

Abstract
Human induced pluripotent stem cells (hiPSC) hold great promise in diagnostic and therapeutic applications. However, translation of hiPSC technology depends upon a means of assessing hiPSC quality that is quantitative, high‐throughput, and can decipher malignant teratocarcinoma clones from normal cell lines. These attributes are lacking in current approaches such as detection of cell surface makers, RNA profiling, and/or teratoma formation assays. The latter remains the gold standard for assessing clone quality in hiPSCs, but is expensive, time‐consuming, and incompatible with high‐throughput platforms. Herein, we describe a novel method for determining hiPSC quality that exploits pluripotent cells’ documented hypersensitivity to the topoisomerase inhibitor etoposide (CAS No. 33419‐42‐0). Based on a study of 115 unique hiPSC clones, we established that a half maximal effective concentration (EC50) value of <300 nM following 24 hours of exposure to etoposide demonstrated a positive correlation with RNA profiles and colony morphology metrics associated with high quality hiPSC clones. Moreover, our etoposide sensitivity assay (ESA) detected differences associated with culture maintenance, and successfully distinguished malignant from normal pluripotent clones independent of cellular morphology. Overall, the ESA provides a simple, straightforward method to establish hiPSC quality in a quantitative and functional assay capable of being incorporated into a generalized method for establishing a quality control standard for all types of pluripotent stem cells. Stem Cells Translational Medicine 2017;6:1829–1839
Keywords: Pluripotent stem cells, Etoposide, Functional, Quantification, Hypersensitivity
Significance Statement.
A quantitative measure of human induced pluripotent stem cell (hiPSC) pluripotency has yet to be standardized, an issue that must be remedied if widespread translational application of this technology is to occur. Secreto et al. provide an innovative method for quantifying hiPSC pluripotency based upon exploiting the hypersensitivity pluripotent cells exhibit to the DNA damaging agent etoposide. Their etoposide sensitivity assay determines hiPSC suitability by quantifying the amount of etoposide required to kill the treated cells, and comparing that amount to a standard value known to be associated with high quality clones.
Introduction
Human induced pluripotent stem cell (hiPSCs) hold great promise as a regenerative platform for an array of clinical maladies and in identifying underlying pathological mechanisms of disease 1. Production of large repositories of hiPSCs is underway 2, 3; however, a standardized method for establishing hiPSC suitability for clinical and/or diagnostic applications remains elusive. Polymerase chain reaction (PCR)/microarray‐based assays and immunostaining for cell surface markers are popular techniques for determining hiPSC quality, while teratoma assays remain the standard by which all other methods are judged 4, 5, 6. PSC colony morphology can be used to gauge overall clone quality, with good colonies exhibiting clean, delineated borders containing tightly packed cells 7. However, morphology assessment requires a significant amount of experience, and a quantitative appraisal has only recently been described 8. All of these approaches either do not provide a functional readout, or are not adaptable to high‐throughput screening.
Hypersensitivity to DNA damage is a hallmark of pluripotent stem cells (PSCs). It was initially demonstrated in mouse embryonic stem (ES) cells which, when exposed to DNA‐damaging compounds in conjunction with either γ‐irradiation or ribonucleoside tri‐phosphate depletion, rapidly underwent p53‐independent apoptosis 9. Similar results were obtained in human ES cells and hiPSCs when exposed to γ‐irradiation, resulting in capase‐3 cleavage within four hours of treatment 10. Interestingly, PSCs exhibit a response to DNA damage that is remarkably similar regardless of lineage, whereas the DNA damage response elicited by differentiated somatic cells is highly cell‐type dependent 11. Recent data also demonstrated that the degree of pluripotency contributes to the DNA damage response elicited by irradiation, with iPSCs exhibiting a greater response than neonatal stromal cells, both of which are more sensitive than adult stromal cells 12.
While hiPSC hypersensitivity to DNA damage clearly correlates with pluripotent potential, better control and consistency are needed to translate this observation to clinical and diagnostic settings. Therefore, an assay using a small molecule‐based DNA‐damaging agent should be superior to γ‐irradiation. In addition, the ideal compound would demonstrate significantly more toxicity in PSCs compared to differentiated cells, thus providing a means to distinguish pluripotent clones from those contaminated with spontaneously differentiated cells. Chemotherapeutic agents designed to target rapidly proliferating, poorly differentiated cancerous cells fit these criteria, given that PSCs and cancer cells share many phenotypic characteristics 13. Inhibitors of the DNA unwinding enzyme topoisomerase II create double‐stranded DNA breaks while simultaneously inhibiting topoisomerase II‐mediated DNA repair 14, 15, 16. One such compound is etoposide (VP 16), which has been used as a chemotherapeutic agent for nearly three decades 17. Hypersensitivity to etoposide was initially described in human ESCs; unlike γ‐irradiation, etoposide‐induced apoptosis was dependent upon p53 [18]. Subsequent studies using mouse and human iPSCs by our laboratory and others demonstrated that iPSC hypersensitivity to etoposide was similar to that observed in human ESCs 18, 19. Remarkably, terminally differentiated cells were consistently far more etoposide‐resistant than PSCs 18, 19, 20. Our results support the underlying hypothesis that a rapid loss of etoposide sensitivity is directly coupled to the initiation of cellular differentiation, allowing us to exploit etoposide sensitivity as a means to quantitatively assess PSC quality.
Materials and Methods
Generation of hiPSCs
Skin biopsies from de‐identified healthy individuals and patients afflicted with hypoplastic left heart syndrome were undertaken in accordance with institutional regulations (Mayo Clinic IRB 10‐006845, Clinical Trials Identifier NCT01860898). hiPSCs were generated from primary human fibroblasts originally isolated from donor skin biopsies by ReGen Theranostics (Rochester, MN). Lentiviral‐based reprogramming and clonal selection were performed as previously described 21, except without the addition of L‐uridine to the media. Sendai reprogramming and clonal selection was achieved using CytoTune‐iPS Sendai Reprogramming Kits (Invitrogen, Carlsbad, CA, http://www.thermofisher.com/us/en/home/brands/invitrogen.html) according to manufacturer's instructions.
Cardiac Differentiation of hiPSCs
Human iPSCs (250,000) were seeded in wells of a 24‐well plate and cultured in PSGro media (StemRD, Burlingame, CA, http://www.stemrd.com/) until they achieved ∼95% confluence. Directed cardiac differentiation was achieved using the StemRD PSdif‐Cardio cardiomyocyte differentiation kit according to manufacturer instructions. At 1, 2, 3, 4, 6, and 9 days of differentiation, a subset of cells were subjected to ESA analyses as described below.
Human Fibroblast Cell Culture and Staining
Primary human fibroblasts were cultured in DMEM (Thermo Fisher, Waltham, MA, http://www.thermofisher.com/us/en/home.html) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin and 1× GlutaMAX (Thermo Fisher). Media was changed every 3 days and ESA analyses were conducted as described above. Human fibroblasts were stained with carboxyfluorescein succinimidyl ester (CFSE) using the CellTrace CFSE Cell Proliferation kit (Thermo Fisher) according to manufacturer's instructions.
