

Abstract
Objectives
Over the past decade an intriguing connection between cell polarity and tumorigenesis has emerged. Multiple core components of the junction complexes that help to form and maintain cell polarity display both pro‐ and anti‐tumorigenic functions in a context‐dependent manner, with the underlying mechanisms poorly understood.
Materials and Methods
With transgenic fly lines that overexpress or knock down specific signalling components, we perform genetic analysis to investigate the precise role of the polarity protein Canoe (Cno) in tumorigenesis and the downstream pathways.
Results
We show that overexpression of cno simultaneously activates JNK and Ras‐MEK‐ERK signalling, resulting in mixed phenotypes of both overproliferation and cell death in the Drosophila wing disc. Moderate alleviation of JNK activation eliminates the effect of Cno on cell death, leading to organ overgrowth and cell migration that mimic the formation and invasion of tumours. In addition, we find that the Hippo pathway acts downstream of JNK and Ras signalling to mediate the effect of Cno on cell proliferation.
Conclusions
Our work reveals an oncogenic role of Cno and creates a new type of Drosophila tumour model for cancer research.
Keywords: Canoe/Afadin, Hippo signalling, JNK signalling, Ras signalling, tumorigenesis
1. INTRODUCTION
Cell polarity is the morphological and functional asymmetry that helps to form tissue structures such as tubes and alveoli.1, 2 This type of asymmetric distribution of intracellular proteins is established and maintained mainly via cell‐cell junctions and adhesions that are formed by a diversity of polarity proteins and adhesion molecules.3, 4 Among these are three groups of core regulators of cell polarity, including the apical Crumbs polarity complex that consists of Crumbs (Crb)/Pals/Patj, the basolateral Par polarity complex that consists of Cdc42/Par3/Par6/aPKC and the Scribble polarity complex that consists of Scribble (Scrib)/Discs large (Dlg)/Lethal (2) giant larvae (Lgl).5, 6 Over the last decade, evidence has been accumulating for the link between tumorigenesis and defects in cell polarity. For instance, in mammary epithelial cells, depleting Crb or Pals caused dephosphorylation and nuclear translocation of YAP, which is a crucial step for the inhibition of the Hippo signalling pathway and results in increased cell proliferation.7, 8 In a DMBA/TPA‐induced skin cancer model, the loss of Par3, aPKC or both, strongly reduced tumour size and multiplicity, mainly via impairing the activation of ERK1/2 and Akt by Ras.9 In mammary epithelia, dysregulation of Scrib prevented Myc‐induced apoptosis and promoted epithelial‐mesenchymal transition (EMT) and tumorigenesis.10, 11 The effects of cell polarity disruption on tumour growth rely on the association and regulation of downstream signalling pathways.12, 13 Besides, the roles of polarity proteins in tumorigenesis seem complicated, displaying either pro‐ or anti‐tumorigenic functions and largely depending on the context of the cells.5, 13, 14
The adhesion molecule Afadin (AF‐6), encoded by theMLLT4 gene, is localised at cell‐cell adhesion sites in epithelial cells and fibroblasts to help the formation of adherens junctions (AJs) and maintain cell polarity.15, 16, 17 Originally identified as a fusion partner of the MLL gene in acute myeloid leukaemia with chromosome translocation,18 AF‐6 is also associated with initiation and progression of solid tumours. Low levels of AF‐6 expression was reported in 15% of breast cancer patients and linked to adverse prognosis,19 and the loss of AF‐6 was found to promote pancreatic cancer metastasis by inducing Snail expression,20 suggesting that AF‐6 might be a tumour suppressor. In another study, however, elevated AF‐6 expression was closely related to adverse outcomes of breast cancer patients.21 Furthermore, phosphorylation of AF‐6 by Akt induced its translocalisation from AJs to the nucleus and increased breast cancer cell migration,22 revealing the pro‐tumorigenic role of AF‐6. Therefore, the precise roles of Afadin in tumorigenesis and the underlying mechanisms still remain unclear.
As a classic model organism for the research of developmental biology, Drosophila has been recently used in cancer studies and provides great insights into the understanding of tumour initiation and progression.23, 24 Indeed, the first in vivo evidence for the contributions of polarity proteins to tumorigenesis came from the study of Drosophila brains, in which the cells with mutant polarity genes dlg or lgl displayed overgrown and invasive behaviours.25, 26 Subsequent studies demonstrated that loss of scrib, dlg or lgl in the larval eye disc accelerated the growth and metastasis of rasV12‐induced benign tumours, which was largely dependent on the activation of the JNK signalling pathway.27, 28 In addition to this rasV12‐polarity defects model, other Drosophila models for tumorigenesis have also been developed. For example, co‐overexpression of EGFR and PI3K in larval glia induced neoplasia in Drosophila and mimicked glioma.29, 30 Besides, elevating the levels of the Src kinase in cells along the anterior‐posterior (A/P) compartment boundary of Drosophila wing disc produced a metastatic phenotype, providing a model for genetic screening of genes involved in cancer metastasis.31, 32 Significantly, the core components of the Hippo signalling pathway, which plays vital roles in organ size control and exhibits various mutations in a plethora of tumours, were initially identified using Drosophila genetic models, highlighting the reliability of Drosophila models for cancer research.8, 33
In this study, we show that the overexpression of Cno, the Drosophila homolog of AF‐6, induces cell proliferation, cell death and cell migration in the larval wing disc. We find that these mixed effects result from strong activation of JNK signalling and Ras‐MAPK signalling. Moderately reducing the activation levels of JNK signalling suppresses the effect of Cno on cell death, thereby inducing massive cell overproliferation and disc overgrowth. In addition, we demonstrate that Hippo signalling acts as a downstream effect or to mediate Cno‐induced proliferation, revealing the underlying mechanism for the pro‐tumorigenic function of Cno in Drosophila.
2. MATERIALS AND METHODS
2.1. Fly stocks
Flies are raised at 25°C under standard conditions. The fly stocks used in this study are as follows: ptc‐Gal4, ap‐Gal4, nub‐Gal4, gmr‐Gal4, UAS‐GFP/cyo, UAS‐GFP/TM6b, UAS‐cno‐IR (NIG 2534R‐3), UAS‐ras‐IR (VDRC 106642), UAS‐mek‐IR (VDRC 107276). UAS‐cno is a gift from Prof. Ulrike Gaul. UAS‐hep‐IR, UAS‐bsk‐IR, UAS‐Egr, UAS‐Scrib‐IR, puc‐lacZ are gifts from Prof. Lei Xue. UAS‐yki‐IR, ex‐lacZ, ban‐lacZ, diap1‐lacZ are gifts from Prof. Lei Zhang.
2.2. TUNEL staining
Staining was carried out according to the protocol provided by the manufacturer (MK1021, Boster). Briefly, wing discs from third instar larva were dissected and fixed with 4% formaldehyde in 10 m mol L−1 PBS at room temperature for 30 minutes and washed with PBS and distilled water, respectively. Discs were then incubated with labelling buffer in a wet box at 37°C for 2 hours and washed with 0.01 M TBS. After blocking at room temperature for 30 minutes, discs were incubated with anti‐Digoxin at 37°C for 30 minutes. Washed with TBS, discs were reacted with diluted SABC at 37°C for 30 minutes. After washing with TBS, discs were mounted for microscopy.
2.3. Immunostaining and image acquisition
Staining was carried out using standard protocols. Briefly, wing discs from third instar larva were dissected and fixed in PBS‐T containing 4% formaldehyde for 20 minutes at room temperature. After washing with PBS‐T, they were blocked with 5% BSA in PBS‐T for 30 minutes and then incubated with primary antibodies overnight at 4°C. Washed discs were subsequently incubated with secondary antibodies. The following primary antibodies were used: mouse anti‐MMP1 (3A6B4, 1:300) and mouse anti‐Wg (4D4, 1:500) were purchased from Developmental Studies Hybridoma Bank. Mouse anti‐β‐galactosidase (sc‐65670, 1:500) was from Santa Cruz. Rabbit anti‐Caspase3 (#9661, 1:1000) and rabbit anti‐p‐Histone H3 (#9701, 1:1000) was from Cell Signaling Technology. Secondary antibodies goat anti‐mouse Alexa Fluor594 (A11012, 1:1000) and goat anti‐rabbit Alexa Fluor594 (A11005, 1:1000) were from Invitrogen. Fluorescence images were recorded using a Nikon DS‐Ri1 fluorescence microscope and a Zeiss LSM 880 confocal microscope. Images of the adult eyes were acquired using a Nikon SMZ‐745T trinocular stereo microscope. Images were then analysed using Zeiss Zen, Image J and Adobe Photoshop software.
3. RESULTS
3.1. Overexpression of cno induces mixed effects in the wing disc
To explore the role of Drosophila AF‐6 in cell proliferation, we overexpressed cno at the A/P compartment boundary of Drosophila wing disc by ptc‐Gal4. The expression domain of cno, labelled by GFP‐positive cells, became broader and irregular than the control, with some cells migrating into the posterior compartment of the disc (Figure 1A,B, Supporting Information Figure S1D). We monitored the signal of the mitosis marker, phosphorylated Histone H3 (pH3) and observed a significant increase in the number of pH3‐positive cells in the region of cno overexpression in the wing disc (Supporting Information Figure S1 A‐C). The activation of JNK signalling has a well‐established role in inducing cell migration in Drosophila wing disc.34, 35, 36 We therefore examined the expression of JNK target genes in these wing discs. Significantly elevated expression of JNK targets, including the matrix metalloproteinase MMP1 and an enhancer‐trapped lacZ reporter of the JNK inhibitor puckered (puc), was observed in cno expressing domain (Figure 1A‐D). The activation of JNK signalling can either promote cell proliferation or trigger cell death, depending on the levels of activation. Whereas, mild to moderate JNK activation is linked to proliferation, strong JNK activation additionally causes cell death.37, 38, 39, 40 Indeed, we observed increased cell death in the wing discs overexpressing cno, as indicated by the immunostaining of cleaved caspase3 and TUNEL staining (Figure 1E‐H). Thus, overexpression of cno in Drosophila wing disc induces mixed effects on cell proliferation, cell death and cell migration, which are connected to strong activation of JNK signalling.
Figure 1.
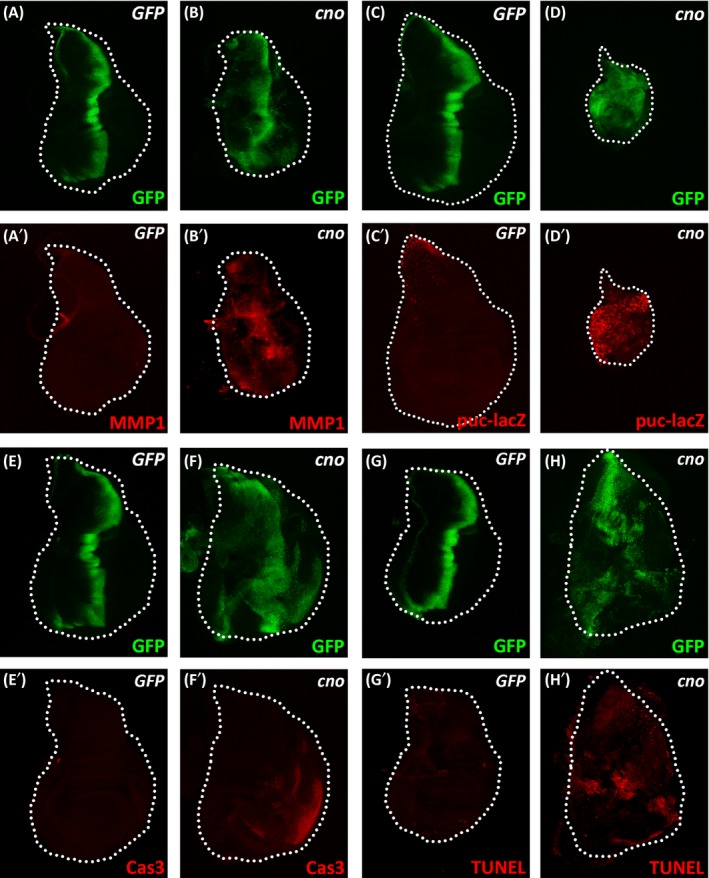
Overexpression of cno induces mixed effects. (A‐H) Immnostaining of MMP1, β‐gal, cleaved caspase3, or TUNEL staining in wing discs expressing ptc‐Gal4 UAS‐GFP/+; UAS‐GFP/+ (A, E and G), ptc‐Gal4 UAS‐GFP/+; UAS‐cno/+ (B, F and H), ptc‐Gal4 UAS‐GFP/+; UAS‐GFP/puc‐lacZ (C) or ptc‐Gal4 UAS‐GFP/+; UAS‐cno/puc‐lacZ (D). White dashed lines indicate the edges of the wing discs. Note the reduced disc size in the presence of enhancer‐trapped puc‐lacZ, which additionally increases JNK activity due to the removal of one copy of puc
3.2. Moderate alleviation of JNK activation augments Cno‐mediated proliferation
To investigate the roles of the JNK pathway in Cno‐mediated phenotypes in Drosophila wing disc, we examined the effects of differentially reducing the levels of Cno‐induced JNK activation. hemipterous (hep) and basket (bsk) encode the Drosophila homolog of JNKK and JNK, respectively. The knockdown of hep or bsk displayed different levels of suppression on activated JNK signalling (Supporting Information Figure S2). In a scrib knockdown induced mild JNK activation model, both hep knockdown and bsk knockdown effectively suppressed the activated expression of MMP1 (Supporting Information Figure S2A‐D). In contrast, in an eiger (egr, the Drosophila tumour necrosis factor superfamily ligand) overexpression‐induced small eye model, the knockdown of hep only weakly recovered the size of the eye, whereas, the knockdown of bsk largely rescued the eye phenotype (Supporting Information Figure S2E‐H). These data indicate that the knockdown of hep moderately inhibits the activation of JNK signalling, while the knockdown of bsk inhibits it more effectively. As suggested by the expression domain of GFP, the knockdown of hep or bsk at the A/P compartment boundary had no effect on cell proliferation or migration (Supporting Information Figure S3). Interestingly, cno overexpression in the presence of hep knockdown at the A/P compartment boundary induced massive cell proliferation and disc overgrowth, with the GFP‐positive cells spreading throughout the wing disc (Figure 2A,B, Supporting Information Figure S4). The combination with bsk knockdown displayed similar but weaker phenotype (Figure 2C). Similar effects by the knockdown of hep or bsk were also observed when cno was overexpressed in other compartments of the wing disc (Supporting Information Figure S5). The induction of MMP1 expression by cno overexpression was weakened by bsk knockdown but barely affected by hep knockdown, suggesting the requirement for the activation of JNK signalling to mediate the function of Cno in massive cell proliferation and migration (Figure 2A‐C). In addition, either hep knockdown or bsk knockdown largely eliminated the cell apoptosis induced by cno overexpression (Figure 2D‐F). Therefore, moderate alleviation of Cno‐induced JNK activation by hep knockdown predominantly displays growth‐promoting and pro‐migration effects, which can serve as a new type of Drosophila tumour model. In contrast, strong inhibition of Cno‐induced JNK activation by bsk knockdown only moderately promotes cell proliferation and migration. Importantly, the knockdown of cno in the presence of hep knockdown did not stimulate cell proliferation or migration, indicating that our observations above were not caused by dominant negative effects (Supporting Information Figure S6).
Figure 2.
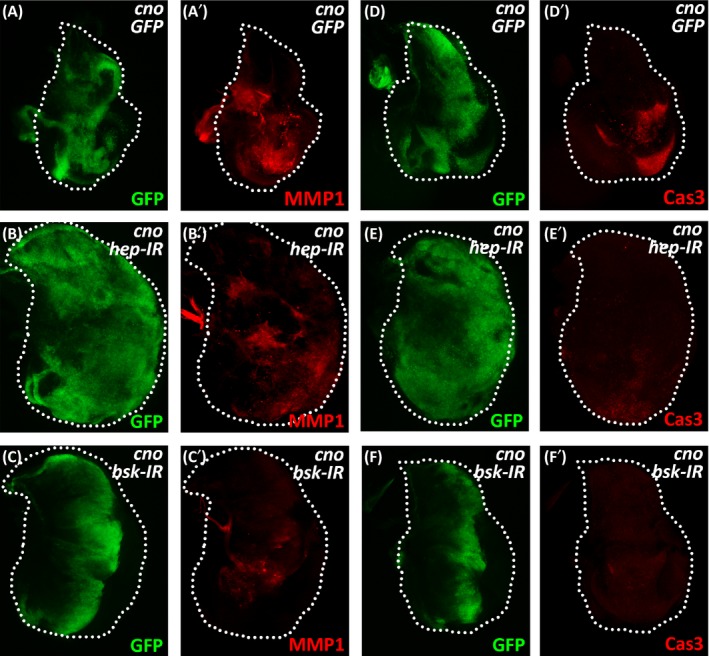
The levels of JNK activation on Cno‐induced proliferation. (A‐F) Immunostaining of MMP1 and cleaved caspase3 in wing discs expressing ptc‐Gal4 UAS‐GFP/UAS‐GFP; UAS‐cno/+ (A and D), ptc‐Gal4 UAS‐GFP/+; UAS‐cno/UAS‐hep‐IR (B and E) or ptc‐Gal4 UAS‐GFP/+; UAS‐cno/UAS‐bsk‐IR (C and F). White dashed lines indicate the edges of the wing discs
3.3. Cno induces proliferation via the Hippo signalling pathway
The Hippo signalling pathway is an evolutionarily conserved signal transduction pathway that plays vital roles in cell proliferation and apoptosis to control cell fate, organ size and tissue homeostasis.8 It has been reported that multiple components of the Hippo pathway are regulated directly by the cell adhesion and polarity machinery, and defects in cell polarity may result in tumorigenesis via dysfunction of the Hippo pathway.41 To investigate whether Cno‐induced overgrowth is mediated by the Hippo pathway, we examined the expression of target genes of Yorkie (Yki), the key transcriptional coactivator of Hippo signalling. We observed that the expression of Yki target gene expanded (ex) was prominently activated upon the overexpression of cno, which was further enhanced in the presence of hep knockdown or bsk knockdown (Figure 3A‐D). Noticeably, the knockdown of bsk was less potent in enhancing the expression of ex than the knockdown of hep, revealing the requirement of JNK activation for the induction of Yki targets (Figure 3C,D). Similarly, activated expression of other Yki target genes such as bantam (ban) and diap1 was also observed in this tumour model (Figure 3E‐H). Importantly, the knockdown of yki greatly reduced Cno‐induced cell overproliferation, even in the presence of hep knockdown (Figure 4). Consistently, Cno‐induced expression of Yki target gene wingless (wg) was largely suppressed by the knockdown of yki (Figure 4). These results confirm that Cno‐induced cell overproliferation depends on Hippo signalling and requires the induction of Yki target gene expression.
Figure 3.
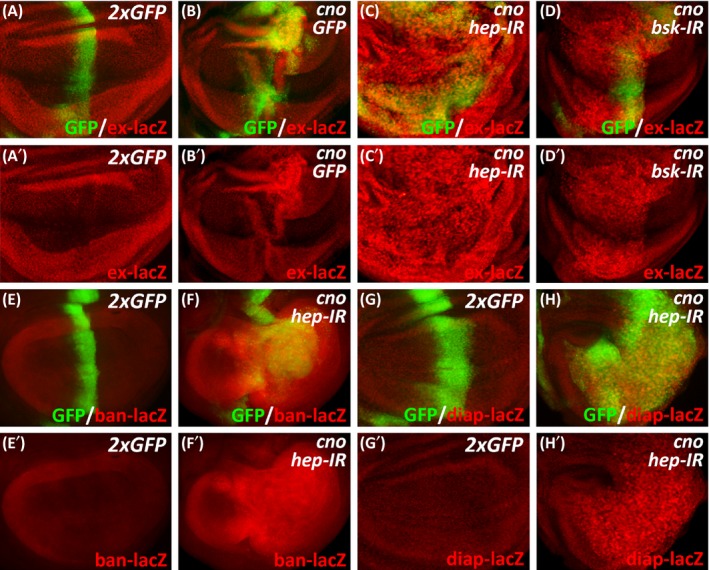
Cno‐mediated overgrowth depends on the Hippo signalling pathway. (A‐H) β‐galstaining of wing discs expressing ptc‐Gal4 UAS‐GFP/ex‐lacZ; UAS‐GFP/UAS‐GFP (A), ptc‐Gal4 UAS‐GFP/ex‐lacZ; UAS‐cno/UAS‐GFP (B), ptc‐Gal4 UAS‐GFP/ex‐lacZ; UAS‐cno/UAS‐hep‐IR (C), ptc‐Gal4 UAS‐GFP/ex‐lacZ; UAS‐cno/UAS‐bsk‐IR (D), ptc‐Gal4 UAS‐GFP/ban‐lacZ; UAS‐GFP/UAS‐GFP (E), ptc‐Gal4 UAS‐GFP/ban‐lacZ; UAS‐cno/UAS‐hep‐IR (F), ptc‐Gal4 UAS‐GFP/UAS‐GFP; UAS‐GFP/diap‐lacZ (G) or ptc‐Gal4 UAS‐GFP/+; UAS‐cno UAS‐hep‐IR/diap‐lacZ (H)
Figure 4.
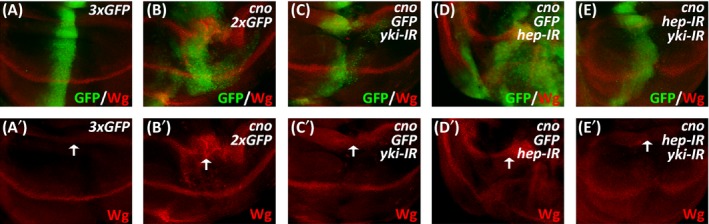
The effects of yki knockdown on Cno‐mediated overgrowth. (A‐E) Immunostaining of Wg in wing discs expressing ptc‐Gal4 UAS‐GFP/UAS‐GFP; UAS‐GFP/UAS‐GFP (A), ptc‐Gal4 UAS‐GFP/UAS‐GFP; UAS‐cno/UAS‐GFP (B), ptc‐Gal4 UAS‐GFP/UAS‐yki‐IR; UAS‐cno/UAS‐GFP (C), ptc‐Gal4 UAS‐GFP/UAS‐GFP; UAS‐cno/UAS‐hep‐IR (D) or ptc‐Gal4 UAS‐GFP/UAS‐yki‐IR; UAS‐cno/UAS‐hep‐IR (E). White arrows indicate Wg signals at the hinge region of the wing discs
3.4. The Ras‐MAPK signalling pathway regulates Cno‐induced cell proliferation
As strong inhibition of JNK activation still effectively activated the expression of Yki target genes, we speculated that Cno might affect additional signalling, in addition to JNK signalling, to regulate the Hippo pathway. The Ras‐MEK‐ERK pathway is one branch of the Mitogen‐activated protein kinase (MAPK) pathways. It transduces extracellular signals into the nucleus through sequential activation of the MEKK‐MEK‐MAPK kinase cascade, regulates multiple physiological processes such as cell proliferation, differentiation and migration, and participates in the regulation of Hippo signalling.34, 42, 43, 44, 45 To investigate whether the Ras‐MAPK pathway is involved in Cno‐induced overproliferation, we examined the effects of ras knockdown in above genetic setups. As controls, the knockdown of ras at the A/P compartment boundary or the dorsal compartment of the wing disc showed no effect on cell proliferation (Supporting Information Figure S7). In contrast, cell overproliferation induced by the overexpression of cno, in the absence or presence of hep knockdown, was strongly inhibited by the knockdown of ras (Figure 5A‐H). Intriguingly, despite the effective inhibition of cell overproliferation, the knockdown of ras seemed unable to block cell migration induced by cno overexpression as was achieved by bsk knockdown (Figure 2C and Figure 5A‐H). These data suggest that Cno‐induced phenotypes can be partially separated and are mediated by distinct pathways. Whereas, the Ras‐MAPK pathway specifically contributes to the regulation of cell proliferation, the JNK pathway regulates both cell proliferation and migration. It has been reported that the Ras‐MAPK signalling influences cell growth through an Yki‐dependent mechanism.46 We thus examined whether the effects of ras knockdown on Cno‐induced proliferation were dependent on the Hippo pathway. Indeed, the knockdown of ras significantly impaired Cno‐induced expression of Yki target genes (diap1 and wg) in the wing disc (Figure 5I‐L). In addition, the knockdown of MEK, another core component of Ras‐MAPK signalling, exhibited similar effects as the knockdown of ras on Cno‐induced phenotypes in the wing disc, including cell proliferation, cell migration and Yki target gene expression (Supporting Information Figure S8). Together, our results suggest that Cno can activate both JNK and Ras‐MAPK signalling and regulate cell proliferation via the downstream Hippo signalling.
Figure 5.
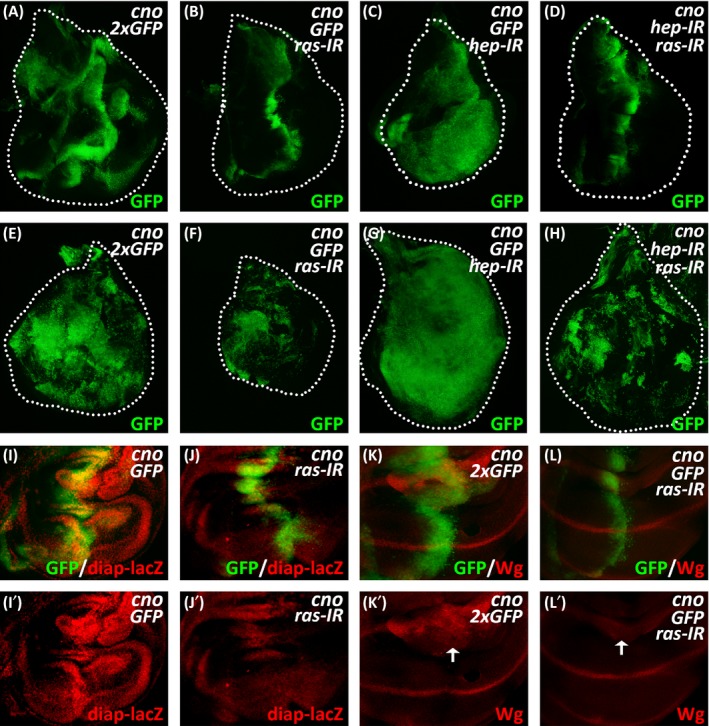
Ras‐MAPK signalling acts between Cno and Hippo signalling. (A‐D) The proliferation of GFP‐positive cells at the A/P compartment boundary of the wing disc expressing ptc‐Gal4 UAS‐GFP/UAS‐GFP; UAS‐cno/UAS‐GFP (A), ptc‐Gal4 UAS‐GFP/UAS‐ras‐IR; UAS‐cno/UAS‐GFP (B), ptc‐Gal4 UAS‐GFP/UAS‐GFP; UAS‐cno/UAS‐hep‐IR (C) or ptc‐Gal4 UAS‐GFP/UAS‐ras‐IR; UAS‐cno/UAS‐hep‐IR (D). (E‐H) The proliferation of GFP‐positive cells in the dorsal region of the wing discs expressing ap‐Gal4 UAS‐GFP/UAS‐GFP; UAS‐cno/UAS‐GFP (E), ap‐Gal4UAS‐GFP/UAS‐ras‐IR; UAS‐cno/UAS‐GFP(F), ap‐Gal4 UAS‐GFP/UAS‐GFP; UAS‐cno/UAS‐hep‐IR (G) or ap‐Gal4 UAS‐GFP/UAS‐ras‐IR; UAS‐cno/UAS‐hep‐IR (H). White dashed lines display the edges of the wing discs. (I‐L) Imunnostaining of β‐gal or Wg in wing disc expressing ptc‐Gal4 UAS‐GFP/UAS‐GFP; UAS‐cno/diap‐lacZ (I), ptc‐Gal4 UAS‐GFP/UAS‐ras‐IR; UAS‐cno/diap‐lacZ (J), ptc‐Gal4 UAS‐GFP/UAS‐GFP; UAS‐cno/UAS‐GFP (K) or ptc‐Gal4 UAS‐GFP/UAS‐ras‐IR; UAS‐cno/UAS‐GFP (L). White arrows indicate Wg signals at the hinge region of the wing discs
4. DISCUSSION
Although dual regulation of cell proliferation by polarity proteins have been observed and studied for quite a period, controversies about the precise roles of cell polarity in such an important cellular process still exist.13 For instance, the adhesion molecular Afadin, which contributes to forming and sustaining cellular junctions and cell polarity, has been reported to have pro‐ or anti‐tumorigenic functions.47 In this work, by expressing Cno, the Drosophila homolog of Afadin, we observed mixed phenotypes of cell death, growth and migration. Since the JNK signalling pathway is well known to participate in all of these cellular processes,34 we thus checked the activity of the JNK pathway and indeed found elevated expression of the JNK target genes. However, when we attempted to dissect the mixed phenotypes by knocking down key kinases of the JNK pathway, we observed unexpected outcomes. Particularly, knocking down hep greatly promoted Cno‐induced overgrowth, while knocking down bsk had a milder effect. Given the stronger inhibition of the JNK activation by the knockdown of bsk than the knockdown of hep, we conclude that moderately alleviating the levels of JNK activation can largely assist Cno‐mediated cell proliferation.
Strong activation of the JNK pathway mainly triggers cell death and migration instead of the mixture of cell death and proliferation, implying that additional mechanisms beyond the JNK pathway underlie the effects ofcno overexpression. Since the Hippo signalling pathway is one of the most well‐known regulators that control the process of cell proliferation and is frequently associated with tumour initiation and progression,8, 41 we also examined the downstream readouts of this pathway in wing disc with cno overexpression, and confirmed the involvement of the Hippo pathway. Essentially, knocking down hep substantially increased the expression of Hippo target genes induced by cno overexpression, while knocking down bsk exerted lesser effects on them, suggesting that moderate activation of the JNK pathway also promotes cell growth via the regulation of Hippo signalling.
It is proposed that the contribution of the JNK signalling to cell proliferation is converted from anti‐ to pro‐tumorigenic by Ras,48 the core component of the Ras‐MAPK signalling pathway. We thus investigated the roles of the Ras‐MAPK pathway in Cno‐mediated phenotypes. We found that the knockdown of either ras or its downstream kinase MEK significantly inhibited Cno‐mediated cell proliferation, especially in the context of the massive overgrowth induced by concurrent hep knockdown. Notably, neither ras nor mek knockdown was capable of reducing Cno‐induced cell death and migration, implying that there exist multiple mechanisms that are responsible for the mixed phenotypes induced by cno overexpression. Importantly, by examining the expression of several Hippo targets, we could determine that the effects of ras or mek knockdown were mediated by the regulation of Hippo signalling.
Based on the observations above, we propose a model for the mechanisms underlying Cno‐mediated phenotypes (Figure 6). Overexpression of cno in Drosophila wing disc activates both the JNK and Ras‐MAPK signalling pathways. While the Ras‐MAPK pathway mainly promotes cell growth via Hippo signalling, the effects of the JNK pathway are more complicated. When the JNK pathway is strongly activated by cno overexpression, it predominantly triggers cell death and cell migration. Combined with the overgrowth induced by the Ras‐Hippo axis, the cells display mixed phenotypes of death and growth. However, when the levels of JNK activation are moderately reduced, it mostly promotes cell growth rather than cell death via the Hippo pathway. Under this context, cooperating with the Ras‐MAPK signalling, the cells display massive overproliferation.
Figure 6.
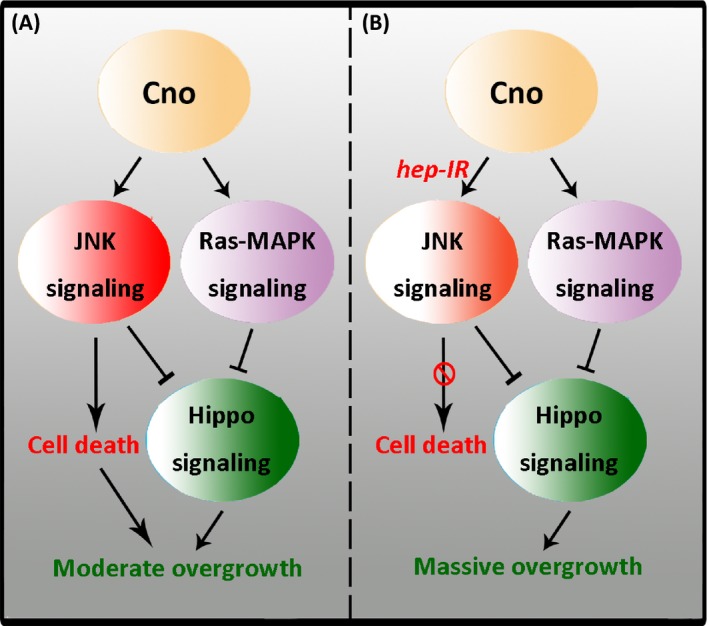
The working model for Cno‐mediated effects on cell proliferation. A, Overexpression of cno simultaneously activates JNK and Ras‐MAPK signalling, inducing both cell proliferation and cell death. B, Moderately reducing the activation levels of JNK signalling suppresses the effect of Cno on cell death, inducing massive cell overproliferation via Hippo signalling
In conclusion, the influences of polarity proteins on cell fate determination are complicated and context‐dependent in many occasions. As illustrated in this study, the polarity protein Cno activates both the JNK pathway and the Ras‐MAPK pathway. Importantly, polarity proteins affect cell fate not only by disruption of cell polarity, but also by acting as signal transducing molecules to participate in signalling pathways that regulate cell death and proliferation. Therefore, the use of anti‐cancer drugs or treatment targeting polarity proteins must consider their complicated roles in cell proliferation and the signalling pathways they are associated with. In addition, the combination of cno expression and hep knockdown in Drosophila wing disc provides a new model for studying cell proliferation and tumour formation in this classic model organism.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
HS designed the research; ZM, PL and XH performed the experiments; HS and ZM analysed the data and wrote the paper.
Supporting information
ACKNOWLEDGEMENTS
We thank the Developmental Studies Hybridoma Bank, the Bloomington Drosophila Stock Center, the Vienna Drosophila RNAi Center and the NIG‐Fly Stock Center for fly strains, and also thank Prof. Ulrike Gaul, Prof. Lei Xue and Prof. Lei Zhang for fly strains and their conceptual advices. This research was supported by the National Natural Science Foundation of China (31571498 and 81773434).
Ma Z, Li P, Hu X, Song H. Polarity protein Canoe mediates overproliferation via modulation of JNK, Ras‐MAPK and Hippo signalling. Cell Prolif. 2018;52:e12529 10.1111/cpr.12529
REFERENCES
- 1. Nelson WJ, Dickinson DJ, Weis WI. Roles of cadherins and catenins in cell‐cell adhesion and epithelial cell polarity. Prog Mol Biol Transl Sci. 2013;116:3‐23. [DOI] [PubMed] [Google Scholar]
- 2. Carthew RW. Adhesion proteins and the control of cell shape. Curr Opin Genet Dev. 2005;15(4):358‐363. [DOI] [PubMed] [Google Scholar]
- 3. McCaffrey LM, Macara IG. Epithelial organization, cell polarity and tumorigenesis. Trends Cell Biol. 2011;21(12):727‐735. [DOI] [PubMed] [Google Scholar]
- 4. Martin‐Belmonte F, Mostov K. Regulation of cell polarity during epithelial morphogenesis. Curr Opin Cell Biol. 2008;20(2):227‐234. [DOI] [PubMed] [Google Scholar]
- 5. Ellenbroek SI, Iden S, Collard JG. Cell polarity proteins and cancer. Semin Cancer Biol. 2012;22(3):208‐215. [DOI] [PubMed] [Google Scholar]
- 6. Warner SJ, Yashiro H, Longmore GD. The Cdc42/Par6/aPKC polarity complex regulates apoptosis‐induced compensatory proliferation in epithelia. Curr Biol. 2010;20(8):677‐686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Varelas X, Samavarchi‐Tehrani P, Narimatsu M, et al. The Crumbs complex couples cell density sensing to Hippo‐dependent control of the TGF‐beta‐SMAD pathway. Dev Cell. 2010;19(6):831‐844. [DOI] [PubMed] [Google Scholar]
- 8. Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19(4):491‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vorhagen S, Kleefisch D, Persa OD, et al. Shared and independent functions of aPKClambda and Par3 in skin tumorigenesis. Oncogene. 2018; 10.1038/s41388-018-0313-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhan L, Rosenberg A, Bergami KC, et al. Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell. 2008;135(5):865‐878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial‐mesenchymal transitions in development and disease. Cell. 2009;139(5):871‐890. [DOI] [PubMed] [Google Scholar]
- 12. Keder A, Rives‐Quinto N, Aerne BL, Franco M, Tapon N, Carmena A. The hippo pathway core cassette regulates asymmetric cell division. Curr Biol. 2015;25(21):2739‐2750. [DOI] [PubMed] [Google Scholar]
- 13. Halaoui R, McCaffrey L. Rewiring cell polarity signaling in cancer. Oncogene. 2015;34(8):939‐950. [DOI] [PubMed] [Google Scholar]
- 14. Balakrishnan S, Bhat FA, Raja Singh P, et al. Gold nanoparticle‐conjugated quercetin inhibits epithelial‐mesenchymal transition, angiogenesis and invasiveness via EGFR/VEGFR‐2‐mediated pathway in breast cancer. Cell Prolif. 2016;49(6):678‐697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mandai K, Nakanishi H, Satoh A, et al. Afadin: a novel actin filament‐binding protein with one PDZ domain localized at cadherin‐based cell‐to‐cell adherens junction. J Cell Biol. 1997;139(2):517‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Takai Y, Ikeda W, Ogita H, Rikitake Y. The immunoglobulin‐like cell adhesion molecule nectin and its associated protein afadin. Annu Rev Cell Dev Biol. 2008;24:309‐342. [DOI] [PubMed] [Google Scholar]
- 17. Komura H, Ogita H, Ikeda W, Mizoguchi A, Miyoshi J, Takai Y. Establishment of cell polarity by afadin during the formation of embryoid bodies. Genes Cells. 2008;13(1):79‐90. [DOI] [PubMed] [Google Scholar]
- 18. Prasad R, Gu Y, Alder H, et al. Cloning of the ALL‐1 fusion partner, the AF‐6 gene, involved in acute myeloid leukemias with the t(6;11) chromosome translocation. Cancer Res. 1993;53(23):5624‐5628. [PubMed] [Google Scholar]
- 19. Letessier A, Garrido‐Urbani S, Ginestier C, et al. Correlated break at PARK2/FRA6E and loss of AF‐6/Afadin protein expression are associated with poor outcome in breast cancer. Oncogene. 2007;26(2):298‐307. [DOI] [PubMed] [Google Scholar]
- 20. Xu Y, Chang R, Peng Z, et al. Loss of polarity protein AF6 promotes pancreatic cancer metastasis by inducing Snail expression. Nat Commun. 2015;6:7184. [DOI] [PubMed] [Google Scholar]
- 21. Charpin C, Tavassoli F, Secq V, et al. Validation of an immunohistochemical signature predictive of 8‐year outcome for patients with breast carcinoma. Int J Cancer. 2012;131(3):E236–E243. [DOI] [PubMed] [Google Scholar]
- 22. Elloul S, Kedrin D, Knoblauch NW, Beck AH, Toker A. The adherens junction protein afadin is an AKT substrate that regulates breast cancer cell migration. Mol Cancer Res. 2014;12(3):464‐476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miles WO, Dyson NJ, Walker JA. Modeling tumor invasion and metastasis in Drosophila . Dis Model Mech. 2011;4(6):753‐761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tipping M, Perrimon N. Drosophila as a model for context‐dependent tumorigenesis. J Cell Physiol. 2014;229(1):27‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Woodhouse E, Hersperger E, Stetler‐Stevenson WG, Liotta LA, Shearn A. Increased type IV collagenase in lgl‐induced invasive tumors of Drosophila . Cell Growth Differ. 1994;5(2):151‐159. [PubMed] [Google Scholar]
- 26. Woodhouse E, Hersperger E, Shearn A. Growth, metastasis, and invasiveness of Drosophila tumors caused by mutations in specific tumor suppressor genes. Dev Genes Evol. 1998;207(8):542‐550. [DOI] [PubMed] [Google Scholar]
- 27. Vaccari T, Bilder D. At the crossroads of polarity, proliferation and apoptosis: the use of Drosophila to unravel the multifaceted role of endocytosis in tumor suppression. Mol Oncol. 2009;3(4):354‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Igaki T, Pagliarini RA, Xu T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila . Curr Biol. 2006;16(11):1139‐1146. [DOI] [PubMed] [Google Scholar]
- 29. Read RD, Cavenee WK, Furnari FB, Thomas JB. A drosophila model for EGFR‐Ras and PI3K‐dependent human glioma. PLoS Genet. 2009;5(2):e1000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Witte HT, Jeibmann A, Klambt C, Paulus W. Modeling glioma growth and invasion in Drosophila melanogaster. Neoplasia. 2009;11(9):882‐888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vidal M, Larson DE, Cagan RL. Csk‐deficient boundary cells are eliminated from normal Drosophila epithelia by exclusion, migration, and apoptosis. Dev Cell. 2006;10(1):33‐44. [DOI] [PubMed] [Google Scholar]
- 32. Vidal M, Salavaggione L, Ylagan L, et al. A role for the epithelial microenvironment at tumor boundaries: evidence from Drosophila and human squamous cell carcinomas. Am J Pathol. 2010;176(6):3007‐3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu S, Huang J, Dong J, Pan D. hippo encodes a Ste‐20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell. 2003;114(4):445‐456. [DOI] [PubMed] [Google Scholar]
- 34. Huang C, Jacobson K, Schaller MD. MAP kinases and cell migration. J Cell Sci. 2004;117(Pt 20):4619‐4628. [DOI] [PubMed] [Google Scholar]
- 35. Tang Y, Liu L, Wang P, Chen D, Wu Z, Tang C. Periostin promotes migration and osteogenic differentiation of human periodontal ligament mesenchymal stem cells via the Jun amino‐terminal kinases (JNK) pathway under inflammatory conditions. Cell Prolif. 2017;50(6):e12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Q, Deng S, Sun K, et al. MMP‐2 and Notch signal pathway regulate migration of adipose‐derived stem cells and chondrocytes in co‐culture systems. Cell Prolif. 2017;50(6):e12385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Solinas G, Becattini B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol Metab. 2017;6(2):174‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roulston A, Reinhard C, Amiri P, Williams LT. Early activation of c‐Jun N‐terminal kinase and p38 kinase regulate cell survival in response to tumor necrosis factor alpha. J Biol Chem. 1998;273(17):10232‐10239. [DOI] [PubMed] [Google Scholar]
- 39. Ventura JJ, Hubner A, Zhang C, Flavell RA, Shokat KM, Davis RJ. Chemical genetic analysis of the time course of signal transduction by JNK. Mol Cell. 2006;21(5):701‐710. [DOI] [PubMed] [Google Scholar]
- 40. Putcha GV, Le S, Frank S, et al. JNK‐mediated BIM phosphorylation potentiates BAX‐dependent apoptosis. Neuron. 2003;38(6):899‐914. [DOI] [PubMed] [Google Scholar]
- 41. Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 2015;163(4):811‐828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Santarpia L, Lippman SM, El‐Naggar AK. Targeting the MAPK‐RAS‐RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16(1):103‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Y, Jia Z, Diao S, et al. IGFBP5 enhances osteogenic differentiation potential of periodontal ligament stem cells and Wharton's jelly umbilical cord stem cells, via the JNK and MEK/Erk signalling pathways. Cell Prolif. 2016;49(5):618‐627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang J, Chen H, Cao P, et al. Inflammatory cytokines induce caveolin‐1/beta‐catenin signalling in rat nucleus pulposus cell apoptosis through the p38 MAPK pathway. Cell Prolif. 2016;49(3):362‐372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Niu Z, Mu H, Zhu H, Wu J, Hua J. p38 MAPK pathway is essential for self‐renewal of mouse male germline stem cells (mGSCs). Cell Prolif. 2017;50(1):e12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reddy BV, Irvine KD. Regulation of Hippo signaling by EGFR‐MAPK signaling through Ajuba family proteins. Dev Cell. 2013;24(5):459‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mandai K, Rikitake Y, Shimono Y, Takai Y. Afadin/AF‐6 and canoe: roles in cell adhesion and beyond. Prog Mol Biol Transl Sci. 2013;116:433‐454. [DOI] [PubMed] [Google Scholar]
- 48. Enomoto M, Kizawa D, Ohsawa S, Igaki T. JNK signaling is converted from anti‐ to pro‐tumor pathway by Ras‐mediated switch of Warts activity. Dev Biol. 2015;403(2):162‐171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials