivalent volume of phenol/chloroform/isoamyl alcohol. At this point, starting with 300 ml of cells, there is ~10 ml of DNA distributed over two 15-ml conical tubes with ~5 ml of DNA per tube.
-
(xiv)Combine the aqueous phases in one Sorvall tube on ice. Per 15 ml of the original culture, add 4 μg of glycogen and mix. Add 2.1 volumes of ice-cold (100% (vol/vol)) ethanol (e.g.,21 ml of ethanol to 10 ml of aqueous phase) to each tube, mix well, and incubate at −20 °C for 1 h.
-
(xv)Pellet with a Sorvall centrifuge at 16,800g for 30 min at 4 °C and discard the supernatant.
-
(xvi)Add 10 ml of 70% ethanol, centrifuge at 16,800g for 30 min and discard the supernatant.
-
(xvii)Air-dry pellet, and resuspend the pellet in a total of 128 μl of nuclease-free H2O. Mix with 32 μl of 5× reaction buffer.
-
(iv)
- PAUSE POINT The samples can be stored at −20 °C until further use.
Box 2 |. Phenol extraction and ethanol precipitation of DNA Timing 2 h.
This procedure describes isolation of the excised oligomers.
Procedure
-
1
Add 1 volume of phenol/chloroform/isoamyl alcohol (25:24:1) and pulse-vortex for 10 s.
-
2
Spin at 16,800g for 5 min at room temperature and transfer the upper layer to a new 1.5-ml tube.
-
3
Add 2.5 volumes of 100% (vol/vol) ethanol, 0.10 volume of 3 M NaAc (pH 5.2) and 1 μl of glycogen to the tube, mix the solution well by inverting ten times, and place the tube at −20 °C for 1h.
-
4
Spin at 16,800g for 30 min at 4 °C to pellet the DNA, and then discard the supernatant.
-
5
Add 190 μl of 70% ethanol to the tube, spin at 16,800g for 5 min at 4 °C, and discard the supernatant.
- CRITICAL STEP It is important to avoid discarding the small DNA pellet.
-
6
Wash the pellet again by repeating step 5.
-
7Remove the residual liquid carefully and air-dry the pellet for 3–5 min.
-
CRITICAL STEP It is important to avoid over drying of the DNA pellet.
-
-
8
Dissolve the pellet with an appropriate volume of TE or nuclease-free H2O.
Box 3 |. DNA purification using a MicroSpin G-50 column Timing 10 min.
This procedure describes purification of the excised oligomers.
Procedure
-
1
Resuspend the resin in the MicroSpin G-50 column by pulse-vortexing for 5 s.
-
2
Loosen the cap, twist off the bottom closure, and place the column into a new collection tube.
-
3
Spin at 735g for 1 min at room temperature, and place the column into a new 1.5-ml tube.
-
4Slowly pipette the 50 μl of solution containing the excised oligomers onto the top-center of the column resin.
-
CRITICAL STEP It is important to avoid touching the resin bed with the pipette tip.
-
-
5
Spin at 735g for 2 min at room temperature to collect the purified DNA.
Immunoprecipitation with anti-TFIIH/XPG or anti-DNA damage–specific antibodies Timing 5 h plus overnight and ~1 h the next day
-
3Purify the excised oligomers using immunoprecipitation with anti-TFIIH or anti-XPG antibodies (options A and B, respectively) or anti-DNA damage-specific antibodies (options C and D):
-
(A)Immunoprecipitation with anti-XPG (human cells)
-
(i)Add 10 μl of anti-XPG antibody (2 μg) and 6 μl of RNase A (~200 μg) per 500 μl of non-hromatin fraction from Step 2A(viii) or 2B(vi), and mix well by pipetting.
-
(ii)Rotate the tube for 3–5 h on a rotating wheel at 4 °C.
-
(iii)To prepare the protein A/G PLUS-agarose, transfer 1 ml of protein A/G PLUS-agarose slurry, which is supplied pre-blocked with BSA, to a new 1.5-ml tube, spin at 750g for 2 min at 4 °C, and discard the supernatant. Resuspend the protein A/G PLUS-agarose in 1 ml of buffer A, spin at 750g for 2 min at 4 °C, and discard the supernatant. Repeat the washing step once with 1 ml of buffer A. Resuspend the protein A/G PLUS-agarose as a 50% slurry in 500 μl of buffer A.
-
(iv)Add 10 μl of the resuspended protein A/G PLUS-agarose 50% slurry (5 μl bed volume) per 500 μl of non-chromatin fraction and rotate the tube at 4 °C overnight.
-
(v)Spin the tube at 750g for 2 min at 4 °C and discard the supernatant.
- (vi) To wash the antibody-coated protein A/G PLUS-agarose, add 1 ml of buffer A to the tube and rotate it for 5 min on a rotating wheel at 4 °C. Spin the tube at 750g for 2 min at 4 °C and discard the supernatant. Repeat the wash with buffer A one time and with buffer B two times. Proceed immediately to the relevant option of Step 4 of the protocol.
-
(i)
-
(A)
-
(B)Immunoprecipitation with anti-TFIIH (mice)
-
(i)Add 150 μl of anti-p89 antibody (30 μg), 75 μl of anti-p62 antibody (15 μg) and 60 μl of RNase A (~1,000 μg) to the non-chromatin fraction from Step 2C(ix), and mix well by pipetting.
-
(ii)Perform the immunoprecipitation steps as described in Step 3A(ii-vi).
-
(i)
-
(C)Immunoprecipitation with DNA damage-specific antibodies (A. thaliana or S. cerevisiae)
-
(i)Incubate Dynabeads with DNA damage-specific antibodies, following the protocol described in Box 4.
-
(ii)Add 90 μl of antibody-coated beads slurry to a 1.5-ml tube and transfer 10 μl of purified excised oligomers from Step 2D(vii) or 2E(xiv) to the tube.
-
(iii)Rotate the tube on a rotating wheel at 4 °C overnight.
- CRITICAL STEP It is extremely important to ensure that the bead slurry moves up and down in the tube during the rotation.
-
(iv)Quickly spin the tube at 2,000g for 2 s at room temperature and place it on a magnetic rack.
-
(v)Discard the supernatant when the solution becomes clear, and remove the tube from the magnetic rack.
-
(vi)Wash the beads: add 200 μl of wash buffer I to the tube and rotate it on a rotating wheel for 2 min at room temperature. Spin the tube at 2,000g for 2 s at room temperature and place it on a magnetic rack. Discard the solution when it becomes clear. Repeat the wash one time sequentially with wash buffer II, wash buffer III, wash buffer IV and TE buffer. Proceed immediately to the relevant option of Step 4 of the protocol.
-
(i)
-
(D)Immunoprecipitation with DNA damage-specific antibodies (E. coli)
-
(i)Incubate Dynabeads with DNA damage–specific antibodies, following the protocol described in steps 1–7 of Box 4.
