

Abstract
Background
Phosphatidylethanols (PEths) are specific, direct alcohol biomarkers that can be determined in human blood to distinguish between heavy and social drinking. PEth 16:0/18:1 is among the most predominant PEth homologues in human blood. The aim of the study was to develop a high throughput and sensitive UHPLC‐MS/MS method for the determination of PEth 16:0/18:1 in whole blood.
Methods
Whole blood samples were prepared by 96‐well supported liquid extraction (SLE). Extracted samples were analyzed for PEth 16:0/18:1 by reversed phase UHPLC‐MS/MS.
Results
The developed UHPLC‐MS/MS method was fully validated in whole blood with PEth 16:0/18:1‐D5 as internal standard. Intermediate precision and intermediate accuracy were within ≤± 12% and ≤± 17%, respectively, at PEth 16:0/18:1 concentrations of 1.4‐2112 ng/mL (2.0‐3004 nmol/L). Limit of quantification (LOQ) was 1.7 ng/mL (2.4 nmol/L).
Conclusion
For the first time, 96‐well SLE was used for preparation of a PEth homologue in biological samples. A mixture of tert‐butyl methyl ether and 2‐propanol (5:1, v:v) was chosen as organic eluent based on an evaluation of extraction recovery, purity of extracts, and evaporation time. The developed UHPLC‐MS/MS method can be used for high throughput analyses and sensitive determinations of PEth 16:0/18:1 in whole blood.
Keywords: 96‐well supported liquid extraction, Alcohol, LC‐MS/MS, PEth 16:0/18:1, phosphatidylethanol
1. INTRODUCTION
Alcohol use is ranked as one of the top five highest risk factors for disease, injury, and death, and alcohol abuse over time can aggravate and accelerate the development of many diseases and lead to complications.1 In addition, use of alcohol is associated with social and economic costs of society.1 Better assessment of the patients` alcohol use will have great impact on both clinical practice and epidemiological studies related to alcohol use. Phosphatidylethanols (PEths) are non‐oxidative direct alcohol biomarkers, with a substantially longer half‐life than ethanol, which can be determined in human blood to distinguish between heavy and social drinking.2, 3, 4, 5 PEths have a common phosphoethanol “head” linked to two fatty acid chains of variable lengths and degree of saturation. More than 40 PEths have been detected in blood from heavy drinkers,6 and among the most predominant ones in human blood are PEth 16:0/18:1, PEth 16:0/18:2, and PEth 18:1/18:1.7, 8, 9, 10, 11 The distribution of different PEths in human blood shows inter‐ and intra‐individual variations, and alcohol habits and other factors such as a person`s diet and diseases might influence the distribution of PEth homologues.12, 13 Nalesso et al10 identified 17 different PEths in blood from alcohol‐dependent subjects, whereas only PEth 16:0/18:1 and PEth 16:0/18:2 were found in blood from social drinkers. The average half‐life of PEth is approximately 3‐5 days for heavy drinkers and 10‐12 days for persons with no or low alcohol consumption.14, 15 A blood concentration of PEth ≥211 ng/mL (300 nmol/L) indicates alcohol abuse, whereas a PEth concentrations ≤21 ng/mL (≤30 nmol/L) indicate low alcohol consumption.14
LC‐MS/MS is commonly used for PEth analysis due to high sensitivity and due to the possibility to distinguish between the different PEth homologues.6, 8, 13, 16 A review published in 2016 by Oppolzer et al16 shows that most LC‐MS/MS methods used for the determination of PEths in blood employ sample preparation by liquid–liquid extraction (LLE) using 2‐propanol and hexane as organic solvents. Aradottir and Olsson17 reported PEth recovery to be 25% higher when 2‐propanol was added before hexane compared to when a mixture of the two compounds was added, probably due to the phospholipids from the cell membranes becoming more available after the lipid‐protein linkages are broken by adding 2‐propanol. Protein precipitation (PPT) and solid phase extraction (SPE) have also been used for sample preparation of PEths from biological samples.10, 18, 19 Recently, Andreassen et al20 described a high throughput UHPLC‐MS/MS method for determination of PEth 16:0/18:1 in whole blood using 96‐well PPT with 2‐propanol as organic solvent. Supported liquid extraction (SLE) is another technique that that is fast and easy which can be used for isolation of different compounds from biological samples, and a technique that often generates cleaner extracts than what is obtained by PPT.21, 22, 23, 24, 25, 26, 27 By 96‐well SLE, the sample preparation time and the need for manual interventions can be significantly reduced compared to single vial LLE, SPE, SLE, and PPT.28, 29, 30, 31 For laboratories doing large volume PEth analyses, a method combining a high throughput sample preparation by 96‐well SLE with a sensitive and selective determination by UHPLC‐MS/MS can be of great interest.
To the best of our knowledge, we present the first UHPLC‐MS/MS method for the determination of PEth in whole blood using SLE for sample preparation. Different organic mixtures of MTBE/2‐propanol and of heptane/ethyl acetate were investigated for the 96‐well SLE to find the best compromise between getting a high recovery for PEth 16:0/18:1, clean extracts and short evaporation time. Recently, Nguyen et al32 report a high recovery of PEth prepared from red blood cell stroma by LLE using MTBE as the organic solvent in their UHPLC‐MS/MS method. For our 96‐well SLE, we had to include 2‐propanol in addition to MTBE to get a satisfactory recovery for PEth 16:0/18:1. Figure 1 shows the molecular structure of PEth 16:0/18:1 and the internal standard PEth 16:0/18:1‐D5.
Figure 1.
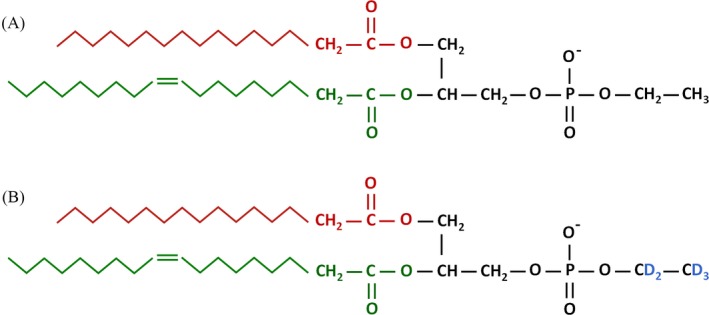
Molecular structure of PEth 16:0/18:1 (A) and PEth 16:0/18:1‐D5 (B)
2. MATERIALS AND METHODS
2.1. Chemicals and materials
PEth 16:0/18:1, 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphoethanol, used for calibrators and control samples (QCs), was purchased from Avanti Polar Lipids (Alabaster, AL, USA). The internal standard, PEth 16:0/18:1‐D5, was purchased from Chiron AS (Trondheim, Norway). Methanol (MeOH) of LC‐MS grade was acquired from Honeywell (Seelze, Germany), and acetonitrile (ACN) of HPLC Far UV grade was purchased from JT. Baker (Deventer, the Netherlands). MTBE, 2‐propanol, ethyl acetate and n‐heptane were purchased from Merck (Darmstadt, Germany). Formic acid (98%) was acquired from VWR International AS (Oslo, Norway). Type 1 water (18.2 MΩ) purified with a Synthesis A 10 milli‐Q system from Millipore (Billerica, MA, USA) was used. Human whole blood was obtained from the Blood bank at Oslo University Hospital Ullevaal (Oslo, Norway).