Etoposide Sensitivity Assay
Human iPSCs were mechanically dissociated (colonies physically cut into ∼1 mm × 1 mm squares, media changed, incubated for 15 minutes at 37°C, scraped off the plate and distributed to a new plate) from a 60 mm dish (∼50% confluent) and seeded onto a 24‐well plate, where they were maintained in mTeSR1 medium (STEMCELL Technologies, Vancouver, BC, http://www.stemcell.com) for 3–5 days in GelTrex (Thermo Fisher, purity >98%) coated plates (5.5 µg/mm2). Cells were then treated with etoposide (Sigma‐Aldrich, St. Louis, MO, https://www.sigmaaldrich.com/united-states.html) at varying concentrations (1–1,000 nM) or dimethyl sulfoxide (DMSO), in triplicate, for 24 hours. Conditioned media (DMSO or etoposide supplemented media isolated from each well following 24 hours of exposure to hiPSCs) was collected into 12 × 75 mm flow tubes, and 200 µl TrypLE Express (Thermo Fisher) was added to each well for 5–10 minutes at 37°C. Subsequently, 1 ml of prewarmed (37°C) FACs buffer (Dulbecco's phosphate‐buffered saline [DPBS] pH 7.4 without Ca2+, Mg2+ containing 4 mM Ethylenediaminetetraacetic acid (EDTA) and 0.5% FBS) was added to each well, and total well contents were transferred to the corresponding flow tubes containing the conditioned media. Cells were collected by centrifugation at 400g for 5 minutes, washed once with ice‐cold annexin binding buffer (ABB; 10 mM HEPES, 2.5 mM CaCl2, 140 mM NaCl, pH 7.4) and stained for 30 minutes on ice with 100 µl annexin‐FITC (BD Biosciences, Franklin Lakes, NJ, http://www.bdbiosciences.com) diluted 1:20 in ABB. Cells were washed once in ABB and resuspended in 400 µl ABB containing 2.5 µg/ml propidium iodide (Sigma‐Aldrich). Samples were immediately assayed by flow cytometry and percent viability was assessed using Kaluza flow cytometry analysis software (Beckman Coulter, Brea, CA, http://www.beckman.com).
RT‐PCR Analysis
In order to assess the pluripotency of specific human iPSC clones, semi‐quantitative real‐time PCR (RT‐PCR) was performed on total RNA isolated from nine individual human iPSC clones. Total RNA was isolated by extraction with Trizol (Invitrogen) followed by column purification using a Qiagen (Germantown, MD, http://www.qiagen.com) RNeasy kit. RNA (1–2 µg) was reverse transcribed using an iScript cDNA synthesis kit (Bio‐Rad, Hercules, CA), and 15 ng of resulting cDNA was used per RT‐PCR reaction in a 384‐well plate. All primers were purchased from IDT (Coralville, IA, http://www.idtdna.com) and are listed in Table I, Supporting Information. PCR amplification was conducted using TaqMan universal PCR master mix (Applied Biosystems, Foster City, CA, http://www.thermofisher.com/us/en/home/brands/applied-biosystems.html), a ViiA7 thermocycler (Applied Biosystems) and an epMotion 5070 robotic pipettor (Eppendorf, Hauppauge, NY, http://www.eppendorf.com/US-en/about-us/eppendorf-north-america). All biological data points were generated from technical triplicates in order to ensure well‐to‐well reproducibility. The ΔΔCt method was used to calculate relative expression, with final represented values corresponding to fold change calculated according to the formula 2−ΔΔCt.
Immunocytochemistry
Human iPSCs and differentiated cardiomyocytes were fixed on glass coverslips coated with GelTrex or fibronectin (Sigma Aldrich), respectively, in 4% paraformaldehyde for 5 minutes, washed three times with PBS and stored in PBS at 4°C. Prior to staining, cells were permeabilized in 1% Triton X‐100 for 30 minutes, washed three times with PBS and blocked for 3 hours at room temperature in Super Block (Thermo Fisher). After removal of blocking solution, hiPSCs were incubated with primary antibodies against SSEA‐3 (09‐0014; Stemgent, Lexington, MA, http://www.stemgent.com) and TRA‐1–60 (09–0010; Stemgent), and cardiomyocytes were incubated with antibodies against cTnT (MAB1874; Biotechne, Minneapolis, MN, http://www.bio-techne.com) and cTnI (MAB6887; Biotechne). The final concentration of all antibodies was 0.5 mg/ml in PBS containing 10% Super Block and 0.1% Tween. After overnight incubation at 4°C on a rotator, cells were washed three times in PBS/0.1% Tween, then incubated with FITC or Texas Red ‐conjugated secondary antibodies (anti‐rat IgG or anti‐mouse IgM; Invitrogen) diluted 1:250 in antibody dilution buffer and incubated for one hour at room temperature protected from light. Cells were washed two times with PBS/0.1% Tween, followed by three washes with PBS. Slides were covered with a 25‐mm cover‐glass slip and treated with 1–2 drops of DAPI mounting medium (hiPSCs: Prolong Gold Antifade, Invitrogen; cardiomyocytes: VECTASHIELD, Vector Laboratories, Burlingame, CA, www.vectorlabs.com). Slides were stored in the dark until analysis (40×) using a Zeiss LSM 510 confocal microscope (Oberkochen, Germany, http://www.zeiss.com).
Teratoma Formation Assays
Teratoma formation was assessed in 6‐ to 8‐week‐old athymic nude mice under Mayo Clinic IACUC protocol #A17111. Human iPSCs cultured in 60‐mm plates were treated with 10 µM of the Rho/ROCK inhibitor Y‐27632 (Bio‐Techne) for one hour prior to enzymatic removal using TrypLE Express. Cells were washed once in PBS and resuspended in mTeSR1 medium containing 10 µM Y‐27632 and placed on ice. One million cells were injected beneath the left testicular capsule in anesthetized mice, while corresponding sham injections were performed beneath the right testicular capsule. Each clone was injected into ten mice. Mice were monitored every other day and sacrificed 60 days post injection. Teratoma growth can be practically monitored by external examination. Testicular teratomas typically demonstrate a total mass of 1–2 g, which correlates to approximately 10% of the total body weight at around 8 weeks post cell injection. Testes were excised and fixed in 10% formaldehyde, then dehydrated through a series of alcohol grades to xylene. The tissue was embedded in paraffin and cut serially into 5 µm sections for H&E staining. Sections were observed by light microscopy and photographed.