-
(ii)Add 160 μl of DNA from Step 2F(xvii) to the tube and mix well by pipetting.
-
(iii)Perform the immunoprecipitation steps as described in Step 3C(iii–vi).
-
(i)
Box 4 |. Pre-incubation of Dynabeads with anti-CPD, anti-(6–4)PP and anti-BPDE antibodies Timing 2.5 h.
This procedure describes how to pre-incubate the Dynabeads with anti-CPD, anti-(6–4)PP and anti-BPDE antibodies for one reaction.
Procedure
-
CRITICAL Calculate the total amount of beads and antibodies needed according to the experiment and prepare a mix for all the samples. Keep samples and buffers on ice, except when using the magnetic stand at room temperature.
-
1Transfer 5 μl of Protein G Dynabeads and 5 μl of anti-rabbit Dynabeads per immunoprecipitation reaction to a new 1.5-ml tube, place the tube on a magnetic rack for 2 min, and then discard the original Dynabeads buffer.
-
1
- CRITICAL STEP The Dynabeads should be resuspended before use. They should be shaken during incubations to avoid settling. We generally prepare at least five immunoprecipitation reactions at a time.
-
2
Remove the tube from the magnetic rack. Resuspend the Dynabeads in 200 μl of reaction buffer, place the tube on a magnetic rack for 2 min, and discard the supernatant to wash the beads.
-
3
Repeat step 2 to wash the Dynabeads a total of two times.
-
4
Resuspend the Dynabeads in 20 μl of reaction buffer, which is two times the original volume of Dynabeads.
-
5Perform the pre-incubation by following one of the options (A–D) according to the specific antibody used in the experiment.
-
(A)Pre-incubation with anti-CPD antibody
-
iAdd 1 μl of rabbit anti-mouse IgG and 1 μl of anti-CPD antibody to the Dynabeads suspension.
-
i
-
(B)Pre-incubation with anti-(6–4)PP antibody
-
iAdd 1 μl of rabbit anti-mouse IgG and 1 μl of anti-(6–4)PP antibody to the Dynabeads suspension.
-
i
-
(C)Pre-incubation with anti-BPDE antibody
-
iAdd 1.5 μl of rabbit anti-mouse IgG and 4 μl of anti-BPDE antibody into the Dynabeads suspension.
-
i
-
(A)
-
6
Place the tube on a rotator and incubate the reaction for 2 h at 4 °C.
-
7
Place the tube on a magnetic rack for 2 min, and then discard the supernatant.
-
8
Resuspend the Dynabeads in 90 μl of reaction buffer per immunoprecipitation reaction.
Elution and isolation of the excised oligomers Timing 2.5–6 h
-
4Elute and isolate the excised oligomers by following option A (anti-XPG and anti-TFIIH immunoprecipitation) or option B (anti-DNA damage–specific immunoprecipitation).
-
(A)Elution and isolation of the excised oligomers (anti-TFIIH or anti-XPG immunoprecipitation)
-
(i)Add a volume of elution buffer I equal to that of the original antibody-coated protein A/G PLUS-agarose slurry to the tube from Step 3A(vi) or 3B(ii), and incubate the tube in a digital heating, shaking dry bath at 1,000 r.p.m. for 10 min at 65 °C.
- CRITICAL STEP Use at least 50 μl of elution buffer I if the original volume of protein A/G PLUS-agarose slurry is <50 μl.
-
(ii)Spin the tube at 1,000g for 1 min at room temperature, and transfer the elution solution to a new 1.5 ml tube.
-
(iii)Add 1 μl of proteinase K per 50 μl of elution buffer I to the elution solution, mix it well by pipetting and incubate in a water bath at 55 °C for 10 min.
-
(iv)Repeat the elution (Step 4A(i–iii)) with the same volume of elution buffer I, transfer the elution solution to the tube containing the first elution solution, and then incubate at 55 °C for 10 min.
-
(v)Extract the excised oligomers by following the protocol described in Box 2 and dissolve the pellet in 45 μl of TE buffer.
-
(vi)Add 5 μl of RNase A/T1 mixture per 50 μl of elution buffer I to the tube, mix well by pipetting and incubate at 37 °C for 1 h.
-
(vii)Add 50 μl of phenol/chloroform/isoamyl alcohol to the tube, pulse-vortex it for 10 s, and spin the tube at 16,800g for 4 min at room temperature.
-
(viii)Transfer the supernatant to a MicroSpin G-50 column and purify the excised oligomers as described in Box 3.
-
(ix)Isolate the excised oligomers as described in steps 3–7 of Box 2 and dissolve the pellet in 5.8 μl of nuclease-free H2O.
- PAUSE POINT The excised oligomers can be stored at −20 °C for up to 12 months.
-
(i)
-
(B)Elution and isolation of the excised oligomers (anti-DNA damage–specific immunoprecipitation)
-
(i)Add 60 μl of elution buffer I to the tube containing the beads from Step 3C(vi) or 3D(iii) and incubate in a digital heating, shaking dry bath at 1,000 r.p.m. for 10 min at 65 °C.
-
(ii)Spin the tube at 2,000g for 2 s at room temperature and place it on a magnetic rack. Transfer the elution solution to a new 1.5-ml tube when it becomes clear.
-
(iii)Repeat the elution step with 50 μl of elution buffer I and transfer the elution solution to the tube containing the first elution solution.
-
(iv)Extract the excised oligomers by following the protocol described in Box 2 and dissolve the pellet in 5.8 μl of nuclease-free H2O.
- PAUSE POINT The excised oligomers can be stored at −20 °C for up to 12 months.
-
(i)
-
(A)
Ligation with adapters Timing 1 h plus overnight incubation
-
5
Add 1 μl of 20 μM 5′ adapter (A5), 2 μl of 20 μM 3′ adapter (A3), 1.2 μl of 10× hybridization buffer, and 5.8 μl of the excised oligomers from Step 4 to a PCR tube. Mix well by pulse-vortexing and spin the tube at 2,000g for 2 s at room temperature.
-
6
Place the PCR tube on a thermal cycler and incubate the reaction at 60 °C for 10 min and then at 16°C for 5 min.
-
7
Remove the PCR tube and put it on ice.
-
8
Add 4 μl of 5× T4 DNA ligase buffer, 4 μl of nuclease-free H2O, 1 μl of 50% (wt/vol) PEG 8000 and 1 μl of T4 DNA ligase (5 U/μl) to the PCR tube. Mix well by pipetting up and down at least 15 times.
-
9
Incubate the ligation reaction at 16 °C overnight in a PCR block.
Immunoprecipitation with DNA damage-specific antibodies (second round) Timing 6 h plus overnight and ~6 h the next day
-
10
Transfer the 20 μl of ligation mixture from Step 9 to a new 1.5-ml tube, add 80 μl of nuclease-free H2O to the tube, and mix well by pulse-vortexing.