2.2. Whole blood samples
Human whole blood samples from hospital patients with somatic illness participating in a research study were collected to determine PEth 16:0/18:1 concentrations. The samples were received in 5 mL BD Vacutainer®Plus glass blood collection tubes containing 10 mg sodium fluoride and 8 mg potassium oxalate from BD (Franklin Lake, NJ, USA). An amount of 100 μL blood was transferred to 5 mL polypropylene tubes from Sarstedt AG (Rommelsdorf, Germany) and stored at 4°C before analysis. Blank whole blood obtained from non‐alcohol drinkers or PEth negative whole blood samples were used when the method was validated. The blank blood from non‐alcohol drinkers were collected in the same collection tubes as the samples from the hospital patients with somatic illness. An exception was the recovery test, which was investigated using whole blood containing 2 g sodium fluoride, 6 mL heparin and 10 mL water per 450 mL blood obtained from the Blood Bank at Ullevaal Oslo University Hospital (Oslo, Norway).
2.3. Preparation of calibrators, quality control samples, and internal standards
Stock solutions of PEth 16:0/18:1 and the internal standard PEth 16:0/18:1‐D5 were prepared in 2‐propanol/ACN (1:1, v:v). Calibrator and QC working solutions were prepared in 2‐propanol/ACN (1:1, v:v) by appropriate dilution of the stock solutions. Internal standard working solution containing PEth 16:0/18:1‐D5 was prepared in 2‐propanol/ACN (1:1, v:v) at a concentration of 1129 ng/mL (1606 nmol/L).
2.4. Sample preparation
Calibrator and QC samples were prepared by first adding 100 μL whole blood and 100 μL Type 1 water to plastic tubes; then, 25 μL of the calibrator or QC sample working solutions was added. The unknown blood samples (100 μL blood) were added 100 μL Type 1 water and 25 μL 2‐propanol/ACN (1:1, v:v). Thereafter, 25 μL internal standard working solution was added to all the tubes (calibrators, QC samples, blank samples and blood samples) and vortexed. The samples were then transferred to an Isolute 96‐well SLE+ plate with 400 μL bed volumes from Biotage (Uppsala, Sweden) using an 8‐channel pipette with adjustable spacer from Mettler‐Toledo (Oakland, CA, USA). When necessary, vacuum was applied for approximately 10‐15 seconds to adsorb the sample to the sorbent. Subsequently, 500 μL 2‐propanol/MTBE (1:5, v:v) was added to the wells in the 96‐SLE plate. The elution step was performed twice and vacuum was applied for complete elution of the organic eluent into a 96‐well collection plate. The samples were evaporated to dryness with a N2 pressure of 45 psi at 45°C within approximately 15 minutes using a TurboVap 96‐well plate evaporator from Biotage (Charlotte, NC, USA). Evaporation time was approximately 15 minutes. The residues were reconstituted in 100 μL 2‐propanol/ACN (1:1, v:v). Finally the 96‐well collection plate was sealed with a self‐closing rapid slit seal from BioChromato (Fujisawa‐shi, Japan), vortexed and then placed in the sample organizer for UHPLC–MS/MS analysis.
2.5. Instrumental analysis
UHPLC‐MS/MS analyses were performed on a 1290 Infinity UHPLC and a 6490 triple quadrupole MS, both from Agilent Technologies (Waldbronn, Germany) with an Agilent Jet Stream Electrospray Ionization (ESI) interface in negative mode. Chromatographic separations were performed on a HSS T3 C18‐column (100 mm × 2.1 mm i.d., 1.8 μm particles) from Waters (Milford, MA, USA) at a column temperature of 65°C and a mobile phase flow rate of 0.6 mL/min. Mobile phase was a mixture of 5 mmol/L ammonium formate buffer (pH 5)/ACN (5:1, v:v) as solvent A, and MeOH/ACN (1:5, v:v) as solvent B. The gradient profile was: 0.0‐2.5 minutes, 80%‐100% B; 2.5‐4.7 minutes, 100% B; 4.7‐4.8 minutes, 100%‐80% B; 4.8‐5.5 minutes, 80% B. The injection volume was 5.0 μL. Negative ESI‐MS/MS detection was performed in multiple reaction monitoring (MRM) mode with nitrogen as collision gas. The ESI‐MS/MS detections were performed with a sheath gas heater temperature of 200°C, a sheath gas flow of 11 (L/h), a gas temperature of 200°C, a gas flow of 14 L/min, and a capillary voltage of 3 kV. Data processing was performed using Agilent MassHunter Quantitative Analysis software (version B.07.00 from Agilent Technologies, Santa Clara, CA, USA). Table 1 shows the analyte and internal standard MRM transitions, fragmentation ion voltages, collision energies, and dwell times.
Table 1.
Multiple reaction monitoring (MRM) transitions, fragmentation ion voltages, collision energies, and dwell times
| Time window | MRM transition | MS/MS parameters | |||
|---|---|---|---|---|---|
| Peth 16:0/18:1 | Peth 16:0/18:1‐D5 | Fragmentation voltage (V) | Collision energy (eV) | Dwell times (ms) | |
| 1.5‐3.5 | 701.5 > 281.2a | 706.5 > 281.2a | 380 | 40 | 200 |
| 1.5‐3.5 | 701.5 > 255.2 | 706.5 > 255.2 | 380 | 45 | 200 |
Quantifier MRM transition.
2.6. Method validation
The method validation was performed using whole blood as matrix and included: calibration curves, inter‐assay precision and accuracy, limit of detection (LOD), limit of quantification (LOQ), recovery, matrix effects (ME), carry‐over, and stability. There are many guidelines and recommendations for validation of analytical methods that are published.33, 34, 35, 36, 37, 38, 39 The validation of the method in this study is based on internal guidelines used at Oslo University Hospital for bioanalytical LC‐MS/MS methods, which are partly based on previously published papers by Magnusson and U. Örnemark, by Rivier and by Peters et al34, 35, 38 Whole blood used for calibrators and QC samples was obtained from person's not drinking alcohol and from PEth negative whole blood samples.
3. RESULTS AND DISCUSSION
The aim of the present study was to develop and validate a rapid, sensitive, accurate, precise, and robust UHPLC‐MS/MS method for determination of PEth 16:0/18:1 in whole blood.
3.1. Optimization of sample preparation
It can be difficult to obtain clean extracts when PEths are prepared from whole blood as PEths are phospholipids and therefore difficult to isolate from other phospholipids. At first, we tested PPT with 2‐propanol and then LLE using an organic solvent mixture of hexane/2‐propanol, heptane/2‐propanol, or MTBE/2‐propanol, all mixtures containing 40% of 2‐propanol. The highest PEth 16:0/18:1 responses were observed with LLE using MTBE/2‐propanol as organic solvent (data not shown). To increase sample throughput, 96‐well SLE was investigated using different MTBE/2‐propanol mixtures. In addition, mixtures of ethyl acetate/heptane were examined, as ethyl acetate/heptane (5:1, v:v) has been successfully used in our laboratory to extract many other compounds from whole blood in forensic toxicology analyses.23, 40, 41 Table 2 shows the recovery of PEth 16:0/18:1 obtained in these experiments.
