

Abstract
Human indoleamine 2,3-dioxygenase 1 (hIDO1) is an important heme-containing enzyme that is a key drug target for cancer immunotherapy. Several hIDO1 inhibitors have entered clinical trials, among which BMS-986205 (BMS) stands out as the only suicide inhibitor. Despite its “best-in-class” activity, the action mechanism of BMS remains elusive. Here, we report three crystal structures of hIDO1−BMS complexes that define the complete binding trajectory of the inhibitor. BMS first binds in a solvent exposed surface cleft near the active site in an extended conformation. The initial binding partially unfolds the active site, which triggers heme release, thereby exposing a new binding pocket. The inhibitor then undergoes a large scale movement to this new binding pocket, where it binds by adopting a high energy kinked conformation. Finally, the inhibitor relaxes to a bent conformation, via an additional large scale rearrangement, culminating in the energy minimum state. The structural data offer a molecular explanation for the remarkable efficacy and suicide inhibition activity of the inhibitor. They also suggest a novel strategy that can be applied for drug development targeting hIDO1 and related enzymes.
Human indoleamine 2,3-dioxygenase 1 (hIDO1) catalyzes the first and rate-limiting step of the kynurenine pathway, the dioxygenation of Trp to N-formyl kynurenine.1,2 Its ability to efficiently deprive the essential amino acid Trp and to promote the production of kynurenine pathway metabolites in the tumor microenvironment has been shown to be critical for cancer immune escape.3–7 Accordingly, hIDO1 is identified as a key cancer immunotherapeutic target. A large number of inhibitors targeting hIDO1 have been developed,8,9 four of which have entered clinical trials (Figure 1): epacadostat (Incyte Corp.),10 navoximod (NewLink Genetics),11 PF-06840003 (iTeos Therapeutics/Pfizer),12 and BMS-986205 (Flexus Biosciences, Inc./Bristol-Myers Squibb).13 Among them, BMS-986205 (referred to as BMS hereafter) is unique as it is the only suicide inhibitor (that irreversibly inhibits the enzyme activity) and it exhibits the best cell-based potency.13
Figure 1.
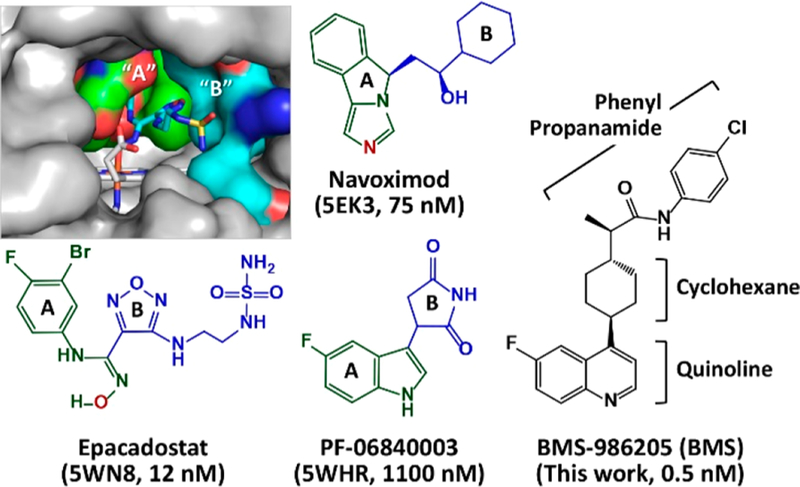
Molecular structures of hIDO1 inhibitors entered into clinical trials. The active site structure of the hIDO1−epcadostat complex is shown in the upper left inset to illustrate the locations of the “A” and “B” pockets in the active site (Sa). The fragments occupying the “A” and “B” pockets in each inhibitor are depicted in green and blue, respectively. The N and O atoms coordinated to the heme iron are labeled in red. The PDB code of hIDO1 in complex with each inhibitor and the cell-based potency of the inhibitor are indicated in the parentheses.
Crystal structures of hIDO1 in complex with a variety of inhibitors, including epacadostat,14 PF-06840003,12 navoximod derivatives,15 imidazothiazoles,16 phenyl imidazole,17 and amino triazole,18 have been reported. All of these inhibitors occupy the active site (Sa) and coordinate to the heme iron via an N atom, except epacadostat, which coordinates to the heme iron via an O atom, and PF-06840003, which sits on top of the heme iron without coordinating to it. Regardless of the heme iron coordination, all the high affinity inhibitors possess two fragments occupying the distinct “A” and “B” pockets in the Sa site (Figure 1). In contrast, inhibitors with a smaller framework, such as phenyl imidazole,17 typically occupy only the “A” pocket and exhibit a significantly lower efficacy.
The scaffold of BMS is analogous to that of PF-06840003, both of which possess a fused aromatic ring (quinoline and indole, respectively) with a side chain group extending out of it. It is tempting to assume that it binds to the Sa site with the quinoline group and the cyclohexane/phenyl propanamide group occupying the “A” and “B” pocket, respectively. However, this scenario is not consistent with the fact that BMS functions as a suicide inhibitor.9,13 A recent study reported by Nelp et al.13 revealed that BMS irreversibly inhibits hIDO1 by binding to the apo-form, instead of the holo-form, of the enzyme (here, the holo- and apo-forms stand for the enzyme with and without the prosthetic heme group, respectively). However, the mechanism by which the enzyme releases the heme and is targeted by the inhibitor remains elusive. In this work, we sought to delineate the action mechanism of BMS by carrying out X-ray crystallographic studies.
We first crystallized the inhibitor-free hIDO1 complex and then soaked it with BMS as a function of time. Through a large scale screening process, we identified three unique forms of the hIDO1−BMS complex and solved their structures (Table S1). Although hIDO1 functions as a monomer in free solution, all the three structures were solved in a dimeric form as reported previously.14,17 In the first structure (C0/C2), one subunit is in an inhibitor-free holo-form (C0), which represents the starting inhibitor-free structure, and the other subunit is trapped in an apo-form (C2), with the inhibitor bound near the “A” pocket of the Sa site. In the second structure (C1/C3), one subunit is trapped in a holo-form (C1), with the inhibitor bound near the “B” pocket of the Sa site, while the other subunit is in a new apo-form (C3), in which the inhibitor occupies both the “A” pocket of the Sa site and a previously identified inhibitory binding site (Si) on the proximal side of the heme.14 In the last structure (C0/C3), one subunit is in the C0 form, while the other is in the C3 form. The C3 structures identified in the C1/C3 and C0/C3 complexes are essentially identical (see Supporting Information). They are similar to that of hIDO1 in complex with a BMS analogue reported by Nelp et al.,13 suggesting that C3 is the energy minimum state of the complex. Taken together, the data suggest that the inhibitor binding reaction follows a sequential C0 → C1 → C2 → C3 mechanism as will be discussed below.
The inhibitor-free C0 structure exhibits an overall protein fold similar to that of the hIDO1-CN-Trp/IDE complex (PDB code: 5WMV),14 where the Sa and Si sites are occupied by the substrate Trp and an effector 3-indole ethanol (IDE), respectively (Figure S1A). However, the JK-Loop connecting the J-helix to the K-helix is completely disordered, as observed in other substrate-free complexes.12,14–18 The F270 side chain moves down to the now empty Si site. In addition, a water molecule enters the Sa site and sits on top of the heme iron.
In the C1 structure (Figures 2 and S1B), BMS binds in a solvent-exposed surface cleft in an extended conformation near the “B” pocket of the Sa site. The quinoline group, which sits perpendicular to the heme, is stabilized by π-stacking with the heme, F226, and F163. The phenyl propanamide moiety extends out via the cyclohexane linker along one side of the DE-hairpin to the roof of the “B” pocket. The binding induces a large scale rotation of the W237 side chain, such that it sterically hinders the DE-hairpin from assuming its native conformation, causing its partial unfolding.
Figure 2.

Initial binding of BMS in the C1 site. (A) C1 structure with the D-helix, DE-hairpin, DE-loop, and E-helix highlighted in cyan. The equivalent structural elements taken from the C0 structure (highlighted in magenta) are superimposed to highlight the large scale rotation of W237 and the partial unfolding of the DE-hairpin during the C0 → C1 transition. The JK-loop connecting the J-helix to the K-helix is disordered and unresolved in the structure. (B) Expanded view of the C1 site and the 2FO−FC map of the bound BMS contoured at 1.0σ. The associated FO−FC omit map and Polder map are shown in Figure S2A. (C) Surface view of the BMS binding site, where the D-helix, DE-hairpin, DE-loop, and E-helix are highlighted in cyan. BMS and the heme are shown as cyan and gray sticks, respectively.
The partial unfolding of the DE-hairpin and/or the close proximity of the bound BMS to the heme plausibly weaken the H346−iron coordination bond, thereby triggering the release of the heme. It leaves a spacious hydrophobic void in the interior of the protein that drives a large-scale movement of the mostly hydrophobic inhibitor into it. In this new C2 site (Figures 3A,B and S1C), BMS adopts an unusual kinked conformation. The phenyl group occupies the “A” pocket of the Sa site, where it is stabilized by π-stacking with Y126 and F163. The quinoline moiety lies in a space equivalent to that occupied by the heme, where it is stabilized by π-stacking with H364 in a T-shaped configuration. The C1 → C2 transition induces the refolding of the DE-hairpin to its native conformation. The significantly smaller size of the quinoline ring with respect to that of the heme, however, leads to imperfect packing that triggers the entry of a glycerol molecule (a cryoprotectant used for the crystal preparation) into the Si site.
Figure 3.

Sequential movement of BMS from the C1, C2, to C3 sites. (A) C1 structure with the D-helix, DE-hairpin, DE-loop, and E-helix (see the definitions in Figure 2) highlighted in cyan. The equivalent structural elements taken from the C2 structure (highlighted in magenta) are superimposed to highlight the refolding of the DE-hairpin during the C1 → C2 transition. The BMS and H346 in the C1 structure are shown as cyan and gray sticks, respectively. The BMS bound in the C2 and C3 sites (magenta and green sticks, respectively) are superimposed in the C1 structure to illustrate the inhibitor binding trajectory. (B) Expanded view of the C2 site and the 2FO−FC map (contoured at 1.0σ) of the bound BMS (magenta sticks) and glycerol (cyan sticks), a cryoprotectant used for the crystal preparation. (C) Expanded view of the C3 site and the 2FO−FC map (contoured at 1.0σ) of the bound BMS (green sticks). The associated FO−FC omit maps and Polder maps are shown in Figure S2B,C. (D) Three BMS conformers identified in this work.
The conformational strain of the BMS molecule and its imperfect packing in the C2 site trigger an additional structural transition. In the new C3 structure (Figures 3A,C and S1D), BMS relaxes to a bent conformation, with its quinoline group extending out into the Si site. The quinoline group is stabilized by π-stacking with F270 and by H-bonding with R343. The phenyl and propanamide moieties move slightly up away from the Si site to establish H-bonds with S167 and H346 via its amine and carbonyl group, respectively.
Comparison of the three distinct binding poses of BMS reveals that the inhibitor in the C3 structure establishes the most extensive interactions with its protein surroundings, underscoring the scenario that it is the energy minimum state of the complex. The overall fold of the enzyme remains almost the same along the full binding trajectory, despite the fact that the DE-loop is transiently unfolded during the C0 → C1 transition and that the inhibitor is translocated/rearranged by up to 15 Å within the protein matrix during each of the C1 → C2 → C3 transitions. The kinetic path by which BMS finds its way to the final binding site highlights the incredible plasticity of the protein and the unusual flexibility of the inhibitor (Figure 3D). The recognition and binding of BMS along the presumably energetically downhill trajectory can be described by a combination of three extremes of possible mechanisms. BMS initially binds to the C1 site in an extended conformation based on a classical lock and key mechanism. It cracks open the active site, allowing the release of the heme and the exposure of the new C2 site, where the inhibitor binds in a high energy kinked conformation via a conformational selection mechanism. Finally, the inhibitor relaxes to a bent conformation in the C3 site through an induced fit mechanism.
It is noteworthy that, in contrast to our sequential mechanism, a previous study performed by Nelp et al. suggests that BMS targets only the apo-form of hIDO1 to form the inhibitory complex C3.13 To differentiate the two mechanisms, we carried out spectroscopic studies of the binding reaction in free solution. As illustrated in Figure S4, when [BMS] is high enough to saturate the enzyme, mixing hIDO1 with BMS leads to heme release with a rate independent of [BMS] (as the reaction is ratelimited by heme release), which is consistent with the data reported by Nelp et al. However, at relatively low [BMS], an increase in [BMS] gradually increases the total amount of heme released, while the apparent heme release rate progressively decreases owing to the facilitated bimolecular heme rebinding reaction. The kinetic data fully support our mechanism, while disagree with the Nelp mechanism.
In conclusion, it is a nontrivial task to define binding trajectories of inhibitors due to the dynamic nature of protein targets. As such inhibitor binding pathways are typically only attainable by molecular dynamics simulations. This work demonstrates for the first time that a full binding trajectory of a drug to its target can be experimentally determined by X-ray crystallography. It calls for additional molecular dynamics simulations to further define the molecular details of each step of the binding reaction. The unprecedented interplay between inhibitor binding, protein unfolding, heme release, and inhibitor migration/rearrangement suggests a novel strategy that can be applied for drug development targeting hIDO1 and related enzymes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. Denis L. Rousseau and Thomas L. Poulos for helpful discussions. The structural data were collected by the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline staff at Sector 31 of the Advanced Photon Source. This research used resources of the Advanced Photon Source, a US Department of Energy (DOE), Office of Science User Facility, operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility. This work is supported by National Institute of Health Grant GM115773 and National Science Foundation Grant CHE-1404929 (to S.-R.Y.).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b07994.
Material and methods; crystallographic data collection and refinement statistics; structures of hIDO1−BMS complexes superimposed with that of hIDO1-CN-Trp/ IDE complex; FO−FC omit maps and Polder maps of BMS in the C1, C2, and C3 sites; structure of BMS in the C0/C3 complex; BMS binding reaction in free solution (PDF)
Accession Codes
Crystallography, atomic coordinates, and structure factors have been deposited in the Protein Data Bank (www.rcsb.org/pdb/home/home.do) under PDB ID codes 6DPQ, 6DPR, and 6MQ6.
Notes
The authors declare no competing financial interest.
Contributor Information
Khoa N. Pham, Department of Physiology and Biophysics, Albert Einstein College of Medicine, Bronx, New York 10461, United States
Syun-Ru Yeh, Department of Physiology and Biophysics, Albert Einstein College of Medicine, Bronx, New York 10461, United States.
REFERENCES
- (1).Sono M; Roach MP; Coulter ED; Dawson JH Heme-Containing Oxygenases. Chem. Rev 1996, 96 (7), 2841–2888. [DOI] [PubMed] [Google Scholar]
- (2).Raven EL A short history of heme dioxygenases: rise, fall and rise again. JBIC, J. Biol. Inorg. Chem 2017, 22 (2–3), 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Friberg M; Jennings R; Alsarraj M; Dessureault S; Cantor A; Extermann M; Mellor AL; Munn DH; Antonia SJ Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int. J. Cancer 2002, 101 (2), 151–5. [DOI] [PubMed] [Google Scholar]
- (4).Uyttenhove C; Pilotte L; Theate I; Stroobant V; Colau D; Parmentier N; Boon T; Van den Eynde BJ Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med 2003, 9 (10), 1269–74. [DOI] [PubMed] [Google Scholar]
- (5).Muller AJ; Prendergast GC Marrying immunotherapy with chemotherapy: why say IDO? Cancer Res 2005, 65 (18), 8065–8. [DOI] [PubMed] [Google Scholar]
- (6).Munn DH; Mellor AL Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Invest 2007, 117 (5), 1147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Prendergast GC Immune escape as a fundamental trait of cancer: focus on IDO. Oncogene 2008, 27 (28), 3889–900. [DOI] [PubMed] [Google Scholar]
- (8).Rohrig UF; Majjigapu SR; Vogel P; Zoete V; Michielin O Challenges in the Discovery of Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitors. J. Med. Chem 2015, 58 (24), 9421–37. [DOI] [PubMed] [Google Scholar]
- (9).Prendergast GC; Malachowski WP; DuHadaway JB; Muller AJ Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res 2017, 77 (24), 6795–6811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Beatty GL; O’Dwyer PJ; Clark J; Shi JG; Bowman KJ; Scherle PA; Newton RC; Schaub R; Maleski J; Leopold L; Gajewski TF First-in-Human Phase I Study of the Oral Inhibitor of Indoleamine 2,3-Dioxygenase-1 Epacadostat (INCB024360) in Patients with Advanced Solid Malignancies. Clin. Cancer Res 2017, 23 (13), 3269–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Nayak A; Hao Z; Sadek R; Dobbins R; Marshall L; Vahanian N; Ramsey J; Kennedy E; Mautino M; Link C; Lin R; Royer-Joo S; Morrissey K; Mahrus S; McCall B; Pirzkall A; Munn D; Janik J; Khleif S Phase 1a study of the safety, pharmacokinetics, and pharmacodynamics of GDC-0919 in patients with recurrent/ advanced solid tumors. Eur. J. Cancer 2015, 51, S69. [Google Scholar]
- (12).Crosignani S; Bingham P; Bottemanne P; Cannelle H; Cauwenberghs S; Cordonnier M; Dalvie D; Deroose F; Feng JL; Gomes B; Greasley S; Kaiser SE; Kraus M; Negrerie M; Maegley K; Miller N; Murray BW; Schneider M; Soloweij J; Stewart AE; Tumang J; Torti VR; Van Den Eynde B; Wythes M Discovery of a Novel and Selective Indoleamine 2,3-Dioxygenase (IDO-1) Inhibitor 3-(5-Fluoro-1H-indol-3-yl)pyrrolidine-2,5-dione (EOS200271/PF-06840003) and Its Characterization as a Potential Clinical Candidate. J. Med. Chem 2017, 60 (23), 9617–9629. [DOI] [PubMed] [Google Scholar]
- (13).Nelp MT; Kates PA; Hunt JT; Newitt JA; Balog A; Maley D; Zhu X; Abell L; Allentoff A; Borzilleri R; Lewis HA; Lin Z; Seitz SP; Yan C; Groves JT Immune-modulating enzyme indoleamine 2,3-dioxygenase is effectively inhibited by targeting its apo-form. Proc. Natl. Acad. Sci. U. S. A 2018, 115 (13), 3249–3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lewis-Ballester A; Pham KN; Batabyal D; Karkashon S; Bonanno JB; Poulos TL; Yeh SR Structural insights into substrate and inhibitor binding sites in human indoleamine 2,3-dioxygenase 1. Nat. Commun 2017, 8 (1), 1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Peng YH; Ueng SH; Tseng CT; Hung MS; Song JS; Wu JS; Liao FY; Fan YS; Wu MH; Hsiao WC; Hsueh CC; Lin SY; Cheng CY; Tu CH; Lee LC; Cheng MF; Shia KS; Shih C; Wu SY Important Hydrogen Bond Networks in Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Design Revealed by Crystal Structures of Imidazoleisoindole Derivatives with IDO1. J. Med. Chem 2016, 59 (1), 282–93. [DOI] [PubMed] [Google Scholar]
- (16).Tojo S; Kohno T; Tanaka T; Kamioka S; Ota Y; Ishii T; Kamimoto K; Asano S; Isobe Y Crystal Structures and Structure-Activity Relationships of Imidazothiazole Derivatives as IDO1 Inhibitors. ACS Med. Chem. Lett 2014, 5 (10), 1119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sugimoto H; Oda S; Otsuki T; Hino T; Yoshida T; Shiro Y Crystal structure of human indoleamine 2,3-dioxygenase: catalytic mechanism of O2 incorporation by a heme-containing dioxygenase. Proc. Natl. Acad. Sci. U. S. A 2006, 103 (8), 2611–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Alexandre JAC; Swan MK; Latchem MJ; Boyall D; Pollard JR; Hughes SW; Westcott J New 4-Amino-1,2,3-Triazole Inhibitors of Indoleamine 2,3-Dioxygenase Form a Long-Lived Complex with the Enzyme and Display Exquisite Cellular Potency. ChemBioChem 2018, 19 (6), 552–561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.