

Abstract
Background
This is an update of a previously published version of the review (Issue 10, 2011).
Epithelial ovarian cancer (EOC) is the seventh most common cause of cancer death among women worldwide. Treatment consists of a combination of surgical debulking and platinum‐based chemotherapy. Between 55% and 75% of women who respond to first‐line therapy experience relapse within two years. Second‐line chemotherapy is palliative and aims to reduce symptoms and prolong survival. Improved understanding about the molecular basis of EOC has led to the development of novel agents, such as epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors and anti‐EGFR antibodies.
Objectives
To compare the effectiveness and harmful effects of interventions that target the epidermal growth factor receptor in the treatment of epithelial ovarian cancer (EOC).
Search methods
We searched the Cochrane Gynaecological Cancer Group Trials Register, the Cochrane Central Register of Controlled Trials (CENTRAL; 2010, Issue 4), MEDLINE, and Embase up to October 2010. We also searched registers of clinical trials, abstracts of scientific meetings, and reference lists of included studies, and we contacted experts in the field. This update includes further searches up to September 2017.
Selection criteria
Randomised controlled trials (RCTs) comparing anti‐EGFR agents with or without conventional chemotherapy versus conventional chemotherapy alone or no treatment in women with histologically proven EOC.
Data collection and analysis
Two review authors independently abstracted data, assessed risk of bias, and performed GRADE assessment.
Main results
From 6105 references obtained through the literature search and an additional 15 references derived from grey literature searches, we identified seven RCTs that met our inclusion criteria and included 1725 participants. Trial results show that after first‐line chemotherapy is provided, maintenance treatment with erlotinib (EGFR tyrosine kinase inhibitor (TKI)) probably makes little or no difference in overall survival (hazard ratio (HR) 0.99, 95% confidence interval (CI) 0.81 to 1.20; one study; 835 participants; low‐certainty evidence) and may make little or no difference in progression‐free survival (HR 1.05, 95% CI 0.90 to 1.23; one study; 835 participants; very low‐certainty evidence). Less than 50% of participants provided quality of life data, and study authors reported these results incompletely. The certainty of evidence is very low, but treatment may reduce quality of life compared to observation.
Treatment with an EGFR TKI (vandetanib) for women with relapsed EOC may make little or no difference in overall survival (HR 1.25, 95% CI 0.80 to 1.95; one study; 129 participants; low‐certainty evidence) and may make little or no difference in progression‐free survival (HR 0.99, 95% CI 0.69 to 1.42; one study; 129 participants; very low‐certainty evidence). In treating patients with relapse, giving EGFR TKI may slightly increase some toxicities, such as severe rash (risk ratio (RR) 13.63, 95% CI 0.78 to 236.87; one study; 125 participants; very low‐certainty evidence). Quality of life data were not available for meta‐analysis.
Anti‐EGFR antibody treatment in relapsed EOC may or may not make a difference to overall survival (HR 0.93, 95% CI 0.74 to 1.18; four studies; 658 participants; moderate‐certainty evidence) and may or may not have any effect on progression‐free survival (HR 0.90, 95% CI 0.70 to 1.16; four studies; 658 participants; low‐certainty evidence). Anti‐EGFR antibody treatment may or may not increase side effects, including severe nausea and/or vomiting (RR 1.27, 95% CI 0.56 to 2.89; three studies; 503 participants; low‐certainty evidence), severe fatigue (RR 1.06, 95% CI 0.66 to 1.73; I² = 0%; four studies; 652 participants; low‐certainty evidence), and hypokalaemia (RR 2.01, 95% CI 0.80 to 5.06; I² = 0%; three studies; 522 participants; low‐certainty evidence). Severe diarrhoea rates were heterogeneous across studies (RR 2.87, 95% CI 0.59 to 13.89; four studies; 652 participants; low‐certainty evidence), and subgroup analysis revealed that severe diarrhoea was more likely with pertuzumab (RR 6.37, 95% CI 1.89 to 21.45; I² = 0%; three studies; 432 participants; low‐certainty evidence) than with seribantumab treatment (RR 0.38, 95% CI 0.07 to 2.23; I² = 0%; one study; 220 participants; very low‐certainty evidence). Quality of life data were incompletely reported, and we were unable to combine them in a meta‐analysis.
Authors' conclusions
Current evidence suggests that an anti‐EGFR single‐agent biological treatment (EGFR TKI or anti‐EGFR antibody) makes little or no difference to survival, either as maintenance treatment after first‐line chemotherapy or in association with chemotherapy in recurrent cancer. Anti‐EGFR therapy may increase some side effects and may or may not reduce quality of life.
Plain language summary
Do epidermal growth factor receptor (EGFR) inhibitors, alone or with chemotherapy, improve outcomes for women with epithelial ovarian cancer (EOC)?
What is the aim of this review? The aim of this review was to find out if medicines that inhibit epidermal growth factor receptors improve the outcomes of women with EOC and to identify the harms of treatment. We sought to collect and analyse results of all relevant studies to answer this question and found seven studies.
What are the key messages of the review? Limited evidence suggests that there is little or no benefit from taking anti‐EGFR agents either alongside chemotherapy at relapse, or as maintenance treatment after first‐line chemotherapy for EOC, and that some side effects may be increased.
What was studied in the review? Approximately a quarter of gynaecological cancers are of ovarian origin, although they account for half of all deaths related to gynaecological cancers. The annual incidence worldwide is about 6.6 cases per 100,000 women, with an annual mortality rate of four deaths per 100,000 women, as three‐quarters of these cases are diagnosed at an advanced stage. Treatment usually consists of a combination of surgery to remove as much of the visible cancer as possible (debulking surgery) and platinum‐based chemotherapy. Most cases of EOC (70% to 80%) respond to chemotherapy. Unfortunately, most women with advanced disease experience relapse and ultimately die because of resistance to chemotherapy.
EGFR is involved in controlling cell growth. High EGFR activity is linked to development of EOC and to poor outcomes. Preventing EGFR activity is an attractive target for novel therapeutic agents. Anti‐EGFR agents have been developed and have been tried in combination with chemotherapy or as maintenance treatment after chemotherapy.
What are the main results of the review? This review found evidence from seven studies on the effects of an anti‐EGFR antibody or an EGFR tyrosine kinase inhibitor (TKI) (erlotinib and vandetanib) in women treated for EOC. This was given either as maintenance treatment, following completion of first‐line chemotherapy, or for EOC that had grown after initial treatment (recurrent or refractory disease) .
We found low‐certainty evidence to suggest that following first‐line chemotherapy, maintenance treatment with erlotinib probably makes little or no difference in overall survival, and very low‐certainty evidence that it makes little or no difference in progression‐free survival (time before cancer starts to grow again). Treatment may reduce quality of life compared to no treatment (observation), but minimal data were available, and we have very low‐certainty about these findings. Data on adverse events were not available for inclusion in the meta‐analysis.
We found low‐certainty evidence to suggest that treatment with vandetanib for women with relapsed EOC probably makes little or no difference in overall survival, and very low‐certainty evidence that it makes little or no difference in progression‐free survival. Vandetanib treatment probably increases the risk of a severe rash, but data on other side effects were of very low‐certainty due to small numbers and very wide confidence intervals.
We found moderate‐certainty evidence to show that treatment with an anti‐EGFR antibody probably makes little or no difference in overall survival, and low‐certainty evidence suggesting that it may make little or no difference in progression‐free survival in cases of relapsed disease. Treatment with the anti‐EGFR antibody pertuzumab probably increases the risk of diarrhoea (low‐certainty), but evidence for its effect on other side effects is of very low‐certainty due to low numbers of events.
Summary of findings
Background
Description of the condition
In 2012, 238,719 women worldwide received the diagnosis of epithelial ovarian cancer (EOC), and 151,917 died, corresponding to an annual age‐standardised incidence of 6.1 cases per 100,000 women, an annual mortality rate of 3.8 deaths per 100,000, and a cumulative lifetime risk of 0.5% (GLOBOCAN 2012). In terms of age‐standardised incidence and mortality, EOC is the seventh most common cancer among women. Onset of the disease is often insidious; symptoms are vague and may mimic other conditions. This may lead to a delay in diagnosis, and three‐quarters of women with EOC receive the diagnosis when the disease has spread throughout the abdomen (stage III or IV) (Shepherd 1989). By this time, five‐year survival is 20% to 30% (Jemal 2008). Epithelial ovarian cancer (EOC), which may arise from the surface of the ovary, accounts for 90% of all ovarian cancers and typically presents in postmenopausal women, with a peak incidence when women are in their early sixties, although it does occur in younger women and is often associated with genetic predispositions (Quinn 2001). More recent data suggest that the site of origin of the most common type of EOC (high grade serous adenocarcinoma) could be the epithelial lining of the fallopian tubes. Intraepithelial precursor lesions (serous tubal intraepithelial carcinoma or serous tubal in situ carcinoma (STIC) lesions) are commonly found at the fimbrial ends of fallopian tubes removed from women at high risk of developing EOC due to BRCA mutations (Erickson 2013).
Description of the intervention
Management of advanced EOC consists of debulking surgery and platinum‐based chemotherapy, with or without the addition of a taxane (Morrison 2012; Stewart 1999). Randomised controlled trials (RCTs) found that, in advanced disease not thought amenable to primary debulking surgery, there was no difference in survival if surgery was performed before or after the first three cycles of chemotherapy (Vergote 2010; Kehoe 2015). Despite good initial response to platinum agents and taxanes, most women will experience relapse, will require further treatment with chemotherapy, and eventually will develop resistance to conventional chemotherapeutic agents.
Conventional chemotherapeutic agents have shown activity on all rapidly dividing cells, hence the common side effects such as bone marrow suppression and mucositis. Increasing knowledge of the genetic basis for cancer has led to the development of novel reagents that target cancer‐specific pathways. It is hoped that these reagents will spare normal cells and will reduce the toxic side effects of chemotherapy, in addition to conferring an enhanced therapeutic effect.
How the intervention might work
Cancer cells, just like normal cells, can respond to external stimulation via growth factor receptors. These pathways are often mutated in cancer and therefore serve as a potential target for control of cancer cell growth.
Epidermal growth factor receptors and EOC
The epidermal growth factor receptor (EGFR or ErbB1) is a cell surface molecule that is normally involved in controlling cell growth. The EGFR is a tyrosine kinase enzyme that is made up of an extracellular ligand‐binding domain, a cell membrane‐spanning region, and an intracellular tyrosine kinase domain (Figure 1A). Following binding of its ligand, epidermal growth factor (EGF), the EGFR is activated; EGFR tyrosine kinase activity phosphorylates tyrosine residues on the EGFR and on other proteins (Figure 1B), causing their activation and precipitating a sequence of downstream events that lead to increased cell growth (Figure 1C). EGFR was first implicated in cancer aetiology when it was discovered that an oncogenic retrovirus encoded a mutated version of EGFR (Downward 1984). Abnormal EGFR activation has been demonstrated in EOC, is associated with a poorer prognosis (Nicholson 2001), and can happen through a variety of mechanisms. EGFR mutation occurs in some cases of EOC (Moscatello 1995); the most common EGFR mutation is seen in the extracellular region and has been shown to result in EGF‐independent activation (Ekstrand 1992). Overexpression of EGFR is common in many cancers (Bartlett 1996; Slamon 1989). EGFR activity can also be stimulated by increased production of EGF by tumour cells (Bandera 2003). EGFR is central to the promotion of cell growth and has a role in the development of cancer. Therefore, preventing EGFR activity could be an attractive target for novel therapeutic agents. Anti‐EGFR agents have been developed to prevent extracellular EGF binding or to inhibit tyrosine kinase activity (Figure 1D). EGFR is a member of a family of similar molecules called 'the epidermal growth factor receptor family'. This family also includes human epidermal growth factor receptor (HER2/neu), Erb3 and Erb4.
1.
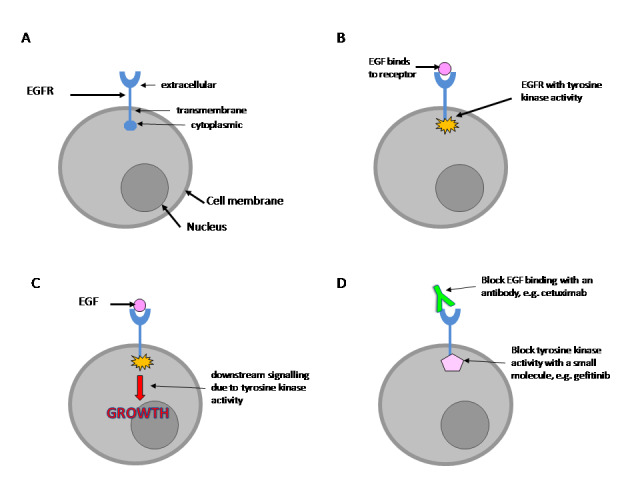
(A) The EGFR is a transmembrane protein. (B) Following binding to its ligand, EGF, the EGFR is stimulated and develops tyrosine kinase activity. (C) Tyrosine kinase activity sets off a sequence of downstream events that lead to stimulation of cell growth. (D) EGFR activity can be blocked by antibodies that prevent EGF binding to the receptor or by use of chemicals that inhibit tyrosine kinase enzyme activity.
EGFR tyrosine kinase inhibitors
Clinical trials have evaluated several small molecule inhibitors of the EGFR tyrosine kinase for treatment of EOC, including gefitinib and erlotinib.
Gefitinib (Iressa/ZD1839) is a small molecule that specifically inhibits EGFR tyrosine kinase activity (Moulder 2001). Preclinical studies have shown antitumour activity (Ciardiello 2001), and a phase I clinical trial showed that the orally active agent was well tolerated by patients with a range of tumour types, including ovarian (Baselga 2002). A phase II study of gefitinib demonstrated poor response rates in women with platinum‐resistant EOC who had not had the EGFR status of their EOC tested (Posadas 2007). However, a 9% response rate reported in participants with EGFR‐positive tumours highlighted the need for selecting patients likely to benefit from treatment (Schilder 2005). Although response rates were modest, treatment was well tolerated, and rash and diarrhoea were the main toxicities.
Erlotinib (Tarceva/OSI‐774), another EGFR tyrosine kinase inhibitor, has been through phase I and II clinical trials. Results from a previous phase II trial suggest that erlotinib may show activity in ovarian carcinoma (Gordon 2005). Researchers who gave erlotinib to 34 participants with EGFR‐positive recurrent, refractory, chemotherapy‐resistant EOC reported that responses were modest, but the treatment was well tolerated. Further phase II and phase III trials for erlotinib have been performed since the first version of this review was performed (NCT00263822; NCT00520013).
Neither gefitinib nor erlotinib has been licenced for use in EOC, outside of clinical trials. However, both agents have shown promise in treating other types of cancer: gefitinib has been licenced by the Food and Drug Administration (FDA) in the USA for use in certain types of non‐small cell lung cancer (NSCLC), and erlotinib has been licenced by both the FDA and the European Medicines Agency (EMEA) for use in some types of NSCLC and in pancreatic cancer.
Antibodies against EGFR
It is also possible to block the EGFR pathway by using specific antibodies.
Monoclonal antibodies have a specific target pattern to which they bind. Monoclonal antibodies have been developed against the extracellular portion of EGFR (Figure 1D). These antibodies can reduce EGFR activity, either by directly blocking EGF binding, or by causing the EGFR to be taken into the cell and degraded, thereby reducing the number of receptors available for stimulation at the cell surface. The monoclonal antibodies developed for clinical trials are humanised. This means that in addition to binding to the EGFR, a portion of these antibodies are identical to normal human antibodies, so they can stimulate the patient's own immune system. When the antibody binds to the EGFR on a cell, it labels the cancer cell so that it is recognised as foreign and is then destroyed by cells of the patient's own immune system. HER‐2/neu is another member of the EGFR family, and overexpression is related to poor outcomes in breast cancer. A monoclonal antibody, trastuzumab (Herceptin), has been developed that binds to HER‐2/neu (Baselga 2001; Cooley 1999). Bookman 2003 reported a 7.3% response rate with no significant toxicity among women with recurrent EOC treated with Herceptin (Bookman 2003). Another monoclonal antibody, pertuzumab, prevents dimerisation of HER‐2 with other HER receptors, and clinical trials have used pertuzumab to treat women with EOC (Gordon 2006; Makhija 2010).
IMC‐C225 (Cetuximab/Erbitux) is a humanised mouse monoclonal antibody against EGFR (HER‐1/ErbB1) that has shown activity in combination with topotecan (a conventional chemotherapeutic agent) in preclinical studies (Ciardiello 1999; Goldstein 1995). Cetuximab binds to the EGFR and blocks EGF binding, thereby preventing downstream signalling and growth stimulation, as well as antibody‐directed cell killing by the immune system.
Why it is important to do this review
Novel types of treatment strategies work in different ways when compared with conventional chemotherapeutic agents. Therefore, it is important to establish whether adding these new drugs to conventional chemotherapy regimens yields added benefit in terms of survival, and, if so, at what cost, in terms of additional harmful effects. Furthermore, these compounds may be less toxic than conventional chemotherapy agents; therefore it may be possible to give these newer treatments to women who are not currently taking chemotherapy (so called maintenance treatment) to reduce the chance of, or to delay, recurrence of their EOC.
Objectives
To compare the effectiveness and harmful effects of interventions that target the epidermal growth factor receptor in the treatment of epithelial ovarian cancer (EOC).
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs).
Types of participants
Adult women with histologically proven EOC. We excluded women with other concurrent malignancies.
Types of interventions
Anti‐EGFR agents (tyrosine kinase inhibitors and/or monoclonal antibodies) + conventional chemotherapy versus conventional chemotherapy
Anti‐EGFR agents (tyrosine kinase inhibitors and/or monoclonal antibodies) versus no treatment
Types of outcome measures
Primary outcomes
Overall survival: survival until death from all causes
Secondary outcomes
Progression‐free survival
Quality of life, measured by a validated scale
-
Toxicity: grades of toxicity to be extracted and grouped as follows ‐ see CTEP 2017
Haematological (leucopenia, anaemia, thrombocytopenia, neutropaenia, haemorrhage)
Gastrointestinal (nausea, vomiting, anorexia, diarrhoea, liver, proctitis)
Genitourinary
Skin (stomatitis, mucositis, alopecia, allergy)
Neurological (peripheral and central)
Other side effects not categorised above.
Search methods for identification of studies
We searched for papers written in all languages and carried out translations when necessary.
Electronic searches
We searched the following electronic databases.
Cochrane Central Register of Controlled Trials (CENTRAL; 2017, Issue 8), in the Cochrane Library;
MEDLINE via Ovid (October 2010 to August week 4, 2017);
Embase via Ovid (October 2010 to 2017, week 36).
We have presented the CENTRAL, MEDLINE, and Embase search strategies related to the review topic in Appendix 1, Appendix 2, and Appendix 3, respectively.
We searched the databases from 1990 until September 2017. These novel agents have been developed recently, and so searches before 1990 would not have been relevant.
We identified all relevant articles on PubMed and, by using the 'related articles' feature, we carried out a further search for newly published articles.
Searching other resources
We searched Physicians Data Query, ISRCTN registrywww.clinicaltrials.gov, National Cancer Institute Clinical Trials Information, and the National Research Register (NRR) for ongoing trials. We sought details of ongoing or unpublished trials from the FDA (Food and Drug Administration, the regulatory body for medicines within the USA) and EMEA (European Medicines Agency, the drug regulatory body within Europe), and from pharmaceutical company sources.
We searched the reference lists of all included trials for further relevant trials.
Correspondence
We contacted the authors of relevant trials to ask for clarification and for further data, which may or may not have been published, and we requested further information from pharmaceutical companies involved in the development of anti‐EGFR agents.
Data collection and analysis
Selection of studies
We downloaded all titles and abstracts retrieved by electronic searching to the reference management database Endnote and removed duplicates. At least two review authors (a combination from KG, CT, KH, TL, RG, and JM) independently examined the remaining 6105 unique references (4103 in searches for the original review and 2002 from updated searches). We identified another 15 records from the grey literature. We excluded studies that clearly did not meet the inclusion criteria, and we obtained full‐text copies of 54 potentially relevant references (20 from the original review). At least two review authors (a combination of KG, KH, RG, TL, JM, and CT) independently assessed the eligibility of retrieved papers and excluded 42 references. The two review authors resolved disagreements by discussion and, when necessary, by consultation with a third review author (JM). We have documented reasons for exclusion in the Characteristics of excluded studies table. For the original review, we identified five ongoing studies through searches of the grey literature, but all had been published in the most recent search, at least in abstract form. In the original review, three studies were ongoing; all were subsequently published and are now among the Included studies in this updated review. We have included a total of seven studies in the review, and we have detailed study characteristics in the Characteristics of included studies table.
Data extraction and management
For included studies, we abstracted data as follows.
Author, year of publication, and journal citation (including language).
Country.
Setting.
Inclusion and exclusion criteria.
Study design and methods.
-
Study population.
Total number enrolled.
Characteristics.
Age.
Comorbidities.
Previous treatment.
Total study duration.
Total number of intervention groups.
-
EOC details at diagnosis.
International Federation of Obstetrics and Gynecology (FIGO) stage.
Histological cell type.
Tumour grade.
Extent of disease.
-
Intervention details.
Type of EGFR inhibitor.
Dose.
Duration of treatment.
Consolidation treatment or treatment of active disease.
-
Comparison details.
Type of control: conventional chemotherapy or no treatment.
Dose (if appropriate).
Duration (if appropriate).
Deviations from protocol.
Risk of bias in the study (see below).
Duration of follow‐up.
-
Outcomes: overall survival, progression‐free survival, quality of life, toxicity.
For each outcome: outcome definition (with diagnostic criteria if relevant).
Unit of measurement (if relevant).
For scales: upper and lower limits, and whether high or low score is good.
Results: number of participants allocated to each intervention group.
For each outcome of interest: sample size; missing participants.
We extracted data on outcomes as below.
For time‐to‐event (overall survival and progression‐free survival) data, we extracted the log of the hazard ratio [log(HR)] and its standard error from trial reports; if these were not reported, we attempted to estimate them from other reported statistics using the methods of Parmar 1998.
For dichotomous outcomes (toxicity), we extracted the number of participants in each treatment arm who experienced the outcome of interest and the number of participants assessed at endpoint, to estimate a risk ratio (RR).
We extracted both unadjusted and adjusted statistics, if reported. When we extracted adjusted results, we recorded the variables that were adjusted.
When possible, all extracted data were those relevant to an intention‐to‐treat analysis, in which participants were analysed in the groups to which they were assigned.
We noted the time points at which outcomes were collected and reported.
Two review authors (KH and KG) abstracted data independently onto a data abstraction form specially designed for the review. We resolved differences by discussion, or by appeal to a third review author (JM or SN) when necessary.
Assessment of risk of bias in included studies
We used the Cochrane tool to assess risk of bias in included RCTs. This included assessment of:
sequence generation;
allocation concealment;
blinding of participants, treatment providers, and outcome assessors;
incomplete outcome data: we recorded the proportion of participants whose outcomes were not reported at the end of the study; we noted whether loss to follow‐up was not reported. We coded a satisfactory level of loss to follow‐up for each outcome as:
yes, if less than 20% of participants were lost to follow‐up and reasons for loss to follow‐up were similar in both treatment arms;
no, if more than 20% of participants were lost to follow‐up or reasons for loss to follow‐up differed between treatment arms; and
unclear; if loss to follow‐up was not reported
selective reporting of outcomes; and
other possible sources of bias.
Two review authors (CT and KG) independently applied the risk of bias tool and resolved differences by discussion or by appeal to a third review author (JM). We have presented these results in a risk of bias summary table. We have interpreted these results in the light of findings with respect to risk of bias.
Measures of treatment effect
We used the following measures of the effects of treatment.
For time‐to‐event data, we used the hazard ratio (HR).
For dichotomous outcomes, we used the risk ratio (RR). However, we were unable to estimate an RR for comparison of treatments if one or both treatment groups experienced no events, as in skin toxicity and congestive heart failure outcomes.
When adjusted results were available, we preferred to use them; otherwise we used unadjusted results.
Unit of analysis issues
The units of analysis were the participants receiving interventions of interest.
Dealing with missing data
We did not impute missing outcome data for any outcomes. If data were missing or only imputed data were reported, we contacted trial authors to request data on outcomes only among participants who were assessed.
Assessment of heterogeneity
We assessed heterogeneity between studies by visually inspecting forest plots, by estimating the percentage of heterogeneity between trials that cannot be ascribed to sampling variation (Higgins 2003), by performing a formal statistical test of the significance of the heterogeneity (Deeks 2001), and, when possible, by conducting subgroup analyses (see below). If we found evidence of substantial heterogeneity, we investigated and reported possible reasons for this.
Assessment of reporting biases
We had planned to use funnel plots to investigate possible reporting bias and the presence of small‐study effects. However, we did not produce funnel plots due to the limited number of studies per outcome (i.e. fewer than 10) (Guyatt 2011).
Data synthesis
We pooled results of clinically similar studies in meta‐analyses.
For time‐to‐event data, we pooled HRs by using the generic inverse variance facility of RevMan 5.
For dichotomous outcomes, we calculated and pooled the RR for each study.
For continuous outcomes, we would have pooled mean differences between treatment arms at the end of follow‐up if trials had measured the outcome on the same scale; otherwise we would have pooled standardised mean differences.
Assessing the certainty of the evidence
The GRADE method was introduced following publication of the initial protocol, and we included it in this update as an a priori analysis to meet current MECIR guidelines (GRADE Working Group). Two review authors independently exported data into the GRADEpro Guideline Development Tool and analysed them using GRADE methods, with reference to a third review author to reach consensus when disagreements arose (GRADEpro GDT; Schünemann 2014).
We downgraded the evidence from 'high' certainty by one level for serious (or by two levels for very serious) concerns for each limitation.
High‐certainty: we are very confident that the true effect lies close to that of the estimate of the effect.
Moderate‐certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different.
Low‐certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect.
Very low‐certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect.
Certainty of evidence could be upgraded for large magnitude of effect if all plausible confounding would reduce or increase the demonstrated effect, if no effect was observed, or if a dose‐response effect was noted.
We summarised data in a 'Summary of findings' table that included the most important outcome data: overall survival and progression‐free survival for primary disease and recurrent disease, and any G3‐4 toxicity (subdivided by treatment type). As the current GRADEpro software does not allow data reported as hazard ratios, we analysed survival at time points for overall survival and progression‐free survival, when data allowed.
Subgroup analysis and investigation of heterogeneity
We had not originally planned to conduct subgroup analyses. However, we did perform subgroup analyses based on type of anti‐EGFR treatment (tyrosine kinase inhibitor vs monoclonal antibody) because these treatments may have involved different activities and varying side effect profiles, given their different mechanisms of action. We performed analyses separately for primary treatment and for treatment of recurrent disease. We also performed subgroup analyses based on platinum resistance/sensitivity in recurrent disease because biologically these often yield response rates that are different from those seen with conventional treatment. When visual inspection of forest plots revealed obvious heterogeneity between individual anti‐EGFR agents for the same type of anti‐EGFR treatment, we also performed subgroup analyses (see Differences between protocol and review section).
Sensitivity analysis
We performed no sensitivity analyses (see Differences between protocol and review section) due to the small number of studies included in each group.
Results
Description of studies
Results of the search
Original review: up to 2011
The original search (October 2010) for the previously published version of this review revealed 4103 unique references. Through title and abstract screening of these references, review authors identified 20 trials as potentially eligible for inclusion in the review. Upon full‐text screening of these 20 references, 19 studies were excluded for the reasons described in the Characteristics of excluded studies table. A single RCT met review inclusion criteria, and this study was described in the Characteristics of included studies table (Makhija 2010).
Searches of the grey literature at that stage yielded three relevant ongoing trials and eight other studies that, for the reasons described, were included in the Characteristics of excluded studies table.
Review update: 2011 to 2017
We updated the search in August 2017. We de‐duplicated the results of this search in Endnote and uploaded the references into Covidence systematic review software to aid sifting of titles and abstracts. We initially identified an additional 2002 references and five from the grey literature. Upon title and abstract screening, we excluded all but 34 references. We screened the full‐text articles and excluded 23 for reasons given in the Characteristics of excluded studies table. We identified eight references that were duplicates of studies identified in the previous search. We included seven studies from the remaining 28 references (Characteristics of included studies). Three of these studies corresponded with the three references identified as ongoing studies in the original review.
In summary, we screened a total of 6120 references (6105 from searches and 15 from grey literature). We excluded 6039 studies on title and abstract screening, and another 42 unique references after review of the full texts. We included seven studies (32 references ‐ 24 unique and eight duplicates) that met our inclusion criteria (Figure 2).
2.
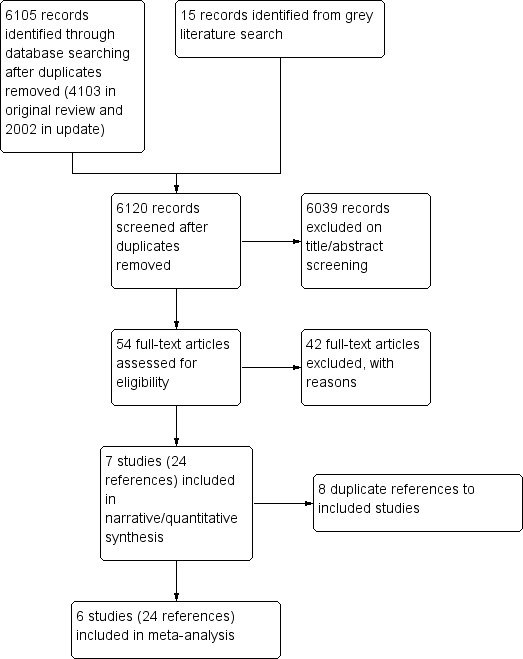
Study flow diagram.
Included studies
Seven studies (24 references) including 1725 women met the inclusion criteria, and we included them in our analysis (Chekerov 2017; Coleman 2014; Kaye 2013; Kurzeder 2016; Lui 2016; Makhija 2010; Vergote 2014).
Participants
All studies were limited to adult women with histologically proven epithelial ovarian, primary peritoneal, or fallopian tube cancer.
First‐line treatment
Vergote 2014 included 835 women who had completed first‐line chemotherapy within six weeks and was therefore a maintenance and consolidation study after first‐line treatment. This trial excluded women with platinum‐refractory disease (growth of cancer despite platinum‐based chemotherapy). Participants were not selected for tumour EGFR expression. Median age was 59 years (range 19 to 85) in the erlotinib group, and 59 years (range 27 to 84) in the observation arm.
Treatment for recurrent disease
The other six studies enrolled 890 women with relapsed disease (one patient was subsequently excluded) (Chekerov 2017; Coleman 2014; Kaye 2013; Kurzeder 2016; Lui 2016; Makhija 2010).
Chekerov 2017 was an open‐label study that randomised 102 women and included 96 in the final analysis. Study authors have presented data in a conference proceedings abstract only, and so reasons for the loss of six participants are unclear. Eligible women had platinum‐sensitive relapsed epithelial ovarian/fallopian or peritoneal cancer and had received no more than two prior treatments for this disease. Inclusion criteria included a requirement to have measurable disease or elevated cancer antigen (CA)‐125 and to have KRAS wild type on tumour biopsy.
Coleman 2014 included 129 women with recurrent EOC who were previously treated with at least one cycle of platinum‐based chemotherapy and could have received up to three previous cycles of chemotherapy, including previous antiangiogenic agents. Other inclusion criteria included a performance status showing Zubrod score 0 to 2 and adequate haematological, renal, liver, and cardiovascular function. Median age was 61.7 years (range 32.6 to 80) in the control group and 61.9 years (range 34.3 to 82.5) in the study group.
Kaye 2013 included 149 women at first relapse, although all women had platinum‐sensitive disease (progression‐free interval greater than 6 months after completion of a platinum‐based regimen). Other requirements included Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1 and adequate haematological, renal, hepatic, and cardiac function. Median age was 58 years (range 18 to 81) in the pertuzumab group and 58 years (range 18 to 85 years) in the placebo group.
Kurzeder 2016 included 156 women with platinum‐resistant or platinum‐refractory EOC and low tumour HER‐3 mRNA expression. Originally 324 women were enrolled, but researchers excluded 156 due to ineligibility, including failure of testing and central review of pathology or after HER‐3 tumour testing.
Lui 2016 included 223 women with platinum‐resistant or ‐refractory advanced EOC. Participants underwent mandatory pretreatment core needle biopsy and submitted archived tumour samples, as available, for biomarker analysis (heregulin (HRG); human epidermal growth factor receptor (HER‐3 (ErbB3); HER‐2; EGFR; and betacellulin (BTC)). Median age was 58.5 years (range 30 to 82) for seribantumab plus paclitaxel (S + P) and 60.6 years (range 28 to 85) for paclitaxel only (P).
Makhija 2010 included 131 women and assessed 130 (99%) of them at the end of the trial. All women had previously been treated with at least one platinum‐containing chemotherapy regimen and were now platinum‐resistant. All women had ECOG performance status of 0 or 1, left ventricular ejection fraction 50% or higher, and adequate haematological, renal, and hepatic function. All women had platinum‐resistant or platinum‐refractory cancer and measurable disease as per Response Evaluation Criteria in Solid Tumours (RECIST), or clinically or radiologically detectable disease with two consecutive rising CA‐125 levels before treatment. None had received more than one prior treatment for platinum‐resistant disease, but participants could have received any number of platinum‐containing regimens before becoming platinum‐resistant. No participants had received prior treatment with any HER‐2 pathway inhibitors or gemcitabine.
Interventions
First‐line treatment
Vergote 2014 was an open‐label phase III RCT that randomised women to either maintenance erlotinib (EGFR tyrosine kinase inhibitor) 150 mg orally daily for two years (or until disease progression) or observation (not placebo controlled) following response to first‐line platinum‐based chemotherapy.
Treatment for recurrent disease
Chekerov 2017 was an open‐label study of carboplatin AUC4 and gemcitabine 1000 mg/m² or carboplatin AUC5 and pegylated doxorubicin 40 mg/m²; trialists randomised women to panitumumab 6 mg/kg day 1 and day 15, every three or four weeks. Panitumumab is a fully human monoclonal antibody specific to the epidermal growth factor receptor (EGFR).
Coleman 2014 was an open‐label randomised phase II study that compared a combination of docetaxel and vandetanib (D + V) versus docetaxel (D) alone in women with recurrent EOC. Women whose condition progressed on docetaxel (D) were allowed to cross over to single‐agent vandetanib (V). Vandetanib is an oral tyrosine kinase inhibitor that is not a 'pure' EGFR inhibitor but inhibits several tyrosine kinases involved in malignancy: vascular epithelial growth factor receptor 2/3 (VEGFR 2/3), EGFR, and EGFR rearranged during transfection transmembrane receptor tyrosine kinase, which is known as RET. The rationale for vandetanib is that the action of VEGFR inhibitors is thought to be blunted by EGFR signalling, and combined VEGFR/EGFR inhibitors had demonstrated activity in preclinical models of EOC (Wedge 2002).
Kaye 2013 was an open‐label phase II RCT that randomised women to either chemotherapy (carboplatin with paclitaxel or gemcitabine) or chemotherapy and pertuzumab (840‐mg loading dose followed by 420 mg three times weekly). After completion or withdrawal of chemotherapy (if toxicity), pertuzumab was continued three times weekly for another 11 cycles (17 in total) but could be continued for up to a maximum of 52 cycles. No cross‐over was allowed at any stage.
Kurzeder 2016 was a double‐blind, placebo‐controlled, randomised phase III trial that compared chemotherapy plus either pertuzumab (840‐mg loading dose followed by 420 mg every three weeks) or placebo.
Makhija 2010 examined the activity of pertuzumab, an antibody that prevents human EGFR‐2 (HER‐2) dimerisation with other EGFR monomers ‐ a process required for activation and signalling. This randomised double‐blind phase II study randomly assigned women to receive gemcitabine (800 mg/m² on days 1 and 8 of a 21‐day cycle) plus either placebo or pertuzumab (840‐mg loading dose administered intravenously followed by 420 mg every three weeks). Treatment was administered until disease progression or unacceptable toxicity was evident. Kaplan‐Meier plots show that maximum length of follow‐up was 30 months.
Lui 2016 was an open‐label study of seribantumab, a fully human immunoglobulin G2 monoclonal antibody that binds to human epidermal growth factor receptor (HER)‐3 (ErbB3). Antibody binding blocks heregulin (HRG)–mediated ErbB3 signalling and induces ErbB3 receptor downregulation. Women with platinum‐resistant or ‐refractory disease received paclitaxel plus/minus seribantumab. Paclitaxel was given weekly (80 mg/m² during cycle one, with optional modification in subsequent cycles) once per week for three weeks, followed by one week of rest. Seribantumab was given as a 40‐mg/kg loading dose, then at 20 mg/kg once weekly.
Outcomes
Progression‐free survival and overall survival were the primary or secondary outcomes in all studies. Most studies also reported adverse events using Common Terminology Criteria for Adverse Events version 5.0 (CTEP 2017) or an earlier version. Other frequently reported outcomes included overall response rate (ORR), complete response rate (CR), and partial response rate.
Vergote 2014 evaluated quality of life as a secondary outcome by using the European Organization for Research and Treatment of Cancer core quality of life questionnaire (EORTC QLQ‐C30) (Giesinger 2016).
In a separate conference abstract (Lalla 2008, in Makhija 2010), trial investigators also reported on quality of life, as measured by the FOSI questionnaire (Functional Assessment of Cancer Therapy – Ovarian (FACT‐O) Symptom Index) (Jensen 2011). Similarly, Kurzeder 2016 reported quality of life data outcomes in a separate abstract (Hilpert 2016, in Kurzeder 2016). Lui 2016 incompletely reported quality of life data that were not available for analysis.
Excluded studies
After obtaining full‐text articles, we excluded 42 references for the following reasons.
Two references were narrative review articles and did not include any study that met our inclusion criteria (Dinh 2008; Palayekar 2008).
Nine references were non‐randomised phase I studies of EGFR antagonists conducted to establish maximum tolerated dose and toxicity profiles (Bauman 2012; Campos 2010; Harter 2013; Jhaveri 2012; Kimball 2008; Koolen 2011; Nimeiri 2008; Vasey 2008; Vlahovic 2012).
Twenty‐nine references were non‐randomised studies of single‐agent EGFR antagonists undertaken to assess response in women with EOC with or without combination conventional chemotherapy (Annunziata 2010; Blank 2010; Bookman 2003; Campos 2005; Chambers 2010; Ciunci 2014; Garcia 2012; Gordon 2005; Gordon 2006; Guastalla 2007; Hariprasad 2006; Hariprasad 2009; Hirte 2010; Joly 2009; Konner 2008; Krasner 2005; Lheureux 2012; NCT00861120; NCT01296035; Pautier 2010; Posadas 2007; Ray‐Coquard 2008; Schilder 2005; Schilder 2009; Secord 2008; Seiden 2007; Steffensen 2013; Wagner 2007; Weroha 2011).
One reference was a protocol for an RCT that compared erlotinib (an EGFR inhibitor) plus bevacizumab (Avastin; a monoclonal antibody that inhibits the vascular endothelial growth factor pathway ‐ another target for novel anticancer drugs) versus bevacizumab alone as first‐line consolidation chemotherapy for women with advanced EOC (Campos 2011 (NCT00520013)). Although this was an RCT, the comparison is not between an EGFR inhibitor and either standard chemotherapy or no treatment, and thus it did not fulfil our inclusion criteria.
One reference was a randomised study of lapatinib monotherapy in a range of cancers; only three randomised participants had EOC, and the study was closed early due to lack of efficacy (Galsky 2012).
For further details of all excluded studies, see the Characteristics of excluded studies table.
Risk of bias in included studies
See Figure 3 for visual representation of risk of bias assessments.
3.

Methodological quality summary: review authors' judgements about each methodological quality item for each included study.
Allocation
Sequence generation
Four studies did not report the method of generation of the sequence of random numbers used to allocate women to treatment arms (Chekerov 2017; Kurzeder 2016; Lui 2016; Makhija 2010). Therefore they were at unclear risk of bias.
The other three studies provided details on sequence generation and were at low risk of bias (Coleman 2014; Kaye 2013; Vergote 2014).
Allocation concealment
Three studies were at low risk of bias because researchers explained concealment of allocation and it appeared robust (Coleman 2014; Kaye 2013; Vergote 2014). Four studies did not mention whether an effort was made to conceal allocation from participants and healthcare professionals involved in the trial (Chekerov 2017, Kurzeder 2016; Lui 2016; Makhija 2010).
Blinding
Blinding of participants and personnel
Makhija 2010 initially blinded participants and healthcare professionals, but subsequently all but one participant discontinued blinded treatment. It is unclear how this may have affected outcomes liable to bias through lack of blinding. Kurzeder 2016 was a double‐blind study but did not describe methods used to ensure blinding. The other five studies were open‐label or included a no treatment control group (Chekerov 2017; Coleman 2014; Kaye 2013; Lui 2016; Vergote 2014). Therefore they are at high risk of bias for subjective outcomes, such as toxicity and progression‐free survival, but at low risk of bias for overall survival outcomes.
Blinding of outcome assessors
Kurzeder 2016 was a double‐blind study but did not describe methods used to ensure blinding. In Makhija 2010, it is unclear whether or not outcome assessors were blinded, although this is unlikely to have affected overall survival, which is at low risk of bias. Five studies were open‐label or included a no treatment control group (Chekerov 2017; Coleman 2014; Kaye 2013; Lui 2016; Vergote 2014). Therefore we deemed these studies to be at high risk of bias for subjective outcomes, such as toxicity and progression‐free survival, but at low risk of bias for overall survival outcomes.
Incomplete outcome data
All studies adequately accounted for all women initially included. Quality of life data in Vergote 2014 were limited by low response rates (85% at baseline, ranging from 72% to 51% during the first year and < 50% during the second year), which could suggest high risk of bias, although these data were not available in a form suitable for inclusion in the meta‐analysis. Generally, data on quality of life were poorly reported, making comparison in a meta‐analysis challenging.
Selective reporting
Review authors identified five of the included studies before publication and included them as ongoing studies in the previous version of this review (Chekerov 2017; Coleman 2014; Kurzeder 2016; Kaye 2013; Vergote 2014). Data reported were as specified before study completion, although not all outcome data were made available in an analysable form either in the publication or following contact with study authors. Overall, we deemed these studies to be at low or unclear risk of selective reporting bias.
Other potential sources of bias
All studies were industry sponsored with links to industry declared by some of the trial authors. However, given that results demonstrated no significant effect, this is unlikely to have had an effect on trial results, especially as the outcomes reported were prespecified.
In Coleman 2014, 33 of 63 women in the control group crossed over to receive vandetanib without chemotherapy on progression of disease. Given that vandetanib appeared to have minimal efficacy, we are unsure how this may have affected overall survival data, although progression‐free survival would not have been affected by cross‐over.
Effects of interventions
See: Table 1; Table 2; Table 3
Summary of findings for the main comparison. EGFR tyrosine kinase inhibitor (TKI) compared to observation alone for maintenance treatment of epithelial ovarian cancer after first‐line chemotherapy.
| EGFR tyrosine kinase inhibitor (TKI) compared to observation alone for maintenance treatment of epithelial ovarian cancer after first‐line chemotherapy | ||||||
| Patient or population: maintenance treatment of epithelial ovarian cancer after first‐line chemotherapy Setting: hospital outpatient treatment of women with ovarian/fallopian tube/primary peritoneal cancer after response to first‐line chemotherapy Intervention: EGFR tyrosine kinase inhibitor (TKI) Comparison: observation alone | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with observation alone | Risk with EGFR tyrosine kinase inhibitor (TKI) | |||||
| Overall survival | Median 50.8 months in the EGFR TKI group vs 59.1 months in the observation arm See comment |
HR 0.99 (0.81 to 1.20) | 835 (1 RCT) | ⊕⊕⊝⊝ LOWa | Outcome unlikely to be affected by blinding. due to the way HRs are calculated, the assumed and corresponding risks were not estimated. | |
| Progression‐free survival | Median PFS of 12.7 months in the EGFR TKI group vs 12.4 months in the observation arm See comment |
HR 1.05 (0.90 to 1.23) | 835 (1 RCT) | ⊕⊝⊝⊝ VERY LOWb | due to the way HRs are calculated, the assumed and corresponding risks were not estimated. | |
| Quality of life assessed with EORTC QLQ‐C30 and OV28 (Ovarian Cancer Module) questionnaires | "Global health/QOL scores showed a significant overall difference between the two treatment arms during the first year (P 0.0102) in favour of the observation arm. In addition, the QLQ‐C30 found statistically significant differences at the 5% level in symptom levels for diarrhoea, loss of appetite, nausea/vomiting, and fatigue, with worse symptom scores for the erlotinib arm. None of the scales, however,reported differences of 10 points except for the diarrhoea [sic] scale in which differences of more than 20 points were observed at most assessments during the first year. Sensitivity analyses by means of imputation revealed similar results". | ‐ | 835 (1 RCT) | ⊕⊝⊝⊝ VERY LOWc | Analysable data were not provided in the published paper or after communication with the study author, and so we were unable to analyse or exclude selective reporting bias. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; EGFR: epidermal growth factor receptor; HR: hazard ratio; RR: risk ratio; OR: odds ratio; PFS: progression‐free survival; QOL: quality of life; QLQ‐C30: Quality of Life Questionnaire C30; RCT: randomised controlled trial; TKI: tyrosine kinase inhibitor. | ||||||
| GRADE Working Group grades of evidence High‐certainty: we are very confident that the true effect lies close to that of the estimate of the effect Moderate‐certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low‐certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low‐certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
aDowngraded by two levels for imprecision (confidence intervals that cross zero and single study) and inability to assess inconsistency, as results were based on a single study. bDowngraded by three levels due to risk of bias (unblinded study); imprecision (confidence intervals that cross zero and single study); and inability to assess inconsistency, as results were based on a single study. cDowngraded by three levels due to the possibility of selective reporting bias (incompletely reported predefined outcome, so possibility of selective outcome reporting); imprecision; and risk of bias (unblinded), as no data were available to analyse effect and outcome was highly likely to be affected by lack of blinding.
Summary of findings 2. EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer.
| EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer | ||||||
| Patient or population: treatment of relapsed epithelial ovarian cancer Setting: hospital outpatient treatment of women with relapsed ovarian/fallopian tube/primary peritoneal cancer Intervention: EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy Comparison: chemotherapy alone | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with chemotherapy alone | Risk with EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy | |||||
| Overall survival | Median OS for chemotherapy alone was 18 months compared to 14 months in the chemotherapy plus EGFR TKI arm. | HR 1.25 (0.80 to 1.95) | 129 (1 RCT) | ⊕⊕⊝⊝ LOWa,b | Outcome unlikely to be affected by blinding. due to the way HRs are calculated, the assumed and corresponding risks were not estimated. | |
| Progression‐free survival | Median PFS for chemotherapy only was 3.5 months compared to a median PFS of 3.0 months in the chemotherapy plus EGFR TKI arm. | HR 0.99 (0.69 to 1.42) | 129 (1 RCT) | ⊕⊝⊝⊝ VERY LOWc | due to the way HRs are calculated, the assumed and corresponding risks were not estimated. | |
| Toxicity: grade 3 or 4 rash Assessed with National Cancer Institute Common Toxicity Criteria version 3 (CTCv3.0) or CommonTerminology Criteria for Adverse Events (CTCAE) | Study population | RR 13.63 (0.78 to 236.87) | 125 (1 RCT) | ⊕⊝⊝⊝ VERY LOWc | ||
| 1 per 100 | 11 per 100 (1 to 100) | |||||
| Toxicity: grade 3 or 4 nausea ± vomiting Assessed with National Cancer Institute Common Toxicity Criteria version 3 (CTCv3.0) or Common Terminology Criteria for Adverse Events (CTCAE) | Study population | RR 0.63 (0.16 to 2.52) | 125 (1 RCT) | ⊕⊝⊝⊝ VERY LOWc | ||
| 8 per 100 | 5 per 100 (1 to 20) | |||||
| Toxicity: grade 3 or 4 fatigue Assessed with National Cancer Institute Common Toxicity Criteria version 3 (CTCv3.0) or Common Terminology Criteria for Adverse Events (CTCAE) | Study population | RR 0.87 (0.28 to 2.72) | 125 (1 RCT) | ⊕⊝⊝⊝ VERY LOWc | ||
| 9 per 100 | 8 per 100 (3 to 26) | |||||
| Toxicity: cardiac toxicity (any grade) | Study population | RR 5.24 (0.26 to 107.02) | 125 (1 RCT) | ⊕⊕⊝⊝ LOWb | ||
| 16 per 1000 | 82 per 1000 (4 to 1000) | |||||
| Quality of life: not measured | ‐ | ‐ | ‐ | ‐ | ‐ | No QoL data included in the publication |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CTCAE: Common Terminology Criteria for Adverse Events; EGFR: epidermal growth factor receptor; HR: hazard ratio; PFS: progression‐free survival; QoL: quality of life; RCT: randomised controlled trial; RR: risk ratio; TKI: tyrosine kinase inhibitor | ||||||
| GRADE Working Group grades of evidence High‐certainty: we are very confident that the true effect lies close to that of the estimate of the effect Moderate‐certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low‐certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low‐certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
aOutcome unlikely to be affected by lack of blinding. bDowngraded by two levels due to imprecision (one small study, wide confidence intervals that cross zero, and too few events for adequate power). cDowngraded by three levels due to risk of bias (blinding absent or unclear); imprecision; and inability to assess inconsistency (one small study, wide confidence intervals that cross zero, and too few events for adequate power).
Summary of findings 3. Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer.
| Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer | ||||||
| Patient or population: treatment of relapsed epithelial ovarian cancer Setting: hospital outpatient treatment of women with ovarian/fallopian tube/primary peritoneal cancer after response to first‐line chemotherapy Intervention: anti‐EGFR antibody plus chemotherapy Comparison: chemotherapy alone | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with chemotherapy alone | Risk with anti‐EGFR antibody plus chemotherapy | |||||
| Overall survival | Median OS by study in intervention and placebo groups, respectively Chekerov 2017: "overall survival are not yet evaluable" Kaye 2013: 28.2 months vs median overall survival not reached Kurzeder 2016: median OS 10.2 months vs 8.4 months Lui 2016: median OS 13.7 months vs 10.12 months Makhija 2010: median OS 13 months and 13.1 months |
HR 0.93 (0.74 to 1.18) | 658 (4 RCTs) | ⊕⊕⊕⊝ MODERATEa | Outcome unlikely to be affected by blinding. due to the way HRs are calculated, the assumed and corresponding risks were not estimated. | |
| Progression‐free survival | Median progression‐free survival (PFS) by study in intervention and placebo groups, respectively Chekerov 2017: median PFS 9.5 months vs 10.7 months Kaye 2013: median PFS 34.1 weeks vs 40.0 weeks Kurzeder 2016: median PFS 4.3 months vs 2.6 months Lui 2016: median PFS 3.75 months vs 3.68 months Makhija 2010: median PFS 2.9 months and 2.6 months |
HR 0.90 (0.70 to 1.16) | 658 (4 RCTs) | ⊕⊕⊝⊝ LOWb | due to the way HRs are calculated, the assumed and corresponding risks were not estimated. | |
| Toxicity: grade 3 to 4 anaemia Assessed with National Cancer Institute Common Toxicity Criteria version 3 (CTCv3.0) or Common Terminology Criteria for Adverse Events (CTCAE) | Study population | RR 0.84 (0.47 to 1.49) | 652 (4 RCTs) | ⊕⊕⊝⊝ LOWb | ||
| 78 per 1000 | 65 per 1000 (37 to 116) | |||||
| Toxicity: grade 3 or 4 diarrhoea Assessed with National Cancer Institute Common Toxicity Criteria version 3 (CTCv3.0) or Common Terminology Criteria for Adverse Events (CTCAE) | Study population | RR 2.87 (0.59 to 13.89) | 652 (4 RCTs) | ⊕⊝⊝⊝ VERY LOWc | Diarrhoea differential side effect dependent upon type of anti‐EGFR antibody | |
| 20 per 1000 | 58 per 1000 (12 to 283) | |||||
| Toxicity: grade 3 or 4 diarrhoea ‐ pertuzumab Assessed with National Cancer Institute Common Toxicity Criteria version 3 (CTCv3.0) or Common Terminology Criteria for Adverse Events (CTCAE) | Study population | RR 6.37 (1.89 to 21.45) | 432 (3 RCTs) | ⊕⊕⊝⊝ LOWb | Diarrhoea was a more consistent side effect with pertuzumab when separated from trials of other anti‐EGFR inhibitors | |
| 14 per 1000 | 89 per 1000 (26 to 299) | |||||
| Toxicity: grade 3 or 4 diarrhoea ‐ seribantumab Assessed with National Cancer Institute Common Toxicity Criteria version 3 (CTCv3.0) or Common Terminology Criteria for Adverse Events (CTCAE) | Study population | RR 0.38 (0.07 to 2.23) | 220 (1 RCT) | ⊕⊝⊝⊝ VERY LOWd | ||
| 38 per 1000 | 14 per 1000 (3 to 84) | |||||
| Toxicity: grade 3 or 4 nausea ± vomiting Assessed with National Cancer Institute Common Toxicity Criteria version 3 (CTCv3.0) or Common Terminology Criteria for Adverse Events (CTCAE) | Study population | RR 1.27 (0.56 to 2.89) | 503 (3 RCTs) | ⊕⊕⊝⊝ LOWb | ||
| 41 per 1000 | 52 per 1000 (23 to 118) | |||||
| Toxicity: grade 3 or 4 fatigue Assessed with National Cancer Institute Common Toxicity Criteria version 3 (CTCv3.0) or Common Terminology Criteria for Adverse Events (CTCAE) | Study population | RR 1.06 (0.66 to 1.73) | 652 (4 RCTs) | ⊕⊕⊝⊝ LOWb | ||
| 95 per 1000 | 101 per 1000 (63 to 164) | |||||
| Toxicity: grade 3 or 4 hypokalaemia Assessed with National Cancer Institute Common Toxicity Criteria version 3 (CTCv3.0) or Common Terminology Criteria for Adverse Events (CTCAE) | Study population | RR 2.01 (0.80 to 5.06) | 522 (3 RCTs) | ⊕⊕⊝⊝ LOWb | ||
| 26 per 1000 | 52 per 1000 (21 to 132) | |||||
| Quality of life | Quality of life (QoL) (Hilpert data 2016 ‐ see additional references for Kurzeder 2016 study): abdominal/gastrointestinal QoL (QLQ‐OV28) score 3.9 (95% CI ‐3.3 to 11.2); diarrhoeal symptoms QoL score worse on pertuzumab; score difference 21.2 (95% CI 10.1 to 32.3; P = 0.0003). Makhija 2010 (reported only in conference abstract form ‐ see Lalla 2008 in subsidiary references for Makhija 2010): "The median time to symptom deterioration was 1.7 months in the gemcitabine+placebo arm vs. 3.8 months in the gemcitabine+pertuzumab arm (HR = 0.62, 95% CI: 0.36‐1.05). Symptom improvement (≥ 3 point increase in FOSI) occurred in 28 women (43%) given gemcitabine+pertuzumab, compared to 18 (28%) in those receiving gemcitabine+placebo". ClinicalTrials.gov identifier: NCT00096993 This outcome was included in the original version of this review; no new results have been identified. |
‐ | (1 RCT) | ⊕⊝⊝⊝ VERY LOWe | Quality of life was not reported consistently; narrative description of data is provided in review text, as data could not be added to meta‐analysis. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CTCAE: Common Terminology Criteria for Adverse Events; EGFR: epidermal growth factor receptor; HR: hazard ratio; OS: overall survival; PFS: progression‐free survival; QLQ‐OV28: European Organization for Research and Treatment of Cancer module for ovarian cancer; QoL: quality of life; RCT: randomised controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High‐certainty: we are very confident that the true effect lies close to that of the estimate of the effect Moderate‐certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low‐certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low‐certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
aOutcome unlikely to be affected by lack of blinding. Downgraded by one level due to imprecision (wide confidence intervals that cross zero). bDowngraded by two levels due to lack of blinding in studies or unclear method of randomisation in studies and imprecision. cDowngraded by three levels for inconsistency between studies of different anti‐EGFR antibodies; imprecision; and lack of blinding or unclear method of randomisation in studies. dDowngraded by three levels due to imprecision (wide confidence intervals that cross zero and too few events for adequate power); lack of blinding or unclear method of randomisation in studies; and inconsistency (one study). eDowngraded by 3+ levels due to risk of bias (lack of blinding); inability to gauge inconsistency (only one study); minimal data presented and inability to assess adequately but wide confidence intervals; selective reporting bias as data collected but not presented in final publication; and risk of indirectness as symptom may be due to progression of disease rather than to treatment.
First‐line treatment
EGFR tyrosine kinase inhibitor (TKI) compared to observation alone for maintenance treatment for epithelial ovarian cancer after first‐line chemotherapy
One study examined the role of a tyrosine kinase inhibitor, erlotinib, against EGFR as maintenance/consolidation treatment following response to first‐line chemotherapy (Vergote 2014).
Overall survival (risk of death)
See Analysis 1.1; Table 1.
1.1. Analysis.

Comparison 1 EGFR tyrosine kinase inhibitor (TKI) compared to observation alone for maintenance treatment of epithelial ovarian cancer after first‐line chemotherapy, Outcome 1 Overall survival.
There is probably no or little difference in overall survival between erlotinib and observation arms (hazard ratio (HR) 0.99, 95% confidence interval (CI) 0.81 to 1.20; P = 0.90; one study; 835 participants; low‐certainty evidence) (Vergote 2014). Trial authors adjusted both this and progression‐free survival for stratification parameters. Median survival was 50.8 months for participants receiving erlotinib versus 59.1 months for those in the observation arm. There was probably no or little difference in death within 36 months between the two groups (40/100 in control group vs 39/100 in erlotinib group; risk ratio (RR) 0.96, 95% CI 0.82 to 1.14: P = 0.67).
Progession‐free survival (risk of disease progression)
See Analysis 1.2; Table 1
1.2. Analysis.

Comparison 1 EGFR tyrosine kinase inhibitor (TKI) compared to observation alone for maintenance treatment of epithelial ovarian cancer after first‐line chemotherapy, Outcome 2 Progression‐free survival.
This study was powered for a primary outcome of progression‐free survival, and there may be no or little difference between treatment groups (HR 1.05, 95% CI 0.90 to 1.23; one study; 835 participants; P = 0.53; very low‐certainty evidence) with median progression‐free survival of 12.7 months in the erlotinib group versus 12.4 months in the observation arm. Two participants died before clinical progression was observed and are included in these figures (clarification obtained from study authors). There may be no or little difference in risk of progression within 12 months between groups (50 episodes of progression in the observation arm vs 48 in the erlotinib arm per 100 women; RR 0.96, 95% CI 0.84 to 1.11; P = 0.61).
Quality of life and adverse effects
Researchers recorded quality of life data, but reporting was incomplete and was limited by low compliance (85% at baseline, 72% to 51% during the first year, and less than 50% during the second year; one study; 835 participants; very low‐certainty evidence). Study authors stated that there was a significant difference in global health/quality of life scores during the first year (P = 0.010) and "QLO‐C30 found statistically significant differences at the 5% level in symptom levels for diarrhoea [sic], loss of appetite, nausea/vomiting, and fatigue, with worse symptom scores for the erlotinib arm", but further data were not made available, despite requests to the study author; therefore, we could not include these in the meta‐analysis, nor could we include toxicity data, because only data for the erlotinib arm were published and comparison group data have not been provided, despite communication with study authors. In terms of erlotinib toxicity, the main side effects were rash (grade 1 to 2, 67%; grade 3 to 4, 12.8%) and diarrhoea (grade 1 to 2, 55.2%; grade 3 to 4, 4.8%) (Table 1).
Treatment for recurrent disease
EGFR TKI plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer
One study, which included 129 participants, examined the addition of vandetanib to docetaxel chemotherapy for treatment of recurrent disease (Coleman 2014). The primary outcome was progression‐free survival, and the study was powered to detect an increase in median progression‐free survival of two months. Overall survival and adverse effects were secondary outcomes; researchers reported and graded adverse effects by National Cancer Institute Common Toxicity Criteria version 2.0 (CTEP 2017). Post hoc analysis revealed no subgroups that benefited from erlotinib. It was noted that a positive fluorescence in situ hybridisation (FISH) score for EGFR expression was a marker of poor prognosis in both arms (overall survival 46.1 months vs negative FISH EGFR score 67.0 months; HR, 1.56, 95% CI 1.01 to 2.40; P = .044).
Overall survival
See Analysis 2.1; Table 2.
2.1. Analysis.
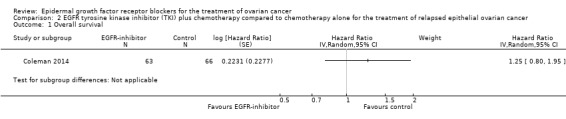
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 1 Overall survival.
There is probably no or little difference in overall survival between docetaxel plus or minus vandetanib (HR 1.25, 95% CI 0.80 to 1.95; one study; 125 participants; low‐certainty evidence), although the study was not powered to find a difference in overall survival. Another confounder was that 33 of 63 women in the control group crossed over to receive vandetanib without chemotherapy on progression. However, lack of effect on progression‐free survival suggests that these data are valid, as the addition of vandetanib appeared to have minimal efficacy.
Progression‐free survival
See Analysis 2.2; Table 2.
2.2. Analysis.
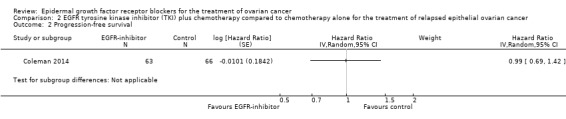
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 2 Progression‐free survival.
There may be little or no difference in progression‐free survival (HR 0.99, 95% CI 0.69 to 1.42; P = 0.49; one study; 125 participants; very low‐certainty evidence). Median progression‐free survival time was three months in the docetaxel/vandetanib group versus 3.5 months in the docetaxel‐only control group.
Toxicity
See Analysis 2.3; Analysis 2.22; Table 2.
2.3. Analysis.
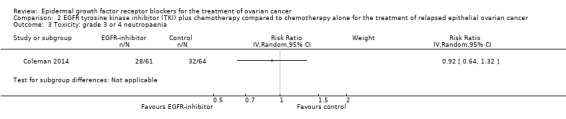
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 3 Toxicity: grade 3 or 4 neutropaenia.
2.22. Analysis.
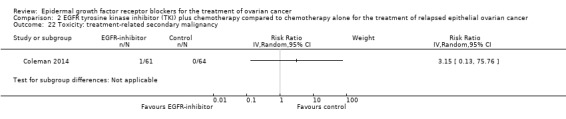
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 22 Toxicity: treatment‐related secondary malignancy.
Rash was the main side effect in the vandetanib group, with six (10%) women experiencing grade 3 to 4 rash compared to 0 (0%) in the control group, although this result had very wide confidence intervals due to the small number of events (RR 13.63, 95% CI 0.78 to 236.87; I² = 0%; one study; 125 participants; very low‐certainty evidence). For rash of any grade, there were 11 cases (18%) in the control group versus 30 (49%) in the docetaxel/vandetanib group.
Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer
We found three studies evaluating the anti‐HER‐2 monoclonal antibody, pertuzumab (Kaye 2013; Kurzeder 2016 ;Makhija 2010); one study evaluating panitumumab, an anti‐EGFR antibody (Chekerov 2017); and one study evaluating an anti‐HER‐3 antibody (seribantumab) (Lui 2016), all in the context of relapsed/recurrent disease.
Kaye 2013 was an open‐label RCT of 149 women that compared six cycles of chemotherapy (carboplatin and either paclitaxel (Taxol) or gemcitabine) with or without pertuzumab in a platinum‐sensitive setting.
Three studies evaluated anti‐EGFR antibodies in platinum‐refractory or ‐resistant disease. Makhija 2010, which included 130 women with platinum‐resistant disease, was a double‐blinded placebo‐controlled RCT that reported data on gemcitabine plus pertuzumab versus gemcitabine plus placebo. Kurzeder 2016 included 156 women with platinum‐resistant or ‐refractory disease who received chemotherapy plus either pertuzumab or placebo. Lui 2016 included 223 women with platinum‐refractory or ‐resistant disease.
Chekerov 2017 was also conducted in a platinum‐sensitive setting and recruited 103 women but analysed 96 with KRAS (gene) wild‐type, platinum‐sensitive recurrent EOC. Women were treated with carboplatin plus gemcitabine or pegylated doxorubicin plus or minus panitumumab ‐ a fully human antibody to EGFR. Researchers provided data only in abstract form, and we were not able to further evaluate them as part of the meta‐analysis. We have presented a narrative description of study results (see Characteristics of included studies).
Overall survival
See Analysis 3.1; Table 3
3.1. Analysis.
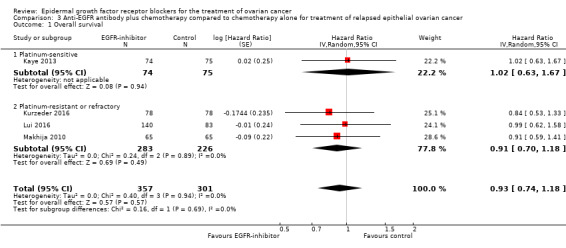
Comparison 3 Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer, Outcome 1 Overall survival.
There may be no or little difference in overall survival between women who received an anti‐EGFR antibody and those given placebo/chemotherapy alone (HR 0.93, 95% CI 0.74 to 1.18; four studies; 658 participants; I² = 0%) (the percentage of variability in effect estimates that is due to heterogeneity rather than to sampling error (chance) is not important); moderate‐certainty evidence; Analysis 3.1). There was little or no difference in effect depending on whether the study was conducted in a platinum‐sensitive (HR 1.02, 95% CI 0.63 to 1.67; one study; 149 participants; I² = not applicable) or a platinum‐resistant/refractory setting (HR 0.91, 95% CI 0.70 to 1.18; I² = 0% (the percentage of variability in effect estimates that is due to heterogeneity rather than to sampling error (chance) is not important); three studies; 509 participants; Analysis 3.1).
Kaye 2013 mentioned no adjustment, although randomisation was stratified by treatment‐free interval (TFI; six to 12 months vs more than 12 months), measurable versus non‐measurable disease, chemotherapy regimen (carboplatin–paclitaxel vs carboplatin–gemcitabine), and territory (Eastern Europe vs Western Europe and Canada). Both studies were exploratory studies and were not powered to detect a difference in overall survival. The median overall survival, after two years of follow‐up, was 28.2 weeks in the pertuzumab group but was not reached in the control group.
In Kurzeder 2016, overall survival was 10.2 months (95% CI 6.7 to 15.2 months) in the pertuzumab group versus 8.4 months (95% CI 6.1 to 12.0 months) in the placebo group.
Makhija 2010 reported 91 (70%) deaths and adjusted estimates of survival outcomes for important prognostic factors, including ECOG score and measurable disease. The median overall survival was 13 months and 13.1 months in the intervention and placebo groups, respectively.
In Lui 2016, median overall survival for S + P was 13.7 months versus 10.12 months for paclitaxel alone (HR 0.99, 95% CI 0.62 to 1.584; P = 0.972).
Chekerov 2017 stated in its conference proceedings abstract that data on overall survival were not yet evaluable and provided no further data.
Progression‐free survival
See Analysis 3.2; Table 3.
3.2. Analysis.
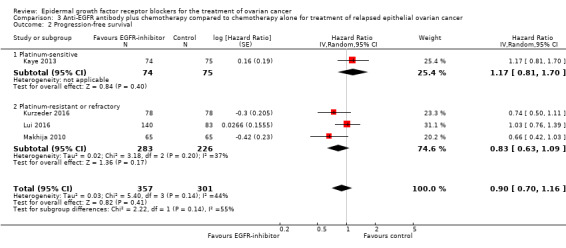
Comparison 3 Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer, Outcome 2 Progression‐free survival.
There may be no or little difference in risk of disease progression among women who received an anti‐EGFR antibody (HR 0.90, 95% CI 0.70 to 1.16; four studies; 658 participants; low‐certainty evidence; Analysis 3.2). There may or may not be a difference in effect, depending on whether this study was conducted in a platinum‐sensitive (HR 1.17, 95% CI 0.81 to 1.70; one study; 149 participants; Analysis 3.2) or a platinum‐resistant/refractory setting (HR 0.82, 95% CI 0.63 to 1.09; three studies; 509 participants; Analysis 3.2).
Chekerov 2017 stated: "progression‐free survival in the intention‐to‐treat population (N = 96) was 9.5 versus 10.7 months (95% CI [of] 8.5 to 11.6 months versus 8.5 to 13.1 months) for the experimental versus [the] standard arm; P = 0.45". Researchers did not state how many participants were included in each group, and review authors were unable to extract data for inclusion in the meta‐analysis. Study authors provided an HR of 0.829 but no CI. Therefore, it was not possible to include these data in the meta‐analysis.
In Kaye 2013, the median progression‐free survival in platinum‐sensitive relapsed disease was 34.1 months in the chemotherapy/pertuzumab group versus 40 months in the chemotherapy alone group.
Makhija 2010 reported 103 (79%) cases of disease progression and median progression‐free survival of 2.9 months and 2.6 months in intervention and placebo groups, respectively.
In Lui 2016, the median progression‐free survival for seribantumab plus paclitaxel was 3.75 months versus 3.68 months for paclitaxel alone (HR 1.027, 95% CI 0.741 to1.425; P = 0.864).
Grade 3 or 4 adverse events
See Analysis 3.3 through Analysis 3.18; Table 3.
3.3. Analysis.
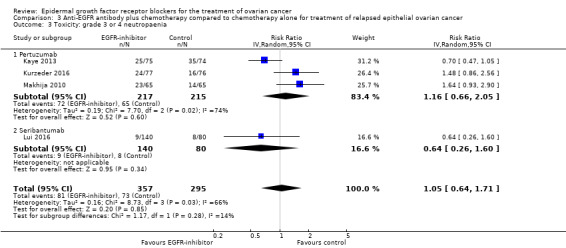
Comparison 3 Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer, Outcome 3 Toxicity: grade 3 or 4 neutropaenia.
3.18. Analysis.
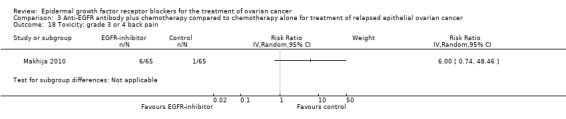
Comparison 3 Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer, Outcome 18 Toxicity: grade 3 or 4 back pain.
Combining pertuzumab and seribantumab may or may not have an effect on the incidence of severe diarrhoea (RR 2.87, 95% CI 0.59 to 13.89; I² = 59%; four studies; 652 participants; very low‐certainty evidence; Analysis 3.8). However, data show heterogeneity of effect between the different antibodies, and so we analysed these separately. Women who received the anti‐EGFR antibody pertuzumab probably reported an increase in diarrhoea, with a six‐fold increased risk of severe diarrhoea (Analysis 3.8.1; three studies; 432 participants; RR 6.37, 95% CI 1.89 to 21.45; I² = 0%; low‐certainty evidence). Seribantumab may or may not have an effect on diarrhoea (Analysis 3.8.2; one study; 220 participants; RR 0.38, 95% CI 0.07 to 2.23; I² = 0%; very low‐certainty evidence).
3.8. Analysis.
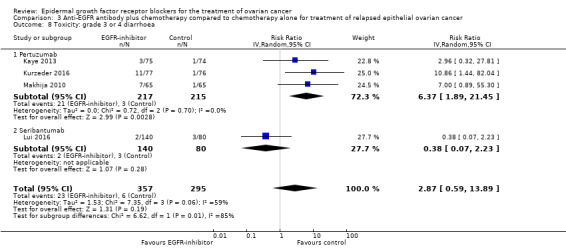
Comparison 3 Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer, Outcome 8 Toxicity: grade 3 or 4 diarrhoea.
Due to the small numbers of participants included in the studies and low numbers of grade 3 and 4 events in either arm, confidence intervals for all grade 3 and 4 toxicities were extremely wide, and there is low‐certainty about whether pertuzumab or seribantumab had an effect on other toxicities.
Chekerov 2017 stated: "The most common treatment related grade 3 plus toxicities included hematologic toxicity (54%), skin reactions (18%), and gastrointestinal events (16%)". It is unclear whether this was so in the panitumumab group, but it is not possible to include these data in the meta‐analysis, as numbers in each group are not stated and there appear to be no control group data. We have contacted trial authors for clarification, but they have not yet provided data.
Quality of life
Kurzeder 2016 presented quality of life data (Hilpert 2016, in Kurzeder 2016). Overall abdominal and gastrointestinal quality of life QLQ‐OV28 (EORTC module for ovarian cancer) scores showed no differences between groups (profile difference 3.9, 95% CI ‐3.3 to 11.2; P value not given in abstract). However, diarrhoeal symptoms were worse with pertuzumab, with a quality of life score profile difference of 21.2 (95% CI 10.1 to 32.3; P = 0.0003). Lui 2016 reported "no significant changes from baseline", although study authors provided no specific data.
Discussion
Summary of main results
First‐line treatment
We included one study that treated women after completion of first‐line chemotherapy for epithelial ovarian cancer (EOC) (Vergote 2014). All women had demonstrated a response to conventional first‐line platinum‐based chemotherapy and to erlotinib, a tyrosine kinase inhibitor, which was given as maintenance treatment until demonstration of disease progression. There was probably little or no difference in either overall or progression‐free survival when erlotinib was added as maintenance following conventional first‐line chemotherapy. Researchers did not report quality of life data in a format amenable to inclusion in a meta‐analysis, but they reported a significant increase in symptom levels in the erlotinib group, especially for diarrhoea, loss of appetite, nausea and vomiting, and fatigue, compared to the non‐treatment arm, although lack of blinding and placebo reduces the certainty of this finding, which is subjective and prone to bias without a blinded placebo control group. Development of a rash was more common in the treatment group (Table 1).
Treatment for recurrent disease
We included six studies in the context of recurrent EOC ‐ one comparing vandetanib, an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (Coleman 2014); three comparing pertuzumab, an anti‐EGFR antibody (Kaye 2013; Kurzeder 2016; Makhija 2010); one comparing panitumumab, the anti‐EGFR antibody (Chekerov 2017); and one comparing seribantumab, the anti‐EGFR antibody (Lui 2016). None of these studies demonstrated benefit in terms of overall or progression‐free survival. Diarrhoea was probably more common in the pertuzumab‐treated group, but other than that, the confidence intervals for adverse events were very wide and our certainty of findings was low (downgraded due to high risk of bias, lack of or unclear blinding, and imprecision) (Table 2; Table 3).
Overall completeness and applicability of evidence
With the exception of Vergote 2014, all of the included studies were small randomised phase II studies, and escalation to phase III is not planned or was abandoned due to lack of demonstrated efficacy in these studies. Studies were largely powered for progression‐free survival outcomes, rather than overall survival.
Although we specified quality of life as an outcome of interest, none of the included studies fully reported this outcome in a form that could be included in the meta‐analysis. Quality of life data collection was poor, with low compliance rates in Vergote 2014, perhaps reflecting the burden of completing questionnaires for the women involved in the studies. These data were also at high risk of bias in Vergote 2014 due to lack of blinding and placebo control. Quality of life after treatment for cancer, especially in a palliative setting (as is the case for advanced and recurrent EOC), is an extremely important outcome, as treatment‐related morbidity very often degrades the quality of the time patients may gain as a result of treatment.
Quality of the evidence
The certainty of the evidence overall was low to very low (downgraded due to small numbers of participants and events, resulting in imprecision and lack of/unclear blinding) (GRADE Working Group), although evidence for overall survival outcomes showed moderate to low‐certainty because this outcome was unlikely to have been biased by lack of blinding in some studies.
EGFR tyrosine kinase inhibitor (TKI) compared to observation alone for maintenance treatment of epithelial ovarian cancer after first‐line chemotherapy
Evidence for overall survival results and death from any cause by 36 months is of low‐certainty, and we downgraded it by one level for imprecision (confidence intervals that cross zero and a single study) and by a further level for inability to assess consistency, because only one study was included. Lack of placebo or blinding is unlikely to have affected this outcome. We judged evidence for progression‐free survival and progression by 12 months to be of very low‐certainty. We downgraded certainty by three levels because of inability to test for inconsistency, imprecision (confidence intervals that cross zero and a single study), and risk of bias (unblinded). We judged quality of life data to be of very low‐certainty and downgraded certainty by at least three levels due to the possibility of selective reporting bias (incompletely reported predefined outcome, so possibility of selective outcome reporting); inability to test for inconsistency because a single study was included; risk of bias (unblinded) as no data are available by which to analyse effect and outcomes are highly likely to be affected by lack of blinding; and imprecision (Table 1).
EGFR TKI plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer
We judged evidence for overall survival results to be of very low‐certainty, and we downgraded it by three levels due to imprecision (one small study, wide confidence intervals that cross zero, and too few events for adequate power) and inability to test for inconsistency, as only one study was included in this analysis. Lack of placebo or blinding is unlikely to have affected this outcome. We judged evidence for progression‐free survival and adverse effects to be of very low‐certainty. We downgraded certainty by three levels due to imprecision and inability to test for inconsistency, and by another level for risk of bias (unblinded). Data on quality of life were not available (Table 2).
Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer
Evidence for overall survival results was limited to pertuzumab, and we judged it to be of moderate certainty; we downgraded it by one level due to imprecision (wide confidence intervals that cross zero and too few events for adequate power). Lack of placebo or blinding is unlikely to have affected this outcome. We judged evidence for progression‐free survival and adverse effects to be of low to very low‐certainty. We downgraded certainty by two levels due to imprecision, by another level for risk of bias (unblinded), and by a further level when we noted inconsistency in outcomes between different EGFR antibodies. Data on quality of life were not available for analysis (Table 3).
Potential biases in the review process
We performed a comprehensive search, including a thorough search of the grey literature, and two or more review authors sifted all studies and extracted data independently. We restricted the included studies to randomised controlled trials (RCTs), as they provide the strongest level of evidence. Several studies were open‐label or included an observation control arm, which presents major potential for bias. This is likely to primarily affect less robust outcome measures, such as toxicity and quality of life, for which side effects would be more likely to be reported by participants taking an active drug than by those undergoing observation only.
The greatest threat to the validity of the review is likely to be the possibility of publication bias (i.e. studies that did not find anti‐EGFR treatment for EOC to have been effective may not have been published). We were unable to assess this possibility formally, although three of the studies included in this review update were identified before data were available, and none of the included studies demonstrated a positive effect.
Agreements and disagreements with other studies or reviews
Several review articles have noted the disappointing results of anti‐EGFR agents in clinical trials compared to promising results in preclinical studies.
Wilken 2012 reviewed the results of clinical trials of anti‐EGFR treatment, including non‐randomised phase I and II studies (this group included trials by Makhija 2010 and Kaye 2013) and noted that trial results did not demonstrate improvement compared to conventional treatment. The phase I toII trials reported low response rates for many anti‐EGFR agents, including cetuximab (anti‐EGFR antibody); trastuzumab (anti‐human epidermal growth factor receptor (HER)‐2 antibody); matuzumab (anti‐EGFR antibody); leflunomide (platelet‐derived growth factor receptor (PDGFR)/fibroblast growth factor receptor (FGFR)/EGFR tyrosine kinase inhibitor (TKI)); vandetanib (EGFR/VEGFR‐2/transmembrane receptor tyrosine kinase (RET) TKI); canertinib (EGFR/HER‐2/HER‐4/EGFRvIII TKI); lapatinib (EGFR/HER‐2 TKI); erlotinib (EGFR TKI); and gefitinib (EGFR TKI). Review authors commented that "a stark contrast exists between the in vitro evaluation of HER‐targeted therapeutics and predicted clinical outcomes in women with EOC treated with HER‐targeted therapeutics", and discussed how preclinical assessment of cell lines in vivo did not correlate well with the clinical outcomes observed, and that better preclinical models were required.
Teplinsky 2015 reviewed clinical studies, including Kaye 2013, Makhija 2010, and Vergote 2014, and concluded that due to "discouraging results...at this point, there seems to be little role for anti‐EGFR or HER‐2 directed therapies in EOC outside of clinical trials".
Authors' conclusions
Implications for practice.
Data demonstrate little or no difference between treatment and observation/control arms. These data suggest that alone, inhibition of the epidermal growth factor receptor (EGFR) pathway has minimal, if any, beneficial effect and may cause harm due to side effects that erode quality of life.
Implications for research.
Although the concept of targeted therapy and a 'magic bullet' in cancer treatment is highly attractive, control of cell growth is, from an evolutionary point of view, key to cell survival. Cells have evolved a highly complex and interconnected set of pathways and cross‐talk with their environment, which have a high level of redundancy, to maintain cell survival. Targeting a single point of failure within a cancer cell, which has high genetic instability due to tumour suppressor gene function loss, presents a greater challenge once the cell is placed within a complex tumour microenvironment rather than in a petri dish. However, EGFR tyrosine kinase inhibitors (TKIs) have been shown in recent clinical trials to be effective for some lung cancers (Okamoto 2018).
Biological therapies, similar to conventional chemotherapy, may be more effective when given in combination to attack complex pathways at several points simultaneously. This is likely to also produce greater toxicity, as normal cells are less likely to be protected due to reduced specificity. Candidates for the combination approach include agents that inhibit mammalian target of rapamycin (mTOR) and phosphatidylinositol 3‐kinase (PI3K)‐Akt pathways (Glaysher 2013).
In addition, subtleties in cell‐cell communication and interaction with tissue stroma, the medium in which cancer cells exist, are not well reproduced in conventional monocultures in vitro. More complex models of the tumour environment, including non‐cancerous cells (e.g. connective tissue components such as collagen, cells of the immune system, and cells from small blood vessels), may be needed for the complexity of the cancer cell environment to be fully understood.
'Individualised therapy' ‐ the challenge and opportunity for modern oncology treatment ‐ remains a holy grail outside of a few specific tumour types. Tailoring treatment to specific cancer mutations will require a radical change in large phase III study design. It would be useful if studies of different agents, with different tumour biology targets, could be conducted in parallel, so that eligible women could be allocated to appropriate trial comparison arms depending on their individual tumour biology. This will require the co‐operation of researchers and pharmaceutical companies and will present a challenge to regulatory authorities, but this approach would more rapidly generate meaningful data for women than would conventionally designed studies.
Presenting data on median survival along with standard deviations would be useful for subsequent meta‐analyses. This would also aid decision‐making because HR data presented within a summary of findings table are more difficult to interpret and weigh than is the time benefit offered by treatment. This is the information women often seek, and presenting these items as a visual 'decision aid' would prove valuable to consumers.
Of primary importance, no treatment for advanced epithelial ovarian cancer (EOC), which is of palliative intent from the outset for the majority, should be provided, nor its results interpreted, without full disclosure of toxicity and quality of life analyses. The benefit of a modest improvement in progression‐free overall survival needs to be balanced carefully against any increase in unpleasant and debilitating side effects. This balance is likely to be different for each woman, but the information should be made available so women can make informed decisions about their care. This approach would lead us towards true 'individualised medicine' based on patient decisions made with full knowledge of harms and benefits, with consideration of patient preferences, values, and goals ‐ not just targeting treatment to tumour biology. In addition, for expensive treatments, which may provide only modest effects in select groups, cost‐benefit analyses would be important to aid decision‐makers as they assess the value of new treatments to a population/society.
Further updates of this review are not planned because it is unlikely that additional studies that meet the review inclusion criteria will be performed.
What's new
| Date | Event | Description |
|---|---|---|
| 5 December 2018 | Review declared as stable | No longer for update as intervention shows no evidence of efficacy. |
History
Protocol first published: Issue 3, 2009 Review first published: Issue 10, 2011
| Date | Event | Description |
|---|---|---|
| 2 July 2018 | New search has been performed | Literature search updated 2017 |
| 30 April 2018 | New citation required and conclusions have changed | Six new studies added |
Acknowledgements
We would like to thank the entire team at Musgrove Park Hospital Library, especially Natalie Parsley, Denise Manning, Carol‐Ann Regan, and Laura Hamilton, for their help and support in sourcing papers for this review.
We thank Krish Haldar (KH), Sean Kehoe (SK), and Shibani Nicum (SN), who contributed to the original version of the protocol and review. Many thanks to Chris Williams (in the original review) and Marcia Hall (in the update) for their expert clinical and editorial advice; to Gail Quinn and Clare Jess, Managing Editors for Cochrane Gynaecological, Neuro‐oncology, and Orphan Cancers, for their contribution to the editorial process; and to Jo Platt for help in designing search strategies and running the searches. We thank Heather Dickinson for many helpful suggestions and for acting as a coauthor for the protocol. We also thank the referees at each stage of the process for their helpful suggestions.
This project was supported by the National Institute for Health Research via Cochrane Infrastructure funding to the Cochrane Gynaecological, Neuro‐oncology, and Orphan Cancer Group. The views and opinions expressed therein are those of the review authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS, or the Department of Health.
We would like to thank the referees, including Nick Reed and Christina Annunziata, for many helpful suggestions and comments.
Appendices
Appendix 1. CENTRAL search strategy
CENTRAL; 2010, Issue 4
ovar*
cancer* or carcinoma* or neoplasm* or tumor* or tumour* or malignan*
(#1 AND #2)
MeSH descriptor Ovarian Neoplasms explode all trees
(#3 OR #4)
epidermal growth factor receptor
EGFR near/5 inhibit*
MeSH descriptor Antibodies, Monoclonal explode all trees
monoclonal antibodies
trastuzumab or herceptin
cetuximab or IMC‐C225 or erbitux
EMD 72000 or matuzumab
panitumumab or ABX‐EGF or vectibix
pertuzumab or rhumab 2C4 or omnitarg
MeSH descriptor Protein‐Tyrosine Kinases explode all trees
tyrosine kinase near/5 inhibit*
gefitinib or ZD1839 or iressa
erlotinib or OSI‐774 or tarceva
lapatinib or gw572016 or tykerb
canertinib or CI‐1033
EKB‐569
PKI‐166
BMS 599626
vandetanib or rinn or zactima or ZD6474
(#6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 OR 14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24)
(#5 AND #25)
Appendix 2. MEDLINE search strategy
Medline Ovid 1950 to October week 3 2010
randomized controlled trial.pt.
controlled clinical trial.pt.
randomized.ab.
placebo.ab.
drug therapy.fs.
randomly.ab.
trial.ab.
groups.ab.
1 or 2 or 3 or 4 or 5 or 6 or 7 or 8
(animals not (humans and animals)).sh.
9 not 10
ovar*.mp.
(cancer* or carcinoma*or neoplasm* or tumor*or tumour*or malignan*).mp.
12 and 13
exp Ovarian Neoplasms/
14 or 15
epidermal growth factor receptor.mp.
(EGFR adj5 inhibit*).mp.
exp Antibodies, Monoclonal/
monoclonal antibodies.mp.
(trastuzumab or herceptin).mp.
(cetuximab or IMC‐C225 or erbitux).mp.
(EMD 72000 or matuzumab).mp.
(panitumumab or ABX‐EGF or vectibix).mp.
(pertuzumab or rhumab 2C4 or omnitarg).mp.
exp Protein‐Tyrosine Kinases/
(tyrosine kinase adj5 inhibit*).mp.
(gefitinib or ZD1839 or iressa).mp.
(erlotinib or OSI‐774 or tarceva).mp.
(lapatinib or gw572016 or tykerb).mp.
(canertinib or CI‐1033).mp.
EKB‐569.mp.
PKI‐166.mp.
BMS 599626.mp.
(vandetanib or rinn or zactima or ZD6474).mp.
17 or 18 or 19 or 20 or 21 or 22 or 23 or 24 or 25 or 26 or 27 or 28 or 29 or 30 or 31 or 32 or 33 or 34 or 35
11 and 16 and 36
key: mp=title, original title, abstract, name of substance word, subject heading word, pt=publication type, ab=abstract, fs=floating subheading
Appendix 3. Embase search strategy
Embase Ovid 1950 to week 43 2010
exp Controlled Clinical Trial/
randomized.ab.
placebo.ab.
dt.fs.
randomly.ab.
trial.ab.
groups.ab.
1 or 2 or 3 or 4 or 5 or 6 or 7
exp Animal/
Human/
9 not (9 and 10)
8 not 11
(ovar* and (cancer* or carcinoma* or neoplasm* or tumor* or tumour* or malignan*)).mp.
exp Ovary Tumor/
13 or 14
exp Epidermal Growth Factor Receptor/
(EGFR adj5 inhibit*).mp.
exp Monoclonal Antibody/
monoclonal antibodies.mp.
(trastuzumab or herceptin).mp.
(cetuximab or IMC‐C225 or erbitux).mp.
(EMD 72000 or matuzumab).mp.
(panitumumab or ABX‐EGF or vectibix).mp.
(pertuzumab or rhumab 2C4 or omnitarg).mp.
exp Protein Tyrosine Kinase/
(tyrosine kinase adj5 inhibit*).mp.
(gefitinib or ZD1839 or iressa).mp.
(erlotinib or OSI‐774 or tarceva).mp.
(lapatinib or gw572016 or tykerb).mp.
(canertinib or CI‐1033).mp.
EKB‐569.mp.
PKI‐166.mp.
BMS 599626.mp.
(vandetanib or rinn or zactima or ZD6474).mp.
16 or 17 or 18 or 19 or 20 or 21 or 22 or 23 or 24 or 25 or 26 or 27 or 28 or 29 or 30 or 31 or 32 or 33 or 34
12 and 15 and 35
key: mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name, ab=abstract, sh=subject heading, fs=floating subheading
Data and analyses
Comparison 1. EGFR tyrosine kinase inhibitor (TKI) compared to observation alone for maintenance treatment of epithelial ovarian cancer after first‐line chemotherapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 1 | Hazard Ratio (Random, 95% CI) | Subtotals only | |
| 2 Progression‐free survival | 1 | Hazard Ratio (Random, 95% CI) | Subtotals only |
Comparison 2. EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 1 | Hazard Ratio (Random, 95% CI) | Subtotals only | |
| 2 Progression‐free survival | 1 | Hazard Ratio (Random, 95% CI) | Subtotals only | |
| 3 Toxicity: grade 3 or 4 neutropaenia | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 4 Toxicity: grade 3 or 4 febrile neutropaenia | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 5 Toxicity: grade 3 or 4 leucopaenia | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 6 Toxicity: grade 3 or 4 leucocytosis | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 7 Toxicity: grade 3 or 4 thrombocytopaenia | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 8 Toxicity: grade 3 or 4 anaemia | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 9 Toxicity: grade 3 or 4 nausea ± vomiting | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 10 Toxicity: grade 3 or 4 constipation | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 11 Toxicity: grade 3 or 4 rash | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 12 Toxicity: grade 3 or 4 allergic reaction/drug hypersensitivity | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 13 Toxicity: grade 3 or 4 skin toxicity (other) | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 14 Toxicity: grade 3 or 4 fatigue | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 15 Toxicity: cardiac toxicity (any grade) | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 16 Toxicity: grade 3 or 4 hypokalaemia | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 17 Toxicity: grade 3 or 4 hypocalcaemia | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 18 Toxicity: grade 3 or 4 hypomagnesaemia | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 19 Toxicity: grade 3 or 4 anorexia | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 20 Toxicity: grade 3 or 4 neuropathy | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 21 Toxicity: grade 3 or 4 oedema | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 22 Toxicity: treatment‐related secondary malignancy | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only |
2.4. Analysis.
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 4 Toxicity: grade 3 or 4 febrile neutropaenia.
2.5. Analysis.
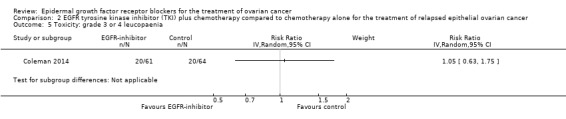
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 5 Toxicity: grade 3 or 4 leucopaenia.
2.6. Analysis.
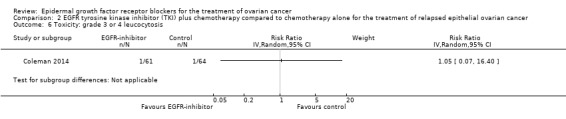
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 6 Toxicity: grade 3 or 4 leucocytosis.
2.7. Analysis.
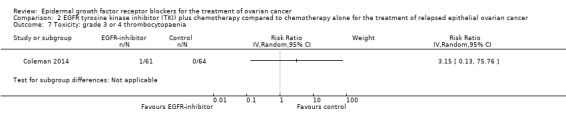
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 7 Toxicity: grade 3 or 4 thrombocytopaenia.
2.8. Analysis.
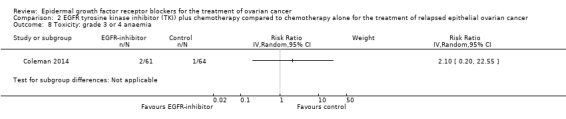
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 8 Toxicity: grade 3 or 4 anaemia.
2.9. Analysis.
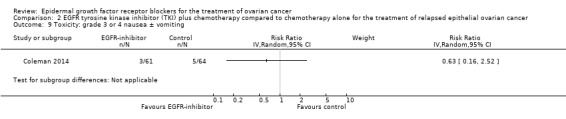
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 9 Toxicity: grade 3 or 4 nausea ± vomiting.
2.10. Analysis.
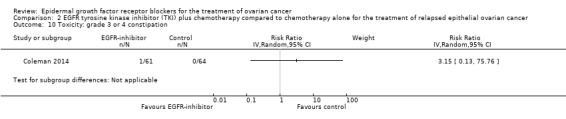
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 10 Toxicity: grade 3 or 4 constipation.
2.11. Analysis.
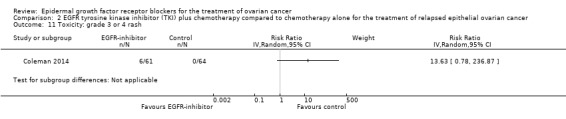
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 11 Toxicity: grade 3 or 4 rash.
2.12. Analysis.
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 12 Toxicity: grade 3 or 4 allergic reaction/drug hypersensitivity.
2.13. Analysis.
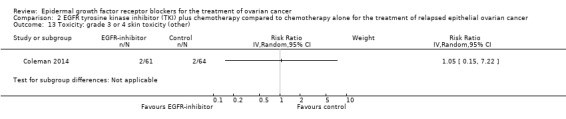
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 13 Toxicity: grade 3 or 4 skin toxicity (other).
2.14. Analysis.
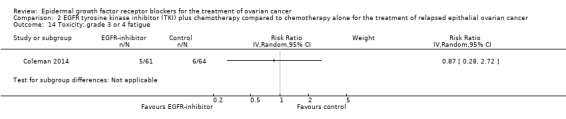
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 14 Toxicity: grade 3 or 4 fatigue.
2.15. Analysis.
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 15 Toxicity: cardiac toxicity (any grade).
2.16. Analysis.
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 16 Toxicity: grade 3 or 4 hypokalaemia.
2.17. Analysis.
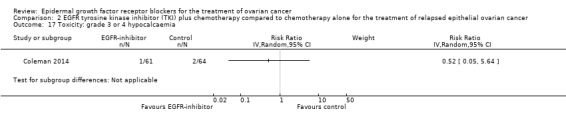
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 17 Toxicity: grade 3 or 4 hypocalcaemia.
2.18. Analysis.
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 18 Toxicity: grade 3 or 4 hypomagnesaemia.
2.19. Analysis.
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 19 Toxicity: grade 3 or 4 anorexia.
2.20. Analysis.
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 20 Toxicity: grade 3 or 4 neuropathy.
2.21. Analysis.
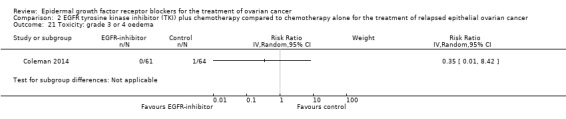
Comparison 2 EGFR tyrosine kinase inhibitor (TKI) plus chemotherapy compared to chemotherapy alone for the treatment of relapsed epithelial ovarian cancer, Outcome 21 Toxicity: grade 3 or 4 oedema.
Comparison 3. Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 4 | 658 | Hazard Ratio (Random, 95% CI) | 0.93 [0.74, 1.18] |
| 1.1 Platinum‐sensitive | 1 | 149 | Hazard Ratio (Random, 95% CI) | 1.02 [0.63, 1.67] |
| 1.2 Platinum‐resistant or refractory | 3 | 509 | Hazard Ratio (Random, 95% CI) | 0.91 [0.70, 1.18] |
| 2 Progression‐free survival | 4 | 658 | Hazard Ratio (Random, 95% CI) | 0.90 [0.70, 1.16] |
| 2.1 Platinum‐sensitive | 1 | 149 | Hazard Ratio (Random, 95% CI) | 1.17 [0.81, 1.70] |
| 2.2 Platinum‐resistant or refractory | 3 | 509 | Hazard Ratio (Random, 95% CI) | 0.83 [0.63, 1.09] |
| 3 Toxicity: grade 3 or 4 neutropaenia | 4 | 652 | Risk Ratio (IV, Random, 95% CI) | 1.05 [0.64, 1.71] |
| 3.1 Pertuzumab | 3 | 432 | Risk Ratio (IV, Random, 95% CI) | 1.16 [0.66, 2.05] |
| 3.2 Seribantumab | 1 | 220 | Risk Ratio (IV, Random, 95% CI) | 0.64 [0.26, 1.60] |
| 4 Toxicity: grade 3 or 4 febrile neutropaenia | 2 | 302 | Risk Ratio (IV, Random, 95% CI) | 0.38 [0.10, 1.43] |
| 5 Toxicity: grade 3 or 4 leucopaenia | 2 | 302 | Risk Ratio (IV, Random, 95% CI) | 0.66 [0.28, 1.57] |
| 6 Toxicity: grade 3 or 4 thrombocytopaenia | 3 | 432 | Risk Ratio (IV, Random, 95% CI) | 0.99 [0.42, 2.34] |
| 7 Toxicity: grade 3 or 4 anaemia | 4 | 652 | Risk Ratio (IV, Random, 95% CI) | 0.84 [0.47, 1.49] |
| 7.1 Pertuzumab | 3 | 432 | Risk Ratio (IV, Random, 95% CI) | 0.71 [0.33, 1.51] |
| 7.2 Seribantumab | 1 | 220 | Risk Ratio (IV, Random, 95% CI) | 1.06 [0.44, 2.55] |
| 8 Toxicity: grade 3 or 4 diarrhoea | 4 | 652 | Risk Ratio (IV, Random, 95% CI) | 2.87 [0.59, 13.89] |
| 8.1 Pertuzumab | 3 | 432 | Risk Ratio (IV, Random, 95% CI) | 6.37 [1.89, 21.45] |
| 8.2 Seribantumab | 1 | 220 | Risk Ratio (IV, Random, 95% CI) | 0.38 [0.07, 2.23] |
| 9 Toxicity: grade 3 or 4 nausea ± vomiting | 3 | 503 | Risk Ratio (IV, Random, 95% CI) | 1.27 [0.56, 2.89] |
| 9.1 Pertuzumab | 2 | 283 | Risk Ratio (IV, Random, 95% CI) | 1.48 [0.54, 4.06] |
| 9.2 Seribantumab | 1 | 220 | Risk Ratio (IV, Random, 95% CI) | 0.95 [0.23, 3.88] |
| 10 Toxicity: grade 3 or 4 dyspepsia | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 11 Toxicity: grade 3 or 4 abdominal pain | 2 | 373 | Risk Ratio (IV, Random, 95% CI) | 1.30 [0.49, 3.44] |
| 11.1 Pertuzumab | 1 | 153 | Risk Ratio (IV, Random, 95% CI) | 0.99 [0.14, 6.83] |
| 11.2 Seribantumab | 1 | 220 | Risk Ratio (IV, Random, 95% CI) | 1.43 [0.46, 4.41] |
| 12 Toxicity: grade 3 or 4 rash | 1 | 130 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 13 Toxicity: grade 3 or 4 allergic reaction/drug hypersensitivity | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 14 Toxicity: grade 3 or 4 skin toxicity (other) | 1 | 130 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 15 Toxicity: grade 3 or 4 fatigue | 4 | 652 | Risk Ratio (IV, Random, 95% CI) | 1.06 [0.66, 1.73] |
| 15.1 Pertuzumab | 3 | 432 | Risk Ratio (IV, Random, 95% CI) | 0.97 [0.57, 1.66] |
| 15.2 Seribantumab | 1 | 220 | Risk Ratio (IV, Random, 95% CI) | 1.57 [0.52, 4.77] |
| 16 Toxicity: cardiac toxicity (any grade) | 3 | 421 | Risk Ratio (IV, Random, 95% CI) | 0.96 [0.54, 1.72] |
| 16.1 Pertuzumab | 3 | 421 | Risk Ratio (IV, Random, 95% CI) | 0.96 [0.54, 1.72] |
| 17 Toxicity: grade 3 or 4 hypokalaemia | 3 | 522 | Risk Ratio (IV, Random, 95% CI) | 2.01 [0.80, 5.06] |
| 17.1 Pertuzumab | 2 | 302 | Risk Ratio (IV, Random, 95% CI) | 1.52 [0.47, 4.93] |
| 17.2 Seribantumab | 1 | 220 | Risk Ratio (IV, Random, 95% CI) | 3.14 [0.71, 13.83] |
| 18 Toxicity: grade 3 or 4 back pain | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only |
3.4. Analysis.
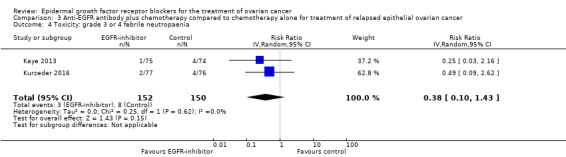
Comparison 3 Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer, Outcome 4 Toxicity: grade 3 or 4 febrile neutropaenia.
3.5. Analysis.
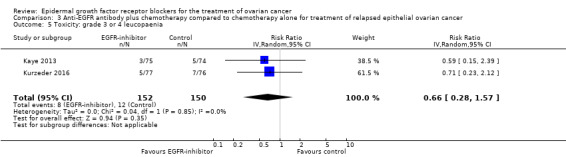
Comparison 3 Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer, Outcome 5 Toxicity: grade 3 or 4 leucopaenia.
3.6. Analysis.
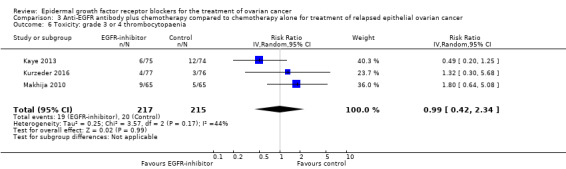
Comparison 3 Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer, Outcome 6 Toxicity: grade 3 or 4 thrombocytopaenia.
3.7. Analysis.
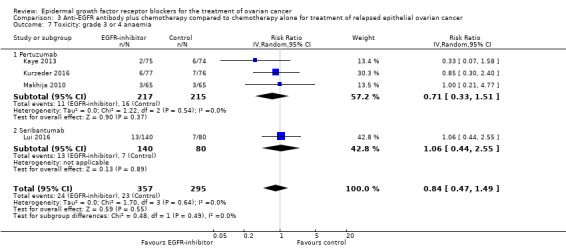
Comparison 3 Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer, Outcome 7 Toxicity: grade 3 or 4 anaemia.
3.9. Analysis.
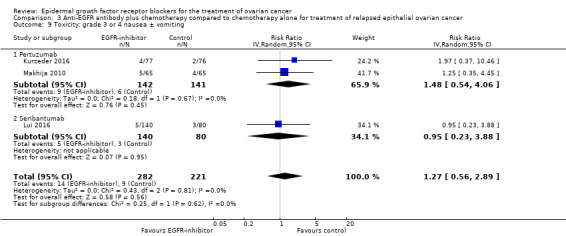
Comparison 3 Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer, Outcome 9 Toxicity: grade 3 or 4 nausea ± vomiting.
3.10. Analysis.
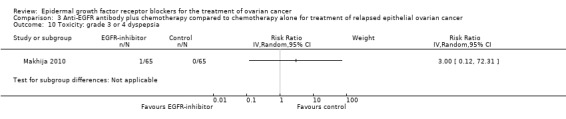
Comparison 3 Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer, Outcome 10 Toxicity: grade 3 or 4 dyspepsia.
3.11. Analysis.
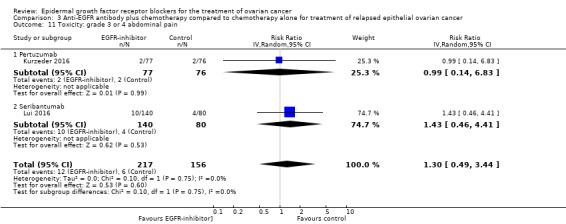
Comparison 3 Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer, Outcome 11 Toxicity: grade 3 or 4 abdominal pain.
3.12. Analysis.
Comparison 3 Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer, Outcome 12 Toxicity: grade 3 or 4 rash.
3.13. Analysis.
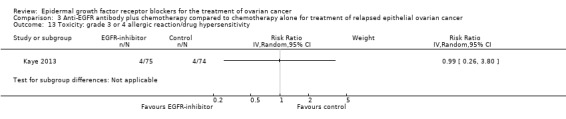
Comparison 3 Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer, Outcome 13 Toxicity: grade 3 or 4 allergic reaction/drug hypersensitivity.
3.14. Analysis.
Comparison 3 Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer, Outcome 14 Toxicity: grade 3 or 4 skin toxicity (other).
3.15. Analysis.
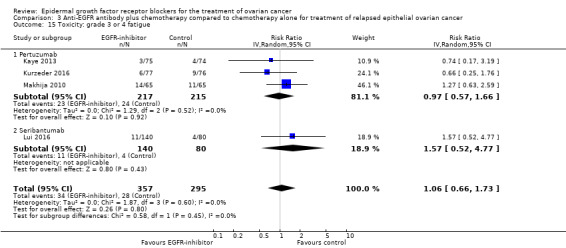
Comparison 3 Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer, Outcome 15 Toxicity: grade 3 or 4 fatigue.
3.16. Analysis.
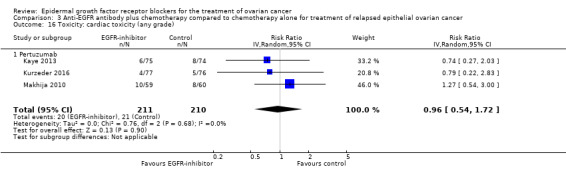
Comparison 3 Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer, Outcome 16 Toxicity: cardiac toxicity (any grade).
3.17. Analysis.
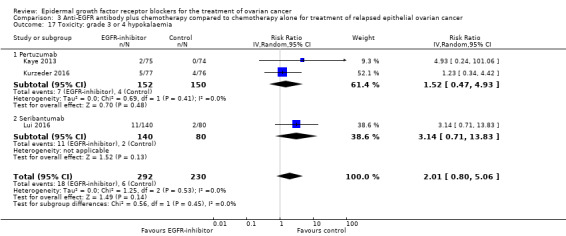
Comparison 3 Anti‐EGFR antibody plus chemotherapy compared to chemotherapy alone for treatment of relapsed epithelial ovarian cancer, Outcome 17 Toxicity: grade 3 or 4 hypokalaemia.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Chekerov 2017.
| Methods | Open‐label randomised multi‐centre phase II study of cytotoxic chemotherapy with vs without panitumumab | |
| Participants | Women 18 years of age or older with histologically proven epithelial ovarian cancer, fallopian tube cancer, or primary peritoneal cancer. Women must have pretreated platinum‐sensitive ovarian cancer with recurrence more than 6 months after completion of a platinum‐containing regimen and wild‐type KRAS status | |
| Interventions | Arm A (intervention): cytotoxic chemotherapy (carboplatin plus either gemcitabine or pegylated liposomal doxorubicin, specified by investigator before randomisation) plus panitumumab Arm B (comparison): cytotoxic chemotherapy (as per intervention arm) alone |
|
| Outcomes |
Outcomes planned from clinicatrials.gov website Primary PFS rate after 12 months Secondary Duration of tumour response PFS at end of follow‐up (up to 1 year) Overall survival Toxicity Tumour response rate Outcomes reported in conference proceedings abstract in 2017 No update on clinicaltrials.gov website since 2013 despite anticipated completion date of 2014 102 participants randomised; 96 enrolled for final analysis Progression‐free survival in the intention‐to‐treat population (N = 96) was 9.5 months vs 10.7 months (HR 0.829, 95% CI 8.5 to 11.6 months vs 8.5 to 13.1 months) for experimental vs standard arm; P = 0.45 "Data of overall survival are not yet evaluable" Grade 3+ toxicities included Haematological toxicity (54%) Skin reactions (18%) Gastrointestinal events (16%) |
|
| Notes | Other study ID numbers: GMIHO‐008/2009_AG56, 2010‐018849‐59 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Findings presented in abstract only ‐ no data provided |
| Allocation concealment (selection bias) | Unclear risk | Findings presented in abstract only ‐ no data provided |
| Blinding of participants and personnel (performance bias) Objective outcome measures (Overall Survival) | Unclear risk | Likely to be at low risk, as data unlikely to be affected by blinding. OS data not yet evaluable |
| Blinding of participants and personnel (performance bias) Subjective outcome measures (PFS; Toxicity; QoL) | High risk | Unblinded open‐label study |
| Blinding of outcome assessment (detection bias) Objective outcome measures (overall survival) | Unclear risk | Likely to be at low risk, as data unlikely to be affected by blinding |
| Blinding of outcome assessment (detection bias) Subjective outcome measures (PFS, Toxicity, QoL) | High risk | Unblinded open‐label study |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | "102 participants were randomised and 96 enrolled for the final analysis" ‐ data in abstract only ‐ unclear if this constitutes a source of bias |
| Selective reporting (reporting bias) | Unclear risk | Abstract only presented, so incomplete data available at this stage; data not updated on clinicaltrials.gov website since 2013 |
| Other bias | Unclear risk | Abstract, so limited details for assessment |
Coleman 2014.
| Methods | Open‐label multi‐centre phase II RCT with cross‐over (137 study locations in the USA). Recruitment from March 2010 to August 2011 | |
| Participants | 129 women with histologically confirmed epithelial ovarian cancer, fallopian tube cancer, or primary peritoneal carcinoma. Participants had to have recurrent, refractory, or progressive/persistent disease (following 1 prior platinum‐based course of chemotherapy for primary disease, and up to 3 additional courses of cytotoxic therapy for recurrent disease) that was measurable or, if not measurable by RECIST criteria, evaluable by imaging. Other inclusion criteria included a performance status of Zubrod 0 to 2 and adequate haematological, renal, liver, and cardiovascular function. Recent cancer (within 5 years from diagnosis for ovarian cancer) was an exclusion criterion, except preinvasive disease and early‐stage endometrial cancer. Study author confirmed that, to his knowledge, no participants had concurrent malignancies. Baseline characteristics Single‐agent docetaxel (D)
Docetaxel and vandetanib (D + V)
|
|
| Interventions |
Intervention Docetaxel (75 mg/m² IV, day 1 only) with vandetanib (100 mg PO daily, days 1 to 21) (21‐day course, repeated until disease progression, unacceptable toxicity, or withdrawal of consent) Comparison Docetaxel (75 mg/m² IV, once every 21 days) (continued until disease progression, unacceptable toxicity, or withdrawal of consent) Note: participants randomised to single‐agent docetaxel were allowed to cross over to single‐agent vandetanib (100 mg PO daily) upon documented progression. |
|
| Outcomes |
Primary
Secondary
|
|
| Notes | ClinicalTrials.gov identifier: NCT00872989 This study was an ongoing trial in the original version of this review; results have since been published. Sponsorship source: Financial Support: R Coleman, A Sood. "This investigation was supported by the Marcus Foundation, the Investigator‐Sponsored Study Program of AstraZeneca, Wilmington, DE, and in part by the following PHS Cooperative Agreement grant numbers awarded by the National Cancer Institute US: DHHS: CA32102, CA38926, CA105409, CA35431, CA45560,CA20319, CA13612, CA46441, CA45808, CA45461,CA67575, CA58882, CA35128, CA37981, CA46282,CA35421, CA76132, CA58723, CA16385, and CA42777; as well as P50 CA083639, OCRP‐OC0931146, and the Blanton‐Davis Ovarian Cancer Research Program. A.K.S. is supported by the Betty Anne Asche Murray Distinguished Professorship. Research support to R.L.C.is by the Ann Rife Cox Chair in Gynecology". Country: United States of America Setting: multi‐centre study Author's name: Robert L. Coleman Institution: University of Texas, MD Anderson Cancer Center, Houston, TX, USA Email: rcoleman@mdanderson.org Address: MD Anderson Cancer Center, Department of Gynecologic Oncology & Reproductive Medicine, 1155 Herman Pressler Drive, CPB6.3271, Houston, TX 77030, USA |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "participants were randomised centrally 1:1 using a dynamic balancing algorithm with stratification" |
| Allocation concealment (selection bias) | Low risk | Comment: randomisation was performed centrally at the SWOG Statistical Center. Thus, it is unlikely that intervention allocations could have been foreseen in advance by those recruiting participants. |
| Blinding of participants and personnel (performance bias) Objective outcome measures (Overall Survival) | Low risk | Outcome unlikely to be affected by blinding |
| Blinding of participants and personnel (performance bias) Subjective outcome measures (PFS; Toxicity; QoL) | High risk | Open‐label study |
| Blinding of outcome assessment (detection bias) Objective outcome measures (overall survival) | Low risk | Outcome unlikely to be affected by blinding |
| Blinding of outcome assessment (detection bias) Subjective outcome measures (PFS, Toxicity, QoL) | High risk | Open‐label study |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attritions/exclusions were well reported |
| Selective reporting (reporting bias) | Low risk | We compared reported outcomes to those intended (from the online protocol): the published paper covered the main planned primary outcomes from the protocol. Exploratory analyses, which had not been prespecified, were generally indicated as such, and these data were not extracted for the review. |
| Other bias | Unclear risk | Conflicts of interest of study authors are reported: one study author served as an uncompensated scientific advisor for other projects of the drug manufacturer; other study authors declare no conflicts. |
Kaye 2013.
| Methods | Open‐label multi‐centre phase II RCT (27 institutions in 9 countries in Western Europe, Eastern Europe, and Canada). Recruitment from February 2006 to November 2006 | |
| Participants | 149 women with a first relapse of histologically confirmed ovarian, fallopian, or primary peritoneal carcinoma (defined by at least 1 measurable lesion according to RECIST or elevated CA‐125 (GCIG criteria of ≥ 2 × upper limit of normal (ULN)), documented on 2 occasions > 1 week and < 3 months apart) Participants were required to have platinum‐sensitive disease (defined by initial response to platinum and a progression‐free interval of > 6 months after completion of a platinum‐based regimen). Other requirements included Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1 and adequate haematological, renal, hepatic, and cardiac function. Median age (range) CT + P = 58.1 (26 to 76); CT 55.3 (19 to 83) WHO PS = 0 CT + P = 41/75 (55%); CT alone = 39/74 (52) WHO PS = 1: CT + P = 33/75 (45%); CT alone = 36/74 (48%) |
|
| Interventions |
Comparison (cytotoxic chemotherapy (CT)) Carboplatin (AUC 5/q3wk with paclitaxel or AUC 4/q3wk with gemcitabine) with either paclitaxel (Taxol; 175 mg/m² q3wk) or gemcitabine (1000 mg/m² days 1 and 8, q3wk) Chemotherapy was administered for a maximum of 6 cycles. Intervention (chemotherapy + pertuzumab (CT + P)) Cytotoxic chemotherapy (CT) (as per comparison group) + pertuzumab (P) (840‐mg loading dose followed by 420 mg q3wk). After completion of chemotherapy or withdrawal of chemotherapy due to toxic effects, pertuzumab was administered for a further 11 cycles q3wk (resulting in a total of 17 cycles); pertuzumab treatment could be continued up to a maximum of 52 cycles. No participants were allowed to cross over to the intervention (chemotherapy + pertuzumab) arm at any stage. |
|
| Outcomes |
Progression‐free survival (PFS) Median PFS (weeks) CT + P = 34.1 weeks vs C = 40.0 weeks; HR 1.17 (80% CI 0.92 to 1.49); P= 0.39727 Overall survival (OS) Median OS CT + P = 28.2 months vs CT= not reached; HR 1.02 (80% CI 0.74 to 1.41); P = 0.9262 ORR (overall response rate, as defined by RECIST or CA‐125) Toxicity (G3 or more) during chemotherapy
|
|
| Notes | ClinicalTrials.gov identifier: NCT02004093 This study was mentioned as ongoing in the original version of this review; results have been published since that time. Funding source This study was sponsored by F. Hoffmann‐La Roche. Declarations of Interest Disclosure statement from the published paper "GR and VMN are employees of F. Hoffmann‐La Roche, and hold stocks or stock options in Roche Products. AS and MM are employees of Roche Diagnostics, and hold stocks or stock options in Roche Diagnostics. SBK and CJP have participated in Roche advisory boards and served as a consultant for F. Hoffmann‐La Roche. AD‐B, LG, GDC, VG, EN, and IV have no conflicts to declare". Survival data The published paper gives results for PFS and OS for both a 'Primary analysis' (conducted 1 year after the last participant was enrolled), and a 'Final analysis' (conducted 2 years after the last treatment was administered). We have used the data from the Final analysis in this review. Toxicity data This trial allowed participants in the intervention (pertuzumab + chemotherapy) arm to continue with pertuzumab beyond cycle 6 (the point at which both arms finished chemotherapy). For the main toxicity comparison analyses in this review, we have used toxicity data up to cycle 6 (although separate data for toxicity after cycle 6 are also reported in the published paper). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was stratified by treatment‐free interval (TFI; 6–12 months versus > 12 months), measurable versus non‐measurable disease, chemotherapy regimen (carboplatin–paclitaxel versus carboplatin–gemcitabine) and territory (Eastern Europe versus Western Europe and Canada)". |
| Allocation concealment (selection bias) | Low risk | Personal communication with the Lead Project Statistician confirmed allocation concealment. |
| Blinding of participants and personnel (performance bias) Objective outcome measures (Overall Survival) | Low risk | Outcome unlikely to be affected by blinding |
| Blinding of participants and personnel (performance bias) Subjective outcome measures (PFS; Toxicity; QoL) | High risk | Open‐label study |
| Blinding of outcome assessment (detection bias) Objective outcome measures (overall survival) | Low risk | Outcome unlikely to be affected by blinding |
| Blinding of outcome assessment (detection bias) Subjective outcome measures (PFS, Toxicity, QoL) | High risk | Open‐label study |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition and exclusions were reported. Participants were excluded from analyses if they did not receive at least 1 dose of the allocated trial medication (1/75 in the intervention group; 2/77 in the control group). |
| Selective reporting (reporting bias) | Low risk | Results for all major expected outcomes were reported. Seconday and exploratory outcomes that were not described in the publication are included in the ClinicalTrials.gov entry of this study (personal communication with the Lead Project Statistician). |
| Other bias | Unclear risk | This study was sponsored by F. Hoffmann‐La Roche. Several of the study investigators have financial links to either F. Hoffmann‐La Roche or Roche Diagnostics, which have been declared in the published paper (and are detailed in the Notes section of the study table above). |
Kurzeder 2016.
| Methods | Double‐blind placebo‐controlled randomised phase III trial | |
| Participants | 156 women with platinum‐resistant or platinum‐refractory epithelial ovarian, primary peritoneal, or fallopian tube carcinoma (progression during platinum therapy or within 6 months of completing 4 or more cycles of platinum‐containing therapy and no more than 2 prior lines of chemotherapy) and low tumour HER‐3 mRNA expression. 324 participants enrolled but 168 excluded due to ineligibility, including failure of testing/central review, or after HER‐3 tumour testing. 3 participants did not receive study treatment, so 153 were available for safety analysis. Age
FIGO stage
Concomitant chemotherapy
Histology
|
|
| Interventions | Chemotherapy plus either pertuzumab (840 mg loading dose followed by 420 mg every 3 weeks) or placebo | |
| Outcomes |
Primary Median PFS = 4.3 months (95% CI 3.7 to 6.0) (pertuzumab) vs 2.6 months (2.1 to 4.3) (placebo); HR 0.74 (95% CI 0.5 to 1.11; P = 0.14) Secondary Overall survival
Clinical benefit rate (CBR; complete or partial response or stable disease maintained for > 42 days)
Toxicity (G3 or more)
3 participants did not receive randomised treatment, so were excluded from safety outcomes. QoL (Hilpert data 2016 ‐ see additional references for study) Abdominal/gastrointestinal QoL (QLQ‐OV28 score 3.9, 95% CI ‐3.3 to 11.2) Diarrhoeal symptoms ‐ QoL score worse on pertuzumab ‐ score difference 21.2 (95% CI 10.1 to 32.3; P = 0.0003) |
|
| Notes | Potential conflicts listed ‐ pharmaceutical companies involved in the production of pertuzumab funded parts of the study and had staff consulting on the trial OS data updated in later abstract (Lorusso et al 2017, IGCS) QoL data from Abstract (Hilpert et al 2016) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | States only "were randomly assigned"; doesn't state the method of randomisation |
| Allocation concealment (selection bias) | Unclear risk | No mention of methods used to conceal allocation from participants and healthcare professionals involved in the trial |
| Blinding of participants and personnel (performance bias) Objective outcome measures (Overall Survival) | Low risk | Outcome unlikely to have been affected by blinding |
| Blinding of participants and personnel (performance bias) Subjective outcome measures (PFS; Toxicity; QoL) | Low risk | Blinding mentioned only in the title = double‐blind multi‐national randomised phase III trial; no other mention of how blinding was achieved/risk of bias was minimised |
| Blinding of outcome assessment (detection bias) Objective outcome measures (overall survival) | Low risk | Outcome unlikely to have been affected by blinding |
| Blinding of outcome assessment (detection bias) Subjective outcome measures (PFS, Toxicity, QoL) | Low risk | Blinding mentioned only in the title = double‐blind multi‐national randomised phase III trial; no other mention of how blinding was achieved/risk of bias was minimised; independent assessment panel to assess PFS endpoints ‐ likely to be at low risk |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study accounted for all participants initially included. Intention‐to‐treat analysis performed |
| Selective reporting (reporting bias) | Low risk | All outcomes specified in protocol have been published in the study. |
| Other bias | Unclear risk | Potential conflicts listed that pharmaceutical companies involved in the production of pertuzumab funded parts of the study and had staff consulting on the trial. |
Lui 2016.
| Methods | Multi‐centre open‐label phase II randomised controlled trial | |
| Participants | 223 women with advanced or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer that was platinum resistant or refractory per Gynecologic Oncology Group criteria. Participants underwent mandatory pretreatment core needle biopsy and submitted archived tumour samples as available. Age Seribantumab + Paclitaxel (S + P) 58.5 years (30 to 82); Paclitaxel (P) = 60.6 years (28 to 85) Histology Seribantumab + Paclitaxel (S + P): serous = 103 (73.6); endometrioid = 7 (5.0%); clear cell = 6 (4.3%); transitional cell = 2 (1.4%); mixed epithelial = 0 (0%); undifferentiated = 7 (5.0%); other = 12 (8.6%) Paclitaxel (P): serous = 55 (66.2%); endometrioid = 5 (6.0%); clear cell = 2 (2.4%); transitional cell = 0 (0%); mixed epithelial = 1 (1.2%); undifferentiated = 0 (0%); other = 18 (21.7%) Number of platinum‐based therapies S + P = 1 cycle = 25 (17.9%); 2 or more = 114 (81.4%) P = 1 cycle = 17 (20.5%); 2 or more = 65 (78.3%) |
|
| Interventions | Seribantumab + Paclitaxel (S + P) vs Paclitaxel (P) Seribantumab = 40 mg/kg loading dose, then 20 mg/kg once per week Paclitaxel = 80 mg/m² during cycle 1, with optional modification in subsequent cycles to 80 mg/m² once per week for 3 weeks followed by 1 week of rest |
|
| Outcomes | 260 participants enrolled; 223 eligible S + P = 140 participants; P = 83 participants OS Median OS = S + P = 13.7 months; P = 10.12 months (HR 0.991, 95% CI 0.62 to 1.584; P = 0.972) PFS Median PFS = S + P = 3.75 months vs P = 3.68 months (HR 1.027, 95% CI 0.741 to 1.425; P = 0.864) QoL "no significant changes from baseline"; data not provided Overall response rate S + P 13.6% (95% CI 19.9 to 19.6%) (n = 140) P = 18.1% (95% CI 9.8 to 26.4%) (n = 83) Toxicity (G3 or more)
|
|
| Notes | Trial was supported by Merrimack Pharmaceuticals, and several trial authors were employees of the company. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details about methods for randomisation provided in the paper |
| Allocation concealment (selection bias) | Unclear risk | No details about methods for allocation concealment provided in the paper |
| Blinding of participants and personnel (performance bias) Objective outcome measures (Overall Survival) | Low risk | Unlikely to be affected by lack of blinding |
| Blinding of participants and personnel (performance bias) Subjective outcome measures (PFS; Toxicity; QoL) | High risk | Open‐label |
| Blinding of outcome assessment (detection bias) Objective outcome measures (overall survival) | Unclear risk | Unlikely to be affected by lack of blinding |
| Blinding of outcome assessment (detection bias) Subjective outcome measures (PFS, Toxicity, QoL) | High risk | Open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Intention‐to‐treat analysis was undertaken, and all participants were accounted for in the CONSORT diagram and for PFS and OS on the basis of ITT, but reasons for withdrawal from toxicity outcomes for 3 participants are not clear. |
| Selective reporting (reporting bias) | Unclear risk | All expected outcomes reported, although specific data for QoL not reported |
| Other bias | Unclear risk | Several trial authors are employees of the drug company supporting the trial, although no significant effect can be seen. |
Makhija 2010.
| Methods | Phase II randomised placebo‐controlled double‐blind multi‐centre clinical trial | |
| Participants | 131 participants were enrolled at 30 US sites. A total of 130 participants were treated ‐ 65 per arm. Baseline characteristics were similar for participants in the 2 arms. One‐third of all participants were 65 years old, and 65% had an ECOG score of 0. Of the 130 treated participants, 115 had measurable disease at baseline. All participants received at least 1 prior platinum‐containing regimen. Fifteen participants in the gemcitabine + placebo arm and 14 participants in the gemcitabine + pertuzumab arm had received 1 non‐platinum‐containing regimen, most commonly doxorubicin. Most participants had received only 1 prior platinum‐based regimen and had a tumour‐free interval of less than 6 months. Placebo group: median age 61 years, range 18 to 85 years. Pertuzumab group: median age 58 years, range 18 to 81 years Placebo group Ovarian cancer: 59 (91%) Of which, epithelial (papillary serous or other): 51 (86%) Of which, other (clear cell, mucinous, or high‐grade adenocarcinoma): 8 (14%) Peritoneal carcinoma: 5 (8%) Fallopian tube carcinoma: 1 (2%) Pertuzumab group Ovarian cancer: 57 (88%) Of which, epithelial (papillary serous or other): 54 (95%) Of which, other (clear cell, mucinous, or high‐grade adenocarcinoma): 3 (5%) Peritoneal carcinoma: 5 (8%) Fallopian tube carcinoma: 3 (5%) ECOG performance status (all participants ECOG 0 or 1) Placebo group: 42 (65%) participants ECOG PS 0 Pertuzumab group: 43 (66%) participants ECOG PS 0 Measurable disease Placebo group: 58 (89%) Pertuzumab group: 57 (88%) Placebo group Prior treatment with platinum‐based chemotherapy: 65 (100%) 1 prior platinum‐based regimen: 42 (65%) 2 or more prior platinum‐based regimens: 23 (35%) Prior treatment for platinum‐resistant disease: 15 Doxorubicin: 9 (60%) Topotecan: 2 (13%) Taxane based: 2 (13%) Other: 3 (20%) Pertuzumab group Prior treatment with platinum‐based chemotherapy: 65 (100%) 1 prior platinum‐based regimen: 48 (74%) 2 or more prior platinum‐based regimens: 17 (26%) Prior treatment for platinum‐resistant disease: 14 Doxorubicin: 11 (79%) Topotecan: 1 (7%) Taxane based: 2 (14%) Other: 5 (36%) Baseline CA‐125 U/mL Placebo group: median 249, range 11 to 2292 Pertuzumab group: median 195, range 11 to 4287 |
|
| Interventions |
Intervention Gemcitabine (800 mg/m² on days 1 and 8 of a 21‐day cycle) plus pertuzumab (administered intravenously as an 840‐mg loading dose followed by 420 mg every 3 weeks) Treatment was administered until either tumour progression or unacceptable toxicity. Participants with non‐progressing disease after 17 cycles of treatment and acceptable toxicity were eligible to continue pertuzumab. Comparison Gemcitabine (800 mg/m² on days 1 and 8 of a 21‐day cycle) plus placebo. Treatment was administered until either tumour progression or unacceptable toxicity. Cross‐over to pertuzumab ± gemcitabine for a total of 17 cycles, administered every 3 weeks for up to 1 year, was allowed for participants randomly assigned to placebo who had objective evidence of disease progression. |
|
| Outcomes |
Cox regression for survival outcomes stratified by baseline ECOG performance status, number of prior regimens for platinum‐resistant disease, and disease measurability was used to estimate hazard ratio (HR). Unstratified Cox regression was used to explore pertuzumab treatment benefit in patient subsets. |
|
| Notes | Length of follow‐up not reported, but Kaplan‐Meier plots show that for overall survival, the maximum length of follow‐up was about 30 months. PFS and objective response analyses were performed after 103 PFS events were observed. All other data presented reflect the final analysis, which occurred after 91 deaths were observed. An open‐label cross‐over was allowed from placebo to pertuzumab in the presence of objective evidence of disease progression. Archival tumour tissue was also taken for correlative gene mRNA expression studies, to look at HER‐1, HER‐2, HER‐3, betacellulin, amphiregulin, and G6‐PDH. In the intervention group, all 65 participants received allocated treatment, all of whom discontinued blinded treatment (53 due to disease progression; 8 due to adverse events; 1 due to the participant's decision; 1 due to physician’s decision; 1 due to non‐compliance; 1 due to other reasons). In the comparison group, only 1 participant out of the 66 who were randomised did not receive allocated intervention (unclear why not). 65/66 participants did receive allocated intervention. Of these, 1/65 completed 17 cycles of blinded treatment and 64/65 discontinued blinded treatment (1 due to death; 56 due to disease progression; 2 due to adverse events; 3 due to the participant's decision; 2 due to physician’s decision). The 1 participant who did not receive the allocated control intervention was not analysed. All other participants were analysed as intention‐to‐treat. Median time‐to‐event data were estimated via the Kaplan‐Meier method. Median PFS was 2.9 months and 2.6 months in intervention and placebo groups, respectively. Median OS was 13 months and 13.1 months in intervention and placebo groups, respectively. Adverse events were recorded as the number of women who experienced an event (N = 65 in each group). Haematological toxicity Placebo group: neutropaenia: grade 3 or 4: 14, any grade: 28; thrombocytopaenia: grade 3 or 4: 5, any grade: 15; anaemia: grade 3 or 4: 3, any grade: 34 Pertuzumab group: neutropaenia: grade 3 or 4: 23, any grade: 32; thrombocytopaenia: grade 3 or 4: 9, any grade: 19; anaemia: grade 3 or 4: 3, any grade: 31 Gastrointestinal toxicity Placebo group: nausea: grade 3 or 4: 4, any grade: 43; diarrhoea: grade 3 or 4: 1, any grade: 23; dyspepsia: grade 3 or 4: 0, any grade: 8 Pertuzumab group: nausea: grade 3 or 4: 5, any grade: 49; diarrhoea: grade 3 or 4: 7, any grade: 44; dyspepsia: grade 3 or 4: 1, any grade: 14 Skin toxicity Placebo group: rash: grade 3 or 4: 0, any grade: 9; stomatitis: grade 3 or 4: 0, any grade: 7 Pertuzumab group: rash: grade 3 or 4: 0, any grade: 26; stomatitis: grade 3 or 4: 0, any grade: 19 Neurological toxicity Placebo group: headache: grade 3 or 4: 1, any grade: 17 Pertuzumab group: headache: grade 3 or 4: 1, any grade: 24 Cardiac toxicity Placebo group: congestive heart failure: grade 3 or 4: 0, any grade: 0; left ventricular ejection fraction decline ≥ 10 points (N = 59): 10 Pertuzumab group: congestive heart failure: grade 3 or 4: 1, any grade: 1; left ventricular ejection fraction decline ≥ 10 points (N = 60): 7 Other Placebo group: fatigue: grade 3 or 4: 11, any grade: 44; back pain: grade 3 or 4: 1, any grade: 15; epistaxis: grade 3 or 4: 0, any grade: 1; rhinorrhoea: grade 3 or 4: 0, any grade: 4 Pertuzumab group: fatigue: grade 3 or 4: 14, any grade: 51; back pain: grade 3 or 4: 6, any grade: 27; epistaxis: grade 3 or 4: 0, any grade: 15; rhinorrhoea: grade 3 or 4: 0, any grade: 10 Fatal adverse events occurred in 2 participants in the gemcitabine + placebo arm: sepsis and infection. Four participants in the gemcitabine + pertuzumab arm had fatal adverse events: acute renal failure; pneumonitis; haemolytic‐uraemic syndrome; sepsis, Clostridium difficile colitis, and hydronephrosis. Quality of life (reported only in conference abstract form ‐ see Lalla 2008 in subsidiary references) "The median time to symptom deterioration was 1.7 months in the gemcitabine+placebo arm vs. 3.8 months in the gemcitabine+pertuzumab arm (HR = 0.62, 95% CI: 0.36‐1.05). Symptom improvement (≥ 3 point increase in FOSI) occurred in 28 women (43%) given gemcitabine+pertuzumab, compared to 18 (28%) in those receiving gemcitabine+placebo". ClinicalTrials.gov identifier: NCT00096993 This study was included in the original version of this review; no new results have been identified. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomly assigned in a 1:1 ratio to gemcitabine ... plus either placebo or pertuzumab". |
| Allocation concealment (selection bias) | Unclear risk | Not clear from the study |
| Blinding of participants and personnel (performance bias) Objective outcome measures (Overall Survival) | Low risk | Outcome unlikely to be affected by blinding |
| Blinding of participants and personnel (performance bias) Subjective outcome measures (PFS; Toxicity; QoL) | Unclear risk | Study was described as double‐blinded, but all but 1 participant discontinued blinded treatment. |
| Blinding of outcome assessment (detection bias) Objective outcome measures (overall survival) | Low risk | Outcome unlikely to be affected by blinding |
| Blinding of outcome assessment (detection bias) Subjective outcome measures (PFS, Toxicity, QoL) | Unclear risk | Study was described as double‐blinded, but it was unclear whether the outcome assessor was masked. Trial protocol was consulted but did not provide further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | % analysed: 130/131 (99%) |
| Selective reporting (reporting bias) | Low risk | All pertinent outcomes appear to have been reported, and exploratory analyses, unplanned beforehand, are clearly labelled as such. |
| Other bias | Unclear risk | Industry involvement and funding (from Genentech) were reported. |
Vergote 2014.
| Methods | Open‐label international phase III RCT (125 institutions in 10 countries). Recruitment from October 2005 to February 2008, with median follow‐up time of 51 months | |
| Participants | 835 women with EOC or primary peritoneal or fallopian tube cancer (of whom 420 were allocated to receive erlotinib, and 415 to observation) Participants were not selected for EGFR expression. Women had to have histologically confirmed high‐risk FIGO stage I (grade 3, or aneuploid grade 1 or 2, or clear cell) or stage II to IV disease. Women also had to have completed first‐line therapy within the past 6 weeks (consisting of 6 to 9 cycles of a platinum derivative alone or in combination with other agents), and had to have achieved complete response, partial response, or stable disease (according to RECIST or GCIG criteria). Baseline characteristics Erlotinib
Observation
Inclusion criteria: eligible women had histologically confirmed high‐risk International Federation of Gynecology and Obstetrics (FIGO) stage I (grade 3, or aneuploid grade 1 or 2, or clear cell) or stage II to IV epithelial ovarian, primary peritoneal, or fallopian tube cancer (women with adenocarcinoma of unknown origin were not eligible). Exclusion criteria: other malignancy within the past 5 years (except adequately treated basal cell or squamous cell skin cancer or cone biopsied carcinoma in situ of the cervix) Pretreatment: participants were randomly assigned to maintenance erlotinib 150 mg orally daily for 2 years (or until disease progression) or to observation. No significant differences between study populations were noted in terms of FIGO stage, tumour histology, serum CA‐125 at entry, and response to first‐line chemotherapy/prior surgery. |
|
| Interventions |
Intervention Erlotinib 150 mg orally daily for 2 years (or until disease progression). If adverse events occurred, the dose could be reduced to 100 mg/d or 75 mg/d, or treatment could be stopped; dose re‐escalation was not allowed. Comparison Observation |
|
| Outcomes |
Primary: PFS Secondary: OS, toxicity, occurrence of rash, quality of life Toxicity was graded according to National Cancer Institute Common Toxicity Criteria version 3.0. Quality of life was defined by the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire C30 (EORTC QLQ‐C30) and OV28 (Ovarian Cancer Module) questionnaires. Overall survival (OS) erlotinib vs observation arms HR = 0.99 (95% CI 0.81 to 1.20; P = 0.903), adjusted for stratification parameters Progression‐free survival (PFS) erlotinib vs observation arms HR = 1.05 (95% CI 0.90 to 1.23; P = 0.525), adjusted for stratification parameters Median PFS 12.7 months in the erlotinib group vs 12.4 months in the observation arm QoL "QOL results were limited by low compliance (85% at baseline, ranging from 72% to 51% during the first year and < 50% during the second year); therefore, longitudinal modelling was limited to the first year only. QOL compliance was similar between the two treatment arms. Global health/QOL scores showed a significant overall difference between the two treatment arms during the first year (P = 0.0102) in favour of the observation arm. In addition, the QLQ‐C30 found statistically significant differences at the 5% level in symptom levels for diarrhoea, loss of appetite, nausea/vomiting, and fatigue, with worse symptom scores for the erlotinib arm. None of the scales, however, reported differences of ≥ 10 points except for the diarrhoea scale, in which differences of more than 20 points were observed at most assessments during the first year. Sensitivity analyses by means of imputation revealed similar results". Toxicity data Adverse events have been reported only for the intervention (erlotinib) arm, not for the observation arm, and thus cannot be included in comparison analyses. Investigators were contacted, but no further data were provided. 420 were originally assigned to erlotinib; of these, 415 received the allocated treatment. 107/415 (25.8%) participants stopped treatment because of unacceptable adverse events. Detailed toxicities for the intervention arm are listed below, with details of toxicity grade if known. Gastrointestinal toxicity Diarrhoea: grade 1: 142 (34.2%), grade 2: 87 (21.0%), grade 3: 19 (4.6%), grade 4: 1 (0.2) Abdominal pain: grade 3 or 4: 10 (2.4%) Increased γ‐glutamyl transpeptidase: grade 3 or 4: 14 (3.4%) Skin Rash: grade 1: 120 (28.9%), grade 2: 154 (37.1%), grade 3: 51 (12.3%), grade 4: 2 (0.5%) Dry skin: grade 3 or 4: 7 (1.7%) Haematological, genitourinary, neurological, or other: not reported Median follow‐up: 51 months |
|
| Notes | ClinicalTrials.gov identifier: NCT00263822 This study was mentioned as an ongoing trial (MRC OV07, the Tarceva trial) in the original review; results have been published since that time. Funding source Financial support statement from the published paper "Supported by a donation from the Vlaamse Liga Tegen Kanker through the European Organisation for Research and Treatment of Cancer (EORTC) Charitable Trust, by Grant No. SCIE2006‐29 from the Stichting Tegen Kanker of Belgium (to the Katholieke Universiteit Leuven [sic], and by an unrestricted educational grant from F. Hoffmann‐La Roche (Basel, Switzerland) to the EORTC". Declarations of interest Study authors' disclosures of potential conflicts of interest from the published paper "Although all authors completed the disclosure declaration, the following author(s) and/or an author’s immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO’s conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors". Employment or leadership position: none Consultant or advisor role: Ignace B. Vergote, Roche (C); Marcia Hall, Roche (C); Christopher B. Steer, Janssen Pharmaceutica (C), Roche (C); John Green, Roche (C); Eric Pujade‐Lauraine, Roche (C) Stock ownership: none Honoraria: Ignace B. Vergote, Roche; Christopher B. Steer, Eli Lilly; Nick Simon Reed, Roche Research funding: Ignace B. Vergote, Roche; Evelyn Despierre, European Organisation for Research and Treatment of Cancer Network of Core Institutions Expert testimony: none Patents: none Other remuneration: Christopher B. Steer, Janssen Pharmaceutica, Eli Lilly Country: Belgium Setting: multi‐centre RCT Comments: funding from European Organisation for Research and Treatment of Cancer Network of Core Institutions Author's name: Ignace Vergote, MD, PhD Institution: University Hospital Leuven Email: ignace.vergote@uzleuven.be Address: University Hospital Leuven, Katholieke Universiteit Leuven, Herestraat49, B‐3000 Leuven, Belgium, European Union |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratified randomisation by algorithm (details kindly provided by study investigators) |
| Allocation concealment (selection bias) | Low risk | Stratified randomisation by algorithm (details kindly provided by study investigators). Allocation should not have been predictable by investigators. |
| Blinding of participants and personnel (performance bias) Objective outcome measures (Overall Survival) | Low risk | Outcome unlikely to be affected by blinding |
| Blinding of participants and personnel (performance bias) Subjective outcome measures (PFS; Toxicity; QoL) | High risk | Open‐label study |
| Blinding of outcome assessment (detection bias) Objective outcome measures (overall survival) | Low risk | Outcome unlikely to be affected by blinding |
| Blinding of outcome assessment (detection bias) Subjective outcome measures (PFS, Toxicity, QoL) | High risk | Open‐label study |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low attrition |
| Selective reporting (reporting bias) | Low risk | Prespecified and expected outcomes were reported. |
| Other bias | Unclear risk | Several investigators involved in the study received compensation from the company producing the investigated study drug (Roche, erlotinib); conflicts of interest have been declared in the study report (and are detailed in the 'Notes' section of the study table above). |
AUC: area under the curve CA‐125: cancer antigen‐125 CBR: clinical benefit rate CI: confidence interval CT: cytotoxic chemotherapy D: docetaxel ECOG: Eastern Cooperative Oncology Group EGFR: epidermal growth factor receptor EOC: epithelial ovarian cancer EORTC QLQ‐C30: European Organization for Research and Treatment of Cancer core quality of life questionnaire FACT‐O: Functional Assessment of Cancer Therapy – Ovarian FIGO: International Federation of Obstetrics and Gynecology FOSI: FACT‐O Symptom Index G6‐PDH: G6‐phosphate dehydrogenase GCIG: Gynecologic Cancer Intergroup HER: human epidermal growth factor receptor HR: hazard ratio ITT: intention‐to‐treat NOS: not otherwise specified. ORR: overall response rate OS: overall survival OV28: Ovarian Cancer Module P: pertuzumab or paclitaxel PFS: progression‐free survival QLQ‐OV28: EORTC Module for Ovarian Cancer QoL: quality of life RCT: randomised controlled trial RECIST: Response Evaluation Criteria in Solid Tumours S: seribantumab SWOG: Southwest Oncology Group TFI: treatment‐free interval ULN: upper limit of normal V: vandetanib VTE: venous thromboembolism WHO PS: World Health Organization Performance Status
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Annunziata 2010 | Not a randomised controlled study. The aim of the study was to evaluate clinical activity and target modulation of vandetanib in women with recurrent ovarian cancer. |
| Bauman 2012 | Not a randomised controlled study. This was a single‐arm, non‐randomised phase I study of erlotinib in combination with escalating doses of 5‐azacytidine, evaluating safety and toxicity in women with solid tumour malignancies. |
| Blank 2010 | Not a randomised controlled study. The purpose of this study was to determine whether adding erlotinib to carboplatin/paclitaxel improved pathological complete response at reassessment surgery in epithelial ovarian, fallopian tube, or primary peritoneal cancers. |
| Bookman 2003 | Not a randomised controlled study. The aim of this study was to evaluate the feasibility, toxicity, and efficacy of single‐agent trastuzumab in ovarian and primary peritoneal carcinoma. |
| Campos 2005 | Not a randomised controlled study. A phase II open‐label clinical trial that evaluated CI‐1033 (canertinib) in women with platinum‐refractory or ‐resistant ovarian cancer |
| Campos 2010 | Not a randomised controlled study. A phase I study of ZD1839 (Iressa) in combination with escalating doses of liposomal doxorubicin evaluating safety and toxicity in women with recurrent gynaecological or metastatic breast cancer |
| Campos 2011 (NCT00520013) | A study evaluating the response rate and progression‐free survival of carboplatin/paclitaxel/bevacizumab followed by either bevacizumab alone or bevacizumab + erlotinib in women with ovarian, fallopian tube, or peritoneal cancer. This trial was excluded because the comparison of the study did not meet our inclusion criteria, as erlotinib is being compared to an agent (bevacizumab) that is neither standard cytotoxic chemotherapy nor a placebo. |
| Chambers 2010 | Not a randomised controlled study. A single‐arm phase II study of the safety and efficacy of erlotinib and bevacizumab, in which all participants were allocated both agents |
| Ciunci 2014 | Dose escalation study with cetuximab in both arms ‐ not an RCT |
| Dinh 2008 | A review of the literature |
| Galsky 2012 | A phase II randomised‐discontinuation study of lapatinib monotherapy evaluating efficacy and safety in women with treatment‐refractory HER‐2 amplified solid tumours. This study was excluded because participants had diverse underlying tumours selected on amplified HER‐2 expression, only 10/32 of ovarian origin, of which only 3 were eligible for randomisation. All participants initially received lapatinib; only those with stable disease were randomised to either continue lapatinib or switch to placebo. Study was closed early due to low stable disease rates. |
| Garcia 2012 | Not a randomised controlled study. A phase II study of lapatinib evaluating activity and tolerability in women with recurrent or persistent EOC or primary peritoneal carcinoma |
| Gordon 2005 | Not a randomised controlled study. The aim of this phase II study was to estimate the tumour response rate and safety profile of erlotinib. |
| Gordon 2006 | Not a randomised controlled study. This study primarily assessed response to pertuzumab among women with relapsed ovarian cancer. |
| Guastalla 2007 | Not a randomised controlled study. This study assessed the effect of adding trastuzumab to paclitaxel and carboplatin in women with resistant advanced ovarian cancer and HER‐2 gene amplification. |
| Hariprasad 2006 | Not a randomised controlled study. This study aimed to evaluate the role of gefitinib among women with advanced and recurrent ovarian cancer to prolong progression‐free survival. |
| Hariprasad 2009 | Not a randomised controlled study. The aim of this study was to assess the safety and toxicity of gefitinib in women with advanced epithelial ovarian cancer. |
| Harter 2013 | Not a randomised controlled study. This started as a phase I trial to investigate the safety/tolerability of adding vandetanib to pegylated liposomal doxorubicin (PLD). A planned subsequent phase II study was not started, as the drug combination was poorly tolerated. |
| Hirte 2010 | Not a randomised controlled study. The primary objective of this study was to assess the response rate to the addition of erlotinib in women with recurrent ovarian cancer who were receiving carboplatin. |
| Jhaveri 2012 | Not a randomised controlled study. A phase I study of trastuzumab and alvespimycin to determine the maximum tolerated dose (MTD) in women with advanced solid tumour malignancies. |
| Joly 2009 | Not a randomised controlled study. This study evaluated the efficacy of lapatinib and topotecan in non‐HER‐screened women who failed first‐line platinum‐based chemotherapy within 12 months. |
| Kimball 2008 | Not a randomised controlled study. A phase I study to assess maximum tolerated dose, toxicity profile, clinical activity, and pharmacokinetics of carboplatin in combination with lapatinib in platinum‐sensitive relapsed ovarian cancer |
| Konner 2008 | Not a randomised controlled study. A phase II study to determine the safety and efficacy of cetuximab plus paclitaxel and carboplatin as initial treatment for stage III/IV ovarian cancer |
| Koolen 2011 | Not a randomised controlled study. A study of LY2334737 (a gemcitabine prodrug) with or without erlotinib to determine MTD and dose‐limiting toxicities among women with advanced solid tumours |
| Krasner 2005 | Not a randomised controlled study. A single‐arm phase II study of ZD1839 (Iressa/gefitinib) and anastrozole (Arimidex) in women with relapsed ovarian cancer, in which all participants were allocated to receive both agents |
| Lheureux 2012 | Not a randomised controlled study. Multi‐centre study of topotecan and lapatinib assessing efficacy in women with EOC relapsed after a first line of chemotherapy |
| NCT00861120 | Non‐randomised phase II study of panitumumab and pegylated liposomal doxorubicin for platinum‐resistant epithelial ovarian cancer with kirsten rat sarcoma viral oncogene homolog (KRAS) wild‐type. No results posted on clinicaltrials.gov as of March 2018 |
| NCT01296035 | Not a randomised study. Phase II evaluation of panitumumab and gemcitabine as treatment for women with recurrent epithelial ovarian cancer ‐ enrolled 8 women only and was terminated early due to lack of response |
| Nimeiri 2008 | Not a randomised controlled study. The objectives of this phase II trial were to assess the activity and tolerability of the combination of bevacizumab and erlotinib in women with recurrent ovarian, primary peritoneal, or fallopian tube cancer. |
| Palayekar 2008 | A review of the literature |
| Pautier 2010 | Not a randomised controlled study. This phase II study investigated the efficacy and tolerability of gefitinib in combination with paclitaxel and carboplatin for second‐line treatment of women with ovarian, tubal, or peritoneal adenocarcinoma. |
| Posadas 2007 | Not a randomised controlled study. This study aimed to assess the biochemical effects of gefitinib on its target signal‐transduction pathways in women with recurrent ovarian tumours. |
| Ray‐Coquard 2008 | Not a randomised controlled study. This study aimed to assess the activity and toxicity of a regimen of paclitaxel, carboplatin, and trastuzumab in women with resistant advanced ovarian cancer and HER‐2 gene amplification. |
| Schilder 2005 | Not a randomised controlled study. A phase II trial that assessed activity and tolerability of oral gefitinib in women with recurrent or persistent ovarian or primary peritoneal cancers |
| Schilder 2009 | Not a randomised controlled study. A phase II trial of cetuximab to determine if dose escalation to grade 2 rash correlates with antitumour activity |
| Secord 2008 | Not a randomised controlled study. A phase II trial that assessed activity and tolerability of cetuximab and carboplatin in women with relapsed platinum‐sensitive ovarian and primary peritoneal cancer |
| Seiden 2007 | Not a randomised controlled study. A phase II study to determine the rate of response to matuzumab in women with recurrent ovarian or primary peritoneal cancers |
| Steffensen 2013 | Not a randomised controlled study. A phase II study of pegylated liposomal doxorubicin (PLD) and panitumumab, investigating response rates among women with relapsed, platinum‐resistant ovarian, fallopian, or peritoneal cancer |
| Vasey 2008 | Not a randomised controlled study. A phase Ib study to assess the safety and maximum tolerated dose of erlotinib with docetaxel/carboplatin in women with ovarian cancer |
| Vlahovic 2012 | Not a randomised controlled study. A phase I study of bevacizumab, everolimus, and panitumumab evaluating MTD, safety, and tolerability in women with solid malignancies |
| Wagner 2007 | Not a randomised controlled study. A phase II trial of gefitinib and tamoxifen in women with ovarian cancer refractory or resistant to first‐line chemotherapy |
| Weroha 2011 | Not a randomised controlled study. A phase II study of topotecan and lapatinib evaluating the efficacy and adverse event profile in women with histologically confirmed, platinum‐refractory, or resistant primary or relapsed, surgically debulked EOC or primary peritoneal carcinoma |
EOC: epithelial ovarian cancer HER: human epidermal growth factor receptor MTD: maximum tolerated dose PLD: pegylated liposomal doxorubicin RCT: randomised controlled trial
Differences between protocol and review
We updated background and supporting references from the original protocol version.
Quality of life
Only one trial reported quality of life, but researchers did not make analysable data available, so we removed the following sections in the protocol, which discussed handling of data for continuous outcomes, as they were unnecessary.
Data extraction and management
For continuous outcomes (e.g. quality of life measures), we will extract the final value and standard deviation of the outcome of interest and the number of participants assessed at endpoint in each treatment arm at the end of follow‐up, in order to estimate the mean difference between treatment arms and its standard error.
Measures of treatment effect
For continuous outcomes, we will use the mean difference between treatment arms if all trials measured the outcome on the same scale, otherwise standardised mean differences will be used.
Data synthesis
We had not originally planned to conduct subgroup analyses. However, we did perform subgroup analyses based on type of anti‐EGFR treatment (tyrosine kinase inhibitor vs monoclonal antibody) because these treatments may have had different activities and varying side effect profiles, given their different mechanisms of action. We performed analyses separately for primary treatment and for recurrent disease treatment. We also conducted subgroup analysis based on platinum resistance/sensitivity in recurrent disease because biologically these often have different response rates to conventional treatment. When heterogeneity was obvious on visual inspection of forest plots between individual anti‐EGFR agents of the same type for anti‐EGFR treatment, we also performed subgroup analyses.
We included no studies with multiple intervention groups or the need for indirect analysis, so we removed these sections from the original protocol and will use them in updates of the review, if necessary.
If any trials have multiple intervention groups, the control group will be divided between the intervention groups ‐ to prevent double counting of participants in the meta‐analysis ‐ and comparisons between each intervention and a split control group will be treated independently.
Random‐effects models with inverse variance weighting were used for all meta‐analyses (DerSimonian 1986)".
If sufficient data are available, indirect comparisons, using the methods of Bucher 1997 will be used to compare competing interventions that have not been compared directly with each other.
Sensitivity analysis
Sensitivity analyses will be performed (i) excluding studies at high risk of bias and (ii) using unadjusted results.
Contributions of authors
SK and JM provided the initial concept for the title; KH conducted the original search and data extraction in collaboration with KG for the original published version of the review, with JM acting as mediator for disagreements. A combination of the review authors CT, KG, TL, RG, and JM performed the search update and sifted and extracted data, with JM acting as mediator for disagreements. AW and JM performed GRADE assessments, with discussion if disagreements arose. CT, AB, and JM wrote the review.
Sources of support
Internal sources
No sources of support supplied
External sources
-
NIHR, UK.
The original version of this review was prepared with the aid of funding for methodological support from a programme grant provided to the Gynaecological, Neuro‐oncology, and Orphan Cancer Cochrane Review Group by NIHR, UK. The update of this review was prepared without financial support.
Declarations of interest
Jo Morrison: I have no financial conflict of interest in the results of this review. Clemens Thoma: None known. Richard J Goodall: None known. Thomas J Lyons: None known. Kezia Gaitskell: None known. Alison J Wiggans: None known. Andrew Bryant: None known.
Stable (no update expected for reasons given in 'What's new')
References
References to studies included in this review
Chekerov 2017 {published data only}
- Chekerov R, Klare P, Krabisch P, Potenberg J, Heinrich G, Mueller L, et al. Panitumumab in platinum‐sensitive epithelial ovarian cancer patients with KRAS wild‐type: the PROVE‐study, a phase II randomized multicenter study of the North‐Eastern German Society of Gynaecologic Oncology. Journal of Clinical Oncology 2017;35(15):5558. [Google Scholar]
- WiSP Wissenschaftlicher Service Pharma GmbH. PROVE: A Randomized Phase II Trial of Standard Carboplatin‐based Chemotherapy With or Without Panitumumab in Platinum‐sensitive Recurrent Ovarian Cancer. ClinicalTrials.gov May 2011.
Coleman 2014 {published data only}
- Coleman RL, Moon J, Sood A, Branham D, Delmore JE, Bonebrake AJ, et al. Randomized phase II study of docetaxel plus vandetanib (D+V) versus docetaxel followed by vandetanib (D‐V) in patients with persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal carcinoma (OC): SWOG S0904. European Journal of Cancer 2014; Vol. 50, issue 9:1638‐48. [DOI] [PMC free article] [PubMed]
- Southwest Oncology Group, National Cancer Institute. Docetaxel with or without vandetanib in treating patients with persistent or recurrent ovarian epithelial cancer, fallopian tube cancer, or primary peritoneal cancer (NCT00872989). Protocol registered at ClinicalTrials.gov on 31 March 2009.
Kaye 2013 {published data only}
- Kaye SB, Poole CJ, Bidzinski M, Gianni L, Gorbunova V, Novikova E, et al. A randomized phase II study evaluating the combination of carboplatin‐based chemotherapy with pertuzumab versus carboplatin‐based therapy alone in patients with relapsed, platinum sensitive ovarian cancer. Journal of Clinical Oncology 2008;26(Suppl):Abstract 5520. [DOI] [PubMed] [Google Scholar]
- Kaye SB, Poole CJ, Danska‐Bidzinska A, Gianni L, Conte G, Gorbunova V, et al. A randomized phase II study evaluating the combination of carboplatin‐based chemotherapy with pertuzumab versus carboplatin‐based therapy alone in patients with relapsed, platinum‐sensitive ovarian cancer. Annals of Oncology 2013;14(1):145‐52. [DOI] [PubMed] [Google Scholar]
Kurzeder 2016 {published data only}
- Colombo N, Gonzalez‐Martin A, Oestergaard MZ, Rau J, Canzler U, Fabbro M, et al. Biomarker results from PENELOPE, a randomised phase III trial evaluating chemotherapy ± pertuzumab for platinum‐resistant ovarian cancer (PROC) with low tumour HER3 mRNA expression. International Journal of Gynecological Cancer 2015; Vol. 19th International Meeting of the European Society of Gynaecological Oncology, ESGO 2015, Nice, France:415‐7.
- Hilpert F, Greimel E, Ottevanger P, Rau J, Lorusso D, Kroep J, et al. Patient‐reported outcome (PRO) results from the AGO‐OVAR 2.20/ENGOT‐OV14/PENELOPE double‐blind placebo‐controlled randomised phase III trial evaluating chemotherapy ± pertuzumab for platinum‐resistant ovarian cancer (PROC). International Journal of Gynecological Cancer 2015; Vol. 19th International Meeting of the European Society of Gynaecological Oncology, ESGO 2015, Nice, France:465‐6.
- Hilpert F, Gropp‐Meier M, Schmalfeldt B, Rau J, Meier W, Hasenburg A, et al. Patient‐reported outcome (PRO) results from the AGO‐OVAR 2.20/ENGOT‐ov14/PENELOPE double‐blind placebo‐controlled randomised phase III trial evaluating chemotherapy ± pertuzumab for platinum‐resistant ovarian cancer (PROC). Oncology Research and Treatment 2018; Vol. 39:138.
- Hoffmann‐La Roche. A two‐part, randomized phase III, double‐blind, multicenter trial assessing the efficacy and safety of pertuzumab in combination with standard chemotherapy vs. placebo plus standard chemotherapy in women with recurrent platinum‐resistant epithelial ovarian cancer and low HER3 mRNA expression. Clinicaltrials.gov 2012.
- Kurzeder C, Bover I, Marme F, Rau J, Pautier P, Colombo N, et al. Double‐blind, placebo‐controlled, randomized phase III trial evaluating pertuzumab combined with chemotherapy for low tumor human epidermal growth factor receptor 3 MRNA‐expressing platinum‐resistant ovarian cancer (PENELOPE). Journal of Clinical Oncology 2016; Vol. 34, issue 21:2516‐25. [DOI] [PubMed]
- Kurzeder C, Bover I, Marme F, Rau J, Pautier P, Colombo N, et al. Efficacy and safety of chemotherapy (CT) ± pertuzumab (P) for platinum resistant ovarian cancer (PROC): AGOOVAR 2.20/ENGOTov14/ PENELOPE double‐blind, placebo‐controlled, randomized phase III trial. Journal of Clinical Oncology 2015;15:5504. [Google Scholar]
- Kurzeder C, Campo JM, Pautier P. PENELOPE/AGO‐OVAR 2.20: a double‐blind placebo (PLA)‐controlled randomized phase III ENGOT trial evaluating chemotherapy (CT) with or without pertuzumab (P) for platinum‐resistant ovarian cancer. Journal of Clinical Oncology: ASCO Annual Meeting Proceedings 2014;32:abstr TPS5613. [Google Scholar]
- Lorusso D, Gonzalez‐Martin A, Luck HJ, Rau J, Selle F, Colombo N, et al. Final overall survival results from the double‐blind placebo‐controlled randomised phase III PENELOPE trial evaluating pertuzumab in platinum‐resistant ovarian cancer with low tumour HER3 MRNA expression. International Journal of Gynecological Cancer. Conference: 16th biennial meeting of the international gynecologic cancer society. Portugal. Conference start: 20161028. Conference end: 20161031 2017; Vol. 26:11‐2.
- Mahner S, Lueck HJ, Grischke EM, Rau J, Gropp‐Meier M, Schmalfeldt B, et al. Chemotherapy (CT) ± pertuzumab (P) for platinum‐resistant ovarian cancer (PROC): AGO‐OVAR 2.20/engot‐ov14/PENELOPE double‐blind placebo‐controlled randomized phase 3 trial. Oncology Research and Treatment 2018; Vol. 39:S97.
- Pautier P, Pardo B, Hilpert F, Rau J, Gadducci A, Savarese A, et al. Subgroup analysis by chemotherapy cohort in the ago‐ovar 2.20/ENGOT‐OV14/PENELOPE double‐blind placebo‐controlled randomised phase III trial evaluating chemotherapy ± pertuzumab for platinum‐resistant ovarian cancer (PROC). International Journal of Gynecological Cancer 2015; Vol. 25:24‐6.
Lui 2016 {published data only}
- Liu JF, Ray‐Coquard I, Selle D, Poveda AM, Cibula D, Hirte H, et al. Randomized phase II trial of seribantumab in combination with paclitaxel in patients with advanced platinum‐resistant or ‐refractory ovarian cancer. Journal of Clinical Oncology 2016; Vol. 34, issue 36:4345‐53. [DOI] [PMC free article] [PubMed]
Makhija 2010 {published data only (unpublished sought but not used)}
- Glenn D, Ueland F, Bicher A, Dizon D, Gold M, Makhija S, et al. A randomized phase II trial with gemcitabine with or without pertuzumab (rhuMAb 2C4) in platinum‐resistant ovarian cancer (OC): preliminary safety data [abstract]. Journal of Clinical Oncology; 2006 ASCO Annual Meeting Proceedings Part I, 2006;24(Suppl):13001. [Google Scholar]
- Lalla D, Paton V, Lin CY, Dizon DS. Affecting quality of life in ovarian cancer: encouraging results from a randomized phase II trial of gemcitabine ± pertuzumab in women with platinum‐resistant disease [Abstract]. Journal of Clinical Oncology 2008;26(Suppl):Abstract 5550. [Google Scholar]
- Makhija S, Amler LC, Glenn D, Ueland FE, Gold MA, Dizon DS, et al. Clinical activity of gemcitabine plus pertuzumab in platinum‐resistant ovarian cancer, fallopian tube cancer, or primary peritoneal cancer. Journal of Clinical Oncology 2010;28(7):1215‐23. [DOI] [PubMed] [Google Scholar]
- Makhija S, Glenn D, Ueland F, Gold M, Dizon D, Paton V, et al. Results from a phase II randomized, placebo‐controlled, double‐blind trial suggest improved PFS with the addition of pertuzumab to gemcitabine in patients with platinum‐resistant ovarian, fallopian tube or primary peritoneal cancer. Journal of Clinical Oncology 2007;25(Suppl):Abstract 5507. [Google Scholar]
Vergote 2014 {published data only}
- European Organization for Research and Treatment of Cancer. Erlotinib or observation in treating patients who have undergone first‐line chemotherapy for ovarian cancer, peritoneal cancer, or fallopian tube cancer (MRC OV07). Protocol registered at ClinicalTrials.gov on 7 December 2005.
- Vergote IB, Jimeno A, Joly F, Katsaros D, Coens C, Despierre E, et al. Randomized phase III study of erlotinib versus observation in patients with no evidence of disease progression after first‐line platin‐based chemotherapy for ovarian carcinoma: a European Organisation for Research and Treatment of Cancer‐Gynaecological Cancer Group, and Gynecologic Cancer Intergroup study. Journal of Clinical Oncology 2014; Vol. 32, issue 4:320‐6. [DOI] [PubMed]
References to studies excluded from this review
Annunziata 2010 {published data only}
- Annunziata CM, Walker AJ, Minasian L, Yu M, Kotz H, Wood BJ, et al. Vandetanib, designed to inhibit VEGFR2 and EGFR signaling, had no clinical activity as monotherapy for recurrent ovarian cancer and no detectable modulation of VEGFR2. Clinical Cancer Research 2010;16(2):664‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bauman 2012 {published data only}
Blank 2010 {published data only}
- Blank SV, Christos P, Curtin JP, Goldman N, Runowicz CD, Sparano JA, et al. Erlotinib added to carboplatin and paclitaxel as first‐line treatment of ovarian cancer: a phase II study based on surgical reassessment. Gynecologic Oncology 2010;119(3):451‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Cancer Institute. A Phase II Study of OSI 774 In Combination With Carboplatin and Paclitaxel in Patients With Ovarian, Cancer of the Fallopian Tube or Primary Peritoneal Carcinoma. ClinicalTrials.gov identifier: NCT00059787. Protocol registered at ClinicalTrials.gov on 6 May 2003.
Bookman 2003 {published data only}
- Bookman MA, Darcy KM, Clarke‐Pearson D, Boothby RA, Horowitz IR. Evaluation of monoclonal humanized anti‐HER2 antibody, trastuzumab, in patients with recurrent or refractory ovarian or primary peritoneal carcinoma with overexpression of HER2: a phase II trial of the Gynecologic Oncology Group. Journal of Clinical Oncology 2003;21:283‐90. [DOI] [PubMed] [Google Scholar]
Campos 2005 {published data only}
- Campos S, Hamid O, Seiden MV, Oza A, Plante M, Potkul RK, et al. Multicenter, randomized phase II trial of oral CI‐1033 for previously treated advanced ovarian cancer. Journal of Clinical Oncology 2005;23:5597‐604. [DOI] [PubMed] [Google Scholar]
Campos 2010 {published data only}
Campos 2011 (NCT00520013) {published data only}
- Campos A, Atkinson T, Berlin A. STAC: a phase II study of carboplatin/paclitaxel/bevacizumab followed by randomization to either bevacizumab alone or erlotinib and bevacizumab in the upfront management of patients with ovarian, fallopian tube or peritoneal cancer [Meeting abstract]. Gynecologic Oncology 2011;120(Suppl 1):S79. Abstract 181. [Google Scholar]
- Dana‐Farber Cancer Institute. Avastin and Erlotinib as First Line Consolidation Chemotherapy After Carboplatin, Paclitaxel, and Avastin (CTA) Induction Therapy for Advanced Ovarian, Fallopian Tube, and Primary Peritoneal Cancer, and Papillary Serous Mullerian Tumors. ClinicalTrials.gov Identifier: NCT00520013. Protocol registered at ClinicalTrials.gov on 22 August 2007.
Chambers 2010 {published data only}
- Chambers SK, Clouser MC, Baker AF, Roe DJ, Cui H, Brewer MA, et al. Overexpression of tumor vascular endothelial growth factor A may portend an increased likelihood of progression in a phase II trial of bevacizumab and erlotinib in resistant ovarian cancer. Clinical Cancer Research 2010;16(21):5320‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- University of Arizona, Genentech. Phase II Open‐Label Trial of Erlotinib (Tarceva) and Bevacizumab in Women With Advanced Ovarian Cancer. ClinicalTrials.gov Identifier: NCT00130520. Protocol registered at ClinicalTrials.gov on 12 August 2005.
Ciunci 2014 {published data only}
Dinh 2008 {published data only}
- Dinh P, Harnett P, Piccart‐Gebhart MJ, Awada A. New therapies for ovarian cancer: cytotoxics and molecularly targeted agents. Critical Reviews in Oncology/Hematology 2008;67:103‐12. [DOI] [PubMed] [Google Scholar]
Galsky 2012 {published data only}
Garcia 2012 {published data only}
- Garcia AA, Sill MW, Lankes HA, Godwin AK, Mannel RS, Armstrong DK, et al. A phase II evaluation of lapatinib in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecologic Oncology 2012; Vol. 124, issue 3:569‐74. [DOI] [PMC free article] [PubMed]
- Gynecologic Oncology Group, National Cancer Institute. A Phase II Evaluation of Lapatinib (GW572016) (NCI‐Supplied Agent, NSC #727989) in the Treatment of Persistent or Recurrent Epithelial Ovarian or Primary Peritoneal Carcinoma. ClinicalTrials.gov Identifier: NCT00113373. Protocol registered at ClinicalTrials.gov on 7 June 2005.
Gordon 2005 {published data only}
- Gordon AN, Finkler N, Edwards P, Garcia AA, Crozier M, Irwin DH, et al. Efficacy and safety of erlotinib HCl, an epidermal growth factor receptor (HER1/EGFR) tyrosine kinase inhibitor, in patients with advanced ovarian carcinoma: results from a phase II multicenter study. International Journal of Gynecological Cancer 2005;15:785‐92. [DOI] [PubMed] [Google Scholar]
Gordon 2006 {published data only}
- Genentech Inc. A Phase II, Open‐label, Multicenter Study to Evaluate the Effect of Tumor‐based HER2 Activation on the Efficacy of rhuMAb 2C4 (Pertuzumab) in Subjects With Advanced, Refractory or Recurrent Ovarian Cancer. ClinicalTrials.gov identifier: NCT00058552. Protocol registered at ClinicalTrials.gov on 8 April 2003.
- Gordon MS, Matei D, Aghajanian C, Matulonis UA, Brewer M, Fleming GF, et al. Clinical activity of pertuzumab (rhuMAb 2C4), a HER dimerization inhibitor, in advanced ovarian cancer: potential predictive relationship with tumor HER2 activation status. Journal of Clinical Oncology 2006;24:4324‐32. [DOI] [PubMed] [Google Scholar]
Guastalla 2007 {published data only}
- Guastalla JP, Allouache D, Combe M, Weber B, Cretin J, Cure H, et al. HER2 overexpression and amplification in advanced ovarian cancer (AOC): treatment with trustuzumab ‐ a GINECO study. Journal of Clinical Oncology 2007;25(18 Suppl):5559. [Google Scholar]
Hariprasad 2006 {published data only}
- Hariprasad R, Kumar L, Patnaik R, Gupta A, Kumar S. Maintenance therapy in epithelial ovarian cancer: could EGFR‐inhibitor gefitinib be a candidate drug? A pilot study. Journal of Clinical Oncology 2006;24(18 Suppl):15046. [Google Scholar]
Hariprasad 2009 {published data only}
- Hariprasad R, Kumar L, Kumar S, Bhatla N, Thulkar S, Das P. Advanced epithelial ovarian cancer (EOC): is there a place for maintenance therapy in patients with sub‐optimal debulking?. Journal of Clinical Oncology 2009;27(Suppl):abstr e14580. [Google Scholar]
Harter 2013 {published data only}
- Harter P, Sehouli J, Kimmig R, Rau J, Hilpert F, Kurzeder C, et al. Addition of vandetanib to pegylated liposomal doxorubicin (PLD) in patients with recurrent ovarian cancer. A randomized phase I/II study of the AGO Study Group (AGO‐OVAR 2.13). Investigational New Drugs 2013;31(6):1499‐504. [DOI] [DOI] [PubMed] [Google Scholar]
Hirte 2010 {published data only}
- Hirte H, Oza A, Hoskins P, Ellard S, Grimshaw R, Dubuc‐Lissoir J, et al. Phase II study of OSI‐774 given in combination with carboplatin in patients (pts) with recurrent epithelial ovarian cancer (EOC): NCIC CTG IND.149. European Journal of Cancer 2003;1(5 Suppl: ECCO 12 Abstract Book):A‐159, S51. [Google Scholar]
- Hirte H, Oza A, Swenerton K, Ellard SL, Grimshaw R, Fisher B, et al. A phase II study of erlotinib (OSI‐774) given in combination with carboplatin in patients with recurrent epithelial ovarian cancer. Gynecologic Oncology 2010;118(3):308‐12. [DOI] [PubMed] [Google Scholar]
- NCIC Clinical Trials Group. A Phase II Study of OSI‐774 (NSC 718781) Given in Combination With Carboplatin in Patients With Recurrent Epithelial Ovarian Cancer. ClinicalTrials.gov identifier: NCT00030446. Protocol registered at ClinicalTrials.gov on 14 February 2002.
Jhaveri 2012 {published data only}
Joly 2009 {published data only}
- Jolly F, Weber B, Pautier P, Fabbro M, Selle F, Krieger S. Combined topotecan and lapatinib in patients with early recurrent ovarian or peritoneal cancer after first line of platinum based chemotherapy: a French FEDEGYN‐FNCLCC phase II trial. Journal of Clinical Oncology 2009;27(15S):Abstract 5555. [Google Scholar]
Kimball 2008 {published data only}
- Kimball KJ, Numnum TM, Kirby TO, Zamboni WC, Estes JM, Barnes MN, et al. A phase I study of lapatinib in combination with carboplatin in women with platinum sensitive recurrent ovarian carcinoma. Gynecologic Oncology 2008;111:95‐101. [DOI] [PubMed] [Google Scholar]
Konner 2008 {published data only}
- ImClone LLC, Bristol‐Myers Squibb. A Phase II Study of Cetuximab (C225)/Paclitaxel/Carboplatin for the Initial Treatment of Advanced Stage Ovarian, Primary Peritoneal, and Fallopian Tube Cancer. ClinicalTrials.gov Identifier: NCT00063401. Protocol registered at ClinicalTrials.gov on 25 June 2003.
- Konner J, Schilder RJ, DeRosa FA, Gerst SR, Tew WP, Sabbatini PJ, et al. A phase II study of cetuximab/paclitaxel/carboplatin for the initial treatment of advanced‐stage ovarian, primary peritoneal or fallopian tube cancer. Gynecologic Oncology 2008;110:140‐5. [DOI] [PubMed] [Google Scholar]
Koolen 2011 {published data only}
Krasner 2005 {published data only}
- A Phase II Study of ZD1839 (Iressa) Plus Anastrozole (Arimidex) in Patients With Relapsed Ovarian Cancer. ClinicalTrials.gov Identifier: NCT00181688. Protocol registered at ClinicalTrials.gov on 12 September 2005.
- Krasner CN, Debernardo RL, Findley M, Penson R, Matulonis U, Atkinson T, et al. Phase II trial of anastrozole in combination with gefitinib in women with asymptomatic mullerian cancer. Journal of Clinical Oncology 2005;23:Abstract 5063. [Google Scholar]
Lheureux 2012 {published data only}
NCT00861120 {unpublished data only}
NCT01296035 {unpublished data only}
- McCourt C. A Phase II Evaluation of Panitumumab and Gemcitabine as Treatment for Women With Recurrent Epithelial Ovarian Cancer. ClinicalTrials.gov 2011.
Nimeiri 2008 {published data only}
- National Cancer Institute. A Phase II Trial of Bevacizumab (NSC# 704865) and OSI‐774 (NSC# 718781) in Recurrent Ovarian Cancer, Fallopian Tube Carcinoma or Primary Peritoneal Carcinoma. ClinicalTrials.gov Identifier: NCT00126542. Protocol registered at ClinicalTrials.gov on 2 August 2005.
- Nimeiri HS, Oza AM, Morgan RJ, Friberg G, Kasza K, Faoro L, et al. Efficacy and safety of bevacizumab plus erlotinib for patients with recurrent ovarian, primary peritoneal, and fallopian tube cancer: a trial of the Chicago, PMH, and California Phase II Consortia. Gynecologic Oncology 2008;110(1):49‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Palayekar 2008 {published data only}
- Palayekar MJ, Herzog TJ. The emerging role of epithelial growth factor receptor inhibitors in ovarian cancer. International Journal of Gynecological Cancer 2008;18(5):879‐90. [DOI] [PubMed] [Google Scholar]
Pautier 2010 {published data only}
- Pautier P, Joly F, Kerbrat P, Bougnoux P, Fumoleau P, Petit T, et al. Phase II study of gefitinib in combination with paclitaxel and carboplatin as second‐line therapy for ovarian, tubal or peritoneal adenocarcinoma (1839IL/0074). Gynecologic Oncology 2010;116:157‐62. [DOI] [PubMed] [Google Scholar]
Posadas 2007 {published data only}
- Posadas EM, Liel MS, Kwitkowski K, Minasian L, Godwin K, Hussain MM, et al. A phase II and pharmacodynamic study of gefitinib in patients with refractory or recurrent epithelial ovarian tumors. Cancer 2007;109(7):1323‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ray‐Coquard 2008 {published data only}
- Ray‐Coquard I, Guastalla JP, Allouache D, Combe M, Weber B, Cretin J, et al. HER2 overexpression/amplification and trastuzumab treatment in advanced ovarian cancer: a GINECO phase II study. Clinical Ovarian Cancer 2008;1(1):54‐9. [Google Scholar]
Schilder 2005 {published data only}
- Gynecologic Oncology Group, National Cancer Institute. A Phase II Trial of ZD1839 (Iressa) (NSC# 715055) in the Treatment of Persistent or Recurrent Epithelial Ovarian or Primary Peritoneal Carcinoma. ClinicalTrials.gov Identifier: NCT00023699. Protocol registered at ClinicalTrials.gov on 13 September 2001.
- Schilder RJ, Sill MW, Chen X, Darcy KM, Decesare SL, Lewandowski G, et al. Phase II study of gefitinib in patients with relapsed or persistent ovarian or primary peritoneal carcinoma and evaluation of EGFR mutations and immunohistochemical expression: a gynecologic oncology group study. Clinical Cancer Research 2005;11(15):5539‐48. [DOI] [PubMed] [Google Scholar]
Schilder 2009 {published data only}
- ImClone LLC, Bristol‐Myers Squibb. A Phase II Trial of Single‐agent Cetuximab Dose Escalated to Rash in Patients With Persistent or Recurrent Epithelial Ovarian or Primary Peritoneal Carcinoma. ClinicalTrials.gov Identifier: NCT00082212. Protocol registered at ClinicalTrials.gov on 3 May 2004.
- Schilder RJ, Pathak HB, Lokshin AE, Holloway RW, Alvarez RD, Aghajanian C, et al. Phase II trial of single agent cetuximab in patients with persistent or recurrent epithelial ovarian or primary peritoneal carcinoma with the potential for dose escalation to rash. Gynecologic Oncology 2009;113(1):21‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Secord 2008 {published data only}
- Gynecologic Oncology Group. A Phase II Evaluation of Cetuximab (C225, NSC #714692) in Combination With Carboplatin (NSC #241240) in the Treatment of Recurrent Platinum‐Sensitive Ovarian or Primary Peritoneal Cancer. ClinicalTrials.gov Identifier: NCT00086892. Protocol registered at ClinicalTrials.gov on 8 July 2004.
- Secord AA, Blessing JA, Armstrong DK, Rodgers WH, Miner Z, Barnes MN, et al. A phase II trial of cetuximab and carboplatin in relapsed platinum‐sensitive ovarian cancer and evaluation of epidural growth factor receptor expression: a gynecologic oncology group study. Gynecologic Oncology 2008;108(3):493‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Seiden 2007 {published data only}
- EMD Serono. An Open‐Label Phase II Study in Subjects With Recurrent EGFR‐Positive Ovarian Cancer to Investigate the Safety and Efficacy of EMD 72000 Administered as a Single Agent. ClinicalTrials.gov Identifier: NCT00073541. Protocol registered at ClinicalTrials.gov on 24 November 2003.
- Seiden MV, Burris HA, Matulonis U, Hall JB, Armstrong DK, Speyer J, et al. A phase II trial of EMD72000 (matuzumab), a humanised anti‐EGFR monoclonal antibody, in patients with platinum‐resistant ovarian and primary peritoneal malignancies. Gynecologic Oncology 2007;104(3):727‐31. [DOI] [PubMed] [Google Scholar]
Steffensen 2013 {published data only}
- Steffensen KD, Waldstrøm M, Lund B, Bergfeldt K, Keldsen N, Marth C, et al. Panitumumab and pegylated liposomal doxorubicin to platinum‐resistant epithelial ovarian cancer with KRAS wild‐type: an ongoing, nonrandomized, multicenter, phase II trial. Journal of Clinical Oncology 2010;28(15 Suppl):abstr TPS254. [Google Scholar]
- Steffensen KD, Waldstrøm M, Pallisgard N, Lund B, Bergfeldt K, Wihl J, et al. Panitumumab and pegylated liposomal doxorubicin in platinum‐resistant epithelial ovarian cancer with KRAS wild‐type: the PaLiDo study, a phase II nonrandomized multicenter study. International Journal of Gynecological Cancer 2013;23(1):73‐80. [DOI] [PubMed] [Google Scholar]
Vasey 2008 {published data only}
- Vasey PA, Gore M, Wilson R, Rustin G, Gabra H, Guastalla J‐P, et al. A phase Ib trial of docetaxel, carboplatin and erlotinib in ovarian, fallopian tube and primary peritoneal cancers. British Journal of Cancer 2008;98(11):1774‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Vlahovic 2012 {published data only}
- Vlahovic G, Meadows KL, Uronis HE, Morse MA, Blode GC, Riedel RF, et al. A phase I study of bevacizumab, everolimus and panitumumab in advanced solid tumors. Cancer Chemotherapy & Pharmacology 2012; Vol. 70, issue 1:95‐102. [DOI] [PMC free article] [PubMed]
Wagner 2007 {published data only}
- Wagner U, du Bois A, Pfistrerer J, Houber J, Loibl S, Luck HJ, et al. Gefitinib in combination with tamoxifen in patients with ovarian cancer refractory or resistant to platinum‐taxane based therapy ‐ a phase II trial of the AGO ovarian cancer study group (AGO‐OVAR 2.6). Gynecologic Oncology 2007;105(1):132‐7. [DOI] [PubMed] [Google Scholar]
Weroha 2011 {published data only}
- Weroha SJ, Oberg AL, Ziegler KL, Dakhilm SR, Rowland KM, Hartmann LC, et al. Phase II trial of lapatinib and topotecan (LapTop) in patients with platinum‐refractory/resistant ovarian and primary peritoneal carcinoma. Gynecologic Oncology 2011; Vol. 122, issue 1:116‐20. [DOI] [PMC free article] [PubMed]
Additional references
Bandera 2003
- Bandera CA, Tsui HW, Mok SC, Tsui FW. Expression of cytokines and receptors in normal, immortalized, and malignant ovarian epithelial cell lines. Anticancer Research 2003;23(4):3151‐7. [PubMed] [Google Scholar]
Bartlett 1996
- Bartlett JM, Langdon SP, Simpson BJ, Stewart M, Katsaros D, Sismondi P, et al. The prognostic value of epidermal growth factor receptor mRNA expression in primary ovarian cancer. British Journal of Cancer 1996;73(3):301‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Baselga 2001
- Baselga J, Albanell J, Molina MA, Arribas J. Mechanism of action of trastuzumab and scientific update. Seminars in Oncology 2001;28(5 Suppl 16):4‐11. [DOI] [PubMed] [Google Scholar]
Baselga 2002
- Baselga J, Rischin D, Ranson M, Calvert H, Raymond E, Kieback DG, et al. Phase I safety, pharmacokinetic, and pharmacodynamic trial of ZD1839, a selective oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with five selected solid tumor types. Journal of Clinical Oncology 2002;20(21):4292‐302. [DOI] [PubMed] [Google Scholar]
Bucher 1997
- Bucher HC, Guyatt GH, Griffith LE, Walter SD. The results of direct and indirect treatment comparisons in meta‐analysis of randomized controlled trials. Journal of Clinical Epidemiology 1997;50(6):683‐91. [DOI] [PubMed] [Google Scholar]
Ciardiello 1999
- Ciardiello F, Bianco R, Damiano V, Lorenzo S, Pepe S, Placido S, et al. Antitumor activity of sequential treatment with topotecan and anti‐epidermal growth factor receptor monoclonal antibody C225. Clinical Cancer Research 1999;5(4):909‐16. [PubMed] [Google Scholar]
Ciardiello 2001
- Ciardiello F, Caputo R, Bianco R, Damiano V, Fontanini G, Cuccato S, et al. Inhibition of growth factor production and angiogenesis in human cancer cells by ZD1839 (Iressa), a selective epidermal growth factor receptor tyrosine kinase inhibitor. Clinical Cancer Research 2001;7(5):1459‐65. [PubMed] [Google Scholar]
Cooley 1999
- Cooley S, Burns LJ, Repka T, Miller JS. Natural killer cell cytotoxicity of breast cancer targets is enhanced by two distinct mechanisms of antibody‐dependent cellular cytotoxicity against LFA‐3 and HER2/neu. Experimental Hematology 1999;27(10):1533‐41. [DOI] [PubMed] [Google Scholar]
Covidence
- Covidence systematic review software, Veritas Health Innovation. www.covidence.org. Melbourne, Australia.
CTEP 2017
- Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. https://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf 2017.
Deeks 2001
- Deeks JJ, Altman DG, Bradburn MJ. Statistical methods for examining heterogeneity and combining results from several studies in meta‐analysis. In: Egger M, Davey Smith G, Altman DG editor(s). Systematic Reviews in Health Care: Meta‐Analysis in Context. 2nd Edition. London: BMJ Publication Group, 2001. [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7(3):177‐88. [DOI] [PubMed] [Google Scholar]
Downward 1984
- Downward J, Yarden Y, Mayes E, Scrace G, Totty N, Stockwell P, et al. Close similarity of epidermal growth factor receptor and v‐erb‐B oncogene protein sequences. Nature 1984;307(5951):521‐7. [DOI] [PubMed] [Google Scholar]
Ekstrand 1992
- Ekstrand AJ, Sugawa N, James CD, Collins VP. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N‐ and/or C‐terminal tails. Proceedings of the National Academy of Science USA 1992;89(10):4309‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Erickson 2013
- Erickson BK, Conner MC, Landen CN. The role of the fallopian tube in the origin of ovarian cancer. American Journal of Obstetrics and Gynecology 2013;209(5):409‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Giesinger 2016
- Giesinger JM, Kieffer JM, Fayers PM, Groenvold M, Petersen MA, Scott NW, et al. Replication and validation of higher order models demonstrated that a summary score for the EORTC QLQ‐C30 is robust. Journal of Clinical Epidemiology 2016;69:79‐88. [DOI: 10.1016/j.jclinepi.2015.08.007] [DOI] [PubMed] [Google Scholar]
Glaysher 2013
- Glaysher S, Bolton LM, Johnson P, Atkey N, Dyson M, Torrance C, et al. Targeting EGFR and PI3K pathways in ovarian cancer. British Journal of Cancer 2013;109:1786‐94. [DOI: 10.1038/bjc.2013.529] [DOI] [PMC free article] [PubMed] [Google Scholar]
GLOBOCAN 2012
- Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, et al. GLOBOCAN 2012v1.0, Cancer Incidence and Mortality Worldwide. IARC CancerBase. Lyon: IARCPress, 2013, issue No. 11 [Internet].
Goldstein 1995
- Goldstein NI, Prewett M, Zuklys K, Rockwell P, Mendelsohn J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clinical Cancer Research 1995;1(11):1311‐8. [PubMed] [Google Scholar]
GRADE Working Group
- GRADE Working Group. Grading quality of evidence and strength of recommendations. BMJ 2004;328:1490‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEpro GDT
- GRADEpro GDT: GRADEpro Guideline Development Tool [Software]. Hamilton (ON): McMaster University, 2015. Developed by Evidence Prime, Inc.
Guyatt 2011
- Guyatt GH, Oxman AD, Montori M, Vist G, Kunz R, Brozek J, et al. GRADE guidelines: 5. Rating the quality of evidence ‐ publication bias. Journal of Clinical Epidemiology 2011;64(12):1277‐82. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jemal 2008
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics. Cancer Journal for Clinicians 2008;58(2):71‐96. [DOI] [PubMed] [Google Scholar]
Jensen 2011
- Jensen SE, Rosenbloom SK, Beaumont JL, Abernethy A, Jacobsen PB, Syrjala K, et al. A new index of priority symptoms in advanced ovarian cancer. Gynecologic Oncology 2011;120(2):214‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kehoe 2015
- Kehoe S, Hook J, Nankivell M, Jayson GC, Kitchener H, Lopes T, et al. Primary chemotherapy versus primary surgery for newly diagnosed advanced ovarian cancer (CHORUS): an open‐label, randomised, controlled, non‐inferiority trial. The Lancet 2015;386(9990):249‐57. [DOI] [PubMed] [Google Scholar]
Morrison 2012
- Morrison J, Haldar K, Kehoe S, Lawrie TA. Chemotherapy versus surgery for initial treatment in advanced ovarian epithelial cancer. Cochrane Database of Systematic Reviews 2012, Issue 8. [DOI: 10.1002/14651858.CD005343] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moscatello 1995
- Moscatello DK, Holgado‐Madruga M, Godwin AK, Ramirez G, Gunn G, Zoltick PW, et al. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Research 1995;55(23):5536‐9. [PubMed] [Google Scholar]
Moulder 2001
- Moulder SL, Yakes FM, Muthuswamy SK, Bianco R, Simpson JF, Arteaga CL. Epidermal growth factor receptor (HER1) tyrosine kinase inhibitor ZD1839 (Iressa) inhibits HER2/neu (erbB2)‐over expressing breast cancer cells in vitro and in vivo. Cancer Research 2001;61(24):8887‐95. [PubMed] [Google Scholar]
NCT00263822
- NCT00263822. A Randomized, Multicenter, Phase III Study of Erlotinib versus Observation in Patients With No Evidence of Disease Progression After First Line, Platinum‐Based Chemotherapy for High‐Risk Ovarian Epithelial, Primary Peritoneal, or Fallopian Tube Cancer. MetaRegister of Controlled Trials 2005.
NCT00520013
- NCT00520013. A Randomized Phase II Trial of Avastin (A) or Avastin and Erlotinib (AE) as First Line Consolidation Chemotherapy After Carboplatin, Paclitaxel and Avastin (CTA) Induction Therapy for Newly Diagnosed Advanced Ovarian, Fallopian Tube and Primary Peritoneal Cancer & Papillary Serous Mullerian Tumors. MetaRegister of Controlled Trials 2007.
Nicholson 2001
- Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. European Journal of Cancer 2001;37(Suppl 4):S9‐15. [DOI] [PubMed] [Google Scholar]
Okamoto 2018
- Okamoto I, Morita S, Tashiro N, Imamura F, Inoue A, Seto T, et al. Real world treatment and outcomes in EGFR mutation‐positive non‐small cell lung cancer: long‐term follow‐up of a large patient cohort. Lung Cancer 2018;117:14‐9. [DOI: 10.1016/j.lungcan.2018.01.005] [DOI] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815‐34. [DOI] [PubMed] [Google Scholar]
Quinn 2001
- Quinn M, Babb B, Brock A, Jones J. In: Office for National Statistics, editor(s). Cancer Trends in England and Wales. London: The Stationery Office, 2001. [Google Scholar]
Schünemann 2014
- Schünemann H, Brożek J, Guyatt G, Oxman A, editors. GRADE Handbook for Grading Quality of Evidence and Strength of Recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from guidelinedevelopment.org/handbook.
Shepherd 1989
- Shepherd JH. Revised FIGO staging for gynaecological cancer. British Journal of Obstetrics and Gynaecology 1989;96(8):889‐92. [DOI] [PubMed] [Google Scholar]
Slamon 1989
- Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER‐2/neu proto‐oncogene in human breast and ovarian cancer. Science 1989;244(4905):707‐12. [DOI] [PubMed] [Google Scholar]
Stewart 1999
- Stewart L, Advanced Ovarian Cancer Trialists Group. Chemotherapy for advanced ovarian cancer. Cochrane Database of Systematic Reviews 1999, Issue 1. [DOI: 10.1002/14651858.CD001418] [DOI] [PubMed] [Google Scholar]
Teplinsky 2015
- Teplinsky E, Muggia F. EGFR and HER2: is there a role in ovarian cancer?. Translational Cancer Research 2015;4(1):107‐17. [Google Scholar]
Vergote 2010
- Vergote I, Tropé CG, Amant F, Kristensen GB, Sardi JE, Ehlen T, et al. Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. New England Journal of Medicine 2010;363(10):943‐53. [DOI] [PubMed] [Google Scholar]
Wedge 2002
- Wedge SR, Ogilvie DJ, Dukes M, Kendrew J, Chester R, Jackson JA, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Research 2002;62(16):4645‐55. [PubMed] [Google Scholar]
Wilken 2012
- Wilken JA, Badri T, Cross S, Raji R, Santin AD, Branscum J, et al. EGFR/HER‐targeted therapeutics in ovarian cancer. Future Medicinal Chemistry 2012; Vol. 4, issue 4:447‐69. [DOI] [PMC free article] [PubMed]
References to other published versions of this review
Gaitskell 2009
- Gaitskell K, Martinek I, Nicum S, Kehoe S, Morrison J. Epidermal growth factor receptor blockers for the treatment of ovarian cancer. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD007927] [DOI] [PMC free article] [PubMed] [Google Scholar]
Haldar 2011
- Haldar K, Gaitskell K, Bryant A, Nicum S, Kehoe S, Morrison J. Epidermal growth factor receptor blockers for the treatment of ovarian cancer. Cochrane Database of Systematic Reviews 2011, Issue 10. [DOI: 10.1002/14651858.CD007927.pub3] [DOI] [PubMed] [Google Scholar]