

Summary
The biotrophic maize head smut fungus Sporisorium reilianum is a close relative of the tumour‐inducing maize smut fungus Ustilago maydis with a distinct disease aetiology. Maize infection with S. reilianum occurs at the seedling stage, but spores first form in inflorescences after a long endophytic growth phase. To identify S. reilianum‐specific virulence effectors, we defined two gene sets by genome comparison with U. maydis and with the barley smut fungus Ustilago hordei. We tested virulence function by individual and cluster deletion analysis of 66 genes and by using a sensitive assay for virulence evaluation that considers both disease incidence (number of plants with a particular symptom) and disease severity (number and strength of symptoms displayed on any individual plant). Multiple deletion strains of S. reilianum lacking genes of either of the two sets (sr10057, sr10059, sr10079, sr10703, sr11815, sr14797 and clusters uni5‐1, uni6‐1, A1A2, A1, A2) were affected in virulence on the maize cultivar ‘Gaspe Flint’, but each of the individual gene deletions had only a modest impact on virulence. This indicates that the virulence of S. reilianum is determined by a complex repertoire of different effectors which each contribute incrementally to the aggressiveness of the pathogen.
Keywords: deletion strains, disease rating, effector, gene cluster, smut
Introduction
The ubiquitous head smut disease of maize and sorghum is caused by the biotrophic basidiomycete Sporisorium reilianum. The disease has only recently spread to Europe, with increasing incidences in France (Bernardo et al., 1992) and occurrences in Germany since 1993 (Dutzmann and Duben, 1993). The separation of S. reilianum from other smut fungi, such as Sporisorium scitamineum and Ustilago maydis, followed the separation of maize from sorghum and sugarcane (Munkacsi et al., 2007). Although avirulence (AVR)–resistance (R) gene interactions are common amongst phytopathogenic fungi (Chaudhari et al., 2014; Deller et al., 2011; Dioh et al., 2000 ), and have been described in the barley smut fungus Ustilago hordei (Ali et al., 2014; Linning et al., 2004) and in the S. reilianum–sorghum interaction (Fuyao et al., 2010; Prom et al., 2011), they do not seem to exist in the U. maydis– or S. reilianum–maize interactions (Baumgarten et al., 2007; Lübberstedt et al., 1998, 1999). Instead, existing maize lines seem to vary in their susceptibility towards S. reilianum, which has been used to map numerous quantitative trait loci (QTLs) of S. reilianum resistance in maize (Bernardo et al., 1992; Chen et al., 2008; Lübberstedt et al., 1999). So far, only one maize resistance gene that is effective against S. reilianum has been identified (Zuo et al., 2015).
Sporisorium reilianum is a close relative of the intensively investigated maize smut pathogen, Ustilago maydis (Begerow et al., 2006). Although both species cause disease on maize, they differ in their behaviour during proliferation in planta, and in the site and type of symptom development. Before plant infection, the development of S. reilianum is similar to that of U. maydis. Diploid teliospores undergo meiosis during germination and give rise to sporidia of four different mating types (Schirawski et al., 2005). Sporidia that differ in both a and b mating types are compatible and form conjugation hyphae that fuse at their tips (Schirawski et al., 2005). Hyphal fusion leads to the formation of a filamentous dikaryon (Schirawski et al., 2005) which grows by tip growth on the plant surface. A hyphal swelling (appressorium) denotes the point of plant penetration, and fungal hyphae proliferate to invade the host tissue (Banuett and Herskowitz, 1994; Ghareeb et al., 2011). After entry into the plant, the behaviour of S. reilianum and U. maydis differ. Hyphae of U. maydis locally induce the formation of prominent tumours, in which the fungus heavily proliferates and develops its diploid spores. In contrast, S. reilianum causes only very mild symptoms of chlorosis at the site of penetration (Martinez and Roux, 2002). For most of the plant’s life, the fungus behaves like an endophyte and can be detected in leaves, nodes, stems and ears (Ghareeb et al., 2011; Xu et al., 1999). Only at flowering time do the symptoms become obvious, when the inflorescence is partly or completely replaced by phyllody or sori harbouring masses of fungal spores (Bressman and Barss, 1933; Ghareeb et al., 2011; Stromberg et al., 1984). In spite of the good knowledge concerning S. reilianum disease symptoms, so far the discovery of only one symptom‐affecting effector (Sad1, Ghareeb et al., 2015) and one virulence‐affecting effector (Pit2, Schweizer et al., 2018) has been published.
To identify the virulence genes and factors influencing symptom formation, the genome of S. reilianum has been sequenced and compared with that of U. maydis. This comparison revealed 43 divergence regions of low sequence conservation embedded in otherwise well‐conserved and syntenic genomes (Schirawski et al., 2010). The divergence regions were enriched in genes predicted to encode secreted proteins. Proteins secreted from the fungus have a high probability to interact with the host, and deletion analysis confirmed a role in virulence for nine of the 14 tested low‐similarity regions in U. maydis (Schirawski et al., 2010). It has been suggested that the divergence regions also contribute to the virulence of S. reilianum; however, the contribution of the encoded factors to the virulence of S. reilianum has still to be demonstrated.
The barley smut fungus Ustilago hordei is a close relative of both S. reilianum and U. maydis. However, in terms of symptom development on its host, it is more similar to S. reilianum. After seedling plant infection, U. hordei persists in a symptomless manner in its host for most of the plant's life and causes spore formation exclusively in the inflorescences (Fischer and Holton, 1957). Although the genome of U. hordei has accumulated transposable and repetitive elements, overall synteny to the genomes of U. maydis and S. reilianum is conserved, and loci containing multiple putative effector genes can be identified (Laurie et al., 2012). Ustilago hordei seems to contain species‐specific effectors that do not have obvious counterparts in either U. maydis or S. reilianum (Laurie et al., 2012). On the other hand, a considerable fraction of the putative effectors present in U. maydis or S. reilianum do have homologues in U. hordei, suggesting their presence in the last common ancestor of the three fungi and a functional involvement in basic plant infection processes. Many of the putative effectors that occur in all three species are only weakly conserved and display amino acid identities of below 50% (Laurie et al., 2012), suggesting molecular evolution, possibly guided by adaptation to the specific hosts or tissues colonized by the three fungi.
Inoculation of maize seedlings with a mixture of two compatible S. reilianum strains leads to disease symptoms in the inflorescences of maize. Maize is a special representative of the grass family that develops two types of inflorescence: the tassel and several ears. The tassel develops at the apical growing point of the plant and is a branched inflorescence composed of flowers harbouring male reproductive organs. The ears are composed of flowers carrying female reproductive organs and appear at the apical growing point of side branches. Symptoms of an S. reilianum infection of maize can be observed in both types of inflorescence, and each inflorescence can show different symptoms. Symptoms include the formation of phyllody and sori. Phyllody is caused by the loss of organ and meristem identities of individual flowers (leading to ‘leafy’ ears), and the additional loss of meristem identity (leading to ‘eary’ ears) (Ghareeb et al., 2011). Sori are roundish bodies filled with masses of fungal spores surrounded by a white peridium, which usually replace individual flowers, but occasionally replace complete inflorescences. Sporisorium reilianum disease quantification is complicated by the fact that not all inflorescences of an infected plant, and not all flowers of a colonized inflorescence, display the same symptoms. Some flowers of an inflorescence may display phyllody, whereas others may be replaced by sori. For example, of the four inflorescences of an individual plant, one may be without any symptoms, whereas the remaining three may show various degrees of disease symptoms. In this work, we developed a suitable evaluation system for the quantification of the multitude of disease symptoms of S. reilianum on maize.
In addition, we investigated whether predicted S. reilianum‐specific effector genes contribute to virulence or S. reilianum‐specific symptom development. We defined ‘S. reilianum‐specific’ factors in two different ways: (i) proteins with no counterparts in U. maydis or U. hordei and (ii) proteins present in S. reilianum, U. maydis and U. hordei, but displaying only weak amino acid conservation. To this end, we generated recombinant S. reilianum strains lacking selected candidate genes belonging to either one of the two classes. This analysis identified several genes in each class that incrementally contributed to the virulence of S. reilianum.
Results
Quantification of disease symptoms of S. reilianum on maize
A prerequisite for the identification of virulence factors that only mildly affect the ability of S. reilianum to colonize maize is a sensitive disease quantification system. To quantify the diverse disease symptoms of S. reilianum on maize, we developed an evaluation system that considers symptoms in each inflorescence (healthy, phyllody, spores) by allocating it to one of nine categories (Fig. 1, see Experimental procedures). We then calculated two values for each infection experiment (see Experimental procedures): a disease incidence index [where ‘disease incidence’ correlates with ‘among‐plant disease severity’, as defined by Groth et al. (1976)] and a disease severity index [where ‘disease severity’ correlates with ‘within‐plant disease severity’ (Groth et al., 1976)]. Disease incidence indicates the maximal damage induced by the particular S. reilianum strains. The disease severity gives information on how strongly the average plant is affected by the pathogen. The two measures together allow a robust evaluation of the virulence capacity of S. reilianum.
Figure 1.

Symptom categories of Sporisorium reilianum‐infected maize inflorescences. Disease symptoms were grouped into nine categories according to the severity of the symptoms (from strongest to weakest) on each inflorescence. (A) Tassel with spores. (B) Tassel with phyllody. (C) Ear with >50% kernels filled with spores. (D) Ear with <50% kernels filled with spores. (E) Ear developing >50% leafy kernels. (F) Ear developing <50% leafy kernels. (G) Ear developing elongated kernels. (H) Immature ear, too small to detect disease symptoms. (I) Healthy ear. Scale bar, 0.5 cm. [Color figure can be viewed at wileyonlinelibrary.com]
Prediction of virulence effector candidates by genome comparison
Genome comparison of smut fungi has been proven to be an efficient tool for the prediction of virulence effector genes (Schirawski et al., 2010). We hypothesized that relevant virulence effectors contributing to S. reilianum‐specific symptom development might be among the effector candidates specific to S. reilianum, or among effector candidates that occur in the three sequenced smut fungi S. reilianum, U. maydis and U. hordei, but are only weakly conserved. Weak conservation would indicate an evolutionary adaptation to the species‐specific requirements for host plant colonization or symptom expression, whereas proteins with no counterparts in U. maydis or U. hordei would fulfil S. reilianum‐specific needs.
To identify candidate effectors specific to S. reilianum, we compared the amino acid sequences of the predicted proteins of S. reilianum (Schirawski et al., 2010) with those of U. maydis (Kämper et al., 2006) and U. hordei (Laurie et al., 2012). We identified a set of 320 predicted S. reilianum proteins that did not have any hit in either U. maydis or U. hordei with a SIMAP identity value (Rattei et al., 2010) above 20%. Of these, four proteins were a2 mating type‐specific proteins (Lga2, Rga2, Mfa2.1, Mfa2.3) and 18 encoded transposon or virus‐related proteins (transposase, polymerase polyprotein, reverse transcriptase, ribonuclease), and were disregarded. Of the remaining 298 candidates (set 1), 286 (96%) did not have any functional annotation (hypothetical or conserved hypothetical), and 65 (22%) were predicted to be secreted (TargetP prediction, reliability classes 1–3; Emanuelsson et al., 2007), (Table S1, see Supporting Information). Weakly conserved genes were identified by gene‐for‐gene comparisons between U. maydis and S. reilianum. This led to the identification of 43 divergence regions containing clustered (i.e. neighbouring) weakly conserved genes which, in total, contained 71% of the weakly conserved genes of S. reilianum (Schirawski et al., 2010). Of these divergence regions (set 2), 41 contained S. reilianum genes that also occurred in U. hordei (Tables S2 and S3, see Supporting Information).
As 298 S. reilianum‐specific genes or 41 divergence regions are too large a number to consider for experimental validation of a possible effect on symptom development, we randomly selected a mix of individual genes and genes occurring in clusters. Of the target genes of set 1, we decided to test six randomly selected individual genes predicted to code for secreted proteins and five randomly selected gene clusters. Mutants were generated of the following individual genes: sr10703 (mutant sruni1), sr11815 (sruni2), sr12538 (sruni3), sr13154 (sruni4), sr14797 (sruni5) and sr15769 (sruni6). In addition, mutants in the following five randomly selected gene clusters were generated, containing two (sr13374 and sr13375 [uni5‐1], sr13900 and sr13901 [uni7‐11]), three (sr13675, sr13676 and sr13677 [uni6‐1], sr14791, sr14792 and sr14793 [sruni11‐1]) or five (sr13413, sr13414, sr13415, sr13416 and sr13417 [uni5‐18]) genes. In total, 21 S. reilianum‐specific genes were selected for deletion analysis, 11 of which encoded secreted proteins (Table 1). Of the 41 target regions of set 2, we maximized the number of tested genes by selecting the two largest divergence regions (19‐1 and 6‐10). These regions together contained 29 S. reilianum genes predicted to code for secreted proteins, and 19 genes with homologues in both U. maydis and U. hordei (Tables S2 and S3).
Table 1.
List of Sporisorium reilianum unique genes selected for functional analysis and their virulence phenotype.
| Mutant name | Sr * gene | SP† | Sr* specific | Disease incidence index‡ | P value | Disease severity index‡ | P value | Virulence phenotype |
|---|---|---|---|---|---|---|---|---|
| ∆sruni1 | sr10703 | S | YES | 11.80 | <0.05 | 11.83 | <0.05 | Increased |
| ∆sruni2 | sr11815 | S | YES | 11.89 | <0.05 | 12.39 | <0.05 | Increased |
| ∆sruni3 | sr12538 | S | YES | 11.70 | >0.05 | 10.48 | >0.05 | JS161 |
| ∆sruni4 | sr13154 | S | YES | 7.90 | >0.05 | 7.82 | >0.05 | JS161 |
| ∆sruni5 | sr14797 | S | YES | 7.34 | <0.01 | 6.89 | <0.05 | Reduced |
| ∆sruni6 | sr15769 | S | YES | 9.21 | >0.05 | 9.16 | >0.05 | JS161 |
| ∆uni5‐1 | sr13374 | S | NO | 8.72 | <0.05 | 7.79 | <0.05 | Reduced |
| sr13375 | YES | |||||||
| ∆uni5‐18 | sr13413 | YES | 9.20 | >0.05 | 9.83 | >0.05 | JS161 | |
| sr13414 | YES | |||||||
| sr13415 | S | YES | ||||||
| sr13416 | YES | |||||||
| sr13417 | YES | |||||||
| ∆uni6‐1 | sr13675 | YES | 9.42 | >0.05 | 8.94 | <0.05 | Severity reduced | |
| sr13676 | S | YES | ||||||
| sr13677 | YES | |||||||
| ∆uni7‐11 | sr13900 | S | YES | 9.11 | >0.05 | 9.40 | >0.05 | JS161 |
| sr13901 | S | YES | ||||||
| ∆uni11‐1 | sr14791 | YES | 11.56 | >0.05 | 11.50 | >0.05 | JS161 | |
| sr14792 | YES | |||||||
| sr14793 | YES |
*Sr, S. reilianum.
†TargetP prediction for S. reilianum genes. TargetP prediction was considered if the reliability class was 1, 2 or 3. S, secretory pathway.
Several tested candidates of set 1 of S. reilianum‐specific genes have an impact on virulence
For the generation of gene and gene cluster deletion variants of the target genes of set 1, we used the solopathogenic S. reilianum strain JS161 (Schirawski et al., 2010). This strain is able to penetrate and colonize maize without prior fusion with a mating partner and induces plant developmental changes similar to those induced by wild‐type (WT) infections, except that it rarely forms spores (Schirawski et al., 2010; Schweizer et al., 2018). For each gene or gene cluster deletion, three individual recombinant strains were selected to test for virulence capacity and the ability to form symptoms on the maize cultivar ‘Gaspe Flint’ (Table S4, see Supporting Information). To measure the virulence of S. reilianum on maize, we calculated and statistically analysed the disease incidence and disease severity indices for each mutant (Tables [Link], [Link], [Link] and S9, see Supporting information).
Of the six individual gene deletion mutants, three (Δsruni3, Δsruni4, Δsruni6) did not show significant changes in either disease incidence or disease severity. In contrast, three (Δsruni1, Δsruni2, Δsruni5) resulted in a small (about 20%–30%), but significant, alteration in disease incidence (Tables 1, S5 and S6; Fig. 2). Surprisingly, only one mutant (Δsruni5) showed significantly reduced virulence relative to JS161, whereas two deletion mutants (Δsruni1 and Δsruni2) showed a virulence increase. Disease severity was altered in a similar manner to disease incidence for the three mutants (Tables 1, S5 and S6; Fig. 2A). Of the five analysed cluster deletion mutants, three (Δuni5‐18, Δuni7‐11, Δuni11‐1) did not show significant changes in either disease incidence or disease severity. One (Δuni5‐1) showed reduced (about 10%–20%) disease incidence and disease severity, and one (Δuni6‐1) resulted in reduced (about 10%) disease severity, but no significant change in disease incidence (Tables 1, S5 and S6; Fig. 2). The observed change in virulence was not a consequence of altered growth behaviour or altered stress tolerance (Figs S1 and S2, see Supporting Information). In summary, we tested 21 of 298 S. reilianum‐specific genes by deletion analysis and showed an effect on virulence for five of the 11 deletion mutants analysed. This suggests that set 1 of S. reilianum‐specific genes contains factors that contribute to the virulence of S. reilianum on maize.
Figure 2.
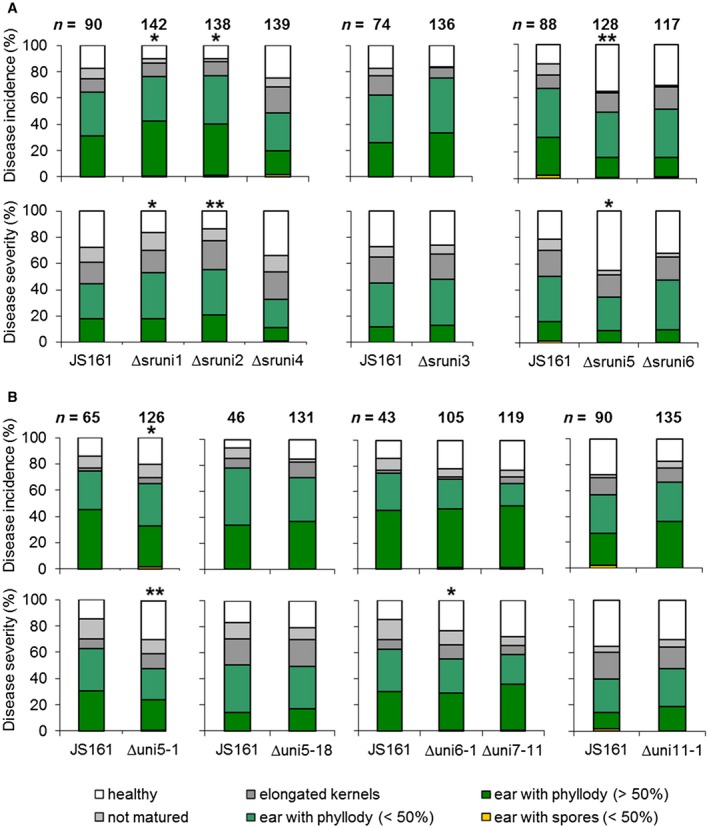
Virulence phenotypes on maize cultivar ‘Gaspe Flint’ of Sporisorium reilianum strains lacking S. reilianum‐specific genes. Disease incidence (top) and disease severity (bottom) of S. reilianum‐specific gene (A) and gene cluster (B) deletion mutants. Data represent the means obtained from three independent mutant strains and three experimental repeats. Asterisks indicate significant differences of disease severity and disease incidence scores calculated from the presented data relative to JS161 (*P < 0.05; **P < 0.01, Student’s t‐test). n, total number of evaluated plants. [Color figure can be viewed at wileyonlinelibrary.com]
The largest divergence cluster 19‐1 of S. reilianum contains multiple virulence effectors
To assess whether set 2 of weakly conserved S. reilianum‐specific genes encoded virulence effectors, we decided to create S. reilianum strains lacking the genes of divergence clusters 6‐10 or 19‐1, as defined by Schirawski et al., (2010). Divergence cluster 6‐10 contains 15 genes, 13 of which are predicted to encode secreted proteins. Six of the genes have weakly conserved homologues in both U. maydis and U. hordei (Tables S2 and S3). Divergence cluster 19‐1 is even larger and encodes at least 30 proteins, 16 of which are predicted to carry a signal sequence for secretion (Fig. 3A; Tables S2 and S3). Nine of the genes have weakly conserved homologues in both U. maydis and U. hordei (Tables S2 and S3). The cluster is split into two parts by the presence of a highly conserved gene (sr10071) encoding a predicted protein with strong sequence identity to the tubulin β‐chains of different fungi. The first part (A1) contains 26 annotated genes, whereas the second part (A2) harbours four genes (Fig. 3A). The delineation of parts A1 and A2 does not correspond to the delineation of the described U. maydis clusters 19A1 and 19A2, which comprise the first half (14 genes) and second half (11 genes), respectively, of cluster 19A of U. maydis (Brefort et al., 2014).
Figure 3.
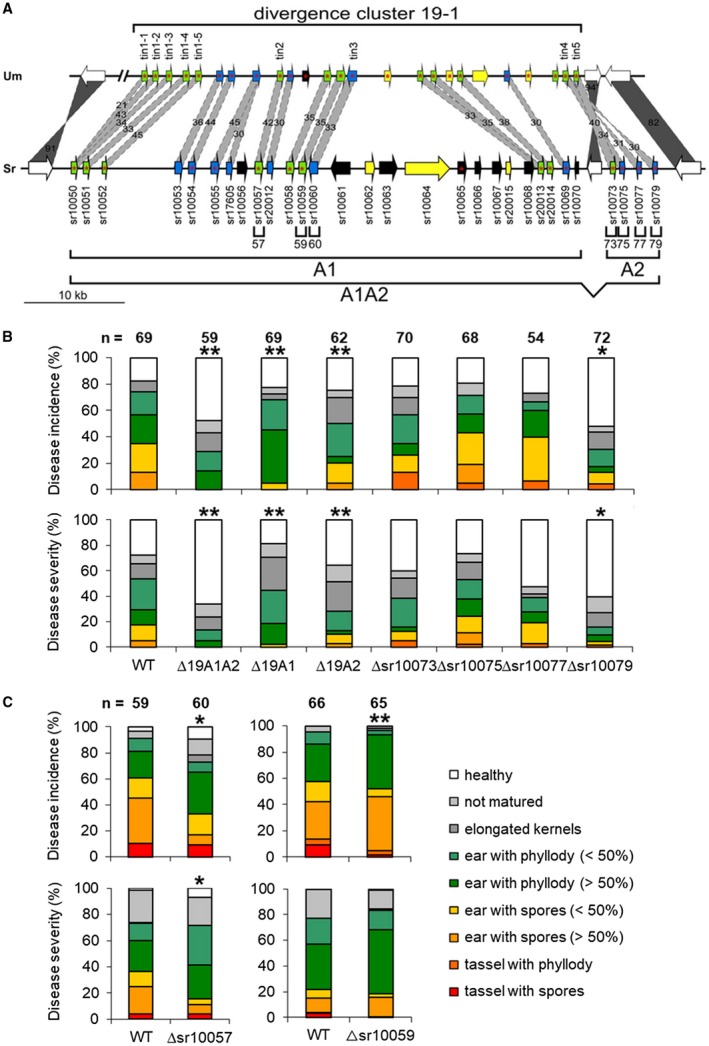
Disease evaluation of divergence cluster 19‐1 deletion strains. (A) Diagram showing the gene organization of divergence cluster 19‐1 of Sporisorium reilianum (Sr, bottom) and Ustilago maydis (Um, top). Cluster genes are represented by coloured arrows oriented in their direction of transcription, and with S. reilianum gene numbers indicated below the arrows. Cluster genes are colour coded: black, species‐specific genes; yellow, homologues only in Ustilago hordei; blue, homologues only in U. maydis and S. reilianum; green, homologues in U. maydis, S. reilianum and U. hordei. Homologues between U. maydis and S. reilianum are connected by bars with amino acid identity (SIMAP) values indicated. Dark grey, highly conserved; light grey, weakly conserved. The delineations of individual gene deletions (57, 59, 60, 73, 75, 77, 79), subcluster deletions (A1 and A2) and complete cluster deletion (A1A2) are indicated by brackets below the scheme. Known virulence effectors of U. maydis are indicated on top of the U. maydis genes. Genes predicted to encode secreted proteins (TargetP, reliability classes 1–3) are marked by a red asterisk in the gene arrow. (B, C) Virulence phenotypes of S. reilianum cluster 19‐1 deletion mutants on maize cultivar ‘Gaspe Flint’. Disease incidence (top) and disease severity (bottom) of A1A2 (∆19A1A2), A1 (∆19A1) and A2 (∆19A2) gene cluster deletion mutants, as well as the individual gene deletion mutants ∆sr10073, ∆sr10075, ∆sr10077, ∆sr10079, ∆sr10057 and ∆sr10059. The data are means of three independent experimental repeats. Asterisks indicate significant differences of disease severity and disease incidence scores calculated from the presented data relative to the wild‐type (WT) (*P < 0.05; **P < 0.01, Student’s t‐test). n, total number of evaluated plants. [Color figure can be viewed at wileyonlinelibrary.com]
We generated constructs for double homologous recombination suitable to replace complete or partial cluster regions by antibiotic resistance cassettes (see Experimental procedures). The constructs were used for protoplast transformation of the two compatible WT strains, SRZ2_5‐1 and SRZ1_5‐2, to generate compatible strains lacking the whole divergence cluster 6‐10 (Δ6‐10), the first (ΔA1), second (ΔA2) or both parts (ΔA1A2) of divergence cluster 19‐1. Successful gene cluster replacements were verified by polymerase chain reaction (PCR) and Southern blot, and two to three independently generated deletion strains of each mating type background were retained for virulence analysis.
Virulence analysis of Δ6‐10 mutants did not reveal significant alterations in disease severity or disease incidence relative to WT infections (Tables S7 and S8, see Supporting Information). In contrast, deletion of the complete cluster 19‐1 led to a severe reduction of both disease incidence and disease severity (ΔA1A2; Fig. 3B; Tables S7 and S8), indicating that one, several or all genes of divergence cluster 19‐1 support the virulence of S. reilianum on maize. We then tested the virulence of the partial cluster deletion mutants ΔA1 and ΔA2. Both deletion mutants were also reduced in both disease incidence and disease severity (Fig. 3B; Tables S7 and S8). The observed change in virulence was not a consequence of altered growth behaviour or altered stress tolerance (Figs S1 and S2), indicating that both parts of the cluster contain factors involved in the virulence of S. reilianum. The overall reduction in disease incidence of the ΔA2 mutant was stronger than that of ΔA1 (Table S7). However, of the three complete or partial divergence cluster 19‐1 deletion variants, the ΔA2 strain led to the highest number of plants with spores (Fig. 3B). This suggests that at least one factor encoded in the second part of cluster 19‐1 contributes to disease incidence, but not to the capacity of spore generation.
In U. maydis, deletion of the divergence cluster 19‐1 (19A) in the solopathogenic strain SG200 leads to a severely reduced capacity for tumour formation on seedling leaves (Kämper et al., 2006). The main effectors responsible for the observed reduction in tumour formation have been mapped to tin2 and tin3 (um05302 and um10556; Brefort et al., 2014). Both genes are located on the first half of cluster 19‐1. The orthologue of Tin2, with 42% overall amino acid identity, is Sr10057 of S. reilianum, and that of Tin3 is the S. reilianum protein Sr10060 (Fig. 3A; Table S2). To test whether the tin2 and tin3 homologous genes, sr10057 and sr10060, respectively, in S. reilianum contribute to the reduced symptom formation rate of the ΔA1 deletion strains, we created individual gene deletions of sr10057 and sr10060 in the two compatible WT strains SRZ2_5‐1 and SRZ1_5‐2 (see Experimental procedures and Table S4). In addition, we generated strains lacking the gene sr10059.
We then tested the virulence of the generated deletion mutants on maize. Deletion of sr10057 led to a significant reduction in disease incidence (about 30%) and disease severity (about 15%) in comparison with WT (Fig. 3C; Tables S7 and S8). Deletion of sr10059 led to a very small (less than 10%), but significant, reduction in disease incidence, but not in disease severity (Fig. 3C; Tables S7 and S8). The observed changes in disease incidence and/or disease severity were not a consequence of altered growth behaviour or altered stress tolerance properties of the deletion strains (Figs S1 and S2). The absence of sr10060 did not affect the virulence of S. reilianum (Tables S7 and S8). This shows that sr10057 and sr10059 have a positive effect on the virulence of S. reilianum on maize. However, their effects are small and, together, do not add up to the effect of the deletion of the complete A1 region, suggesting that they may either exhibit a partially redundant function or that additional genes of the A1 region affect the virulence of S. reilianum on maize.
As the ΔA2 strains showed a strong reduction in virulence, we aimed to identify which of the annotated genes of this region (sr10073, sr10075, sr10077 and sr10079) had the greatest impact on the virulence of S. reilianum. All four potential proteins carry a predicted secretion signal sequence. The first gene sr10073 is homologous to tin5 (um05319) of U. maydis, but with a low amino acid identity (Fig. S3, see Supporting Information). The three following genes (sr10075, sr10077 and sr10079) are homologues of tin4 (um05318) of U. maydis (Fig. S3). The gene sr10077 has recently been identified as being responsible for the induction of subapical ear formation in S. reilianum‐infected maize plants, and was therefore named sad1 for ‘suppressor of apical dominance 1’ (Ghareeb et al., 2015).
We generated individual gene deletion strains in the two mating‐compatible WT strains SRZ1_5‐2 and SRZ2_5‐1 by replacing complete open reading frames with an antibiotic resistance cassette. The deletion strains were verified by PCR and Southern blot, and the phenotypes of two to three independently generated compatible strains of each gene deletion mutant were tested on maize. As the virulence phenotype did not differ between strains carrying the same deletion, we selected one representative pair of compatible deletion strains for each gene to repeat the infection experiments at least twice more.
Deletion of sr10073, sr10075 or sr10077 led to no detectable effect on disease incidence or disease severity on maize (Fig. 3B; Tables S7 and S8), although the expression of these genes during plant colonization could clearly be detected (Ghareeb et al., 2015). In contrast, deletion of sr10079 led to a clear reduction in both disease incidence and disease severity that was almost as strong as the effect of the complete A2 deletion (Fig. 3B; Tables S7 and S8). The reduction in virulence was not a result of altered growth or stress tolerance properties (Figs S1 and S2). This shows that divergence cluster 19‐1 contains multiple virulence effectors, at least three (Sr10059, Sr10057 and Sr10079) of which clearly contribute to disease incidence and disease severity of S. reilianum on maize.
Discussion
In previous attempts to discover S. reilianum resistance loci in maize, spore formation in the inflorescences has been the only measure of disease incidence (Bernardo et al., 1992; Chen et al., 2008; Li et al., 2008; Lübberstedt et al., 1999). To obtain a more sensitive virulence measure of the S. reilianum strains on maize, we developed a disease evaluation system that measures symptom strength by quantifying both disease incidence and disease severity, considering both the phenotype (phyllody or spores) and percentage of the inflorescence displaying the respective phenotype. We used these measures to investigate the impact of potential effector genes on the virulence of S. reilianum on maize deleted in either compatible WT strains or in the weakly virulent solopathogenic S. reilianum strain JS161.
Through genome comparison of smut fungi, many potential pathogen effector genes have been identified which have been suggested to function in virulence, symptom formation and suppression of host defences (Laurie et al., 2012; Schirawski et al., 2010). However, their functional involvement in the virulence of S. reilianum has not yet been addressed. To tackle this task, we generated deletion strains for 13 individual genes (sr10057, sr10059, sr10060, sr10073, sr10075, sr10077, sr10079, sr10703, sr11815, sr12538, sr13154, sr14797, sr15769), as well as nine gene clusters (uni5‐1, two genes; uni7‐11, two genes; uni6‐1, three genes; uni11‐1, three genes; A2, four genes; uni5‐18, five genes; 6‐10, 15 genes; A1, 26 genes; A1A2, 30 genes). We selected target genes encoding potential effectors affecting S. reilianum‐dependent symptom formation based on two different hypotheses. They might be found among the genes specific to S. reilianum with no homologues in U. maydis and U. hordei, or among the genes that are weakly conserved between S. reilianum, U. maydis and U. hordei. In this way, we excluded the analysis of putative effectors necessary for the establishment and maintenance of the biotrophic interaction, which we believe would be found in the set of more highly conserved genes of S. reilianum, U. maydis and U. hordei.
Of the gene set 1 of S. reilianum‐specific genes, we randomly selected 21 that were deleted either individually (six genes; six mutants) or in clusters (15 genes; five mutants) in the weakly virulent solopathogenic strain JS161. Three of the six tested individual gene deletion mutants, and two of the five gene cluster deletion mutants, had a small, but detectable, impact on virulence. This shows that set 1 of S. reilianum‐specific genes contains a high proportion of virulence effectors. Interestingly, two of the mutants with altered virulence phenotype were more virulent than JS161 on the maize variety ‘Gaspe Flint’. Putative smut effectors with a negative impact on virulence have been detected previously. For example, deletion of cluster 2A (Kämper et al., 2006) or cluster 8‐12 (Schirawski et al., 2010) in the solopathogenic U. maydis strain SG200 led to hypervirulence. Hypervirulence may result from the deletion of an effector that is recognized by the plant immune system. In different maize lines in which this effector is not recognized, its deletion may have a negative virulence effect, explaining why it was not lost during the course of fungal evolution.
Putative effectors that did not result in a detectable deletion phenotype could still be involved in virulence: they could have a so far undetected role on virulence in other maize cultivars or other hosts, such as teosinte; their marginal impact on virulence might not be detectable in our assay, but may be relevant in the field; or their function could be complemented by other effectors.
As individual putative effectors were randomly selected for deletion analysis, our analysis suggests that S. reilianum possesses a much higher number of virulence‐reducing effectors than U. maydis, potentially reflecting differences in the lifestyles of the two smut fungi. Sporisorium reilianum has a much longer endophytic phase than U. maydis, and the prevention of excessive fungal growth may be much more important for S. reilianum. Alternatively, it is possible that the efficient detection of hypervirulent mutants is caused by the use of an only weakly virulent solopathogenic strain. Strongly virulence‐attenuated mutants or suboptimal infection conditions have been used previously for the detection of small effects on virulence (Djamei et al., 2011; Fones et al., 2015), and it is possible that the U. maydis strain SG200 is too virulent for the efficient detection of small virulence effects.
Of the 41 divergence clusters of gene set 2 with weak amino acid conservation between U. maydis and S. reilianum, we tested two clusters (6‐10 and 19‐1) that had a high content of potential effectors with homologues in U. hordei. Divergence cluster 6‐10 comprises 15 genes, 12 of which form a weakly conserved effector gene family without any other homologues in the S. reilianum genome. Deletion of the complete 6‐10 divergence cluster in compatible WT strains did not detectably alter the virulence of S. reilianum. In contrast, deletion of the syntenic region in SG200 (cluster 6A; Kämper et al., 2006) led to a severe reduction in virulence of U. maydis. Cluster 6A of U. maydis was described as containing a five‐gene family plus three additional genes. As the dissection of cluster 6A has not been published, it is not known whether one or several factors contribute to the reduced virulence phenotype of U. maydis. However, as every U. maydis gene of cluster 6A has an orthologue in the S. reilianum cluster 6‐10 (Table S2), it was astonishing that the 6‐10 deletion mutant did not show a virulence phenotype. Possibly, deletion of the 6‐10 region has only a mild effect on virulence that is not detectable in WT strains. In this case, we might have detected altered virulence if we had deleted the cluster in the weakly virulent strain JS161. Alternatively, the putative effectors in cluster 6A of U. maydis may have a different function from those of cluster 6‐10 of S. reilianum. Although some effectors have been shown to have conserved functions in smut fungi (e.g. Pep1; Hemetsberger et al., 2015), these show much higher amino acid sequence identity values than those observed between the divergence cluster homologues of U. maydis and S. reilianum (Table S2). An additional possibility to explain why the 6‐10 deletion mutant did not show an altered virulence phenotype is that the cluster might contain effectors with negative as well as positive influences on virulence, resulting in no net alteration in virulence strength if they are deleted together. In this case, individual deletion of effector genes might lead to measurable effects. Alternatively, additional genes without any sequence relatedness, and whose gene products fulfil the same function, might exist elsewhere in the genome and compensate for the loss of the cluster 6‐10 genes in the S. reilianum cluster 6‐10 deletion strains.
Divergence cluster 19‐1 is the largest genomic region of low sequence conservation between S. reilianum and U. maydis with a large number of homologues in U. hordei. This cluster thus represents the most likely region to harbour determinants of the species‐specific biotrophic lifestyle (Schirawski et al., 2010). Congruently, deletion of cluster 19‐1 in S. reilianum led to strongly reduced virulence on maize. We identified three individual genes (sr10057, sr10059, sr10079) that contributed to the virulence of S. reilianum. Interestingly, this list only partially corresponds to the list of the most important virulence effectors of the 19‐1 region in U. maydis. Notably, deletion of sr10060, the orthologue of tin3, or deletion of sr10073, the orthologue of tin5, did not result in decreased virulence. Only deletion of sr10079, the supposed orthologue of tin4 (Fig. S3), resulted in reduced virulence. This could indicate that homologous effectors have different functions in different organisms, that S. reilianum contains unrelated, but functionally redundant, effectors, or that some effectors needed for the virulence of U. maydis are not necessary for the virulence of S. reilianum.
We confirmed the presence of virulence effectors in set 1 of S. reilianum‐specific genes, and in gene set 2 of weakly conserved putative effectors with homologues in S. reilianum, U. maydis and U. hordei. This suggests that both initial hypotheses can be used for experimental effector validation. The fact that we found virulence effectors in both sets in spite of the limited number of investigated genes suggests that both sets contain a high abundance of virulence effectors. The identified virulence effectors only had a small impact on disease severity. This indicates that the virulence of S. reilianum is achieved by multiple factors acting simultaneously.
Experimental Procedures
Maize lines, S. reilianum strains, growth conditions and plant infection
The maize line (Zea mays) cultivar ‘Gaspe Flint’ was used throughout this study and was supplied by Regine Kahmann, Marburg, Germany. Kernels were sown in Type T Fruhstorfer soil and grown in a glasshouse with a 15‐h day period at 28 °C and a 9‐h night period at 22 °C. Seven‐day‐old seedlings were used for plant inoculation, and symptoms were evaluated after the emergence of inflorescences at 7 weeks.
Sporisorium reilianum strains were stocked in 25% glycerol at −80 °C. For plant inoculation experiments, compatible S. reilianum WT strains SRZ1_5‐2 and SRZ2_5‐1 (Schirawski et al., 2005; Zuther et al., 2012) and their deletion derivatives or derivatives of the solopathogenic S. reilianum strain JS161 (Schirawski et al., 2010) were used (Ghareeb et al., 2011). For each mutant, at least three biological repeats were tested on maize plants. The strains used in this study are listed in Table S4.
Symptom evaluation of maize infection by S. reilianum
To quantify the multitude of disease symptoms of S. reilianum on maize, the different symptoms displayed (healthy, phyllody, spores) were considered in each inflorescence of the inoculated plants. Each inflorescence of one plant was individually placed into one of nine symptom categories: (i) tassel with spores; (ii) tassel with phyllody; (iii) ear with more than 50% of flowers replaced by sori; (iv) ear with less than 50% of flowers replaced by sori; (v) ear with more than 50% of flowers displaying phyllody; (vi) ear with less than 50% of flowers displaying phyllody; (vii) ear with elongated kernels; (viii) immature ear that did not show clear symptoms and in which the kernels were not mature enough to be categorized as healthy; (ix) healthy ear (Fig. 1). The categories are listed from the strongest to the weakest symptom, and the stronger category may contain weaker symptoms. For example, if 25% of the flowers of an ear are replaced by sori and 75% display phyllody, the ear belongs in the category ‘less than 50% spores’, as spores is a stronger symptom than phyllody. Tassel with spores is the strongest symptom as, in plants showing spores or phyllody in the tassel, all ears are diseased. Spores are a stronger symptom than phyllody, as phyllody does not support dispersal of the fungus.
To quantify the disease symptoms of S. reilianum, we calculated two values for each infection experiment: a disease incidence index and a disease severity index. To determine the disease incidence, each plant was placed in one of the nine categories that corresponded to the strongest symptom displayed by its inflorescences. We then calculated the percentage of plants in each category relative to the total number of inoculated plants. These data were used to calculate a disease incidence index by multiplying the number of plants sorted into a particular category by the weight of the respective category, defined as 0 (healthy), 1 (not matured), 2 (elongated kernels), 10 (ear with phyllody < 50%), 11 (ear with phyllody > 50%), 15 (ear with spores < 50%), 16 (ear with spores > 50%), 20 (leafy tassel) and 25 (tassel with spores). We added the products and divided the sum by the total number of plants considered. To determine the disease severity, we collected the inflorescences from all plants, sorted them by symptom category and calculated the percentage of inflorescences in each category relative to the total number of inflorescences. To calculate a disease severity index, an analogous calculation as mentioned above was performed considering individual inflorescences sorted into a particular category and the total number of evaluated inflorescences. For easier comparison of disease incidence and disease severity indices, the resulting value for WT was set to 10, and indices of mutants were calculated relative to the score for WT. The statistical significance of the difference between WT and mutants was evaluated by unpaired Student’s t‐test (see Tables [Link], [Link], [Link], [Link]).
Generation of gene deletion strains of S. reilianum
Gene deletion was achieved via replacement of the gene of interest with the hygromycin (pBS‐hhn; Kämper, 2004) or phleomycin (pBS‐Phleo; Brachmann et al., 2004) resistance cassette by double homologous recombination. For the amplification of the right and left flanks of the deletion constructs and amplification of the complete deletion construct, the primers listed in Table S9 were used.
Deletion constructs were used for the transformation of S. reilianum protoplasts as described for U. maydis (Gillissen et al., 1992; Schulz et al., 1990). Putative transformants were selected and verified by PCR and Southern blot using the deletion construct as probe. Up to three independently generated and PCR‐ and Southern blot‐validated strains for each mating background were used for plant inoculation experiments (Table S4).
Growth curves and stress test
For growth rate analysis, cells were grown in YEPS light medium (0.4% yeast extract, 0.4% peptone, 2% sucrose) at 28 °C with shaking with a starting optical density at 600 nm (OD600) of 0.1, and OD600 was measured every 2 h. The doubling times of the strains were estimated from the growth curves. For stress assays, CM‐glucose medium (Holiday, 1974) was used. Solid CM‐glucose medium supplemented with Congo red (100 µm), calcofluor (50 µm), H2O2 (1.5 mm) or sodium chloride (1 m) was used to generate stress conditions. Cells were grown in YEPS light medium to an OD600 of 0.8, washed with H2O and resuspended in H2O to an OD600 of 1.0. Five microlitres of serially diluted (10–1) samples were spotted onto the plates. The plates were incubated for 3 days at 28 °C and photographed.
Multiple sequence alignment
Amino acid sequences of the U. maydis proteins tin4 and tin5 and their homologues in S. reilianum and U. hordei were extracted from the U. maydis, S. reilianum and U. hordei genome databases at https://pedant.gsf.de/genomes.jsp?category=fungal. These were used for multiple sequence alignment with the program MEGA6 (Tamura et al., 2013). Within MEGA, the sequences were aligned using ClustalW with default settings, and the alignment was used to construct a neighbour‐joining tree with the following settings: test of phylogeny, bootstrap method; number of bootstrap replications, 1000; substitution model/method, p‐distance; gaps/missing data treatment, pairwise deletion. The tree was condensed to remove non‐supported nodes, which led to a loss of branch length information.
Supporting information
Fig. S1 Doubling times of selected S. reilianum strains in YEPS light medium. Strains investigated showed a virulence phenotype relative to their progenitor strains JS161 (solopathogenic strain; left block) or SRZ1_5‐2 (wild type; right block), and were of the following mutants: HG40 (Δsruni1), HG43 (Δsruni2), HG49 (Δsruni4), HG57 (Δsruni5), HG21 (Δuni5‐1), HG28 (Δuni6‐1), JS751 (ΔA1), HG19 (ΔA1DA2), HG127 (ΔA2), YZ10 (Δsr10057), YZ121 (Δsr10059), HG99 (Δsr10077), HG92 (Δsr10079). Data of three independent biological replicates is shown. Error bars indicate the standard deviation. Statistical significance relative to the corresponding progenitor strains was evaluated using a Students’s t‐test with P < 0.05. All strains showed the same doubling times as their parental strains.
Fig. S2 Stress response of selected S. reilianum mutants. Serial dilutions of exponentially growing strains were spotted on CM‐glucose plates (CM) or on CMglucose plates supplemented with Calcofluor White (50 \M; Calco), H2O2 (0.5 mM), NaCl (1 M), or Congo red (100 M, Congo). Plates were incubated for three days at 28°C. The solopathogenic strain JS161 and its derivatives HG40 (Δsruni1), HG43 (Δsruni2), HG49 (Δsruni4), HG57 (Δsruni5), HG21 (Δuni5‐1), and HG30 (Δuni6‐1) are indicated in blue, the wild‐type strain SRZ1_5‐2 and its derivatives JS751 (ΔA1), HG127 ΔA2), HG19 (ΔA1ΔA2), YZ10 (Δsr10057), YZ121 (Δsr10059), HG99 (Δsr10077), and HG92 (Δsr10079) are indicated in black.
Fig. S3 Comparison of homologs of Umtin4 and Umtin5 of S. reilianum and U. hordei. A) Amino acid alignment of the three S. reilianum homologs of Umtin4. B) Matrix of amino acid identities of Umtin5 (um05319) and its homologs Sr10073 and UHOR_10033 (left), as well as of Umtin4 (um05318) and its homologs sr10075, sr10077, sr10079 and UHOR_13916 (right). C) Phylogenetic tree built by an alignment of protein sequences showing relatedness of the proteins compared in B with Umtin1.1 (um05294) as outgroup. Bootstrap values above 50 are indicated. The figure was generated as described in the Experimental Procedures section.
Table S1 List of S. reilianum‐specific genes.
Table S2 Diversity cluster genes and their homologs in U. maydis, S. reilianum and U. hordei.
Table S3 Summary of the number of S. reilianum divergence cluster genes with homologs in U. maydis and U. hordei.
Table S4 Strains used in this study.
Table S5 Calculation of the disease incidence index of solopathogenic S. reilianum mutants lacking S. reilianum‐specific genes.
Table S6 Calculation of the disease severity index of solopathogenic S. reilianum mutants lacking S. reilianum‐specific genes.
Table S7 Calculation of the disease incidence index of S. reilianum mutants lacking genes of divergence clusters 6‐10 and 19‐1.
Table S8 Calculation of the disease severity index of S. reilianum mutants lacking genes of divergence clusters 6‐10 and 19‐1.
Table S9 Oligonucleotides used in this study.
Acknowledgements
We thank Helen Hülsmann (Marburg, Germany) for the generation of 6‐10 deletion strains; Theresa Wollenberg (Marburg, Germany) for disease evaluation; Mohammad Tanbir Habib (Göttingen, Germany) for the generation of constructs for the deletion of sr10057 and sr10060; Nils Volkers (Aachen, Germany), Kalle Hüser (Aachen, Germany) and Alana Poloni (Aachen, Germany) for the preparation of growth curves and stress tolerance tests of selected S. reilianum strains; Gertrud Mannhaupt (München, Germany) for annotation of the cluster 19‐1 region in S. reilianum; Elmar Meyer (Marburg, Germany) for technical assistance; Regine Kahmann (Marburg, Germany) for support; and Kilian Smith (Aachen, Germany) for English language editing. This work was supported by the Max Planck Society, Georg‐August‐University through the German Initiative of Excellence (DFG ZUK45/1) (J.S.) and RWTH Aachen University. Funding via the International Max Planck Research School for Environmental, Cellular and Molecular Microbiology (H.G.), the Göttingen Graduate School for Neurosciences, Biophysics, and Molecular Biosciences (GGNB) (H.G.), the FAZIT Foundation (H.G, Y.Z.), the Chinese Scholarship Council (CSC) (Y.Z.) and the German Research Foundation (DFG) (J.S.) is gratefully acknowledged.
References
- Ali, S. , Laurie, J.D. , Linning, R. , Cervantes‐Chávez, J.A. , Gaudet, D. and Bakkeren, G. (2014) An immunity‐triggering effector from the Barley smut fungus Ustilago hordei resides in an Ustilaginaceae‐specific cluster bearing signs of transposable element‐assisted evolution. PLoS Pathog. 10, e1004223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banuett, F. and Herskowitz, I. (1994) Morphological transitions in the life cycle of Ustilago maydis and their genetic control by the a and b loci. Exp. Mycol. 18, 247–266. [Google Scholar]
- Baumgarten, A.M. , Suresh, J. , May, G. and Phillips, R.L. (2007) Mapping QTLs contributing to Ustilago maydis resistance in specific plant tissues of maize. Theor. Appl. Genet. 114, 1229–1238. [DOI] [PubMed] [Google Scholar]
- Begerow, D. , Stoll, M. and Bauer, R. (2006) A phylogenetic hypothesis of Ustilagino mycotina based on multiple gene analyses and morphological data. Mycologia, 98, 906–1016. [DOI] [PubMed] [Google Scholar]
- Bernardo, R. , Bourrier, M. and Olivier, J.L. (1992) Generation means analysis of resistance to head smut (Sporisorium reiliana) in maize. Agronomie, 12, 303–306. [Google Scholar]
- Brachmann, A. , König, J. , Julius, C. and Feldbrügge, M. (2004) A reverse genetic approach for generating gene replacement mutants in Ustilago maydis . Mol. Genet. Genomics, 272, 216–226. [DOI] [PubMed] [Google Scholar]
- Brefort, T. , Tanaka, S. , Neidig, N. , Doehlemann, G. , Vincon, V. and Kahmann, R. (2014) Characterization of the largest effector gene cluster of Ustilago maydis . PLoS Pathog. 10, e1003866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressman, E.N. and Barss, H.P. (1933) Experiments with head smut of corn in western Oregon. Phytopathology, 23, 396–403. [Google Scholar]
- Chaudhari, P. , Ahmed, B. , Joly, D.L. and Germain, H. (2014) Effector biology during biotrophic invasion of plant cells. Virulence, 5, 703–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Chao, Q. , Tan, G. , Zhao, J. , Zhang, M. , Ji, Q. and Xu, M. (2008) Identification and fine‐mapping of a major QTL conferring resistance against head smut in maize. Theor. Appl. Genet. 117, 1241–1252. [DOI] [PubMed] [Google Scholar]
- Deller, S. , Hammond‐Kosack, K.E. and Rudd, J.J. (2011) The complex interactions between host immunity and non‐biotrophic fungal pathogens of wheat leaves. J. Plant Physiol. 168, 63–71. [DOI] [PubMed] [Google Scholar]
- Dioh, W. , Tharreau, D. , Notteghem, J.L. , Orbach, M. and Lebrun, M.‐H. (2000) Mapping of avirulence genes in the rice blast fungus, Magnaporthe grisea, with RFLP and RAPD markers. Mol. Plant‐Microbe Interact., 13, 217–227 [DOI] [PubMed] [Google Scholar]
- Djamei, A. , Schipper, K. , Rabe, F. , Ghosh, A. , Vincon, V. , Kahnt, J. , Osorio, S. , Tohge, T. , Fernie, A.R. , Feussner, I. , Feussner, K. , Meinicke, P. , Stierhof, Y.D. , Schwarz, H. , Macek, B. , Mann, M. and Kahmann, R. (2011) Metabolic priming by a secreted fungal effector. Nature, 478, 395–398. [DOI] [PubMed] [Google Scholar]
- Dutzmann, S. and Duben, J. (1993) Maiskopfbrand zukünftig auch in Deutschland von Bedeutung? mais 21, 140–142. [Google Scholar]
- Emanuelsson, O. , Brunak, S. , von Heijne, G. and Nielsen, H. (2007) Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2, 953–971. [DOI] [PubMed] [Google Scholar]
- Fischer, G.W. and Holton, C.S. (1957) Biology and Control of the Smut Fungi. New York: Ronald Press Company. [Google Scholar]
- Fones, H.N. , Steinberg, G. and Gurr, S.J. (2015) Measurement of virulence in Zymoseptoria tritici through low inoculum‐density assays. Fungal Genet. Biol. 79, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuyao, Z. , Junai, P. , Zhihong, D. , Qingjun, C. and Huang, Y. (2010) Identification of a new race of Sporisorium reilianum and characterization of the reaction of sorghum lines to four races of the head smut pathogen. J. Phytopathol. 159, 342–346. [Google Scholar]
- Ghareeb, H. , Becker, A. , Iven, T. , Feussner, I. and Schirawski, J. (2011) Sporisorium reilianum infection changes inflorescence and branching architectures of maize. Plant Phys. 156, 2037–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghareeb, H. , Drechser, F. , Löfke, C. , Teichmann, T. and Schirawski, J. (2015) SUPPRESSOR OF APICAL DOMINANCE 1 of Sporisorium reilianum modulates inflorescence branching architecture in maize and Arabidopsis. Plant Physiol. 169, 2789–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillissen, B. , Bergemann, J. , Sandmann, C. , Schroeer, B. , Bölker, M. and Kahmann, R. (1992) A two‐component regulatory system for self/non‐self recognition in Ustilago maydis . Cell, 68, 647–657. [DOI] [PubMed] [Google Scholar]
- Groth, J. , Person, C. and Ebba, T. (1976) Relation between two measures of disease expression in barley–Ustilago hordei interactions. Phytopathology, 66, 1342–1347. [Google Scholar]
- Hemetsberger, C. , Mueller, A.N. , Matei, A. , Herrberger, C. , Hensel, G. , Kumlehn, J. , Mishra, B. , Sharma, R. , Thines, M. , Huckelhoven, R. and Doehlemann, G. (2015) The fungal core effector Pep1 is conserved across smuts of dicots and monocots. New Phytol. 206, 1116–1126. [DOI] [PubMed] [Google Scholar]
- Holliday, R. (1974) Ustilago maydis In: King R.C. (Ed.) Handbook of Genetics. 1, Plenum Press NY: New York, pp. 575–595 [Google Scholar]
- Kämper, J. (2004) A PCR‐based system for highly efficient generation of gene replacement mutants in Ustilago maydis . Mol. Genet. Genomics, 271, 103–110. [DOI] [PubMed] [Google Scholar]
- Kämper, J. , Kahmann, R. , Bolker, M. , Ma, L.J. , Brefort, T. , Saville, B.J. , Banuett, F. , Kronstad, J.W. , Gold, S.E. , Müller, O. , Perlin, M.H. , Wosten, H.A. , de Vries, R. , Ruiz‐Herrera, J. , Reynaga‐Pena, C.G. , Snetselaar, K. , McCann, M. , Perez‐Martin, J. , Feldbrugge, M. , Basse, C.W. , Steinberg, G. , Ibeas, J.I. , Holloman, W. , Guzman, P. , Farman, M. , Stajich, J.E. , Sentandreu, R. , Gonzalez‐Prieto, J.M. , Kennell, J.C. , Molina, L. , Schirawski, J. , Mendoza‐Mendoza, A. , Greilinger, D. , Münch, K. , Rössel, N. , Scherer, M. , Vranes, M. , Ladendorf, O. , Vincon, V. , Fuchs, U. , Sandrock, B. , Meng, S. , Ho, E.C. , Cahill, M.J. , Boyce, K.J. , Klose, J. , Klosterman, S.J. , Deelstra, H.J. , Ortiz‐Castellanos, L. , Li, W. , Sanchez‐Alonso, P. , Schreier, P.H. , Hauser‐Hahn, I. , Vaupel, M. , Koopmann, E. , Friedrich, G. , Voss, H. , Schluter, T. , Margolis, J. , Platt, D. , Swimmer, C. , Gnirke, A. , Chen, F. , Vysotskaia, V. , Mannhaupt, G. , Güldener, U. , Münsterkotter, M. , Haase, D. , Oesterheld, M. , Mewes, H.W. , Mauceli, E.W. , DeCaprio, D. , Wade, C.M. , Butler, J. , Young, S. , Jaffe, D.B. , Calvo, S. , Nusbaum, C. , Galagan, J. and Birren, B.W. (2006) Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis . Nature, 444, 97–101. [DOI] [PubMed] [Google Scholar]
- Laurie, J.D. , Ali, S. , Linning, R. , Mannhaupt, G. , Wong, P. , Guldener, U. , Munsterkotter, M. , Moore, R. , Kahmann, R. , Bakkeren, G. and Schirawski, J. (2012) Genome comparison of barley and maize smut fungi reveals targeted loss of RNA silencing components and species‐specific presence of transposable elements. Plant Cell, 24, 1733–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X.H. , Wang, Z.H. , Gao, S.R. , Shi, H.L. , Zhang, S.H. , George, M.L.C. , Li, M.S. and Xie, C.X. (2008) Analysis of QTL for resistance to head smut (Sporisorium reiliana). Field Crop Res. 106, 148–155. [Google Scholar]
- Linning, R. , Lin, D. , Lee, N. , Abdennadher, M. , Gaudet, D. , Thomas, P. , Mills, D. , Kronstad, J.W. and Bakkeren, G. (2004) Marker‐based cloning of the region containing the UhAvr1 avirulence gene from the basidiomycete barley pathogen Ustilago hordei . Genetics, 166, 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lübberstedt, T. , Klein, D. and Melchinger, A. (1998) Comparative QTL mapping of resistance to Ustilago maydis across four populations of European flint‐maize. Theor. Appl. Genet. 97, 1321–1330. [Google Scholar]
- Lübberstedt, T. , Xia, X.C. , Tan, G. , Liu, X. and Melchinger, A.E. (1999) QTL mapping of resistance to Sporisorium reiliana in maize. Theor. Appl. Genet. 99, 593–598. [DOI] [PubMed] [Google Scholar]
- Martinez, C. and Roux, C. (2002) The biological cycle of Sporisorium reilianum f.sp. zeae: an overview using microscopy. Mycologia, 94, 505–514. [PubMed] [Google Scholar]
- Munkacsi, A.B. , Stoxen, S. and May, G. (2007) Domestication of maize, sorghum, and sugarcane did not drive the divergence of their smut pathogens. Evolution, 61, 388–403. [DOI] [PubMed] [Google Scholar]
- Prom, L.K. , Perumal, R. , Erattaimuthu, S.R. , Erpelding, J.E. , Montes, N. , Odvody, G.N. , Greenwald, C. , Jin, Z. , Frederiksen, R. and Magill, C.W. (2011) Virulence and molecular genotyping studies of Sporisorium reilianum isolates in sorghum. Plant Dis. 95, 523–529. [DOI] [PubMed] [Google Scholar]
- Rattei, T. , Tischler, P. , Götz, S. , Jehl, M.A. , Hoser, J. , Arnold, R. , Conesa, A. and Mewes, H.W. (2010) SIMAP–a comprehensive database of precalculated protein sequence similarities, domains, annotations and clusters. Nucleic Acids Res. 38, D223–D226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirawski, J. , Heinze, B. , Wagenknecht, M. and Kahmann, R. (2005) Mating type loci of Sporisorium reilianum: novel pattern with three a and multiple b specificities. Eukaryot. Cell, 4, 1317–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirawski, J. , Mannhaupt, G. , Munch, K. , Brefort, T. , Schipper, K. , Doehlemann, G. , Di Stasio, M. , Rossel, N. , Mendoza‐Mendoza, A. , Pester, D. , Muller, O. , Winterberg, B. , Meyer, E. , Ghareeb, H. , Wollenberg, T. , Munsterkotter, M. , Wong, P. , Walter, M. , Stukenbrock, E. , Guldener, U. and Kahmann, R. (2010) Pathogenicity determinants in smut fungi revealed by genome comparison. Science, 330, 1546–1548. [DOI] [PubMed] [Google Scholar]
- Schulz, B. , Banuett, F. , Dahl, M. , Schlesinger, R. , Schafer, W. , Martin, T. , Herskowitz, I. and Kahmann, R. (1990) The b alleles of U. maydis, whose combinations program pathogenic development, code for polypeptides containing a homeodomain‐related motif. Cell, 60, 295–306. [DOI] [PubMed] [Google Scholar]
- Schweizer, G. , Münch, K. , Mannhaupt, G. , Schirawski, J. , Kahmann, R. and Dutheil, J.Y. (2018) Positively selected effector genes and their contribution to virulence in the smut fungus Sporisorium reilianum . Genome Biol. Evol. 10, 629–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromberg, E.L. , Stienstra, W.C. , Kommedahl, T. , Matyac, C.A. , Windels, C.E. and Geadelmann, J.L. (1984) Smut expression and resistance of corn to Sphacelotheca reiliana in Minnesota. Plant Dis. 68, 880–884. [Google Scholar]
- Tamura, K. , Stecher, G. , Peterson, D. , Filipski, A. and Kumar, S. (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, M.L. , Melchinger, A.E. and Lubberstedt, T. (1999) Species‐specific detection of the maize pathogens Sporisorium reiliana and Ustilago maydis by dot blot hybridization and PCR‐based assays. Plant Dis. 83, 390–395. [DOI] [PubMed] [Google Scholar]
- Zuo, W.L. , Chao, Q. , Zhang, N. , Ye, J.R. , Tan, G.Q. , Li, B.L. , Xing, Y.X. , Zhang, B.Q. , Liu, H.J. , Fengler, K.A. , Zhao, J. , Zhao, X.R. , Chen, Y.S. , Lai, J.S. , Yan, J.B. and Xu, M.L. (2015) A maize wall‐associated kinase confers quantitative resistance to head smut. Nat. Genet. 47, 151–157. [DOI] [PubMed] [Google Scholar]
- Zuther, K. , Kahnt, J. , Utermark, J. , Imkampe, J. , Uhse, S. and Schirawski, J. (2012) Host specificity of Sporisorium reilianum is tightly linked to generation of the phytoalexin luteolinidin by Sorghum bicolor . Mol. Plant–Microbe Interact. 25, 1230–1237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Doubling times of selected S. reilianum strains in YEPS light medium. Strains investigated showed a virulence phenotype relative to their progenitor strains JS161 (solopathogenic strain; left block) or SRZ1_5‐2 (wild type; right block), and were of the following mutants: HG40 (Δsruni1), HG43 (Δsruni2), HG49 (Δsruni4), HG57 (Δsruni5), HG21 (Δuni5‐1), HG28 (Δuni6‐1), JS751 (ΔA1), HG19 (ΔA1DA2), HG127 (ΔA2), YZ10 (Δsr10057), YZ121 (Δsr10059), HG99 (Δsr10077), HG92 (Δsr10079). Data of three independent biological replicates is shown. Error bars indicate the standard deviation. Statistical significance relative to the corresponding progenitor strains was evaluated using a Students’s t‐test with P < 0.05. All strains showed the same doubling times as their parental strains.
Fig. S2 Stress response of selected S. reilianum mutants. Serial dilutions of exponentially growing strains were spotted on CM‐glucose plates (CM) or on CMglucose plates supplemented with Calcofluor White (50 \M; Calco), H2O2 (0.5 mM), NaCl (1 M), or Congo red (100 M, Congo). Plates were incubated for three days at 28°C. The solopathogenic strain JS161 and its derivatives HG40 (Δsruni1), HG43 (Δsruni2), HG49 (Δsruni4), HG57 (Δsruni5), HG21 (Δuni5‐1), and HG30 (Δuni6‐1) are indicated in blue, the wild‐type strain SRZ1_5‐2 and its derivatives JS751 (ΔA1), HG127 ΔA2), HG19 (ΔA1ΔA2), YZ10 (Δsr10057), YZ121 (Δsr10059), HG99 (Δsr10077), and HG92 (Δsr10079) are indicated in black.
Fig. S3 Comparison of homologs of Umtin4 and Umtin5 of S. reilianum and U. hordei. A) Amino acid alignment of the three S. reilianum homologs of Umtin4. B) Matrix of amino acid identities of Umtin5 (um05319) and its homologs Sr10073 and UHOR_10033 (left), as well as of Umtin4 (um05318) and its homologs sr10075, sr10077, sr10079 and UHOR_13916 (right). C) Phylogenetic tree built by an alignment of protein sequences showing relatedness of the proteins compared in B with Umtin1.1 (um05294) as outgroup. Bootstrap values above 50 are indicated. The figure was generated as described in the Experimental Procedures section.
Table S1 List of S. reilianum‐specific genes.
Table S2 Diversity cluster genes and their homologs in U. maydis, S. reilianum and U. hordei.
Table S3 Summary of the number of S. reilianum divergence cluster genes with homologs in U. maydis and U. hordei.
Table S4 Strains used in this study.
Table S5 Calculation of the disease incidence index of solopathogenic S. reilianum mutants lacking S. reilianum‐specific genes.
Table S6 Calculation of the disease severity index of solopathogenic S. reilianum mutants lacking S. reilianum‐specific genes.
Table S7 Calculation of the disease incidence index of S. reilianum mutants lacking genes of divergence clusters 6‐10 and 19‐1.
Table S8 Calculation of the disease severity index of S. reilianum mutants lacking genes of divergence clusters 6‐10 and 19‐1.
Table S9 Oligonucleotides used in this study.