-
11
Extract the ligation product, following the protocol described in Box 2, and dissolve the pellet in 80 μl of nuclease-free H2O.
-
12
Boil the tube containing the ligation product in a water bath for 1 min, place it into ice water immediately, spin the tube at 2,000g for 2 s at room temperature and then add 1 μl of salmon sperm DNA (10 μg/μl) to the tube.
-
13Add buffer to the tube by following option A (UV, BPDE treatment) or option B (cisplatin treatment).
-
(A)UV, BPDE treatment
-
(i)Add 20 μl of 5× reaction buffer to the tube. Mix well by pipetting up and down at least ten times.
-
(i)
-
(B)Cisplatin treatment
-
(i)Add 20 μl of 5× PEXB buffer to the tube. Mix well by pipetting up and down at least ten times.
-
(i)
-
(A)
-
14Pre-incubate Dynabeads with DNA damage-specific antibodies by following option A (anti-CPD, anti-(6–4)PP and anti-BPDE) or option B (anti-cisplatin).
-
15
Resuspend the antibody-coated beads with the 100-μl of the mixture from Step 13.
-
16
Place the tube on a rotator and incubate the reaction at 4 °C overnight.
-
17Perform the bead-washing steps, following the protocols described in option A (UV and BPDE damage) and option B (cisplatin damage).
-
(A)UV and BPDE damage
-
(i)Wash the beads as described in Step 3C(vi).
-
(i)
-
(B)Cisplatin damage
-
(i)Wash the beads: add 200 μl of PEX buffer to the tube from Step 16 and rotate it for 2 min at room temperature. Spin the tube at 2,000g for 2 s at room temperature and place it on a magnetic rack. Discard the solution when it becomes clear. Repeat the wash sequentially with PEX buffer, 1× reaction buffer and TE buffer one time.
-
(i)
-
(A)
-
18
Elute the ligation product as described in Step 4B(i–iii).
-
19
Extract the ligation products by following the protocol described in Box 2 and dissolve the pellet with 10.5 μl of nuclease-free H2O.
-
20
Transfer 0.5 μl of the ligation products to a PCR tube containing 4.5 μl of nuclease-free H2O. This sample serves as a negative control.
- PAUSE POINT The ligation products can be stored at −20 °C for up to 12 months.
Box 5 |. Pre-incubation of Dynabeads with anti-cisplatin antibodies Timing 2.5 h.
This procedure describes how to pre-incubate the Dynabeads with anti-cisplatin antibody.
Procedure
-
1Transfer 25 μl of anti-rat Dynabeads per immunoprecipitation reaction to a new 1.5-ml tube, place the tube on a magnetic rack for 2 min, and then discard the original Dynabeads buffer.
-
CRITICAL STEP It is important to resuspend the Dynabeads by pulse-vortexing before use. We generally prepare at least five immunoprecipitation reactions at a time.
-
-
2
Remove the tube from the magnetic rack. Resuspend the Dynabeads in 200 μl of 1× PEXB buffer, place the tube on a magnetic rack for 2 min, and discard the supernatant to wash the beads.
-
3
Repeat step 2 to wash the Dynabeads two times.
-
4
Resuspend the Dynabeads in 50 μl of 1× PEXB buffer, which is two times the original volume of Dynabeads.
-
5
Add 1 μl of anti-cisplatin antibody to the Dynabeads suspension.
-
6
Place the tube on a rotator and incubate the reaction for 2 h at 4 °C.
-
7
Place the tube on a magnetic rack for 2 min, and then discard the supernatant.
-
8
Resuspend the Dynabeads in 90 μl of 1× PEXB buffer per immunoprecipitation reaction.
DNA damage reversal or TLS Timing 5 h plus overnight and ~6 h the next day
-
21Reverse the DNA damage or perform TLS using one of the following options (A–C). For damage reversal, follow the protocols described in option A (UV damage) and option B (cisplatin damage). For TLS, follow the protocols described in option C (CPD and BPDE damage).
-
(A)CPD and (6–4)PP
-
(i)Add 5 μl of 10× photoreactivation buffer and 34.5 μl of nuclease-free H2O to the 1.5-ml tube containing 10 μl of purified ligation product from Step 19, and then mix it well by pipetting up and down at least ten times.
-
(ii)Add 0.5 μl of CPD photolyase or (6–4)PP photolyase to each of the tubes from Step 19 for CPD or (6–4)PP damage reversal.
-
(iii)Mix well by pipetting up and down at least ten times, lay the tubes on their sides on ice, and then irradiate through a 2-mm-thick glass plate with 1.0 mW/cm2 of black light (366 nm) for 60 min.
-
(iv)Add 50 μl of nuclease-free H2O to the tube and extract the repaired products by following the protocol described in steps 3–7 of Box 2.
-
(v)Dissolve the pellet with 10.5 μl of nuclease-free H2O.
- PAUSE POINT The samples can be stored at −20 °C for up to 12 months.
-
(B)Cisplatin damage
-
(i)Add 2.5 μl of 4 M NaCN and 37.5 μl of TE buffer to the tube containing purified ligation product from Step 19, and then mix well by pipetting up and down for at least ten times.
-
(ii)Incubate the reaction at 65 °C overnight.
-
(iii)Purify the repaired products using a MicroSpin G-50 column as described in Box 3.
-
(iv)Extract the repaired products by following the protocol described in Box 2 and dissolve the pellet with 10.5 μl of nuclease-free H2O.
- PAUSE POINT The samples can be stored at −20 °C for up to 12 months.
-
(C)CPD (by TLS) and BPDE damage
-
(i)Add 15 μl of 2× TLS polymerase buffer and 3 μl of RPIn primer (10 μM; Table 1) to the tube containing purified ligation product from Step 19, and then mix it well by pipetting up and down at least ten times.
-
(ii)Place the tubes on a thermal cycler and perform annealing and primer extension as follows: denature at 98 °C for 3 min, ramp to 65 °C at 0.1 °C/s and hold for 10 min; then ramp to 37 °C at 0.1 °C/s. Then add 2 μl of polymerase η (for CPD damage) or 2 μl of polymerase κ(for BPDE damage) to the reaction, and mix well by pipetting up and down at least ten times; incubate the reaction at 37 °C for 30 min.
-
(iii)Add 70 μl of nuclease-free H2O to the reaction mixture and extract the primer extension products by following the protocol described in Box 2.
-
(iv)Dissolve the pellet with 10.5 μl of nuclease-free H2O.
- PAUSE POINT The samples can be stored at −20 °C for up to 12 months.
-
(i)
-
(A)
PCR and gel purification Timing 8 h plus ~6 h the next day
-
22
Transfer 0.5 μl of the products from Step 21A(v), 21B(iv) or 21C(iv) to a PCR tube containing 4.5 μl of nuclease-free H2O for a pilot PCR.
-
23.
Prepare enough PCR mix for five pilot PCR reactions, as shown below, in a new PCR tube.
| Reagent | Volume (μl) per reaction | Final concentration | |
|---|---|---|---|
| 2× KAPA HiFi HotStart ReadyMix | 5 | 1× | |
| 10 μM Forward primer (RP1; Table 1) | 1.25 | 1.25 μM | |
| 10 μM Reverse primer (RPIn; Table 1) | 1.25 | 1.25 μM | |
-
24
Add 2 μl of nuclease-free H2O per PCR reaction to the PCR mix, and mix it well by pipetting up and down at least ten times.
-
25.
Divide 9.5 μl of the PCR mix from Step 24 among five PCR tubes, add 0.5 μl of nuclease-free H2O (non-template control) to one of the tubes, 0.5 μl of DNA template from Step 20 (negative control) to one of the tubes and 0.5 μl of DNA template from Step 22 (diluted ligation products) to the other three tubes, and then mix well by pipetting up and down at least ten times. The final volume per pilot PCR reaction is 10 μl
-
26.
Perform the pilot PCR in a thermal cycler, using the following PCR program. Remove one tube containing the DNA template from Step 22 immediately after 15 and 18 PCR cycles. Remove the other tubes, including the negative controls, after 21 PCR cycles.
| Step | Temperature | Time | Cycles |
|---|---|---|---|
| Initial denaturation | 95 °C | 3 min | 1 |
| Denaturation | 98 °C | 20 s | 15, 18, 21 |
| Annealing | 62 °C | 15 s | |
| Extension | 72 °C | 15 s | |
-
27.
Add 2 μl of 6× gel-loading dye (with SDS) to each sample and mix it well.
-
28.
Load 6 μl of the mixture from each sample and 2 μl of low-molecular-weight DNA ladder to the wells of a 10% native polyacrylamide gel, and run the gel in 1× TBE buffer at 125 V for 1.5 h.
-
29.
Stain the gel with 20 ml of SYBR gold (1:10,000 dilution) solution on a rocker for 5 min, visualize it using the ChemiDoc XRS+ Imaging System, and estimate the appropriate number of PCR cycles for the following XR-seq library construction (Fig. 2b).
- CRITICAL STEP It is important that the final PCR product be ~50 ng and the number of PCR cycles be ≤15. After choosing the optimal number of PCR cycles from the gel image of the pilot PCR reaction, users should estimate the number of PCR cycles needed for library preparation (Step 30). We calculate the optimal number of PCR cycles as follows: we use only 1/100 of the DNA template in the pilot PCR reaction and 100× more DNA template in the library preparation PCR reaction; in each PCR cycle, the PCR products will double; thus, after 6 PCR cycles, the final PCR products will increase 64-fold (we approximate this to 100-fold). We subtract 6 from the optimal number of cycles in the pilot PCR to account for using 100 times more template in the library PCR reaction to obtain the correct number of PCR cycles to use in Step 30.
 TROUBLESHOOTING
TROUBLESHOOTING
-
30.
Prepare the PCR mix as shown below and divide it into aliquots (20 μl per reaction). Add the remaining 10 μl of the products from Step 21 to each aliquot, and then mix well by pipetting up and down at least ten times.
-
31
Perform the PCR amplification using the PCR program described in Step 26 and use the appropriate number of PCR cycles, determined in Step 29.
-
32
Add 70 μl of nuclease-free H2O to each PCR tube and extract the PCR products by following the protocol described in Box 2.
-
33
Dissolve the pellet with 5 μl of nuclease-free H2O, add 1 μl of 6× gel-loading dye (without SDS) to each sample, and mix well.
-
34
Load 6 μl of the mixture for each sample and 2 μl of low-molecular-weight DNA ladder to the wells of a 10% native polyacrylamide gel, and run the gel in 1× TBE buffer at 125 V for ~2 h.
- CRITICAL STEP It is important to stop running the gel when the cyan dye reaches the bottom of the gel.
-
35
Stain the gel with 20 ml of SYBR gold solution (1:10,000 dilution) on a rocker for 5 min, acquire the image using the ChemiDoc XRS+ Imaging System, and print it out in its original size.
-
36
Sandwich the gel in between the two parts of a sheet protector, place the sandwiched sheet protector on top of the gel image, and make sure that the gel completely overlaps the gel image.
-
37
Use a blade to cut out the band that contains the correct PCR product, cut the excised band into six pieces on the sheet protector, and then transfer the pieces to a new Gel Breaker tube.
-
38
Insert the Gel Breaker tube into a new 1.5-ml tube and centrifuge at 16,800g for 2 min at room temperature.
-
39
Add 200 μl of elution buffer II to the gel fragments and elute the DNA by incubating the sample in a digital heating, shaking dry bath at 1,000 r.p.m. for at least 3 h at 40 °C.
-
40
Transfer the eluate to a 1.5-ml tube, add 100 μl of elution buffer II, and then continue the elution in a digital heating, shaking dry bath at 1,000 r.p.m. for at least 3 h at 40 °C.
-
41
Combine the second eluate with the first one, transfer the 300 μl of eluate to a Spin-X centrifuge tube filter stacked on a new 1.5-ml tube, and then centrifuge at 580g for 2 min at room temperature.
- CRITICAL STEP Store the tubes containing the small gel pieces at 4 °C for additional elution if the elution efficiency is low.
-
42
Discard the Spin-X centrifuge tube filter, extract the XR-seq library as described in Box 2, and dissolve the pellet with 10 μl of buffer EB.
- PAUSE POINT The libraries can be stored at −20 °C until further use for up to 12 months.
Library quality control and sequencing Timing 1 h
-
43
Add 1 μl of the XR-seq library to a new 1.5-ml tube with 2 μl of buffer EB, and mix well by pipetting up and down at least ten times.
-
44
Use 1 μl of the XR-seq library from Step 43 to determine the DNA concentration using a Qubit 3.0 fluorometer and a Qubit dsDNA HS Assay Kit. Perform the quantification, following the manufacturer’s protocol.
TROUBLESHOOTING
-
45
Use 1 μl of the XR-seq library from Step 43 to analyze the size distribution of the library using the Agilent Bioanalyzer and the Agilent High Sensitivity DNA Analysis Kit. Perform the characterization following the manufacturer’s protocol.
TROUBLESHOOTING
-
46
Pool libraries with different index primers based on the results from Steps 44 and 45, and submit them to be sequenced on a HiSeq 2500 platform or any other compatible platform, such as MiSeq and HiSeq 4000.