Flow Cytometric Analysis of SSEA‐4 and TRA‐1‐60
Mechanically disassociated and passaged hiPSCs were seeded onto a 24‐well plate as described above. Confluent cells were washed once in DPBS and enzymatically removed using TrypLE Express as described above. Cells from a single well were divided evenly into two flow tubes, washed once in FACs buffer (0.5% FBS, 2 mM EDTA, 1× DPBS pH 7.4) and incubated for 30 minutes on ice in the dark with 100 µl FACs buffer with or without PE‐conjugated TRA‐1‐60 antibody (clone MA1‐023‐PE; Thermo Fisher) and FITC‐conjugated SSEA‐4 antibody (clone MC813‐70; Fisher Scientific). Cells were then washed and fixed with 5% paraformaldehyde for 15 minutes at room temperature in the dark. Fixed cells were washed once with DPBS and either stored at 4°C or immediately assayed by flow cytometry.
PluriTest Analysis
Teratocarcinoma cell lines were purchased from Sigma‐Aldrich (NTERA‐2, PA‐1, 1156QE/8, 833KE) or ATCC (NCCIT, Cates‐1B), and cultured according to the vendor's instructions on GelTrex coated culture plates. Total RNA was isolated from actively proliferating cells as described above for RT‐PCR analysis. Microarray assays were performed on RNA samples from six teratocarcinoma cell lines and six hiPSC clones at the Roswell Park (Buffalo, NY, http://www.roswellpark.org) Cancer Institute's Genomics Shared Resource using Illumina (San Diego, CA, http://www.illumina.com) human HT‐12v4 Expression Bead chips. Data analysis was performed on IDAT raw data files uploaded to the PluriTest algorithm (pluritest.org).
Statistical Analyses
EC50 values were calculated using a generalized linear model which is fit using the glm function in R with a Gaussian link function, and 95% confidence intervals (CI) were generated. Group comparison of SSEA‐4 and TRA‐1‐60 biomarker data as well as EC50 data from lentiviral‐ and Sendai virus‐reprogrammed hiPSC were performed using the two sample t test in R to assess statistical significance. By default, this test does not assume equal variances between the two groups and estimates variance separately for each group, using the Welch modification to the degrees of freedom. To explore underlying sample structure and similarity, and how the samples related to EC50 or biomarker data, principal components analysis (PCA) was performed on the RT‐PCR data of the seven pluripotent genes analyzed. PCA was carried out in R with the singular value decomposition algorithm. EC50 and biomarker data were then individually mapped to PCA results. Hierarchical analysis using standard R functions was applied to the expression data set to build a dendrogram based on the average distance algorithm and gene expression heatmap.
Results
hiPSC Sensitivity to Etoposide Is Directly Proportional to Clone Quality
We performed a small‐scale experiment using five hiPSC clones to determine if hypersensitivity to DNA damage was capable of quantitatively characterizing PSC quality. A three‐step “weeding out” approach was applied in evaluating hiPSC clone quality using traditional methods. First, a qualitative gradation of hiPSC colony morphology was performed (Fig. 1A). Second, morphology was compared with the presence of glycosphingolipid and podocalyxin‐associated cell surface markers, SSEA‐3 and TRA‐1–60, expressed by pluripotent cells 22, 23 (Fig. 1B). Third, RT‐PCR analyses were conducted to determine if clone‐specific RNA profiles were similar to gene expression patterns exhibited by commercially available hiPSC and hESC cell lines (Fig. 1C). A clear pattern emerged when comparing colony morphology and the compiled gene expression profiles. Colonies originating from RT4.1 and RT6.21 hiPSC cultures exhibited crisp, clean borders comprised of tightly packed cells (Fig. 1A), along with gene expression patterns similar to those observed in hiPSC (IMR90, iPSf2) and hESC (H9, H13) controls (Fig. 1C). Conversely, RT lines producing poorly defined colonies (RT2.1, RT2.3, RT6.1) displayed gene expression patterns highly divergent from the same control cell lines (Fig. 1A–1C). No obvious difference in SSEA‐3/TRA‐1–60 expression was observed among the hiPSC lines, although admittedly, immunofluorescence assays are not quantitative by design (Fig. 1B). RT4.1 and RT6.21 hiPSC clones formed teratomas comprised of all three primary germ layer tissue types (Fig. 1D), confirming that our three‐step approach selected for the highest quality hiPSC clones from the original five tested. We next exposed all of the hiPSC clones to increasing concentrations of etoposide for 24 hours. Clones RT4.1 and RT6.21 displayed the greatest sensitivity to etoposide (Fig. 1E), correlating with the earlier results using traditional methods to characterize hiPSCs. We note here that all etoposide based assays were assessed by flow cytometry and then discarded. No etoposide‐treated cells were ever selected for or used in any subsequent experiments in this manuscript.
Figure 1.
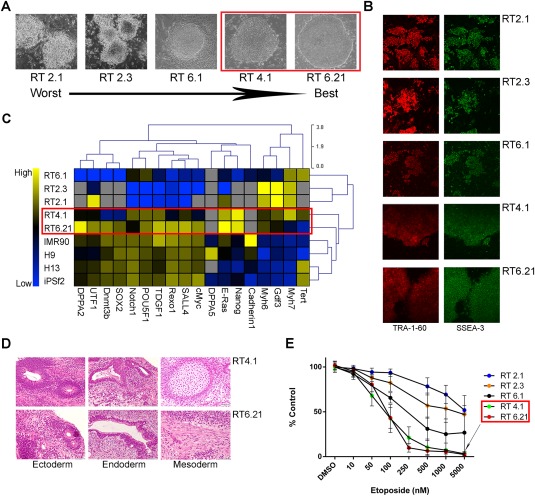
Human induced pluripotent stem cell (hiPSC) clones exhibiting the highest degree of etoposide sensitivity also demonstrated good hiPSC clonal morphology and pluripotent gene expression patterns consistent with pluripotent control cell lines. (A): ×40 images of hiPSC clones. Red outline highlights clones (RT4.1, RT6.21) displaying the best morphology (clean, distinct borders). (B): Immunofluorescence detection (×40) of SSEA‐3 and TRA‐1–60 expression in hiPSCs. (C): qPCR of RNA isolated from five of our hiPSC lines (RT), two control hiPSC lines (IMR90, iPSf2) and two control ESC lines (H9, H13). Red outline highlights the two clones (RT4.1, RT6.21) exhibiting pluripotent gene expression patterns most similar to those displayed by control pluripotent cells. (D): RT4.1 and RT6.21 clones successfully formed teratomas in athymic nude mice. Scan bars = 50 µM. (E): Annexin V/PI staining of hiPSC cells treated w/wo etoposide for 24 hours and plotted as a percent of DMSO control. Red outline highlights two hiPSC lines (RT4.1, 6.21) that demonstrated the greatest sensitivity to etoposide. Data used to calculate the individual means was generated from a minimum of five biological replications. Error bars represent the SD calculated around an individual mean. Abbreviation: DMSO, dimethyl sulfoxide.
Cellular Differentiation Results in a Rapid Loss of Etoposide Sensitivity
Spontaneous differentiation of human PSC cultures results in a loss of pluripotency and is associated with decreased sensitivity to etoposide 18, 24. In agreement with the literature, we determined that four low‐quality hiPSC clones, deemed to contain differentiated cells based upon colony morphology (Fig. 2A), demonstrated a relatively low sensitivity to etoposide (Fig. 2A). However, etoposide sensitivity increased substantially after cultures were subjected to mechanical cleaning to remove differentiated cells (Fig. 2B).
Figure 2.

Poor human induced pluripotent stem cell (hiPSC) clone maintenance and cellular differentiation result in a rapid loss of etoposide sensitivity. (A, B): hiPSC cultures in which spontaneously differentiated cells were removed via MC exhibited increased etoposide sensitivity (B) compared to the same cultures prior to MC (A). (C): Terminally differentiated human fibroblast cells demonstrated no detectable apoptosis or cell death following treatment with etoposide. Data points were derived from the average of ten unique primary human fibroblast cultures and plotted as a percentage of DMSO control. (D): hiPSCs undergoing directed cardiac differentiation display a progressive decrease in etoposide sensitivity. All data points were derived from the average of three technical replicate samples stained with Annexin V/PI, and normalized to DMSO treated cells. Error bars represent the standard deviation calculated around an individual mean. Abbreviations: DMSO, dimethyl sulfoxide; MC, mechanical cleaning.
Furthermore, directed differentiation of a hiPSC clone demonstrated a near‐stepwise decrease in etoposide sensitivity throughout the differentiation time course (Fig. 2D). To the best of our knowledge, this has not been previously demonstrated in hiPSCs. Notably, mature, beating cardiomyocytes obtained at Day 9 displayed a near‐complete lack of sensitivity to etoposide within the dose response range calibrated to PSCs, which mimics results obtained with human fibroblasts (Fig. 2C). These observations demonstrate that etoposide exposure is a highly sensitive means of detecting loss of pluripotency at the earliest stages of hiPSC differentiation. Notably, the presence of feeder cells used in the early stages of hiPSC clonal selection (prior to ESA analysis, all cells evaluated by ESA were plated on GelTrex coated tissue culture ware) (Fig. 1) or lack thereof (Fig. 2A–2C) had no discernible effect on the relationship between colony morphology and sensitivity to etoposide.
Etoposide Sensitivity Is Superior to TRA‐1–60/SSEA4 Detection as a Means of Assessing hiPSC Quality
We next sought to confirm the results of these small‐scale experiments in an expanded population of hiPSC clones obtained from our hypotrophic left heart syndrome cell bank (Supporting Information Fig. 1), with the goal of establishing an etoposide sensitivity assay (ESA) capable of assessing hiPSC quality in a high‐throughput manner. We selected flow‐based detection of SSEA‐4 and TRA‐1‐60 (Fig. 3A) in place of immunocytochemistry due to its inherent quantitative nature. Analysis of 100 hiPSC clones revealed SSEA4/TRA‐1‐60 coexpression detected in an average of 82% of cells (Fig. 3B). The reprogramming strategy selected (lentivirus or Sendai virus) did not significantly affect cell surface expression of either marker (Fig. 3C). These 100 hiPSC clones, along with 15 additional clones, were next exposed to increasing concentrations of etoposide (Fig. 3D). The results were strikingly similar to those obtained in our pilot experiment (Fig. 1E). The mean etoposide EC50 was 63.4 nM across all hiPSC clones (Fig. 3E). Interestingly, in contrast to TRA‐1‐60/SSEA‐4 coexpression data (Fig. 3C), the ESA was able to distinguish between the lentivirus reprogramming strategy hiPSC cohort compared to the Sendai virus cohort, with significantly different EC50 values (53 and 88.1 nM, respectively; p = .0002154) (Fig. 3F). This reprogramming strategy‐dependent difference in etoposide sensitivity is likely due to the higher expression of genes involved in maintaining pluripotency we observed in hiPSC clones derived from lentiviral reprogrammed fibroblasts (Fig. 4A). Taken together with its ability to detect early signs of differentiation (Fig. 2C), ESA analysis provides a substantially more sensitive means of assessing hiPSC quality compared with detection of classic pluripotent cell surface markers.
Figure 3.
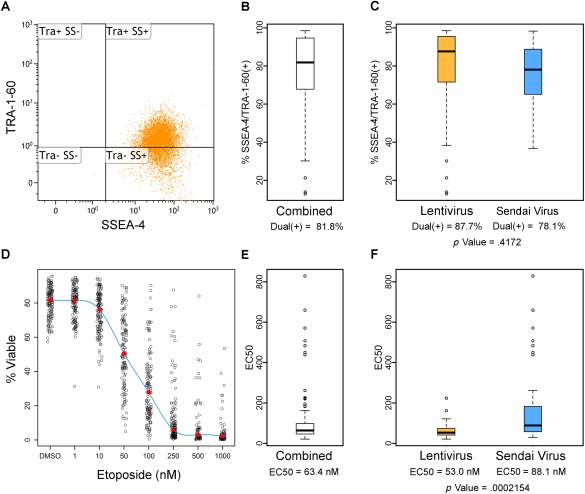
Analysis of 115 unique human induced pluripotent stem cell (hiPSC) clones by etoposide sensitivity assay (ESA); ESA, and not flow‐based TRA‐1–60/SSEA‐4 staining, was able to decipher a significant difference between hiPSC clones originally produced from Sendai or Lentiviral reprogramming methods. (A): Example SSEA‐4/TRA‐1–60 dot plot including Tra+ SS+ values used to determine percent SSEA‐4/TRA‐1–60 expression. (B, C): Mean dual (+) SSEA‐4/TRA‐1–60 value(s) calculated for hiPSC clones (B) combined (n = 100) and (C) separately by lentivirus (n = 57) or Sendai virus (n = 43). (D): A summary graph incorporating 115 hiPSC clones treated with etoposide (or DMSO control) for 24 hours. and subsequently analyzed by ESA (Annexin V/PI). Red dots represent the median value of the associated treatment. (E, F): Mean EC50 value(s) calculated for hiPSC clones (E) regardless of reprogramming strategy (n = 115) and (F) separately by lentivirus (n = 60) or Sendai virus (n = 55). Mean passage numbers for lenti‐ and Sendai‐reprogrammed hiPSC clones were 9.56 ± 2.14 and 8.27 ± 2.21, respectively, with error calculations representing the standard deviation around an individual mean.
Figure 4.
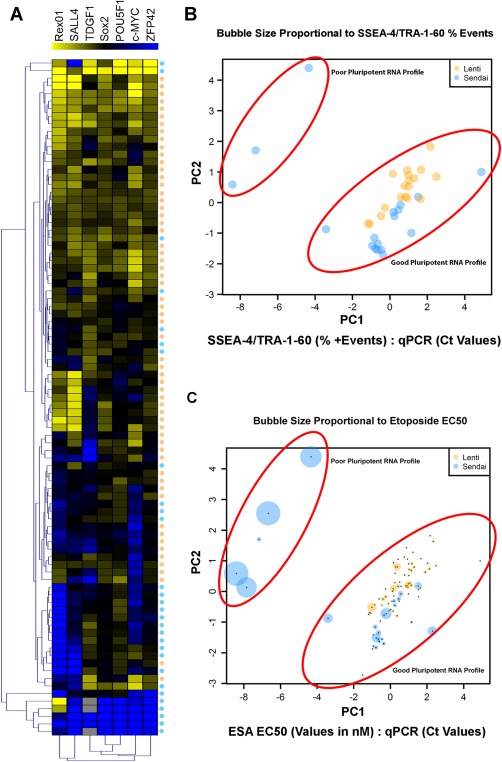
Large scale ESA revealed a positive correlation between ESA derived EC50 values and pluripotent gene expression. (A): Heat map (yellow = RNA expression) of 89 human induced pluripotent stem cell (hiPSC) clones analyzed for seven common markers of pluripotency. Lenti‐ or Sendai virus reprogramming strategy for each clone is indicated by a corresponding orange (lentivirus) or blue (Sendai) dots appearing below heat map. (B): PCA of SSEA‐4/TRA‐1‐60 values from hiPSCs created with lentivirus (n = 18) or Sendai virus (n = 18) compared with qPCR Ct values generated from expression of the seven pluripotency‐related genes listed in Figure 3A. Sphere size is relatively proportional to percent SSEA‐4/TRA‐1‐60 values (i.e., large spheres equate to a high level of SSEA‐4/TRA‐1‐60 coexpression). (C): PCA of ESA EC50 values from hiPSC clones created with lentivirus (n = 60) or Sendai virus (n = 29) compared with Ct values as described in Figure 5B. Sphere size is relatively proportional to EC50 values (i.e., large spheres equate to high EC50 values). Abbreviation: ESA, etoposide sensitivity assay.
ESA Results Are Positively Correlated with Pluripotent Gene Expression Patterns
We subsequently determined whether the ESA results correlated with RNA expression profiling of hiPSCs. RNA expression analysis is a relatively inexpensive, quantifiable means of determining PSC clone quality, with several gene expression platforms commercially available 5, 25, 26. The hiPSC quality control protocol typically carried out in our laboratory incorporates a semiquantitative RT‐PCR based analyses examining the expression of seven genes (Rex01, SALL4, TDGF1, Sox2, POU5F1, c‐MYC, ZFP42) commonly associated with high‐quality clones. As depicted in Figure 4A, we observed differences in gene expression profiles depending on the reprogramming strategy used, with overall gene expression levels typically higher in the lentiviral reprogrammed hiPSCs (Fig. 4A). Changes in pluripotent gene expression patterns dependent upon a particular reprogramming strategy certainly calls into the question the feasibility of using gene expression analysis alone in determining overall hiPSC quality. Therefore, we next determined whether the combination of gene expression and ESA EC50 values can discern high‐ and low‐quality hiPSC populations. PCA of gene expression data mapped to etoposide EC50 values demonstrated a positive correlation between low EC50 values and a “good” RNA expression profile associated with pluripotent gene expression (Fig. 4C). In contrast, PCA mapping of hiPSC gene expression profiles and TRA‐1‐60/SSEA‐4 coexpression demonstrated no significant correlation (Fig. 4B).
ESA can Distinguish Normal hiPSC Clones from Teratocarcinoma Cell Lines in Cases of Indeterminate Gene Expression Analysis
Eliminating potential tumor risk is an essential aspect of PSC quality control. Malignant cells exhibit many of the characteristics shared by noncancerous PSCs; thus, we assessed whether the ESA could distinguish pluripotent teratocarcinoma cell lines from hiPSCs characterized as high‐ or low‐quality based on colony morphology (Supporting Information Fig. 3). We selected six teratocarcinoma cell lines based upon previously established pluripotent characteristics and a demonstrated ability (PA‐1, NCCIT, NTERA‐2) or inability (833KE, Cates‐1B, 1156QE/8) to successfully differentiate into somatic tissue 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38. hiPSC clones were plated as monolayer cultures in order to both mimic the morphology displayed by the teratocarcinoma cell lines and to replicate conditions commonly used when differentiating hiPSCs into cardiomyocytes or adaptation to suspension conditions 39, 40, 41. ESA analysis was conducted on these cell lines in addition to three high‐ and three low‐quality hiPSC clones (Fig. 5A). Notably, only the high‐quality hiPSC clones exhibited a degree of etoposide sensitivity capable of determining an EC50 value (Fig. 5A, inset table).
Figure 5.
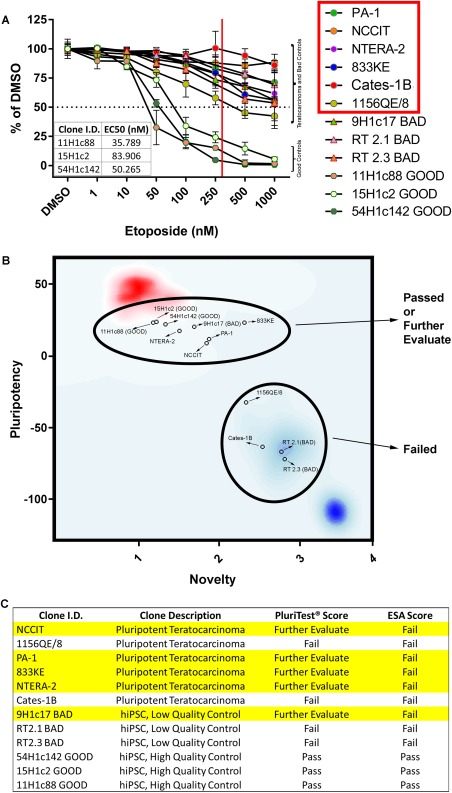
ESA, but not PluriTest, correctly scored all pluripotent teratocarcinoma cell lines assayed. (A): EC50 values were not obtainable from ESA analyses (Annexin V/PI) conducted on all of the pluripotent teratocarcinoma cell lines, as well as all three of the “BAD” hiPSC controls. EC50 values for the “GOOD” hiPSC clones 11H1c88, 15H1c2 and 54H1c142 were 35.8, 84.0, and 50.3 nM, respectively (see inset table). The solid red vertical line approximates the upper cutoff of an acceptable ESA score (EC50 = 300 nM), while the dotted horizontal line represents 50% viability. The red box in the figure legend denotes teratocarcinoma cell lines. (B): PluriTest‐based PCA plot of pluripotency and novelty scores generated from microarray analyses of GOOD (n = 3) and BAD (n = 3) hiPSC RNA, along with RNA isolated from pluripotent teratocarcinoma cell lines (n = 6). The red cluster in the upper left‐hand corner of the plot represents pluripotent cells sharing similar RNA expression profiles as assessed by PluriTest, while cells exhibiting a highly divergent RNA expression profiles and low levels of established pluripotent markers are signified by the blue cluster appearing in the lower right hand corner. (C): A summary table comparing how PluriTest and ESA scored the pluripotent teratocarcinoma cells, along with the GOOD and BAD hiPSC clones. Highlighted rows correspond to clones which PluriTest scored as “Further Evaluate.” Error bars represent the standard deviation calculated around an individual mean. Abbreviations: DMSO, dimethyl sulfoxide; hiPSC, human induced pluripotent stem cell.
We further analyzed these same hiPSC clones and teratocarcinoma cell lines using PluriTest, a commercially available assay used to assess overall PSC quality. PluriTest is a microarray‐based platform that uses total RNA isolated from PSC clones to determine clone quality based on (a) the expression of selected pluripotent genes in comparison to all of the PSC gene expression profiles in the PluriTest database (pluripotency score), and (b) the degree of RNA expression divergence from the same data set (novelty score) 5. The novelty score is critical, as it theoretically identifies pluripotent cells that are otherwise abnormal, at least compared to the majority of pluripotent cells submitted by PluriTest users. The results of our PluriTest analyses are summarized in a PCA plot (Fig. 5B) mapping the pluripotency (Supporting Information Fig. 4) and novelty scores (Supporting Information Fig. 5) from each of the cell lines tested. The red area in the upper left corner represents the mapped position of all PSC data submitted to PluriTest at the time of analysis deemed to be highly pluripotent and sharing similar overall gene expression patterns, while the blue area in the lower right corner denotes the converse relationship. While PluriTest analysis correctly passed all three high‐quality hiPSC clones submitted for analysis, it was unable to fail one of the three low‐quality hiPSC clones, and four of the six teratocarcinoma cell lines (Fig. 5B). In contrast, ESA analyses correctly distinguished all three low‐quality hiPSC clones and all six teratocarcinoma cell lines from the three high‐quality hiPSC controls (Fig. 5C).
Establishment of an ESA‐Based Quantitative Standard for Determination of hiPSC Quality
We sought to create a numerical “ESA score” that would provide a standard representing the quality of a given hiPSC clone in terms of overall quality and lack of tumor risk. The quantitative nature of the ESA permitted us to create a statistical model delineating an EC50 cutoff value corresponding to the minimum etoposide sensitivity exhibited by a hiPSC clone while still maintaining a pluripotent gene expression profile, as exemplified by the hiPSC clones depicted in the lower right oval in Figure 4C. PCA results based on gene expression profiles (Fig. 5B, 5C) combined with EC50 values generated from ESA analyses (Fig. 5C) showed that outliers (top left oval of Fig. 5C) have EC50 values greater than 300 nM. Thus, we concluded that an etoposide EC50 value greater than 300 nM equates to a poor pluripotent RNA expression profile (Fig. 6A), and can be used as a maximum cutoff point for establishing a quality control standard for hiPSC clones.
Figure 6.
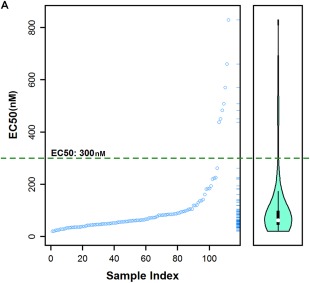
A maximum EC50 cutoff value of 300 nM quantitatively identifies good quality human induced pluripotent stem cell (hiPSC) clones. (A): EC50 maximum cutoff plot generated from the principal components analysis‐based examination of hiPSC gene expression profiles and etoposide sensitivity EC50 data. Outliers are defined as hiPSC clones exhibiting EC50 values greater than 300 nM. Abbreviation: EC, effective concentration.
Discussion
Assessment of hiPSC quality presents a host of issues not encountered with most other primary or immortalized human cell lines. Chief among these is the heterogeneity that exists not only between uniquely derived hiPSCs, but among clones originating from the same parental cell line 42, 43. Additionally, cell surface marker(s) have yet to be discovered that are capable of identifying high‐quality hiPSCs which are rapidly lost following a decrease in hiPSC pluripotency. With the growing number of large hiPSC repositories, there is a pressing need for a standardized means of assessing hiPSC quality that can be adopted by any lab working with hiPSCs. Even more importantly, translation of hiPSC‐based technologies will require a means of establishing the necessary safety and efficacy requirements for gaining approval to conduct clinical trials. This is not simply an academic concern, as a temporary hold was placed on the world's first clinical trial implanting autologous hiPSC‐derived retinal‐pigment epithelial cells, due to a mutation detected in a known oncogene 44.
These criteria fail to be met by the most common methods for determining PSC quality, none of which are able to reliably distinguish between normal and malignant pluripotent cells 45. For example, colony morphology is a very reliable indicator of hiPSC clone quality, and Kato et al. have developed an approach that incorporates high‐quality live cell imaging along with customized algorithms as a means to quantify hiPSC colony morphology 8. However, this method is useless when hiPSCs are maintained as monolayer cultures 39, 46 that lose the morphological features of colonies (Supporting Information Fig. 6), or when they are adapted to suspension cultures for large‐scale production of hiPSCs 41. Flow cytometric analysis, while potentially affordable and high‐throughput, is limited by the lack of known cell surface markers that can reliably report hiPSC quality; even presumptive “stem cell” markers such as TRA‐1‐60, SSEA‐3 and SSEA‐4 can be persistently expressed on low‐quality hiPSC clones with poor colony morphology. RNA expression analysis of specific hiPSC‐associated genes, or indeed the entire hiPSC transcriptome, is another approach to both qualify PSC pluripotency as well as PSC differentiation potential 5, 6. However, this type of data is better suited toward overall clone evaluation, and is insufficient as a standalone quality control assay since it cannot account for a myriad of translational and post‐translational events that may occur during PSC differentiation. In support of this assertion, we found that PluriTest microarray‐based analyses failed to conclusively distinguish pluripotent teratocarcinoma cell lines, nor a hiPSC clone demonstrating a substantial amount of spontaneous differentiation (Fig. 5C; Supporting Information Fig. 3).
In contrast, the ESA demonstrates none of the shortcomings of the aforementioned assays and can be performed on a small aliquot of cells pulled off during cell passaging independent of culturing methodology. Foremost in developing the ESA, we leveraged observations from our laboratory 19 and others 10, 20 that pluripotent cells exhibit a hypersensitivity to DNA‐damaging agents when compared to differentiated cell types, including treatment with etoposide. For example, human fibroblasts treated with etoposide remained nearly unaffected throughout the dose response curve (Fig. 2C), a result also displayed by hiPSC‐derived cardiomyocytes following Day 6 of differentiation (Fig. 2D). Moreover, the early stages of directed cardiac differentiation involve the rapid formation of mesendoderm, followed by the presence of cardioprogenitor cells around Day 5 post induction 47. Despite the lack of terminally differentiated cells present at these early time points, a noticeable loss of etoposide sensitivity was observed within 24 hours of induction (Fig. 2D). Thus, even an incomplete loss of pluripotency within a heterogeneous population of cells can be detected by ESA. We suspect that any type of hiPSC differentiation, directed or otherwise, will rapidly be associated with a concomitant loss of etoposide sensitivity. Naturally, further experimentation using several defined hiPSC‐derived cell types in addition to cardiomyocytes must be conducted prior to accepting any solid conclusions regarding the ubiquitous nature of differentiation‐mediated loss of etoposide sensitivity. However, given our observations of etoposide sensitivity in terminally differentiated cells (human fibroblasts) and hiPSC‐derived cardiomyocytes, as well as spontaneously differentiated hiPSCs (Fig. 2A), we are confident that any future data will support our present findings.
The results shown here confirm that etoposide sensitivity is inversely related to hiPSC quality as measured by colony morphology and pluripotent gene expression patterns. ESA results were also highly repeatable over the course of multiple passages, assuming consistency in culture conditions and maintenance was observed (Fig. 1E). Furthermore, loss of etoposide hypersensitivity in hiPSCs is observable at the earliest stages of cellular differentiation (Fig. 2D). ESA was more sensitive and specific than PluriTest, as it unambiguously identified all teratocarcinoma cell lines and low‐quality hiPSC clones, while missing none of the high‐quality hiPSC clones (Fig. 5C). Based on results with a large number (115) of hiPSC clones (Fig. 6A), we established that an etoposide EC50 > 300 nM can be used as a maximum cutoff point for establishing a quality control standard for hiPSC clones. While this value is certain to be refined in future studies across laboratories, it establishes the likelihood that an “accept/reject” point can be standardized. Thus, the ESA is a quantitative, functional, and potentially high‐throughput reporter of PSC quality that is more sensitive and specific than the use of cell surface markers or gene expression patterns.
Summary
The ESA provides a novel method to assess hiPSC quality in a quantitative, high‐throughput manner, and can be conducted on cells regardless if they are maintained as colonies or monolayers. But perhaps the ESA's most important feature rests in its unique ability to distinguish normal pluripotent hiPSCs from malignant teratocarcinoma cells. However, any single PSC quality control assay has limitations, and the ESA is no exception. For example, we have performed karyotype (G‐banding) analysis on a subset of our hiPSC cohort and determined that ∼15% of our clones do exhibit a karyotypic abnormality and that ESA is not capable of determining whether a clone harbors this type of genetic defect (data not shown). It is becoming abundantly clear that in order to ensure the safety and efficacy of any hiPSC‐derived tissue, assessment of multiple parameters must be undertaken. Such an approach was recently described, incorporating quantification of pluripotent cell surface markers, qPCR analysis of iPSC and embryoid body‐derived RNA and digital karyotyping 48. Since ESA analysis already incorporates pluripotent gene expression, combining ESA with cytogenetic analysis along with our previously described mitochondrial DNA sequencing assay will provide an affordable, high throughput platform capable of mitigating many of the risks posed by hiPSC‐derived tissue.
Author Contributions
F.J.S.: concept and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing; X.L.: concept and design, data analysis and interpretation, software design; A.J.S.: concept and design, collection and/or assembly of data; E.B., E.P‐C., and S.O.: collection and/or assembly of data, data analysis and interpretation; G.H., S.C.H., B.K.A., and E.B.B.: collection and/or assembly of data; D.W.: Provision of study material or patients; T.J.N.: conception and design, financial support, provision of study material or patients, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
Mayo Clinic and author T.J.N. have financial rights to ReGen Theranostics through licensing agreements. Frank Secreto has Intellectual property rights as an inventor or patent holder. All other authors indicated no potential conflicts of interest.
Supporting information
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Acknowledgments
We thank Lois Rowe for her expertise with histological staining, along with the Center for Regenerative Medicine BioTrust for providing access to flow cytometry and RT‐PCR resources. Sherri Biendarra provided essential insight and editorial commentary. High‐throughput hiPSC production and codevelopment of assays were performed in collaboration with ReGen Theranostics, Rochester, MN. This study was supported by the Todd and Karen Wanek Family Program for Hypoplastic Left Heart Syndrome at Mayo Clinic.
References
- 1. Ko HC, Gelb BD. Concise review: Drug discovery in the age of the induced pluripotent stem cell. Stem Cells Translational Medicine 2014;3:500–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scudellari M. How iPS cells changed the world. Nature 2016;534:310–312. [DOI] [PubMed] [Google Scholar]
- 3. Hildreth C. Induced pluripotent stem cells: Competitive analysis of the U.S. Patent landscape. Bioinformant 2015. https://www.bioinformant.com/ips-cell-patents/ [Google Scholar]
- 4. Alisson‐Silva F, de Carvalho Rodrigues D, Vairo L et al. Evidences for the involvement of cell surface glycans in stem cell pluripotency and differentiation. Glycobiology 2014;24:458–468. [DOI] [PubMed] [Google Scholar]
- 5. Muller FJ, Schuldt BM, Williams R et al. A bioinformatic assay for pluripotency in human cells. Nat Methods 2011;8:315–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Avior Y, Biancotti JC, Benvenisty N. TeratoScore: Assessing the differentiation potential of human pluripotent stem cells by quantitative expression analysis of teratomas. Stem Cell Reports 2015;4:967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maherali N, Hochedlinger K. Guidelines and techniques for the generation of induced pluripotent stem cells. Cell Stem Cell 2008;3:595–605. [DOI] [PubMed] [Google Scholar]
- 8. Kato R, Matsumoto M, Sasaki H et al. Parametric analysis of colony morphology of non‐labelled live human pluripotent stem cells for cell quality control. Sci Rep 2016;6:34009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aladjem MI, Spike BT, Rodewald LW et al. ES cells do not activate p53‐dependent stress responses and undergo p53‐independent apoptosis in response to DNA damage. Curr Biol 1998;8:145–155. [DOI] [PubMed] [Google Scholar]
- 10. Momcilovic O, Knobloch L, Fornsaglio J et al. DNA damage responses in human induced pluripotent stem cells and embryonic stem cells. PLoS One 2010;5:e13410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mujoo K, Butler EB, Pandita RK et al. Pluripotent stem cells and DNA damage response to ionizing radiations. Radiat Res 2016;186:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liedtke S, Biebernick S, Radke TF et al. DNA damage response in neonatal and adult stromal cells compared with induced pluripotent stem cells. Stem Cells Translational Medicine 2015;4:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clevers H. The cancer stem cell: Premises, promises and challenges. Nat Med 2011;17:313–319. [DOI] [PubMed] [Google Scholar]
- 14. Chen GL, Yang L, Rowe TC et al. Nonintercalative antitumor drugs interfere with the breakage‐reunion reaction of mammalian DNA topoisomerase II. J Biol Chem 1984;259:13560–13566. [PubMed] [Google Scholar]
- 15. Minocha A, Long BH. Inhibition of the DNA catenation activity of type II topoisomerase by VP16–213 and VM26. Biochem Biophys Res Commun 1984;122:165–170. [DOI] [PubMed] [Google Scholar]
- 16. Ross W, Rowe T, Glisson B et al. Role of topoisomerase II in mediating epipodophyllotoxin‐induced DNA cleavage. Cancer Res. 1984;44:5857–5860. [PubMed] [Google Scholar]
- 17. Hande KR. Etoposide: Four decades of development of a topoisomerase II inhibitor. Eur J Cancer 1998;34:1514–1521. [DOI] [PubMed] [Google Scholar]
- 18. Grandela C, Pera MF, Grimmond SM et al. p53 is required for etoposide‐induced apoptosis of human embryonic stem cells. Stem Cell Res 2007;1:116–128. [DOI] [PubMed] [Google Scholar]
- 19. Smith A, Nelson NG, Oommen S et al. Apoptotic susceptibility to DNA damage of pluripotent stem cells facilitates pharmacologic purging of teratoma risk. Stem Cells Translational Medicine 2012;1:709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Velichko AK, Lagarkova MA, Philonenko ES et al. Sensitivity of human embryonic and induced pluripotent stem cells to a topoisomerase II poison etoposide. Cell Cycle 2011;10:2035–2037. [DOI] [PubMed] [Google Scholar]
- 21. Folmes CDL, Martinez‐Fernandez A, Perales‐Clemente E et al. Disease‐causing mitochondrial heteroplasmy segregated within induced pluripotent stem cell clones derived from A MELAS patient. Stem Cells 2013;31:1296–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Andrews PW, Goodfellow PN, Shevinsky LH et al. Cell‐surface antigens of a clonal human embryonal carcinoma cell line: Morphological and antigenic differentiation in culture. Int J Cancer 1982;29:523–531. [DOI] [PubMed] [Google Scholar]
- 23. Andrews PW, Banting G, Damjanov I et al. Three monoclonal antibodies defining distinct differentiation antigens associated with different high molecular weight polypeptides on the surface of human embryonal carcinoma cells. Hybridoma 1984;3:347–361. [DOI] [PubMed] [Google Scholar]
- 24. Martins AM, Vunjak‐Novakovic G, Reis RL. The current status of iPS cells in cardiac research and their potential for tissue engineering and regenerative medicine. Stem Cell Rev 2014;10:177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tsankov AM, Akopian V, Pop R et al. A qPCR ScoreCard quantifies the differentiation potential of human pluripotent stem cells. Nat Biotech 2015;33:1182–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bock C, Kiskinis E, Verstappen G et al. Reference maps of human ES and iPS cell variation enable high‐throughput characterization of pluripotent cell lines. Cell 2011;144:439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Giovanella BC, Stehlin JS, Williams LJ Jr. Heterotransplantation of human malignant tumors in “nude” thymusless mice. II. Malignant tumors induced by injection of cell cultures derived from human solid tumors. J Natl Cancer Inst 1974;52:921–930. [DOI] [PubMed] [Google Scholar]
- 28. Taylor DD, Taylor CG, Black PH et al. Alterations of cellular characteristics of a human ovarian teratocarcinoma cell line after in vitro treatment with retinoids. Differentiation 1990;43:123–130. [DOI] [PubMed] [Google Scholar]
- 29. Damjanov I, Horvat B, Gibas Z. Retinoic acid‐induced differentiation of the developmentally pluripotent human germ cell tumor‐derived cell line, NCCIT. Lab Invest 1993;68:220–232. [PubMed] [Google Scholar]
- 30. Xia SL, Zhang X, Jing NH. [The induction and differentiation of a human teratocarcinoma cell line (PA‐1) in vitro]. Shi Yan Sheng Wu Xue Bao 1995;28:397–407. [PubMed] [Google Scholar]
- 31. Andrews PW, Damjanov I, Simon D et al. Pluripotent embryonal carcinoma clones derived from the human teratocarcinoma cell line Tera‐2. Differentiation in vivo and in vitro. Lab Invest 1984;50:147–162. [PubMed] [Google Scholar]
- 32. Andrews PW. Retinoic acid induces neuronal differentiation of a cloned human embryonal carcinoma cell line in vitro. Dev Biol 1984;103:285–293. [DOI] [PubMed] [Google Scholar]
- 33. Casper J, Schmoll HJ, Schnaidt U et al. Cell lines of human germinal cancer. Int J Androl 1987;10:105–113. [DOI] [PubMed] [Google Scholar]
- 34. Elliott AY, Cleveland P, Cervenka J et al. Characterization of a cell line from human transitional cell cancer of the urinary tract. J Natl Cancer Inst 1974;53:1341–1349. [DOI] [PubMed] [Google Scholar]
- 35. Mavilio F, Simeone A, Boncinelli E et al. Activation of four homeobox gene clusters in human embryonal carcinoma cells induced to differentiate by retinoic acid. Differentiation 1988;37:73–79. [DOI] [PubMed] [Google Scholar]
- 36. Fogh J. Human Tumor Cells In Vitro. New York: Plenum Press, 1975. [Google Scholar]
- 37. Sun Y. Alterations of SAG mRNA in human cancer cell lines: Requirement for the RING finger domain for apoptosis protection. Carcinogenesis 1999;20:1899–1903. [DOI] [PubMed] [Google Scholar]
- 38. Wang N, Trend B, Bronson DL et al. Nonrandom abnormalities in chromosome 1 in human testicular cancers. Cancer Res 1980;40:796–802. [PubMed] [Google Scholar]
- 39. Burridge PW, Matsa E, Shukla P et al. Chemically defined generation of human cardiomyocytes. Nat Methods 2014;11:855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Burridge PW, Holmstrom A, Wu JC. Chemically defined culture and cardiomyocyte differentiation of human pluripotent stem cells. Curr Protoc Hum Genet: 2015;87:21 23 21–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen VC, Couture LA. The suspension culture of undifferentiated human pluripotent stem cells using spinner flasks. Methods Mol Biol 2015;1283:13–21. [DOI] [PubMed] [Google Scholar]
- 42. Perales‐Clemente E, Cook AN, Evans JM et al. Natural underlying mtDNA heteroplasmy as a potential source of intra‐person hiPSC variability. EMBO J 2016;35:1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hartjes KA, Li X, Martinez‐Fernandez A et al. Selection via pluripotency‐related transcriptional screen minimizes the influence of somatic origin on iPSC differentiation propensity. Stem Cells 2014;32:2350–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kimbrel EA, Lanza R. Current status of pluripotent stem cells: Moving the first therapies to the clinic. Nat Rev Drug Discov 2015;14:681–692. [DOI] [PubMed] [Google Scholar]
- 45. Bulic‐Jakus F, Katusic Bojanac A, Juric‐Lekic G et al. Teratoma: From spontaneous tumors to the pluripotency/malignancy assay. Wiley Interdiscip Rev Dev Biol 2016;5:186–209. [DOI] [PubMed] [Google Scholar]
- 46. Kim MS, Horst A, Blinka S et al. Activin‐A and Bmp4 levels modulate cell type specification during CHIR‐induced cardiomyogenesis. PLoS One 2015;10:e0118670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dambrot C, Passier R, Atsma D et al. Cardiomyocyte differentiation of pluripotent stem cells and their use as cardiac disease models. Biochem J 2011;434:25–35. [DOI] [PubMed] [Google Scholar]
- 48. D'Antonio M, Woodruff G, Nathanson JL et al. High‐throughput and cost‐effective characterization of induced pluripotent stem cells . Stem Cell Reports 2017;8:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.