Table 2.
Recovery of PEth 16:0/18:1 using different elution solvents for 96‐well SLE
| PEth 16:0/18:1 concentration | n | Elution solvent(s) | Recoverya | |||||
|---|---|---|---|---|---|---|---|---|
| (nmol/L) | ng/mL | MTBE (%) | 2‐propanol (%) | Ethylacetate (%) | Heptane (%) | Recovery (%) | RSD (%) | |
| 100 | 70 | 4 | 100 | 0 | 0 | 0 | 3 | 10 |
| 100 | 70 | 4 | 90 | 10 | 0 | 0 | 24 | 10 |
| 100 | 70 | 4 | 80 | 20 | 0 | 0 | 49 | 2 |
| 100 | 70 | 4 | 70 | 30 | 0 | 0 | 58 | 8 |
| 100 | 70 | 4 | 60 | 40 | 0 | 0 | 62 | 7 |
| 100 | 70 | 4 | 0 | 0 | 100 | 0 | 13 | 10 |
| 100 | 70 | 4 | 0 | 0 | 90 | 10 | 10 | 14 |
| 100 | 70 | 4 | 0 | 0 | 80 | 20 | 5 | 13 |
| 100 | 70 | 4 | 0 | 0 | 70 | 30 | 3 | 12 |
| 100 | 70 | 4 | 0 | 0 | 60 | 40 | 2 | 39 |
Recovery for each set was determined as: (Average calculated concentration of samples added PEth before extraction/Average calculated concentration of samples added PEth 16:0/18:1 after extraction) × 100. Internal standard was added after extraction in all samples.
Mixtures of MTBE/2‐propanol gave the highest PEth 16:0/18:1 recoveries. A mixture of MTBE/2‐propanol with 20% 2‐propanol was chosen for the validated method, due to a relatively high PEth 16:0/18:1 recovery, and because the extracts were cleaner (visually observed) and the evaporation time was shorter compared to when 30% and 40% 2‐propanol was used in the mixture.
3.2. UHPLC‐MS/MS analysis
Phosphatidylethanols are lipophilic compounds that normally have a high retention in reversed phase LC‐MS/MS systems, unless the amount of organic solvent in the mobile phase is kept high. The retention of PEths can also be reduced using ACN and/or 2‐propanol as organic solvent instead of using methanol. Preliminary UHPLC‐MS/MS analyses were performed by testing different mobile phase compositions on either an Acquity BEH C18 column (50 mm × 2.1 mm i.d., 1.7 μm particles) or on an Acquity HSS T3 column (100 mm × 2.1 mm i.d., 1.8 μm particles). A mobile phase consisting of ammonium formate (pH 5)/ACN (5:1, v:v) and an organic phase consisting of methanol/ACN (1:5, v:v) was chosen, as this combination gave the highest peak responses and signal/noise (S/N) values of the tested conditions (Figure 2).
Figure 2.
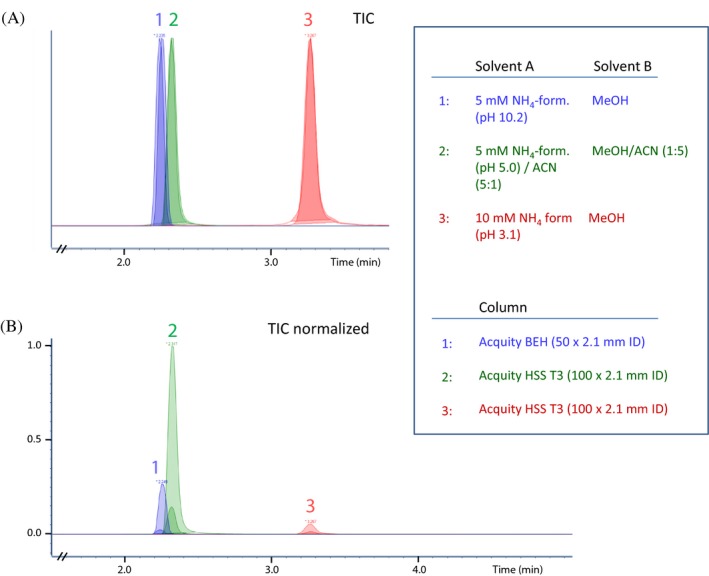
Total ion chromatograms (TICs), not normalized (A), and normalized (B), obtained by UHPLC‐MS/MS analysis of standard samples containing PEth 16:0/18:1 and the internal standard. Gradient profile used was the same as for the validated method. Injection volume was 2 μL. Each of the three numbered sets contains two peaks (overlaid). The two peaks represent injections of two standard samples that had two different concentrations
3.3. Matrix matching of calibrators and QC samples
The scientific working group for forensic toxicology (SWGTOX)42 recommends that calibrators and QC samples are matrix matched. In this study, whole blood was used as matrix for calibrators and QC samples when the method was validated. However, as PEth‐free blood can be difficult to obtain, part of the method was also validated using Type 1 water as surrogate matrix for calibrators and QC samples (see Table S1). Hewavitharana have previously shown that a co‐eluting isotope labeled internal standard can eliminate the need for matrix matching.43 The recent review published in 2016 by Thakare et al36 shows there are a lot of different solvents that are used as surrogate matrixes. In an experiment comparing the use of blank blood and Type 1 water as matrix for standard samples, we observed that peak response ratios of analyte/internal standard were independent of the matrix (Table 3). This indicates that matrix matching may not be necessary. However, if samples studied have a higher concentration than the upper LOQ, dilution of the samples before analysis is required. In such cases, matrix matching is usually necessary as dilution effects may not be tracked by the internal standard that is added after the dilution. As shown in Table 3, also the influence of addition order (whole blood, Type 1 water, analyte working solution, and internal standard working solution) was tested. The addition order had great impact on the PEth peak responses, but peak response ratio (analyte/internal standard) were not affected (Table 3). The low peak responses for the blood samples added working solutions (2‐propanol/ACN, 1:1, v,v) before Type 1 water can be explained by clogging/precipitation of the blood.
Table 3.