Bioinformatics analysis Timing 2–24 h
-
CRITICAL The pipeline is run in a Linux environment. We provide a documented script library for users that sets up the environment and installs the necessary prerequisites (https://github.com/adebali/NGStoolkit). The sequence processing steps (Steps 47–60) are collected in a single. bash script, which can be found at https://github.com/adebali/NGStoolkit/blob/master/stable/XR-seq-basics.sh. To bypass the dependency installation process (e.g., Cutadapt, Bowtie2, SAMtools, BEDTools), we provide a Docker image (see the repository), which provides a Linux container containing all the programs and scripts needed to perform the basic analysis provided here. See the documentation for setup instructions on the repository home page.
-
47
Define variables. If the FASTQ file is named runSample.fastq, the base name of the file would be runSample. The $SAMPLE variable can be defined with
> SAMPLE=runSample
Genome paths can be defined with
> GENOME_DIR=/data/genomes/GRCh38
> BOWTIE2_IND=${GENOME_DIR}/Bowtie2/genome
-
CRITICAL STEP From this point, in the same session, ${SAMPLE} usage will place its value (runSample) until it is redefined or the session is terminated. To retrieve a previously published XR-seq dataset for testing purposes, it is possible to use the SRA Toolkit57 to download the data from Sequence Read Archive57 with the following command.
> fastq-dump --stdout SRR1976056 >${SAMPLE}.fastq && \
fastq-dump --stdout SRR1976057 >>${SAMPLE}.fastq
-
48
Trim the 3′ adapter using Cutadapt58.
> cutadapt -a \
TGGAATTCTCGGGTGCCAAGGAACTCCAGTNNNNNNACGATCTCGTATGCCGTCTTCTGCTTG \
-o ${SAMPLE}_cutadapt.fastq ${SAMPLE}.fastq
-
49
Align sequence reads to the reference genome (e.g., GRCh38) with Bowtie259.
> bowtie2 -p 4 -x $BOWTIE2_IND -U ${SAMPLE}_cutadapt.fastq -S \
${SAMPLE}_cutadapt.sam
-
CRITICAL STEP See the Bowtie2 manual for information on advanced parameters (such as quality control) and how to build the reference genome index. Our repository also has the set of commands needed to download genome files from the Ensembl database60 and prepare the genome files, including the Bowtie2 index as well as the gene list for the GRCh38 genome assembly.
-
50
Convert the alignment to .bed format.
> samtools view -q20 -b -o ${SAMPLE}_cutadapt.bam
${SAMPLE}_cutadapt.sam
> bedtools bamtobed -i ${SAMPLE}_cutadapt.bam > \
${SAMPLE}_cutadapt.bed
-
CRITICAL STEP Standard .bed-formatted files consist of six tab-separated columns: chromosome; start; end; name; score; strand. With a two-step conversion process, the .sam format is first converted (with SAMtools61) to .bam format, which is then converted to .bed format with BEDTools62.
-
51
Sort coordinates by removing duplicates.
> sort -u -k1,1 -k2,2n -k3,3n ${SAMPLE}_cutadapt.bed > \
${SAMPLE}_cutadapt_sorted.bed
-
CRITICAL STEP If the total genome coverage is low (<1×), the likelihood of retrieving the same excised oligomer multiple times would be low as well. The identical oligomers are more likely to be the products of PCR amplification. The same excised oligomer can be represented more than one time as a result of PCR artifacts. Therefore, we remove such artifacts by keeping one of the duplicated reads (regions) in the .bed file. If the genome size is low (e.g., E. coli), the genome coverage will be high, and therefore the deduplication process would remove the real duplicated excised products. In that case, the −u option can be deleted to sort the .bed file without deduplication. A sorted .bed file is necessary for efficient further processing.
-
52
To analyze the data, carry out the following steps. First, count total mapped reads as follows:
> grep -c “^” ${SAMPLE}_cutadapt_sorted.bed > \
${SAMPLE}_cutadapt_sorted_readCount.txt
-
53
Generate the read length distribution by executing the following command:
> awk '{print $3-$2}' ${SAMPLE}_cutadapt_sorted.bed | sort-k1,1n |\
uniq -c | sed 's/\s\s*/ /g' | awk '{print $2”\t”$1}'
-
CRITICAL STEP Read length distribution usually yields one or multiple major populations between 10 and 32 nt. The remaining minor read lengths (e.g., ≥50 nt) are considered unspecific reads rather than excised oligomers. If the unspecific read length ratio is high (>1%) and cannot be ignored, the reads should be filtered to include only the ones between 10 and 32 nt. The command below can be used for filtering.
> awk '{if($3-$2>=10 && $3-$2<=32){print}}' \
${SAMPLE}_cutadapt_sorted.bed > \
${SAMPLE}_cutadapt_sorted_fltrd.bed
-
54
To generate dinucleotide distribution of sequences at a certain read length, first retrieve sequences of a certain length (e.g., n = 26; the most abundant read length can be used) with the following command:
> awk '{ if ($3-$2 == 26) { print } }' \
${SAMPLE}_cutadapt_sorted.bed > \
${SAMPLE}_cutadapt_sorted_26.bed
-
55
Then, to retrieve sequences in FASTA format, type the following:
> bedtools getfasta –fi ${GENOME_DIR}/genome.fa \
-bed ${SAMPLE}_cutadapt_sorted_26.bed -fo \
${SAMPLE}_cutadapt_sorted_26.fa
-
56
Compute the (di)nucleotide content of sequences at a certain length. Unlike other steps, which mostly benefit from simple .bash commands, here we use a custom script retrieved from our repository (https://github.com/adebali/NGStoolkit). The module should be installed as instructed on the repository front page. To retrieve the dinucleotide content, we use the fa2kmerAbundance-Table.py script.
> fa2kmerAbundanceTable.py -i ${SAMPLE}_cutadapt_sorted_26.fa \
-k 2 -o ${SAMPLE}_cutadapt_sorted_26_dinucleotideTable.txt
-k can be defined as 1 to retrieve mononucleotide content.
-
57
To generate BigWig files to visualize on a genome browser using BEDTools and the bedGraphtoBigWig script retrieved from ucsctools63, first separate reads mapping onto two strands with the following commands:
> awk '{if($6==“+”){print}}' ${SAMPLE}_cutadapt_sorted.bed >\
${SAMPLE}_cutadapt_sorted_plus.bed
> awk '{if($6==“-”){print}}' ${SAMPLE}_cutadapt_sorted.bed >\
${SAMPLE}_cutadapt_sorted_minus.bed
-
58
Then, generate BedGraph files by typing the following:
> bedtools genomecov -i ${SAMPLE}_cutadapt_sorted_plus.bed -g \
${GENOME_DIR}/genome.fa.fai -bg -scale \
$(cat ${SAMPLE}_cutadapt_sorted_readCount.txt | \
awk '{print 1000000/$1}') >${SAMPLE}_cutadapt_sorted_plus.bdg
> bedtools genomecov -i ${SAMPLE}_cutadapt_sorted_minus.bed -g \
${GENOME_DIR}/genome.fa.fai -bg -scale \
$(cat ${SAMPLE}_cutadapt_sorted_readCount.txt | \
awk '{print −1000000/$1}') >${SAMPLE}_cutadapt_sorted_minus.bdg
-
59
Generate BigWig files with the following commands:
> bedGraphToBigWig ${SAMPLE}_cutadapt_sorted_plus.bdg \ ${GENOME_-
DIR}\genome.fa.fai \ ${SAMPLE}_cutadapt_sorted_plus.bw
> bedGraphToBigWig ${SAMPLE}_cutadapt_sorted_minus.bdg \ ${GENOME_-
DIR}/genome.fa.fai \ ${SAMPLE}_cutadapt_sorted_minus.bw
-
60
Count the normalized read values for transcribed and nontranscribed strands of the genes separately with the following commands:
> bedtools intersect -sorted -a ${GENOME_DIR}/genes.bed \
-b ${SAMPLE}_cutadapt_sorted.bed -wa -c -S -F 0.5 > \ ${SAMPLE}
_cutadapt_sorted_TScount.txt
> bedtools intersect -sorted -a ${GENOME_DIR}/genes.bed \
-b ${SAMPLE}_cutadapt_sorted.bed -wa -c -s -F 0.5 > \
${SAMPLE}_cutadapt_sorted_NTScount.txt
-
CRITICAL STEP After the gene counts are generated, they should be normalized to RPKM (reads per kilobase per million mapped reads) values for a meaningful comparison across samples. RPKM conversion can be performed with the following formula for each gene:
where n is the read count, 1 is the length of the gene and m is the total number of mapped reads. Note that repair in genic regions provides only a subset of the entire map. For advanced analyses of the entire genome, ${SAMPLE}_cutadapt_sorted.bed should be used as the starting file.
Troubleshooting
Troubleshooting advice can be found in Table 2.
Table 2 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 29 | The band representing the correct size of the XR-seq library is absent on the gel | Photoreactivation has occurred for UV damage in A. thaliana, yeast and E. coli | Repeat Steps 1 and 2 under yellow light |
| The band representing the correct size of the XR-seq library is faint on the gel | Low efficiency of immunoprecipitation | Repeat Steps 3 and 10–20. Use new 1.5-ml tubes if the immunoprecipitation solution does not move up and down on the rotator in the original 1.5-ml tubes | |
| Low dose of DNA-damaging agent | Increase the dose of DNA-damaging agent in Step 1 | ||
| Low number of organs/cells | Increase the number of organs/cells in Step 1 | ||
| Low ligation efficiency of T4 DNA ligase | Use new T4 DNA ligase in Steps 5–9 | ||
| Multiple bands longer than the correct size of the XR-seq library appear on the gel | PCR overamplification | Decrease the number of PCR cycles | |
| 44 | Low DNA concentration | Low DNA elution efficiency in Steps 39–41 | Use the gel pieces from Step 41 and repeat Step 39 with overnight incubation |
| 45 | Extra peak at the −118-bp position | PCR products of adapter dimers are cut off in Step 37 | Precisely cut off the band corresponding to the XR-seq library only. See example in Fig. 2d |
Timing
Step 1, DNA damage induction and repair incubation: 10 min–48 h
Step 2, cell lysis and non-chromatin fraction separation: 1 h, plus overnight and $6 h the next day
Step 3, immunoprecipitation with anti-TFIIH/XPG or anti-DNA damage-specific antibodies: 5 h, plus overnight and $1 h the next day
Step 4, elution and isolation of the excised oligomers: 2.5–6 h
Steps 5–9, ligation with adapters: 1h, plus overnight incubation
Steps 10–20, immunoprecipitation with DNA damage-specific antibodies (second round): 6 h, plus overnight and $6 h the next day
Step 21, DNA damage reversal or TLS: 5 h, plus overnight and $6 h the next day
Steps 22–42, PCR and gel purification: 8 h, plus $6 h the next day
Steps 43–46, library quality control and sequencing: 1 h
Steps 47–60, bioinformatics analysis: 2–24 h
Box 1, excision assay: 3 d
Box 2, phenol extraction and ethanol precipitation of DNA: 2 h
Box 3, DNA purification using a MicroSpin G-50 column: 10 min
Box 4, pre-incubation of Dynabeads with anti-CPD, anti-(6–4)PP, and anti-BPDE antibodies: 2.5 h
Box 5, pre-incubation of Dynabeads with anti-cisplatin antibodies: 2.5 h
Anticipated results
XR-seq libraries constructed by following this protocol typically have a total yield of 5–100 ng. For mammalian cells, gel electrophoresis of a typical XR-seq library after the final PCR amplification shows a major band at the 145-nt position (Fig. 2b,c,e). The length distributions of the excised oligomers from CPD XR-seq and CPD tXR-seq show the same pattern (Fig. 2f). The sizes of major PCR products for E. coli, yeast and A. thaliana cells are 130, 142 and 146 nt, respectively.
High-throughput sequencing of a CPD XR-seq library14 (SRA ID: SRX997094) from human NHF1 cells typically has the features described below. The pipeline starts with $20 million reads. $16 million of the reads (81%) can be mapped to the genome with the quality score criterion (>20) applied. The deduplication process results in $10 million reads corresponding to 52% of the starting read number. 99.95% of the deduplicated reads are ≤32 nt (Fig. 3a). The damage site nucleotides (dipyrimidines for UV damage) are expected to be enriched at 5–6 nt away from the 3′ end of the excised oligomers (Fig. 3b). For well-established transcription-coupled excision repair events in which only one strand is actively transcribed during the repair process, repair asymmetry between the two strands should be observed for expressed genes (Fig. 3c). Because the antisense (noncoding) strand is transcribed by RNAP II, it is expected to have higher levels of repair than the opposite sense (coding) strand (Fig. 3c).
Fig. 3 |. Preliminary analyses of CPD XR-seq data at a 1-h time point from treatment of human NHF1 cells with 10 J/m2 UV.
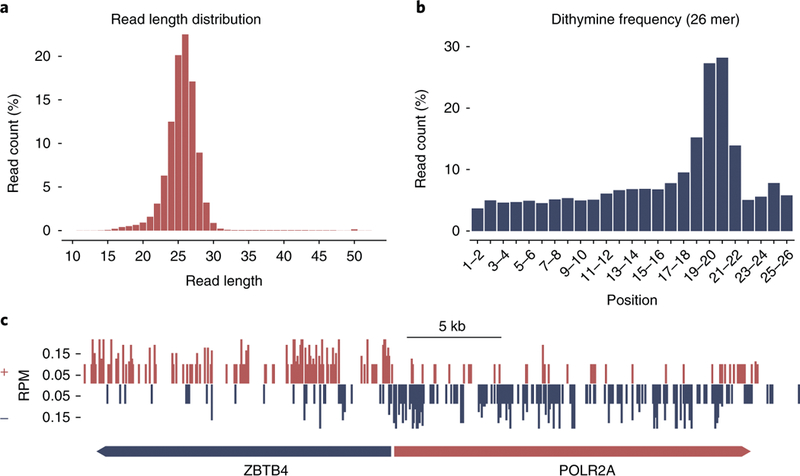
a, The read length distribution shows a single population (no degradation products) following TFIIH immunoprecipitation. b, Dithymine frequency is enriched at two main positions, 19–20 and 20–21, for the 26-nt excised oligomers obtained from CPD XR-seq. c, Screenshot of XR-seq signals in two genes, ZBTB4 and POLR2A, transcribed in opposite directions. Arrows indicate the direction of transcription. Repair asymmetry between strands that is due to transcription-coupled repair in the expressed genes is observed. The transcribed strands are the plus (above) and minus (below) strands for ZBTB4 and POLR2A, respectively. Therefore, those regions are repaired at higher levels because of transcription-coupled repair. The magnitude of the asymmetry varies depending on organism, gene and damage type. Data in c are from Hu et al.13. Histograms were plotted using the R software and the ggplot264 package, and the genomic region-specific repair levels were captured using IGV65.
Table 1 |.
Oligonucleotides used in this protocola
| Name | Purpose and step(s) used in | Sequence (from 5′ to 3′) |
|---|---|---|
| A5F | 5′ adapter, Step 5 | GTTCAGAGTTCTACAGTCCGACGATC |
| A5R | 5′ adapter, Step 5 | NNNNNGATCGTCGGACTGTAGAACTCTGAAC/SpC3/ |
| A3F | 3′ adapter, Step 5 | /phos/TGGAATTCTCGGGTGCCAAGG/SpC3/ |
| A3R | 3′ adapter, Step 5 | CCTTGGCACC CGAGAATTCCANNNNN/SpC3/ |
| RP1 | PCR primer, Steps 23 and 30 | AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA |
| RPIn | PCR primer, Steps 23 and 30 | CAAGCAGAAGACGGCATACGAGATXXXXXXGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA |
All oligonucleotides are custom orders from IDT. Nisa random nucleotide. The bold ‘xref’ indicates different index sequences in accordance with those in the Illumina TrueSeq Small RNA Kit. A5F, RP1 and RPIn are purified by PAGE; A5R, A3F and A3R are purified by HPLC. A5F, A5R, A3F and A3R are diluted to 250 μM in TE buffer. RP1 and RPIn are diluted to 100 μM in TE buffer.
Acknowledgements
We thank A. Kakoki for CRITICAL reading of the manuscript. This work was supported by National Institutes of Health grants GM118102 and ES027255 (to A.S.) and Scientific and Technological Research Council of Turkey grant 118C023 (to O.A.).
Footnotes
Competing interests
The authors declare no competing interests.
Data availability
The sample data we used for the computational pipeline was retrieved from the SRA database with the accessioncodes SRR1976056 and SRR1976057. The stable code release can be accessed at https://doi.org/10.5281/zenodo.1493379.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41596–018-0093–7.
Reprints and permissions information is available at www.nature.com/reprints.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Sancar A Mechanisms of DNA repair by photolyase and excision nuclease (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 55, 8502–8527 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Wood RD Nucleotide excision repair in mammalian cells. J. Biol. Chem. 272, 23465–23468 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Hu J, Selby CP, Adar S, Adebali O & Sancar A Molecular mechanisms and genomic maps of DNA excision repair in Escherichia coli and humans. J. Biol. Chem. 292, 15588–15597 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gong F, Kwon Y & Smerdon MJ Nucleotide excision repair in chromatin and the right of entry. DNA Repair (Amst) 4, 884–896 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Hanawalt PC & Spivak G Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell. Biol. 9, 958–970 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Mao P, Wyrick JJ, Roberts SA & Smerdon MJ UV-induced DNA damage and mutagenesis in chromatin. Photochem. Photobiol. 93, 216–228 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teng Y et al. A novel method for the genome-wide high resolution analysis of DNA damage. Nucleic Acids Res. 39, e10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bryan DS, Ransom M, Adane B, York K & Hesselberth JR High resolution mapping of modified DNA nucleobases using excision repair enzymes. Genome Res. 24, 1534–1542 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Powell JR et al. 3D-DIP-Chip: a microarray-based method to measure genomic DNA damage. Sci. Rep. 5, 7975 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu J, Lieb JD, Sancar A & Adar S Cisplatin DNA damage and repair maps of the human genome at single-nucleotide resolution. Proc. Natl. Acad. Sci. USA 113, 11507–11512 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mao P, Smerdon MJ, Roberts SA & Wyrick JJ Chromosomal landscape of UV damage formation and repair at single-nucleotide resolution. Proc. Natl. Acad. Sci. USA 113, 9057–9062 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia-Nieto PE et al. Carcinogen susceptibility is regulated by genome architecture and predicts cancer mutagenesis. EMBO J. 36, 2829–2843 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu J, Adar S, Selby CP, Lieb JD & Sancar A Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev. 29, 948–960 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adar S, Hu J, Lieb JD & Sancar A Genome-wide kinetics of DNA excision repair in relation to chromatin state and mutagenesis. Proc. Natl. Acad. Sci. USA 113, E2124–E2133 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu J, Adebali O, Adar S & Sancar A Dynamic maps of UV damage formation and repair for the human genome. Proc. Natl. Acad. Sci. USA 114, 6758–6763 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiou YY, Hu J, Sancar A & Selby CP RNA polymerase II is released from the DNA template during transcription-coupled repair in mammalian cells. J. Biol. Chem. 293, 2476–2486 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Y et al. Cisplatin-DNA adduct repair of transcribed genes is controlled by two circadian programs in mouse tissues. Proc. Natl. Acad. Sci. USA 115, E4777–E4785 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oztas O, Selby CP, Sancar A & Adebali O Genome-wide excision repair in Arabidopsis is coupled to transcription and reflects circadian gene expression patterns. Nat. Commun. 9, 1503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li W, Adebali O, Yang Y, Selby CP & Sancar A Single-nucleotide resolution dynamic repair maps of UV damage in Saccharomyces cerevisiae genome. Proc. Natl. Acad. Sci. USA 115, E3408–E3415 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adebali O, Chiou YY, Hu J, Sancar A & Selby CP Genome-wide transcription-coupled repair in Escherichia coli is mediated by the Mfd translocase. Proc. Natl. Acad. Sci. USA 114, E2116–E2125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adebali O, Sancar A & Selby CP Mfd translocase is necessary and sufficient for transcription-coupled repair in Escherichia coli. J. Biol. Chem. 292, 18386–18391 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reardon JT & Sancar A Nucleotide excision repair. Prog. Nucleic Acid Res. Mol. Biol. 79, 183–235 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Truglio JJ, Croteau DL, Van Houten B & Kisker C Prokaryotic nucleotide excision repair: the UvrABC system. Chem. Rev. 106, 233–252 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Canturk F et al. Nucleotide excision repair by dual incisions in plants. Proc. Natl. Acad. Sci. USA 113, 4706–4710 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kemp MG, Reardon JT, Lindsey-Boltz LA & Sancar A Mechanism of release and fate of excised oligonucleotides during nucleotide excision repair. J. Biol. Chem. 287, 22889–22899 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu J et al. Nucleotide excision repair in human cells: fate of the excised oligonucleotide carrying DNA damage in vivo. J. Biol. Chem. 288, 20918–20926 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi JH, Kim SY, Kim SK, Kemp MG & Sancar A An integrated approach for analysis of the DNA damage response in mammalian cells: nucleotide excision repair, DNA damage checkpoint, and apoptosis. J. Biol. Chem. 290, 28812–28821 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baek S, Han S, Kang D, Kemp MG & Choi JH Simultaneous detection of nucleotide excision repair events and apoptosis-induced DNA fragmentation in genotoxin-treated cells. Sci. Rep. 8, 2265 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li W et al. Human genome-wide repair map of DNA damage caused by the cigarette smoke carcinogen benzo[a]pyrene. Proc. Natl. Acad. Sci. USA 114, 6752–6757 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.ENCODE Project Consortium.. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wade JT & Grainger DC Pervasive transcription: illuminating the dark matter of bacterial transcriptomes. Nat. Rev. Microbiol. 12, 647–653 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Thomason MK et al. Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J. Bacteriol. 197, 18–28 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Llorens-Rico V et al. Bacterial antisense RNAs are mainly the product of transcriptional noise. Sci. Adv. 2, e1501363 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang TH, Lindsey-Boltz LA, Reardon JT & Sancar A Circadian control of XPA and excision repair of cisplatin-DNA damage by cryptochrome and HERC2 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 107, 4890–4895 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaddameedhi S, Selby CP, Kaufmann WK, Smart RC & Sancar A Control of skin cancer by the circadian rhythm. Proc. Natl. Acad. Sci. USA 108, 18790–18795 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perera D et al. Differential DNA repair underlies mutation hotspots at active promoters in cancer genomes. Nature 532, 259–263 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Sabarinathan R, Mularoni L, Deu-Pons J, Gonzalez-Perez A & Lopez-Bigas N Nucleotide excision repair is impaired by binding of transcription factors to DNA. Nature 532, 264–267 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Poulos RC et al. Functional mutations form at CTCF-cohesin binding sites in melanoma due to uneven nucleotide excision repair across the motif. Cell Rep. 17, 2865–2872 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Lim B, Mun J, Kim YS & Kim SY Variability in chromatin architecture and associated DNA repair at genomic positions containing somatic mutations. Cancer Res. 77, 2822–2833 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Lavigne MD, Konstantopoulos D, Ntakou-Zamplara KZ, Liakos A & Fousteri M Global unleashing of transcription elongation waves in response to genotoxic stress restricts somatic mutation rate. Nat. Commun. 8, 2076 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klaassen CD, Casarett LJ & Doull J Casarett and Doull’s Toxicology: The Basic Science of Poisons. 8th edn (McGraw-Hill Education, New York, 2013). [Google Scholar]
- 42.Bohr VA, Smith CA, Okumoto DS & Hanawalt PC DNA repair in an active gene: removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell 40, 359–369 (1985). [DOI] [PubMed] [Google Scholar]
- 43.Besaratinia A & Pfeifer GP Measuring the formation and repair of UV damage at the DNA sequence level by ligation-mediated PCR. Methods Mol. Biol. 920, 189–202 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li S, Waters R & Smerdon MJ Low- and high-resolution mapping of DNA damage at specific sites. Methods 22, 170–179 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Yu S et al. Global genome nucleotide excision repair is organized into domains that promote efficient DNA repair in chromatin. Genome Res. 26, 1376–1387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shu X, Xiong X, Song J, He C & Yi C Base-resolution analysis of cisplatin-DNA adducts at the genome scale. Angew. Chem. Int. Ed. Engl. 55, 14246–14249 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang TH, Reardon JT, Kemp M & Sancar A Circadian oscillation of nucleotide excision repair in mammalian brain. Proc. Natl. Acad. Sci. USA 106, 2864–2867 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pfeifer GP, Drouin R, Riggs AD & Holmquist GP In vivo mapping of a DNA adduct at nucleotide resolution: detection of pyrimidine (6–4) pyrimidone photoproducts by ligation-mediated polymerase chain reaction. Proc. Natl. Acad. Sci. USA 88, 1374–1378 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pfeifer GP, Drouin R, Riggs AD & Holmquist GP Binding of transcription factors creates hot spots for UV photoproducts in vivo. Mol. Cell. Biol. 12, 1798–1804 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wellinger RE & Thoma F Taq DNA polymerase blockage at pyrimidine dimers. Nucleic Acids Res. 24, 1578–1579 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wellinger RE & Thoma F Nucleosome structure and positioning modulate nucleotide excision repair in the non-transcribed strand of an active gene. EMBO J. 16, 5046–5056 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kunala S & Brash DE Excision repair at individual bases of the Escherichia coli lacI gene: relation to mutation hot spots and transcription coupling activity. Proc. Natl. Acad. Sci. USA 89, 11031–11035 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirt B Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26, 365–369 (1967). [DOI] [PubMed] [Google Scholar]
- 54.Heffernan TP et al. An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol. Cell. Biol. 22, 8552–8561 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Selby CP & Sancar A A cryptochrome/photolyase class of enzymes with single-stranded DNA-specific photolyase activity. Proc. Natl. Acad. Sci. USA 103, 17696–17700 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Selby CP & Sancar A The second chromophore in Drosophila photolyase/cryptochrome family photoreceptors. Biochemistry 51, 167–171 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kodama Y, Shumway M & Leinonen R, International Nucleotide Sequence Database Collaboration. The Sequence Read Archive: explosive growth of sequencing data. Nucleic Acids Res. 40, D54–D56 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011). [Google Scholar]
- 59.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hubbard T et al. The Ensembl genome database project. Nucleic Acids Res. 30, 38–41 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Quinlan AR BEDTools: the Swiss-Army Tool for genome feature analysis. Curr. Protoc. Bioinformatics 47, 11. 12.1–11.12.34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kuhn RM, Haussler D & Kent WJ The UCSC genome browser and associated tools. Brief. Bioinform. 14, 144–161 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wickham H ggplot2: Elegant Graphics for Data Analysis (Springer, New York, 2016). [Google Scholar]
- 65.Robinson JT et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]