

Abstract
Background
Cytolytic vaginosis (CV) is a common disease that results in pruritus, dyspareunia, and vulvar dysuria. However, the pathological mechanisms of the disease are still unclear. Compared to traditional methods, high‐throughput sequencing can obtain more accurate qualitative and quantitative information on the microbiome.
Methods
We collected 75 samples from 32 healthy women (average age 44 ± 8) and 43 patients with CV (average age 38 ± 8). We used high‐throughput sequencing of the 16S rRNA V3‐V4 region to characterize and compare the vaginal microbiota of patients with CV and healthy women and to identify potential biomarkers for CV.
Results
The vaginal pH of patients with CV was ≤3.8, and the vaginal concentration of H2O2 was ≥2 μmol/L. Colony densities of Lactobacillus spp. in patients with CV ranged from +++ (5‐30) to ++++ (>30) and were significantly higher than those in healthy women. High‐throughput sequencing showed that Lactobacillus was the most prominent genus both in patients with CV and in healthy women, with abundances of 83.8% and 97.2%, respectively (P < 0.001). Lactobacillus crispatus was more abundant in patients with CV, whereas Lactobacillus sp. L‐YJ was more abundant in healthy women, with area under the curve (AUC) values of 0.9375 and 0.8379, respectively.
Conclusion
The abundance of Lactobacillus spp. in CV patients was significantly different from that of healthy patients. Two suitable biomarkers, L. crispatus and Lactobacillus sp. L‐YJ, were identified. These results will be useful for the identification of women at risk of serious illness before they develop obvious symptoms.
Keywords: cytolytic vaginosis, high‐throughput sequencing, vaginal microbiomes
1. INTRODUCTION
Cytolytic vaginosis (CV) has received little attention because it is often confused with aerobic vaginitis (AV),1, 2, 3 bacterial vaginosis (BV),4, 5, 6 vulvovaginal candidiasis (VVC),7, 8, 9 and trichomonas vaginitis (TV).10, 11, 12 Clinical symptoms of CV include pruritus, dyspareunia, vulvar dysuria, and more pronounced cycling during the ovulatory and luteal phases.13, 14, 15, 16 Diagnostic criteria of CV include the absence of Trichomonas spp., Gardnerella vaginalis, Atopobium vaginae, Megasphaera spp., Sneathia spp., and Prevotella spp. and an increase in Lactobacillus spp. Under the influence of estrogen, glycogen is deposited in the vaginal epithelial cells as Lactobacillus converts glucose to lactic acid.17, 18, 19, 20 Large numbers of Lactobacillus spp. also increase hydrogen peroxide (H2O2) and other antibacterial substances and decrease vaginal pH and bacterial diversity.21, 22, 23, 24, 25
High‐throughput sequencing of the region of the bacterial 16S rRNA gene is a powerful tool for assessing and comparing the structure of microbial communities at a high phylogenetic resolution. 16S rRNA sequencing can obtain more accurate qualitative and quantitative information on the microbiome, and it is also Short‐read and cost‐effective, moreover, marker gene analysis is frequently used for broad studies that involve a large number of different samples.26 The goal of this study was to use high‐throughput sequencing to identify biomarkers for CV.
2. MATERIAL AND METHODS
2.1. Specimen collection
Vaginal secretion samples were obtained using aseptic cotton swabs from 43 patients with CV (CV group) and 32 healthy women (CK group). The samples were stored in phosphate‐buffered saline (PBS) at 4°C and were sequenced within 48 hours. Patients were recruited at Women's Hospital School of Medicine Zhejiang University, and the healthy women were recruited at Sir Run Run Shaw Hospital School of Medicine, Zhejiang University, China. Patients were excluded if they used antibiotics. This study was approved by the Institutional Review Board of Sir Run Run Shaw Hospital School of Medicine, Zhejiang University, and Women's Hospital School of Medicine, Zhejiang University. All research was performed in accordance with all relevant guidelines and regulations.
2.2. DNA extractions
DNA was extracted using the E.Z.N.A.® Stool DNA Kit (D4015, Omega Bio‐Tek, Norcross, GA, USA) according to manufacturer's instructions. This kit was designed to recover trace amounts of DNA from samples and has been shown to be effective for the preparation of DNA from most bacteria. Nuclease‐free water was used as the blank. Total DNA was eluted in 50 μL elution buffer and stored at −80°C until PCR assessment by LC‐Bio Technology Co., Ltd (Hangzhou, China).
2.3. PCR amplification and 16S rRNA sequencing
The V3‐V4 region of the 16S rRNA gene was amplified with primers 338F (5′‐ACTCCTACGGGAGGCAGCAG‐3′) and 806R (5′‐GGACTACHVGGGTWTCTAAT‐3′).27 The 5′ ends of the primers were tagged with sample‐specific barcodes and universal sequencing primers. PCR amplification was performed in a 25 μL reaction mixture containing 25 ng of template DNA, 12.5 μL PCR premix, 2.5 μL of each primer, and PCR‐grade water to adjust the volume. The PCR conditions for the amplification of the prokaryotic 16S fragment consisted of an initial denaturation at 98°C for 30 seconds; 35 cycles of denaturation at 98°C for 10 seconds, annealing at 54 or 52°C for 30 seconds, and extension at 72°C for 45 seconds; and a final extension at 72°C for 10 minutes. The PCR products were confirmed by electrophoresis on a 2% agarose gel. Throughout the DNA extraction process, ultrapure water was used in place of template DNA as a negative control to exclude false‐positive PCR results. PCR products were purified using an AxyPrep PCR Cleanup Kit and quantified using Promega QuantiFluor. The pooled amplicons were prepared for sequencing, and the size and quantity of the amplicon library were assessed using an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA) and the Library Quantification Kit for Illumina (Kapa Biosciences, Woburn, MA, USA), respectively. Libraries were sequenced on a 300PE MiSeq.
2.4. Data analysis
Samples were sequenced by LC‐Bio Technology Co., Ltd. Paired‐end reads were assigned to samples based on their unique barcode, and samples were truncated by cutting off the barcode and primer sequences. Paired‐end reads were merged using FLASH (v1.2.8).28 Quality filtering on raw tags was performed using specific filtering conditions to obtain high‐quality clean tags with FastQC. Verseach (v2.3.4)29 was used to filter chimeric sequences and to assign samples with ≥97% sequence similarity to the same operational taxonomic units (OTUs). The representative sequence of each cluster is selected to represent the operational taxonomy units (OTUs).30 Representative sequences were chosen for each OTU, and taxonomic data were assigned to each representative sequence using the Ribosomal Database Project (v11.5)31, 32, 33and NCBI classifier. OTU abundance data were normalized using a standard sequence number corresponding to the sample with the least number of sequences. These indices were calculated for our samples using QIIME (v1.8.0).34 We used R packages to create receiver operating characteristic (ROC)35, 36 curves and other images and conducted functional prediction analyses using PICRUSt.37 We used STAMP (v2.1.3)38, 39for statistical analyses of differences.
3. RESULTS
3.1. OTU Sequence diversity and richness
Characteristics of women from groups CV and CK are shown in Table 1. We obtained 21 665 reads from women in the CK group and 21 798 reads from women in the CV group. Using a cutoff of 97% sequence similarity, we identified 645 OTUs. Because alpha diversity is often used to reflect the diversity of a particular environment or ecosystem based on the richness and uniformity of species and the depth of sequencing, we assessed alpha diversity using seven common indicators40 (Table 1).
Table 1.
Characteristics of the study population
| CK | CV | |
|---|---|---|
| Age (average, median) | 44, 42 | 38, 37 |
| 16S sequences (average, median) | 21 665, 20 650 | 21798, 1629 |
| Observed_species (average ± SD) | 57.31 ± 34.33 | 49.28 ± 26.71 |
| Shannon (average ± SD) | 1.82 ± 1.03 | 1.65 ± 0.5 |
| Simpson (average ± SD) | 0.5 ± 0.22 | 0.51 ± 0.13 |
| Chao1 (average ± SD) | 80.7 ± 37.51 | 79.3 ± 37.63 |
| ACE (average ± SD) | 83.6 ± 38.42 | 82.27 ± 30.69 |
| Goods coverage (average ± SD) | 0.998 ± 0.001 | 0.998 ± 0.001 |
3.2. Taxonomic variation at the phylum, genus, and species levels
Although the vaginal microbiota of all women in the study was characterized by high levels of Firmicutes, their abundance was significantly higher in the CV group (97.53%) than in the CK group (88.85%; P = 0.0054). In contrast, the abundance of Actinobacteria was 4.44% in the CK group and 0.27% in the CV group. Similarly, the abundance of Fusobacteria was 2.28% in the CK group and 0.01% in the CV group (Figure 1. Phylum). Diversity was also higher in the vaginal microbiomes of the CK group, which included Gardnerella (3.19%; P = 0.008), Sneathia (2.26%; P = 0.0001), and Streptophyta (1.44%; P = 0.41) (Fig 1. Genus).
Figure 1.
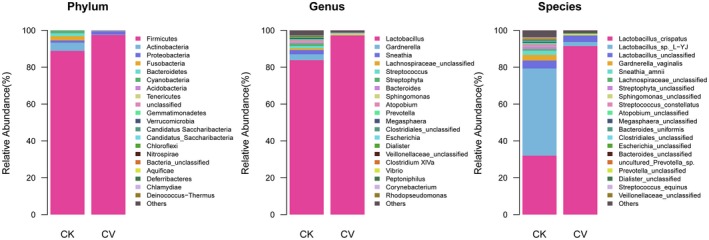
Composition of the vaginal microbiomes of healthy women (CK group) and cytolytic vaginosis (CV) patients (CV group). Relative abundance is shown at the phylum, genus, and species levels
Lactobacillus was the most prominent genus detected in both the CK (83.8%) and CV groups (97.2%; P < 0.001). In addition to the overall difference in the abundance of Lactobacillus between the CK and CV groups, differences were observed in the abundances of specific Lactobacillus species. In the CK group, 60% of Lactobacillus species in the vaginal microbiome were Lactobacillus sp. L‐YJ and 40% were L. crispatus, whereas in the CV group, 97.5% of Lactobacillus species were L. crispatus and 2.5% were Lactobacillus sp. L‐YJ.
Microbial diversity at the species level was also higher in the CK group (Fig 1. Species). Anahtar et al41 demonstrated a strong link between high levels of diversity within cervicovaginal bacterial communities and genital inflammation in vivo in both cross‐sectional and longitudinal studies of young South African women. Using high‐resolution taxonomic identification, they reported several cervicotypes, one of which was characterized by L. crispatus and another by Lactobacillus iners. Virtanen et al sampled 10 Finnish women representing populations with diverse clinical characteristics. Six of these women had Lactobacillus‐dominated microbiota, four had L. iners‐dominated microbiota (community state type III), and two had L. crispatus‐dominated bacteria (community state type I).
3.3. Differential analysis of microbiota using linear discriminant analysis (LDA) effect size (LEfSe)
We investigated differences in the microbiomes of the two groups using linear discriminant analysis (LDA) effect size (LEfSe) with a P value cutoff of 0.05 for Kruskal‐Wallis and pairwise Wilcoxon's tests and a threshold of 4.0 for the logarithmic LDA score. We found that the diversity of the CK group was approximately twice that of the CV group. Differences in the microbial communities of women in the CK and CV groups, including for Lactobacillus sp. L‐YJ and L. crispatus, are shown in Fig 2.
Figure 2.
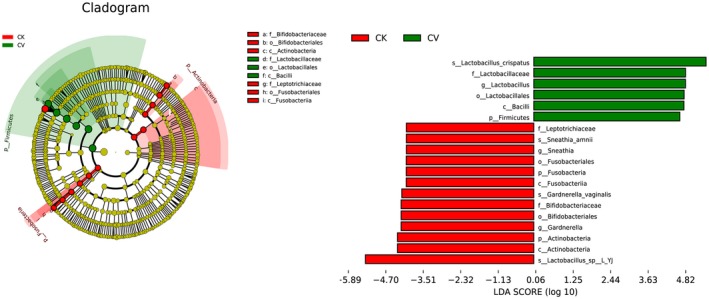
Cladogram and histogram of linear discriminant analysis (LDA) scores computed for differentially abundant genera between cytolytic vaginosis (CV) patients (CV group) and healthy women (CK group). Genera enriched in the CV group are indicated by a positive LDA score, whereas genera enriched in the CK group have a negative score (P < 0.05)
3.4. Suitable biomarkers for CV identified using ROC plots
ROC curves combining sensitivity and specificity with graphic methods and the area under the curve (AUC) were calculated to evaluate the accuracy of diagnoses. Results were considered accurate for AUC values between 0.7 and 0.9 and highly accurate for AUC values >0.9. Using these criteria, we determined that L. crispatus and Lactobacillus sp. L‐YJ could be used as biomarkers to distinguish between the CK and CV groups (Fig 3). Using ROC curve analysis, Ki Ho Hong et al42 assessed the microbiomes of women in South Korea and reported that Lactobacillus spp. had an AUC value of 0.8559, which was slightly higher than our results for Lactobacillus sp. L‐YJ (0.8379) but lower than our results for L. crispatus (0.9375). The reason for the differences between their results and our findings could be related to differences in the vaginal microflora between women from China and South Korea.
Figure 3.
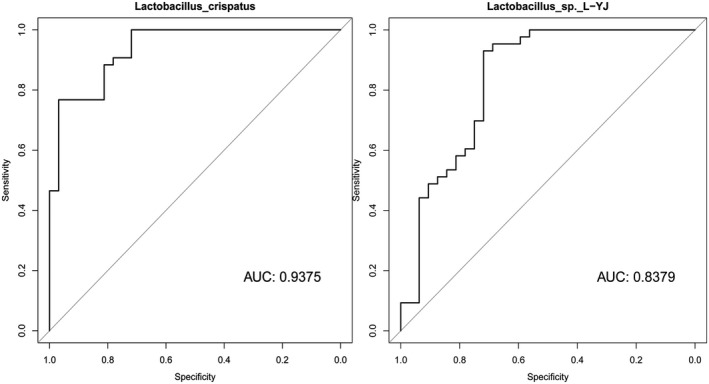
The abundance of two microbial biomarkers (Lactobacilli crispatus and Lactobacillus sp. L‐YJ) differed significantly between cytolytic vaginosis (CV) patients (CV group) and healthy women (CK group). The area under the curve (AUC) values from receiver operating characteristic (ROC) curves were 0.9375 for L. crispatus and 0.8379 for Lactobacillus sp. L‐YJ, respectively
3.5. PICRUSt analysis of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways
Kyoto Encyclopedia of Genes and Genomes (KEGG) orthology identified six level 2 and 3 KEGG categories, including metabolism of cofactors and vitamins, nucleotide metabolism, carbohydrate metabolism, translation, replication and repair, and membrane transport. The most enriched KEGG categories in the CV group were carbohydrate metabolism, which included seven level 3 pathways, and membrane transport, which included two level 3 pathways. In particular, citrate (TCA) cycle, pyruvate metabolism, starch and sucrose metabolism, glycolysis/gluconeogenesis, and phosphotransferase system (PTS) pathways were significantly enriched in the CV group (P < 0.01; Fig 4). These results were supported by PICRUSt analyses and by P values for Welch's t‐tests computed using STAMP.
Figure 4.
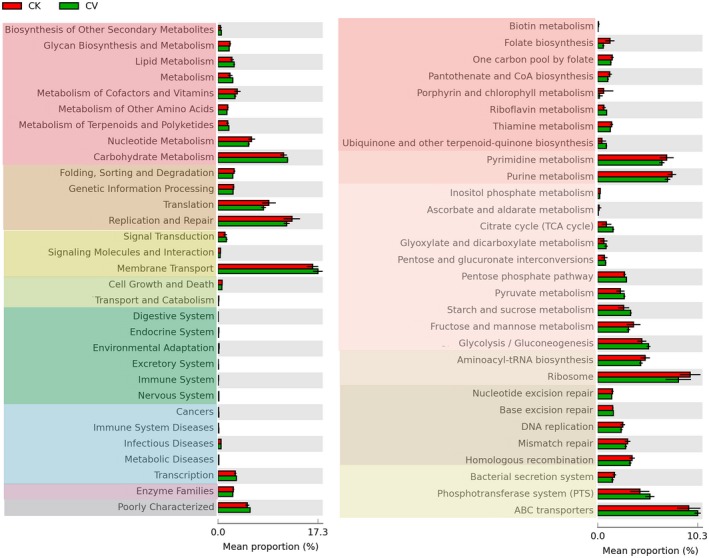
Analysis of differences in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways between cytolytic vaginosis (CV) patients (CV group) and healthy women (CK group) using PICRUSt. KEGG level 2 is shown on the left, and KEGG level 3 is shown on the right
4. DISCUSSIONS
This study aimed at examining the variability between the microbiomes of patients with CV and healthy women to accurately detect CV and avoid inappropriate treatment and provided a comprehensive overview of the vaginal microbiome of patients with CV and healthy women in China using short‐read, high‐throughput sequencing of the V3‐V4 region of the bacterial 16S rRNA gene. In our study, we found that the vaginal microbiomes of patients with CV were significantly less diverse than those of healthy women. Lactobacillus spp. were highly abundant in all women but were more abundant in CV patients than in healthy women (P < 0.05). These results support previous reports that CV is characterized by the abundant growth of Lactobacillus spp., which leads to lysis of vaginal epithelial cells at the molecular level. We also identified two useful biomarkers, L. crispatus and Lactobacillus sp. L‐YJ, to identify CV. Our results indicated that Lactobacillus sp. L‐YJ was the predominant Lactobacillus species in the vagina of healthy women. However, L. crispatus was the major Lactobacillus species in patients with CV. Lactobacillus sp. L‐YJ is a necessary element of the normal vaginal environment of healthy women and may control the overgrowth of other organisms. The overgrowth of organisms can lead to vaginal epithelial tissue damage. Indeed, the abundant growth of L. crispatus appears to be a key factor in the development of CV.
Balanced microbiota in healthy women protects not only against ascending infections or HIV acquisition, but also against prematurity.43, 44, 45 There are more than 200 bacterial species in the normal and the abnormal vaginal microbiota, and the vaginal microbiota is influenced by genes, ethnic background, and environmental and behavioral factors. Only several species of lactobacilli are dominate in healthy vagina. They support a defense system together with antibacterial substances, cytokines, defensins and others against dysbiosis, infections and care for a normal pregnancy without preterm birth. The disturbed vaginal microbiota may cause various vaginal diseases such as bacterial vaginosis (BV), aerobic vaginitis (AV), and CV. BV, which cause by the strains include Atopobium vaginae, Clostridiales and Gardnerella vaginalis develop in different mixtures and mumbers polymicrobial biofilms on the vaginal epithelium, while AV is dominated by aerobic bacteria such as Streptococcus agalactiae and Escherichia coli.46
The vaginal microbiome must be kept in a complicated balance. Internal and external factors that affect this balance and change the composition of the vaginal microbiome may lead to the development of CV. Therefore, assessing the microbiome may help to reduce the incidence of CV.
ETHICAL APPROVAL
This study was approved by the Institutional Review Board of Sir Run Run Shaw Hospital School of Medicine, Zhejiang University, and Women's Hospital School of Medicine, Zhejiang University. All research was performed in accordance with all relevant guidelines and regulations.
INFORMED CONSENT
The human materials used were vaginal swab samples, and all patients provided written informed consent.
Xu H, Zhang X, Yao W, Sun Y, Zhang Y. Characterization of the vaginal microbiome during cytolytic vaginosis using high‐throughput sequencing. J Clin Lab Anal. 2019;33:e22653 10.1002/jcla.22653
Funding information
This study was supported by Zhejiang Provincial Natural Science Foundation of China (Grant No. LY18H040001).
Haihong Xu and Xueying Zhang contributed equally to this work.
REFERENCES
- 1. Vieira‐Baptista P, Lima‐Silva J, Pinto C, et al. Bacterial vaginosis, aerobic vaginitis, vaginal inflammation and major Pap smear abnormalities. Eur J Clin Microbiol Infect Dis. 2016;35(4):657‐664. [DOI] [PubMed] [Google Scholar]
- 2. Geng N, Wu W, Fan A, et al. Analysis of the risk factors for aerobic vaginitis: a case‐control study. Gynecol Obstet Invest. 2015;81:148‐154. [DOI] [PubMed] [Google Scholar]
- 3. Dermendjiev T, Pehlivanov B, Hadjieva K, et al. Epidemiological, clinical and microbiological findings in women with aerobic vaginitis. Akush Ginekol. 2015;54(9):4‐8. [PubMed] [Google Scholar]
- 4. Nasioudis D, Linhares IM, Ledger WJ, et al. Bacterial vaginosis: a critical analysis of current knowledge. BJOG. 2017;124(1):61‐69. [DOI] [PubMed] [Google Scholar]
- 5. Lokken EM, Balkus JE, Kiarie J, et al. Association of recent bacterial vaginosis with acquisition of Mycoplasma genitalium . Am J Epidemiol. 2017;186(2):194‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee S, You HJ, Kwon B, et al. Complete genome sequence of Lactobacillus jensenii strain SNUV360, a probiotic for treatment of bacterial vaginosis isolated from the vagina of a healthy Korean woman. Genome Announc. 2017;5(10):pii:e01757‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Talaei Z, Sheikhbahaei S, Ostadi V, et al. Recurrent vulvovaginal candidiasis: could it be related to cell‐mediated immunity defect in response to Candida antigen? Int J Fertil Steril. 2017;11(3):134‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Seay J, Mandigo M, Hew K, et al. Vaginal infections in Haitian immigrant women living in Miami, Florida. J Health Care Poor Underserved. 2017;28(3):1141‐1150. [DOI] [PubMed] [Google Scholar]
- 9. Nakamura‐Vasconcelos SS, Fiorini A, Zanni PD, et al. Emergence of Candida glabrata in vulvovaginal candidiasis should be attributed to selective pressure or virulence ability? Arch Gynecol Obstet. 2017;296:519‐526. [DOI] [PubMed] [Google Scholar]
- 10. Gatti FA, Ceolan E, Greco FS, et al. The prevalence of trichomoniasis and associated factors among women treated at a university hospital in southern Brazil. PLoS One. 2017;12(3):e0173604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang H, Huang Z, Wu Z, et al. An epidemiological study on vaginitis in 6,150 women of reproductive age in Shanghai. New Microbiol. 2017;40(2):113‐118. [PubMed] [Google Scholar]
- 12. Virtanen S, Kalliala I, Nieminen P, et al. Comparative analysis of vaginal microbiota sampling using 16S rRNA gene analysis. PLoS One. 2017;12(7):e0181477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang S, Zhang Y, Liu Y, et al. Clinical significance and characteristic clinical differences of cytolytic vaginosis in recurrent Vulvovaginitis. Gynecol Obstet Invest. 2017;82(2):137‐143. [DOI] [PubMed] [Google Scholar]
- 14. Suresh A, Rajesh A, Bhat RM, et al. Cytolytic vaginosis: a review. Indian J Sex Transm Dis. 2009;30(1):48‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Paavonen J. Vulvodynia–a complex syndrome of vulvar pain. Acta Obstet Gynecol Scand. 1995;74(4):243‐247. [DOI] [PubMed] [Google Scholar]
- 16. Demirezen S. Cytolytic vaginosis: examination of 2947 vaginal smears. Cent Eur J Public Health. 2003;11(1):23‐24. [PubMed] [Google Scholar]
- 17. Cerikcioglu N, Beksac MS. Cytolytic vaginosis: misdiagnosed as candidal vaginitis. Infect Dis Obstet Gynecol. 2004;12(1):13‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Batashki I, Markova D, Milchev N. Frequency of cytolytic vaginosis–examination of 1152 patients. Akush Ginekol. 2009;48(5):15‐16. [PubMed] [Google Scholar]
- 19. Hills RL. Cytolytic vaginosis and lactobacillosis. Consider these conditions with all vaginosis symptoms. Adv Nurse Pract. 2007;15(2):45‐48. [PubMed] [Google Scholar]
- 20. Hu Z, Zhou W, Mu L, et al. Identification of cytolytic vaginosis versus vulvovaginal candidiasis. J Low Genit Tract Dis. 2015;19(2):152‐155. [DOI] [PubMed] [Google Scholar]
- 21. Mitchell C, Manhart LE, Thomas KK, et al. Effect of sexual activity on vaginal colonization with hydrogen peroxide‐producing lactobacilli and Gardnerella vaginalis . Sex Transm Dis. 2011;38(12):1137‐1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Martin R, Suarez JE. Biosynthesis and degradation of H2O2 by vaginal lactobacilli. Appl Environ Microbiol. 2010;76(2):400‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Agrawal BM, Agrawal S, Rizvi G, et al. Role of non‐H2O2 producing lactobacilli and anaerobes in normal and complicated pregnancy. J Indian Med Assoc. 2002;100(11):652, 654‐655. [PubMed] [Google Scholar]
- 24. Hillier SL, Krohn MA, Klebanoff SJ, et al. The relationship of hydrogen peroxide‐producing lactobacilli to bacterial vaginosis and genital microflora in pregnant women. Obstet Gynecol. 1992;79(3):369‐373. [DOI] [PubMed] [Google Scholar]
- 25. Klebanoff SJ, Hillier SL, Eschenbach DA, et al. Control of the microbial flora of the vagina by H2O2‐generating lactobacilli. J Infect Dis. 1991;164(1):94‐100. [DOI] [PubMed] [Google Scholar]
- 26. Asshauer KP, Wemheuer B, Daniel R, et al. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics. 2015;31(17):2882‐2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fadrosh DW, Ma B, Gajer P, et al. An improved dual‐indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome. 2014;2(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957‐2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rognes T, Flouri T, Nichols B, et al. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Caporaso JG, Lauber CL, Walters WA, et al. Ultra‐high‐throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6(8):1621‐1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cole JR, Wang Q, Cardenas E, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37(Database issue):D141‐D145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Larsen N, Olsen GJ, Maidak BL, et al. The ribosomal database project. Nucleic Acids Res. 1993;21(13):3021‐3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maidak BL, Cole JR, Parker CT Jr, et al. A new version of the RDP (Ribosomal Database Project). Nucleic Acids Res. 1999;27(1):171‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high‐throughput community sequencing data. Nat Methods. 2010;7(5):335‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hand DJ. Evaluating diagnostic tests: the area under the ROC curve and the balance of errors. Stat Med. 2010;29(14):1502‐1510. [DOI] [PubMed] [Google Scholar]
- 36. Pencina MJ, D'Agostino RB Sr, D'Agostino RB Jr, et al. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27(2):157‐172; discussion 207‐112. [DOI] [PubMed] [Google Scholar]
- 37. Langille MG, Zaneveld J, Caporaso JG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31(9):814‐821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kraemer L, Beszteri B, Gabler‐Schwarz S, et al. STAMP: extensions to the STADEN sequence analysis package for high throughput interactive microsatellite marker design. BMC Bioinformatics. 2009;10:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Luck L, Jackson D, Usher K. STAMP: components of observable behaviour that indicate potential for patient violence in emergency departments. J Adv Nurs. 2007;59(1):11‐19. [DOI] [PubMed] [Google Scholar]
- 40. Blaxter M, Mann J, Chapman T, et al. Defining operational taxonomic units using DNA barcode data. Philos Trans R Soc Lond B Biol Sci. 2005;360(1462):1935‐1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Anahtar MN, Byrne EH, Doherty KE, et al. Cervicovaginal bacteria are a major modulator of host inflammatory responses in the female genital tract. Immunity. 2015;42(5):965‐976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hong KH, Hong SK, Cho SI, et al. Analysis of the vaginal microbiome by next‐generation sequencing and evaluation of its performance as a clinical diagnostic tool in vaginitis. Ann Lab Med. 2016;36(5):441‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hoyme UB, Huebner J. Prevention of preterm birth is possible by vaginal pH screening, early diagnosis of bacterial vaginosis or abnormal vaginal flora and treatment. Gynecol Obstet Invest. 2010;70(4):286‐290. [DOI] [PubMed] [Google Scholar]
- 44. Mendling W, Martius J, Hoyme UB. S1‐guideline on bacterial vaginosis in gynecology and obstetrics: long version – AWMF Guideline, registration no. 015/028, July 2013 Langfassung – AWMF‐Register Nr. 015/028, Juli 2013. Geburtshilfe Frauenheilkd. 2014;74(1):51‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lamont RF, Sobel JD, Akins RA, et al. The vaginal microbiome: new information about genital tract flora using molecular based techniques. BJOG. 2011;118(5):533‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mendling W. Vaginal microbiota. Adv Exp Med Biol. 2016;902:83‐93. [DOI] [PubMed] [Google Scholar]