Influence of addition order during sample preparation and a comparison between using whole blood as matrix and Type 1 water as surrogate matrix for standard samples
| PEth 16:0/18:1 conc. (nmol/L) | n | Calc. conc. (nmol/L) | Accuracy (%‐deviation) | Average int. std. response | Matrix | Order of addition during sample preparationa | |||
|---|---|---|---|---|---|---|---|---|---|
| Added first | Added second | Added third | Added fourth | ||||||
| 20 | 4 | 21 | 6 | 994680 | Whole blood | Whole blood | Type 1 water | QC work. sol. | Int. std.b |
| 50 | 4 | 53 | 6 | 983021 | Whole blood | Whole blood | Type 1 water | QC work. sol. | Int. std.b |
| 500 | 4 | 536 | 7 | 901836 | Whole blood | Whole blood | Type 1 water | QC work. sol. | Int. std.b |
| 20 | 4 | 20 | −2 | 914475 | Whole blood | Whole blood | QC work. sol. | Type 1 water | Int. std. |
| 50 | 4 | 48 | −4 | 890457 | Whole blood | Whole blood | QC work. sol. | Type 1 water | Int. std. |
| 500 | 4 | 520 | 4 | 830122 | Whole blood | Whole blood | QC work. sol. | Type 1 water | Int. std. |
| 20 | 4 | 20 | 1 | 80314 | Whole blood | Whole blood | QC work. sol. | Int. std. | Type 1 waterc |
| 50 | 4 | 51 | 2 | 76349 | Whole blood | Whole blood | QC work. sol. | Int. std. | Type 1 waterc |
| 500 | 4 | 512 | 2 | 94785 | Whole blood | Whole blood | QC work. sol. | Int. std. | Type 1 waterc |
| 20 | 4 | 29 | 43 | 386701 | Whole blood | QC work. sol. | Int. std. | Whole blood | Type 1 waterc |
| 50 | 4 | 61 | 23 | 346303 | Whole blood | QC work. sol. | Int. std. | Whole blood | Type 1 waterc |
| 500 | 4 | 535 | 7 | 374464 | Whole blood | QC work. sol. | Int. std. | Whole blood | Type 1 waterc |
| 20 | 4 | 17 | −14 | 48816 | Whole blood | QC work. sol. | Whole blood | Int. std. | Type 1 waterc |
| 50 | 4 | 46 | −8 | 43860 | Whole blood | QC work. sol. | Whole blood | Int. std. | Type 1 waterc |
| 500 | 4 | 500 | 0 | 29220 | Whole blood | QC work. sol. | Whole blood | Int. std. | Type 1 waterc |
| 20 | 4 | 22 | 12 | 961027 | Whole blood | QC work. sol. | Type 1 water | Whole blood | Int. std. |
| 50 | 4 | 56 | 12 | 921780 | Whole blood | QC work. sol. | Type 1 water | Whole blood | Int. std. |
| 500 | 4 | 567 | 13 | 740526 | Whole blood | QC work. sol. | Type 1 water | Whole blood | Int. std. |
| 20 | 4 | 18 | −10 | 1517918 | Type 1 water | Type 1 water | QC work. sol. | Int. std. | — |
| 50 | 4 | 55 | 10 | 1308136 | Type 1 water | Type 1 water | QC work. sol. | Int. std. | — |
| 500 | 4 | 497 | −1 | 1303207 | Type 1 water | Type 1 water | QC work. sol. | Int. std. | — |
Cal. conc., calculated concentration; int. std., internal standard; work. sol., working solution.
Standard sample working solution and internal standard working solution were both prepared in 2‐propanol/ACN (1:1). All samples were vortexed immediately after the addition of internal standard, but not after the addition of the other solvents. Volume of standard sample working solution and internal standard working solution added to each sample was25 μL of each solvent.
Same addition order as for validated method.
Clogging/precipitation was observed during sample preparation.
3.4. Method validation
Method validation of intermediate precision and accuracy, LOD, LOQ, recovery, matrix effects, and stability were investigated using human blood as matrix. In addition, intermediate precision, accuracy, LOD, LOQ were also investigated using Type 1 water as surrogate matrix (Table S1). To be able to correct for variations in sample preparation recovery and/or ion suppression/enhancement effects during LC‐MS/MS analysis, it is important to include stable isotope labeled internal standards in bioanalytical methods.44, 45, 46, 47 PEth 16:0/18:1‐D5 was used as internal standard in this study.
3.4.1. Calibration curves
Seven calibrators with one replicate for each level and with PEth 16:0/18:1 concentrations within 3.5‐2114 ng/mL (5.0‐3007 nmol/L) were used for the calibration curves. Quadratic or linear calibration curves (weighted 1/x) were constructed by plotting the ratio of the peak height of PEth 16:0/18:1 against the peak height of the isotope labeled internal standard. The average coefficient of determination (R 2) value for the eight assays used to determine precision and accuracy was 0.998. The relative standard deviation (RSD) for the R 2 values was 0.25%. In each assay, %‐deviation from theoretical value for the calibrators were <± 20% and the ion ratio for the qualifier MRM transition for both analyte and internal standard was <± 20% from the expected ion ratio value.
3.4.2. Precision and accuracy
Intermediate precision and accuracy were determined from the calculated concentrations of QC samples from eight different assays prepared and analyzed within a time period of two weeks. QC samples with eight different concentrations with one replicate for each QC sample were included in each assay. Accuracy (bias) was determined by calculating the deviation in percent between the mean of the measured PEth 16:0/18:1 concentration in the QC samples and the theoretical PEth 16:0/18:1 concentrations. The inter‐assay precision was determined as the RSD values obtained at each of the eight concentration levels. The inter‐assay precision and accuracy data are provided in Table 4.
Table 4.
Inter‐assay accuracy and precision, recovery, and matrix effects
| PEth 16:0/18:1 concentration | Accuracy and precision (n = 8)a | Recovery (n = 4)b | Matrix effectsc | |||||
|---|---|---|---|---|---|---|---|---|
| (nmol/L) | (ng/mL) | Accuracy (bias %) | Precision (% RSD) | Recovery (%) | RSD (%) | ME | RSD (%) | ME corrected |
| 1.0 | 0.7 | 40 | 23 | |||||
| 2.0 | 1.4 | 17 | 8 | |||||
| 5.0 | 3.5 | 7 | 8 | 90 | 9 | 103 | ||
| 20 | 14 | 11 | 6 | |||||
| 25 | 18 | 90 | 11 | 103 | ||||
| 50 | 35 | 6 | 5 | |||||
| 100 | 70 | 49 | 2 | |||||
| 250 | 176 | 85 | 12 | 99 | ||||
| 505 | 355 | −10 | 6 | |||||
| 1004 | 706 | 3 | 6 | |||||
| 3004 | 2112 | 12 | 12 | |||||
Determination based on extracted whole blood QC samples with eight different concentrations, analyzed on eight series.
Determined in two different human blood samples with two parallels of each blood sample at one concentration.
Determined in four different human blood samples with two parallels of each blood sample at three different Peth 16:0/18:1 concentrations. PEth 16:0/18:1 concentrations are the theoretical concentrations in autosampler vial.
3.4.3. LOD and LOQ
Limit of detection and LOQ were determined from Equations 1 and 2, respectively, and based on data from the same QC samples that were analyzed to determine intermediate accuracy and precision. To avoid unrealistic low values, the standard deviation of a QC sample with low analyte concentration was used instead of using the standard deviation of a blank sample for the calculations, as described by Armbruster et al.48, 49 Additional requests for LOD and LOQ were that the S/N values of both MRM transitions needed to be ≥3 and ≥10, respectively, and that for LOQ the inter‐assay precision and accuracy values (Table 4) both were ≤20%.
| (1) |
| (2) |
The LOD and LOQ values determined were as follows: 0.8 ng/mL (1.2 nmol/L) and 1.7 ng/mL (2.4 nmol/L), respectively. In Figure 3A, the MRM chromatogram of a QC sample with a PEth 16:0/18:1 concentration of 1.4 ng/mL (2 nmol/L) is presented. In Figure 3B, the MRM chromatogram of an extracted blank whole blood sample is shown. The minor PEth 16:0/18:1 peaks seen in Figure 3B (not integrated) at heights corresponding to approximately 1/10 of LOQ are probably caused by to the presence of a little PEth in the blank whole blood.
Figure 3.
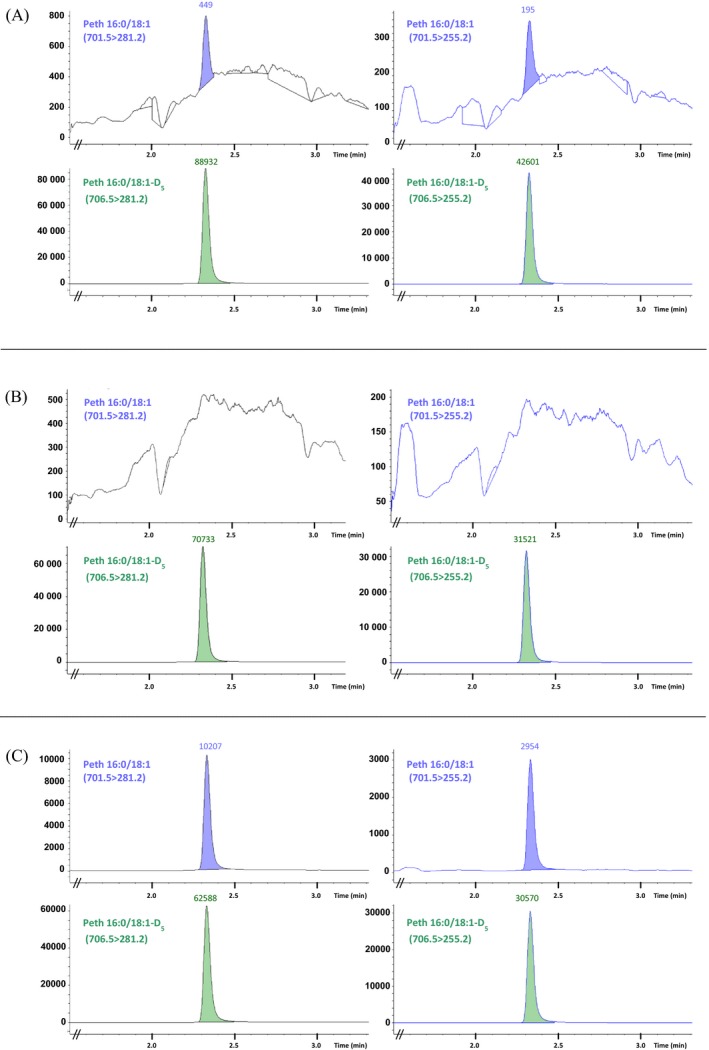
Multiple reaction monitoring chromatograms of PEth 16:0/18:1 and PEth 16:0/18:1‐D5 of an extracted standard samples (whole blood) with PEth 16:0/18:1 concentrations 1.4 ng/mL (2 nmol/L) (A), an extracted blank whole blood sample (B), and an extracted authentic sample (whole blood) with a calculated PEth 16:0/18:1 concentration of 47 ng/mL (67 nmol/L) (C)
3.4.4. Matrix effects
ME were investigated at three concentration levels according to the procedure described by Matuszewski et al.50 Two sets of samples were analyzed at each concentration level. In set 1 (n = 8), human blood samples from four different sources were extracted, evaporated, and added 100 μL reconstitution solvent. The extract (90 μL) was then transferred to autosampler vials and spiked with PEth 16:0/18:1 and internal standard. In set 2 (n = 8), empty autosampler vials were added 90 μL reconstitution solvent and then spiked with PEth 16:0/18:1 and internal standard. Sample solvent composition and total volume of the samples in set 1 and set 2 were the same. All samples were analyzed by the developed UHPLC‐MS/MS method. ME were calculated by Equation 3.
| (3) |
ME = 100 indicates that there were no ME, whereas ME >100 indicates possible matrix enhancement and ME <100 indicates possible matrix suppression. Minor ion suppression effects were observed; however, the co‐eluting isotope labeled internal standard corrected for these effects (Table 4).
3.4.5. Recovery
Recovery was studied by spiking PEth 16:0/18:1 to human blood samples before sample preparation (n = 4 per level) and after sample preparation (n = 4 per level). In both cases, the internal standard was added after sample preparation. A mixture of 2‐propanol/ACN (1:1, v:v, 25 or 50 μL) was added to the samples before the 96‐well SLE to be sure that the sample composition (blood, organic solvents, and Type 1 water) was the same as for ordinary samples extracted by the validated method. Recovery was calculated as mean calculated concentration of samples added analyte prior to preparation, compared to mean calculated concentrations of samples added analyte after sample preparation. The recovery of PEth 16:0/18:1 was determined to be approximately 50% (Table 4). A recovery of 50% was considered enough as the internal standard PEth 16:0/18:1 is expected to correct for recovery variations and possible ion suppression/enhancement effects.
3.4.6. Carry‐over
Carry‐over was investigated by injecting an extracted standard sample with a PEth 16:0/18:1 concentration of 2114 ng/mL (3007 nmol/L) and then three subsequent injections of extracted blank samples. Average carry‐over was 0.03% in the first blank samples (n = 4) and 0.01% in the second blank samples (n = 4) and 0.00% in the third blank samples (n = 4). The same average carry‐over values were obtained for calculations based on peak heights as for calculations based on calculated concentrations. To minimize possible carry‐over, two blank samples have been analyzed after the highest calibrator and two blank samples have been were analyzed after the QC sample with the highest concentration. If an authentic sample with low PEth concentrations is analyzed behind a sample with a high PEth concentration, the sample should be reanalyzed to avoid/minimize possible carry‐over.
3.4.7. Stability
In previous stability studies, Helander and Zheng, and Aradottir and Olsson have found PEth to be stable in blood samples stored at 4°C for at least 5 days and for three weeks, respectively,8, 17 whereas Faller et al51 found PEth to be unstable (concentrations deviated >± 20%) with decreasing concentrations after just a few days storage at 4°C. The developed method is and will be used to determine PEth 16:0/18:1 in whole blood samples stored at 4°C before analysis. Table 5 shows that PEth 16:0/18:1 blood concentrations were quite stable with calculated concentrations from the blood samples stored 36 and 71 days being ≤±30% of the calculated concentrations obtained in the same blood samples analyzed the day of arrival. In addition to stability in whole blood, the stability of PEth 16:0/18:1 in extracted samples placed in autosampler at 10°C was investigated by reanalyzing samples that had been stored in autosampler for 8 days. This test was performed for the same five samples that were analyzed regarding the comparison of concentrations with reference laboratories (Section 3.4.5), and showed that the calculated concentrations obtained after 8 days storage in 96‐well collection plate were ≤ ± 10% of the calculated concentrations obtained the first time the samples were analyzed. An alternative technique that can increase stability of compounds during storage and transportation is dried blood spot (DBS) analysis.52 DBS is also a technique that can offer other advantages, such as less invasive blood sample collection and relatively simple sample preparation. In several of the LC‐MS/MS methods developed to determine PEth in whole blood DBS has been used.53, 54, 55, 56
Table 5.
Stability of PEth 16:0/18:1 in whole blood stored at 4°C and comparison of calculated concentrations obtained for different laboratories
| Sample ID | Stability of PEth 16:0/18:1 in whole blood stored at 4°C | Ethanol (g/kg)b | Comparison of PEth 16:0/18:1 concentrations obtained with concentrations obtained by reference laboratories | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Calculated PEth 16:0/18:1 concentration (stored 0 days) | Stability in whole blood (%) | Sample ID | PEth 16:0/18:1 concentration(ng/mL) | Reference laboratory 1c | Reference laboratory 2 | ||||||
| nmol/L | ng/mL | 36 daysa | 71 daysa | ng/mL | %‐diff.d | ng/mL | %‐diff.d | ||||
| 1 | 1.5 | 1.1 | 93 | 128 | 0.0 | a | 47 | 55 | 18 | 56 | 21 |
| 2 | 3.3 | 2.3 | 97 | 92 | 0.0 | b | 175 | 210 | 20 | 232 | 33 |
| 3 | 5.0 | 3.5 | 106 | 109 | 0.0 | c | 283 | 360 | 27 | 302 | 7 |
| 4 | 6.2 | 4.4 | 105 | 106 | 0.0 | d | 1090 | 1430 | 31 | 1118 | 3 |
| 5 | 11 | 7.5 | 123 | 116 | 0.0 | e | 3395 | 4260 | 25 | 3866 | 14 |
| 6 | 17 | 12 | 98 | 109 | 0.0 | ||||||
| 7 | 22 | 16 | 104 | 90 | 0.0 | ||||||
| 8 | 36 | 25 | 102 | 103 | 0.0 | ||||||
| 9 | 42 | 30 | 108 | 113 | 0.0 | ||||||
| 10 | 52 | 36 | 104 | 96 | 0.0 | ||||||
| 11 | 73 | 51 | 98 | 96 | 0.0 | ||||||
| 12 | 110 | 77 | 104 | 114 | 0.0 | ||||||
| 13 | 124 | 87 | 95 | 92 | 0.0 | ||||||
| 14 | 230 | 162 | 106 | 112 | 0.0 | ||||||
| 15 | 279 | 196 | 130 | 128 | 0.7 | ||||||
| 16 | 430 | 302 | 98 | 96 | 0.0 | ||||||
| 17 | 598 | 420 | 99 | 100 | 0.0 | ||||||
| 18 | 641 | 451 | 96 | 91 | 0.0 | ||||||
| 19 | 765 | 538 | 96 | 94 | 0.2 | ||||||
| 20 | 915 | 643 | 88 | 87 | 0.9 | ||||||
| 21 | 1267 | 891 | 104 | 97 | 0.0 | ||||||
Diff., difference.
Stability was calculated as: Calculated concentration of blood sample stored (36 or 71 days)/calculated concentration of blood sample analyzed the day of arrival at our laboratory. In this test calibrators were prepared in Type 1 water (for all three analyses) and not in whole blood.
Ethanol concentrations were determined by immunological screening as described by Kristoffersen et al.45
PEth 16:0/18:1 concentrations determined by UHPLC‐MS/MS on a Luna Omega 1.6 μm Polar C18 column (30 × 2.1 mm ID) with a sample preparation with PPT using 2‐propanol.
%‐difference for Peth 16:0/18:1 concentrations between reference laboratory and the concentrations obtained by the developed UHPLC‐MS/MS method.
3.4.8. Comparison with a reference laboratory
For comparison with reference laboratories, five unknown blood samples were analyzed in our laboratory by the developed method and by two other laboratories that were using other sample preparation procedures and a different setup for their LC‐MS/MS analyses. Calculated concentrations obtained by reference laboratory 1 and 2 were within 18%‐31% and 3%‐33%, respectively, compared to the concentrations obtained in our laboratory by the developed method (Table 5).
3.4.9. Application of the method
The developed method will be used in a research project to determine PEth 16:0/18:1 in more than 2500 human whole blood samples from hospital patients with somatic illness.
4. CONCLUSIONS
A rapid and sensitive UHPLC‐MS/MS method for determination of PEth 16:0/18:1 in whole blood was developed and fully validated. For the first time, 96‐well SLE was used for the sample preparation of PEth in whole blood.. Based on evaluation of recovery, purity of extracts and evaporation time, a mixture of MTBE/2‐propanol (5:1, v:v) was chosen as organic eluent for the 96‐well SLE. The method was validated using whole blood as matrix. However, we found peak response ratios (analyte/internal standard) to be the same in standard samples prepared in Type 1 water as the ratios for standard samples prepared in whole blood, indicating that matrix matching may not be necessary. The method can be used for high throughput analysis and sensitive determinations of PEth 16:0/18:1 in whole blood, which can be important in clinical practice and epidemiological studies related to alcohol use.
Supporting information
ACKNOWLEDGMENTS
The authors acknowledge Sigurd Hermansson for valuable help during development of the UHPLC conditions and Eline Skadeberg for valuable help during validation of the method. The authors acknowledge Siri Føreid, Ole Martin Fuskevåg, and Christian Walter Thorstensen for great co‐operation regarding comparison of PEth 16:0/18:1 concentrations obtained in reference laboratories. The authors also acknowledge Inger Lise Bogen for valuable comments and critical reading of the manuscript.
Berg T, Eliassen E, Jørgenrud B, Kabashi S, Petukhov A, Bogstrand ST. Determination of phosphatidylethanol 16:0/18:1 in whole blood by 96‐well supported liquid extraction and UHPLC‐MS/MS. J Clin Lab Anal. 2019;33:e22631 10.1002/jcla.22631
REFERENCES
- 1. World Health Organization . Global Status Report on Alcohol and Health. Geneva: World Health Organization; 2014. [Google Scholar]
- 2. Jones AW. Evidence‐based survey of the elimination rates of ethanol from blood with applications in forensic casework. Forensic Sci Int. 2010;200(1‐3):1‐20. [DOI] [PubMed] [Google Scholar]
- 3. Alling C, Gustavsson L, Anggard E. An abnormal phospholipid in rat organs after ethanol treatment. FEBS Lett. 1983;152(1):24‐28. [DOI] [PubMed] [Google Scholar]
- 4. Alling C, Gustavsson L, Mansson JE, Benthin G, Anggard E. Phosphatidylethanol formation in rat organs after ethanol treatment. Biochem Biophys Acta. 1984;793(1):119‐122. [DOI] [PubMed] [Google Scholar]
- 5. Gustavsson L, Alling C. Formation of phosphatidylethanol in rat brain by phospholipase D. Biochem Biophys Res Comm. 1987;142(3):958‐963. [DOI] [PubMed] [Google Scholar]
- 6. Gnann H, Engelmann C, Skopp G, et al. Identification of 48 homologues of phosphatidylethanol in blood by LC‐ESI‐MS/MS. Anal Bioanal Chem. 2010;396(7):2415‐2423. [DOI] [PubMed] [Google Scholar]
- 7. Gunnarsson T, Karlsson A, Hansson P, Johnson G, Alling C, Odham G. Determination of phosphatidylethanol in blood from alcoholic males using high‐performance liquid chromatography and evaporative light scattering or electrospray mass spectrometric detection. Journal of Chromatography B, Biomedical Sciences and Applications. 1998;705(2):243‐249. [DOI] [PubMed] [Google Scholar]
- 8. Helander A, Zheng Y. Molecular species of the alcohol biomarker phosphatidylethanol in human blood measured by LC‐MS. Clin Chem. 2009;55(7):1395‐1405. [DOI] [PubMed] [Google Scholar]
- 9. Holbrook PG, Pannell LK, Murata Y, Daly JW. Molecular species analysis of a product of phospholipase D activation. Phosphatidylethanol is formed from phosphatidylcholine in phorbol ester‐ and bradykinin‐stimulated PC12 cells. The Journal of Biological Chemistry. 1992;267(24):16834‐16840. [PubMed] [Google Scholar]
- 10. Nalesso A, Viel G, Cecchetto G, et al. Quantitative profiling of phosphatidylethanol molecular species in human blood by liquid chromatography high resolution mass spectrometry. J Chromatogr A. 2011;1218(46):8423‐8431. [DOI] [PubMed] [Google Scholar]
- 11. Zheng Y, Beck O, Helander A. Method development for routine liquid chromatography‐mass spectrometry measurement of the alcohol biomarker phosphatidylethanol (PEth) in blood. Clin Chim Acta. 2011;412(15‐16):1428‐1435. [DOI] [PubMed] [Google Scholar]
- 12. Hodson L, Skeaff CM, Fielding BA. Fatty acid composition of adipose tissue and blood in humans and its use as a biomarker of dietary intake. Prog Lipid Res. 2008;47(5):348‐380. [DOI] [PubMed] [Google Scholar]
- 13. Isaksson A, Walther L, Hansson T, Andersson A, Alling C. Phosphatidylethanol in blood (B‐PEth): a marker for alcohol use and abuse. Drug Testing and Analysis. 2011;3(4):195‐200. [DOI] [PubMed] [Google Scholar]
- 14. Aakerøy R, Skråstad RB, Helland A, et al. Nye markører for påvisning av alkoholbruk. Tidsskr Nor Legeforen. 2016;136:1643‐1647. [DOI] [PubMed] [Google Scholar]
- 15. Hahn JA, Anton RF, Javors MA. The formation, elimination, interpretation, and future research needs of phosphatidylethanol for research studies and clinical practice. Alcohol Clin Exp Res. 2016;40(11):2292‐2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oppolzer D, Barroso M, Gallardo E. Bioanalytical procedures and developments in the determination of alcohol biomarkers in biological specimens. Bioanalysis. 2016;8(3):229‐251. [DOI] [PubMed] [Google Scholar]
- 17. Aradottir S, Olsson BL. Methodological modifications on quantification of phosphatidylethanol in blood from humans abusing alcohol, using high‐performance liquid chromatography and evaporative light scattering detection. BMC Biochem. 2005;6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang X, Zheng F, Lin Z, et al. Simultaneous determination of ethanol's four types of non‐oxidative metabolites in human whole blood by liquid chromatography tandem mass spectrometry. Anal Chim Acta. 2017;963:68‐75. [DOI] [PubMed] [Google Scholar]
- 19. Duarte M, Jagadeesan KK, Billing J, Yilmaz E, Laurell T, Ekstrom S. Solid‐phase extraction of the alcohol abuse biomarker phosphatidylethanol using newly synthesized polymeric sorbent materials containing quaternary heterocyclic groups. J Chromatogr A. 2017;1519:1‐8. [DOI] [PubMed] [Google Scholar]
- 20. Andreassen TN, Havnen H, Spigset O, Falch BMH, Skrastad RB. High throughput UPLC(R)‐MSMS method for the analysis of phosphatidylethanol (PEth) 16:0/18:1, a specific biomarker for alcohol consumption, in whole blood. J Anal Toxicol. 2018;42:33‐41. [DOI] [PubMed] [Google Scholar]
- 21. Bylda C, Thiele R, Kobold U, Volmer DA. Recent advances in sample preparation techniques to overcome difficulties encountered during quantitative analysis of small molecules from biofluids using LC‐MS/MS. Analyst. 2014;139(10):2265‐2276. [DOI] [PubMed] [Google Scholar]
- 22. Jiang H, Cao H, Zhang Y, Fast DM. Systematic evaluation of supported liquid extraction in reducing matrix effect and improving extraction efficiency in LC‐MS/MS based bioanalysis for 10 model pharmaceutical compounds. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;891‐892:71‐80. [DOI] [PubMed] [Google Scholar]
- 23. Oiestad EL, Johansen U, Oiestad AM, Christophersen AS. Drug screening of whole blood by ultra‐performance liquid chromatography‐tandem mass spectrometry. J Anal Toxicol. 2011;35(5):280‐293. [DOI] [PubMed] [Google Scholar]
- 24. Sauve EN, Langodegard M, Ekeberg D, Oiestad AM. Determination of benzodiazepines in ante‐mortem and post‐mortem whole blood by solid‐supported liquid‐liquid extraction and UPLC‐MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;883‐884:177‐188. [DOI] [PubMed] [Google Scholar]
- 25. Silvestro L, Savu SR. An update on solid phase‐supported liquid extraction. Bioanalysis. 2015;7(17):2177‐2186. [DOI] [PubMed] [Google Scholar]
- 26. Breiter J, Helger R, Lang H. Evaluation of column extraction: a new procedure for the analysis of drugs in body fluids. For Sci. 1976;7(2):131‐140. [DOI] [PubMed] [Google Scholar]
- 27. Schweizer K, Wick H, Brechbuhler T. An improved method for preparation of samples for the simultaneous assay of some antiepileptic drugs by gas‐liquid chromatography. Clin Chim Acta. 1978;90(3):203‐208. [DOI] [PubMed] [Google Scholar]
- 28. Nguyen L, Zhong WZ, Painter CL, Zhang C, Rahavendran SV, Shen Z. Quantitative analysis of PD 0332991 in xenograft mouse tumor tissue by a 96‐well supported liquid extraction format and liquid chromatography/mass spectrometry. J Pharm Biomed Anal. 2010;53(3):228‐234. [DOI] [PubMed] [Google Scholar]
- 29. Svanstrom C, Hansson GP, Svensson LD, Sennbro CJ. Development and validation of a method using supported liquid extraction for the simultaneous determination of midazolam and 1'‐hydroxy‐midazolam in human plasma by liquid chromatography with tandem mass spectrometry detection. J Pharm Biomed Anal. 2012;58:71‐77. [DOI] [PubMed] [Google Scholar]
- 30. Valen A, Leere Oiestad AM, Strand DH, Skari R, Berg T. Determination of 21 drugs in oral fluid using fully automated supported liquid extraction and UHPLC‐MS/MS. Drug Testing and Analysis. 2017;9(5):808‐823. [DOI] [PubMed] [Google Scholar]
- 31. Zhang Y, Cao H, Jiang H. Supported liquid extraction versus liquid‐liquid extraction for sample preparation in LC‐MS/MS‐based bioanalysis. Bioanalysis. 2013;5(3):285‐288. [DOI] [PubMed] [Google Scholar]
- 32. Nguyen VL, Paull P, Haber PS, Chitty K, Seth D. Evaluation of a novel method for the analysis of alcohol biomarkers: ethyl glucuronide, ethyl sulfate and phosphatidylethanol. Alcohol. 2017;67:7‐13. [DOI] [PubMed] [Google Scholar]
- 33. Cooper GA, Paterson S, Osselton MD. United Kingdom and Ireland Association of Forensic Toxicologists. The United Kingdom and Ireland Association of Forensic Toxicologists Forensic toxicology laboratory guidelines (2010). Sci Justice. 2010;50(4):166‐176. [DOI] [PubMed] [Google Scholar]
- 34. Peters FT, Drummer OH, Musshoff F. Validation of new methods. Forensic Sci Int. 2007;165(2‐3):216‐224. [DOI] [PubMed] [Google Scholar]
- 35. Rivier L. Criteria for the identification of compounds by liquid chromatography‐mas spectrometry and liquid chormatography‐multiple mass spectrometry in forensic toxycology and doping analysis. Anal Chim Acta. 2003;492:69‐82. [Google Scholar]
- 36. Thakare R, Chhonker YS, Gautam N, Alamoudi JA, Alnouti Y. Quantitative analysis of endogenous compounds. J Pharm Biomed Anal. 2016;128:426‐437. [DOI] [PubMed] [Google Scholar]
- 37. Theodorsson E. Validation and verification of measurement methods in clinical chemistry. Bioanalysis. 2012;4(3):305‐320. [DOI] [PubMed] [Google Scholar]
- 38. Magnusson B, Örnemark U. The Fitness for Purpose of Analytical Methods: A Laboratory Guide to Method Validation and Related Topics, 2nd edn http://www.eurachem.org: Eurachem; 2014, 70 p. [Google Scholar]
- 39. Boterman M, Doig M, Breda M, et al. Recommendations on the interpretation of the new European Medicines Agency Guideline on Bioanalytical Method Validation by Global CRO Council for Bioanalysis (GCC). Bioanalysis. 2012;4(6):651‐660. [DOI] [PubMed] [Google Scholar]
- 40. Amundsen I, Oiestad AM, Ekeberg D, Kristoffersen L. Quantitative determination of fifteen basic pharmaceuticals in ante‐ and post‐mortem whole blood by high pH mobile phase reversed phase ultra high performance liquid chromatography‐tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;927:112‐123. [DOI] [PubMed] [Google Scholar]
- 41. Eliassen E, Kristoffersen L. Quantitative determination of zopiclone and zolpidem in whole blood by liquid‐liquid extraction and UHPLC‐MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2014;971:72‐80. [DOI] [PubMed] [Google Scholar]
- 42. Scientific Working Group for Forensic Toxicology . Scientific Working Group for Forensic Toxicology (SWGTOX) standard practices for method validation in forensic toxicology. J Anal Toxicol. 2013;37(7):452‐474. [DOI] [PubMed] [Google Scholar]
- 43. Hewavitharana AK. Matrix matching in liquid chromatography‐mass spectrometry with stable isotope labelled internal standards–is it necessary? J Chromatogr A. 2011;1218(2):359‐361. [DOI] [PubMed] [Google Scholar]
- 44. Berg T, Karlsen M, Oiestad AM, Johansen JE, Liu H, Strand DH. Evaluation of (1)(3)C‐ and (2)H‐labeled internal standards for the determination of amphetamines in biological samples, by reversed‐phase ultra‐high performance liquid chromatography‐tandem mass spectrometry. J Chromatogr A. 2014;1344:83‐90. [DOI] [PubMed] [Google Scholar]
- 45. Berg T, Strand DH. (1)(3)C labelled internal standards–a solution to minimize ion suppression effects in liquid chromatography‐tandem mass spectrometry analyses of drugs in biological samples? J Chromatogr A. 2011;1218(52):9366‐9374. [DOI] [PubMed] [Google Scholar]
- 46. Stokvis E, Rosing H, Beijnen JH. Stable isotopically labeled internal standards in quantitative bioanalysis using liquid chromatography/mass spectrometry: necessity or not? Rapid Commun Mass Spectrom. 2005;19(3):401‐407. [DOI] [PubMed] [Google Scholar]
- 47. Wieling J. LC‐MS‐MS experiences with internal standards. Chromatographia. 2002;55:107‐113. [Google Scholar]
- 48. Armbruster DA, Pry T. Limit of blank, limit of detection and limit of quantitation. Clin Biochem Rev. 2008;29(Suppl 1):S49‐S52. [PMC free article] [PubMed] [Google Scholar]
- 49. Armbruster DA, Tillman MD, Hubbs LM. Limit of detection (LQD)/limit of quantitation (LOQ): comparison of the empirical and the statistical methods exemplified with GC‐MS assays of abused drugs. Clin Chem. 1994;40(7 Pt 1):1233‐1238. [PubMed] [Google Scholar]
- 50. Matuszewski BK, Constanzer ML, Chavez‐Eng CM. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC‐MS/MS. Anal Chem. 2003;75(13):3019‐3030. [DOI] [PubMed] [Google Scholar]
- 51. Faller A, Richter B, Kluge M, Koenig P, Seitz HK, Skopp G. Stability of phosphatidylethanol species in spiked and authentic whole blood and matching dried blood spots. Int J Legal Med. 2013;127(3):603‐610. [DOI] [PubMed] [Google Scholar]
- 52. Jager NG, Rosing H, Schellens JH, Beijnen JH. Procedures and practices for the validation of bioanalytical methods using dried blood spots: a review. Bioanalysis. 2014;6(18):2481‐2514. [DOI] [PubMed] [Google Scholar]
- 53. Bakhireva LN, Leeman L, Savich RD, et al. The validity of phosphatidylethanol in dried blood spots of newborns for the identification of prenatal alcohol exposure. Alcohol Clin Exp Res. 2014;38(4):1078‐1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Beck O, Kenan Moden N, Seferaj S, Lenk G, Helander A. Study of measurement of the alcohol biomarker phosphatidylethanol (PEth) in dried blood spot (DBS) samples and application of a volumetric DBS device. Clin Chim Acta. 2018;479:38‐42. [DOI] [PubMed] [Google Scholar]
- 55. Faller A, Richter B, Kluge M, et al. LC‐MS/MS analysis of phosphatidylethanol in dried blood spots versus conventional blood specimens. Anal Bioanal Chem. 2011;401(4):1163‐1166. [DOI] [PubMed] [Google Scholar]
- 56. Kummer N, Ingels AS, Wille SM, et al. Quantification of phosphatidylethanol 16:0/18:1, 18:1/18:1, and 16:0/16:0 in venous blood and venous and capillary dried blood spots from patients in alcohol withdrawal and control volunteers. Anal Bioanal Chem. 2016;408(3):825‐838. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials