

Abstract
APOBEC3B (A3B) deamination activity on ssDNA is considered a contributing factor to tumor heterogeneity and drug resistance in a number of human cancers. Despite its clinical impact, little is known about A3B ssDNA substrate preference. We have used nuclear magnetic resonance to monitor the catalytic turnover of A3B substrates in real-time. This study reports preferred nucleotide sequences for A3B substrates, including optimized 4-mer oligonucleotides, and reveals a breadth of substrate recognition that includes DNA sequences known to be mutated in drug-resistant cancer clones. Our results are consistent with available clinical and structural data and may inform the design of substrate-based A3B inhibitors.
The human APOBEC3 (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) cytidine deaminase family comprises seven members, which convert C to U on single stranded DNA (ssDNA), among other functions.1,2 APOBEC proteins contain either one (A3A/A3C/A3H) or two domains (A3B/A3DE/A3F/A3G) with similar overall architecture. Although each domain incorporates a zinc binding site, in the case of A3B only the C-terminal domain (CTD) has been associated with catalytic activity.3,4 Activity-modulating functions of the N-terminal domain (NTD) have been proposed;2 however, expression and purification of full-length recombinant wild-type A3B has proven challenging.4 As part of the innate immune system, APOBEC proteins are upregulated in response to viral infection, for example, human immunodeficiency virus type 1 or human papilloma virus, where they deaminate viral genomes and restrict viral replication.1,5 However, several members have also been linked to cancer, and A3B in particular has been associated with increased mutational burden in head and neck, cervical, lung, bladder, and breast cancer.6 Depending on subsequent repair mechanisms, APOBEC-mediated deamination predominantly causes C→T transition and C→G transversion,7 thereby contributing to mutational load, tumor heterogeneity, and the development of drug resistance.8 A3B inhibition with small molecules is therefore considered a promising therapeutic strategy to combat drug resistance in cancer patients. Several studies have analyzed the sequence context of APOBEC-mediated deamination in vitro and from patient sequencing data, identifying a consensus sequence of 5′-TC-3′ (the target C is henceforth underlined) for A3A, A3B, A3F, and A3H, while A3G prefers a 5′-CCC-3′ consensus sequence.2,9−19 A recent study has further evaluated the effect of oligonucleotide length and substrate context on the A3B CTD deamination rate.20 However, a systematic study of A3B substrate sequence preference has not been reported. We use real-time nuclear magnetic resonance (NMR) experiments to determine the impact of substrate sequence and length on turnover rate by wild-type A3B CTD. Through these studies, we further inform the biological and clinical relevance of A3B activity and gain insight into its molecular mechanism.
We found that the 10-mer APOBEC3B substrate sequence 5′-TTATTCATAT-3′ contained in the 30 nucleotide (nt) FAM-labeled ssDNA substrate reported by Fu et al.3 was readily deaminated by A3B CTD in a fluorescence-based deamination assay (Figure S1) and by NMR (Figure 1A, Table S1) with an initial rate of 0.174 ± 0.042 mM·h–1 (n = 3, ±SD). By contrast, no deamination of the mononucleotide 2′-deoxycytidine 5′-monophosphate (5′-dC-3′, dCMP) or the commonly reported A3B preferred motif 5′-TCA-3′10,13 was observed after a 5 h incubation (Figure 1A, Table S1). When we extended the incubation time to 20 h, we detected 20% conversion for 5′-TCA-3′, a deamination rate of 0.007 ± 0.001 mM·h–1 (n = 3, ±SD), 25-fold slower than for the 10-mer 5′-TTATTCATAT-3′. We did not observe any deamination for dCMP even after 50 h incubation.
Figure 1.

Impact of (+) and (−) side nucleotides on substrate turnover. (A) Representative 1H NMR spectra recorded over a time course are shown for the 10-mer 5′-TTATTCATAT-3′, dCMP and 5′-TCA-3′. The intensities of the C peak were used to monitor substrate turnover and to calculate the initial reaction rates. (B) Time course of deamination rates for 10-mer 5′-TTATTCATAT-3′, dCMP, and (+) and (−) side only oligonucleotides. (C) Addition of +1A (left) and two T nucleotides at the (−) side (right) increase turnover of the substrate oligo, whereas a single T on the (−) side appears detrimental. ns not significant, *p-value < 0.05, ***p-value < 0.001, One-way ANOVA and Bonferroni test.
Using the 10-mer oligonucleotide 5′-TTATTCATAT-3′ as a benchmark, we set out to determine the contribution to A3B CTD-mediated deamination of nucleotides both (−) (i.e., 5′) and (+) (i.e., 3′) to C. Dinucleotides, such as 5′-TC-3′ and 5′-CA-3′, were not deaminated by A3B CTD. When we added a second nucleotide on the (+) side, we observed deamination of the trimer 5′-CAT-3′ with an initial rate of 0.023 ± 0.001 mM·h–1 (n = 3, ±SD) (Figure 1B, Figure S2, and Table S1), 8-fold slower than the 10-mer. Notably, 5′-CAT-3′ is the smallest oligonucleotide for which we observed deamination after a 5 h incubation. Shi et al. suggested that aromatic π-stacking of nucleobases positioned +1 to +3 to the substrate C contribute to productive deamination.13 However, in our hands, further extension of the 5′-CAT-3′ oligonucleotide to tetra- and pentanucleotides using a poly-T sequence had little effect (Figure 1B, Figure S2, and Table S1), indicating that nucleotides at positions +3 and +4 do not further enhance substrate turnover rate. A similar study on the (−) side of the substrate C showed lower deamination rate compared with their matched (+) side equivalents (Figure 1B, Figure S2, and Table S1). For example, no deamination was observed for 5′-TTC-3′ after 5 h incubation and a deamination rate could only be obtained with extended incubation time (15 h) [0.009 ± 0.002 mM·h–1 (n = 4, ±SD) (Figure 1B, Figure S2, and Table S1)]. The initial deamination rates of oligonucleotides extended in either the (−) or the (+) direction are depicted as bar charts in Figure S2, and deamination rates are provided in Table S1.
These results prompted us to further investigate the +1 nucleotide. Intriguingly, introduction of A to the +1 position of 5′-TTC-3′ increased the deamination rate to a level similar to the 10-mer rate [5′-TTCA-3′ initial rate = 0.107 ± 0.014 mM·h–1 (n = 3, ±SD)]. This effect was also observed for 5′-ATTC-3′; addition of a +1 A (5′-ATTCA-3′) fully restored the deamination rate to that of the 10-mer (Figure 1C, left, and Table S1). These observations suggest that the +1 nucleotide is important for productive substrate turnover. Analogously, we added T to the −1 position of oligonucleotide 5′-CAT-3′. The resultant 5′-TCAT-3′ sequence showed low catalytic turnover, which could be improved by addition of a second nucleotide on the (−)-side (Figure 1C, right, and Table S1). Thus, only one nucleotide at the (−)-side appears detrimental, whereas addition of both −1 and −2 nucleotides enhances the deamination rate ∼6-fold compared to the 5′-CAT-3′ trinucleotide. Crystal structures of A3A and a chimeric A3A/A3B CTD crystal structure in complex with ssDNA substrates reveal large conformational changes of Tyr132 (A3A), Tyr315 (A3B), and Arg211 (A3B)13 in relation to the apo structures16,21 in order to accommodate T at the −1 position. Our results suggest that the specific interactions of −1 T with Asp314 are not sufficient to stabilize a productive protein conformation, and further interactions of the −2 nucleotide with the protein may be required. This could explain why deamination, albeit weakly, of (+)-side only nucleotides was observed; the turnover of such substrates may not require such conformational adaptation of A3B.
As the shortest oligonucleotide substrate subject to notable deamination was the tetramer 5′-TTCA-3′, we investigated whether the directionality of ssDNA presentation is important for short oligonucleotides. We tested the corresponding 5′-ACTT-3′ sequence (equivalent to 3′-TTCA-5′) and observed no deamination (Figure 2A and Table S1). Using a crystal structure of 5′-TTCA-3′ in complex with the A3A/A3B CTD chimera,13 we modeled 3′-dC-5′ into the catalytic site to understand the difference between 5′-dC-3′ and 3′-dC-5′ binding at an atomic level. Constraining the cytidine base within the catalytic site to a productive binding mode and altering the ssDNA directionality from 5′-dC-3′ to 3′-dC-5′ revealed severe clashes with the protein surface (Figure 2B) due to the fixed chirality of the ribose sugar (Figure 2C).
Figure 2.

Influence of ssDNA directionality on substrate turnover. (A) The 5′ → 3′ sequence TTCA but not the corresponding 3′ → 5′ TTCA (equivalent to 5′-ACTT-3′) oligonucleotide is deaminated by A3B CTD. (B) Binding of 3′ → 5′ deoxycytidine monophosphate (dCMP, orange), but not 5′ → 3′ dCMP (green) results in severe clashes with the A3B protein (blue cartoon/spheres and gray surface). Therefore, only the 5′ → 3′ dCMP is compatible with binding; the crystal structure conformation of the base is fixed in a productive binding mode; PDB entry 5TD5 was used.13 (C) Due to the chirality of the deoxyribose sugar, the 3′ and 5′ ends adopt different vectors in 5′ → 3′ and 3′ → 5′ dCMP; the cytosine base is depicted in the same orientation, which is required for productive deamination (Figure 2B).
Work by Byeon et al.9 suggested that A at the −2 position, that is, 5′-ATCA-3′, is a better A3B substrate than 5′-TTCA-3′, and consistent with their report, we determined an initial rate of 0.205 ± 0.041 mM·h–1 (n = 3, ±SD) for 5′-ATCA-3′ compared to 0.107 ± 0.014 mM·h–1 (n = 3, ±SD) for 5′-TTCA-3′ (p-value = 0.0166, unpaired t test). This observation raised further questions concerning A3B substrate preference, and we therefore systematically evaluated the impact of all possible nucleobases at positions −2, −1, and +1 using the 5′-ATCA-3′ sequence as a benchmark.
Consistent with previous reports,9 only −1 T was compatible with substrate turnover in a 4-mer oligonucleotide substrate. Deamination was reduced with −1 C and absent with −1 A and −1 G (Figure 3A and Table S1); probably due to extensive interactions of −1 T with A3B13 and potential steric restrictions associated with the binding of larger purine bases.22
Figure 3.

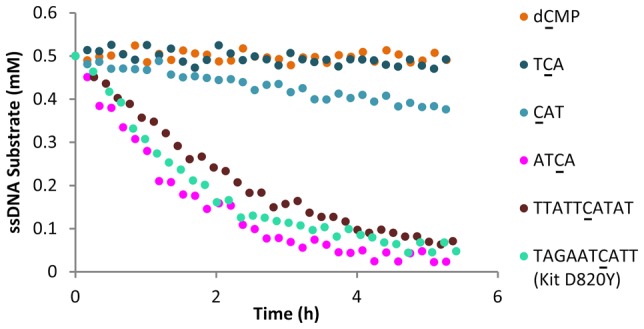
A3B CTD base preference at different substrate positions translates to biologically relevant DNA sequences. (A) Based on the 5′-ATCA-3′ sequence as a benchmark, the effect of different bases at positions −2, −1, and +1 on substrate turnover was evaluated. Preference for G at +1 is also observed with the (+)-side trimer oligonucleotides (p-value = 0.0046, unpaired t test). (B) Oligonucleotides representing commonly observed drug resistance mutations containing identified substrate sequences 5′-TTCA-3′ (PIK3CA E452), 5′-ATCA-3′ (Kit D820), 5′-GTCG-3′ (Smo D473), and 5′-ATCG-3′ (Kit D716) are deaminated at a similar or higher rate to the 10-mer positive control 5′-TTATTCATAT-3′, whereas no turnover was observed for the negative control 5′-TACA-3′ (Kit T670) after 20 h. Notably, all drug resistance mutations except Kit T670I affect the complementary strand. The 10-mer and 5′-CAT-3′ deamination rates from previous experiments are plotted as a reference. ns not significant, *p-value < 0.05, **p-value < 0.01, ***p-value < 0.001, One-way ANOVA and Bonferroni test.
Variation of the −2 nucleobase also influenced substrate turnover, with −2 G representing the best deamination substrate (Figure 3A and Table S1).
Variations at the +1 position of 4-mer substrates had less impact compared to the −1 position with 5′-ATCG-3′ the best substrate (Figure 3A and Table S1). Preference for +1 G was also observed in the (+)-side only trinucleotides, with initial rates of 0.023 ± 0.001 (n = 3, ± SD) vs 0.093 ± 0.021 (n = 3, ± SD) mM·h–1 for 5′-CAT-3′ and 5′-CGT-3′ (Figure 3A and Table S1). Combining these position-dependent results suggested 5′-GTCG-3′ as an optimal A3B substrate; this oligonucleotide was deaminated at a similar rate to 5′-ATCG-3′ and 5′-GTCA-3′. Intriguingly, these 4-mer substrates were deaminated at a similar rate compared to the control 10-mer oligonucleotide 5′-TTATTCATAT-3′ (p-value > 0.05 for 5′-ATCG-3′, 5′-GTCA-3′, and 5′-GTCG-3′, One-way ANOVA and Bonferroni test). Other studies have suggested that longer ssDNA oligo substrates are required for binding22 and deamination by the closely related A3A enzyme;17 however, our data demonstrates proficient A3B substrate turnover with short oligos provided the sequence context is appropriate.
To further investigate the biological relevance of our findings, we tested commonly observed mutations in cancer patients, where mutation of a C base is consistent with the generation of clones known to confer resistance to cancer therapy (Figure 3B and Table S1). For example, mutations within the Kit and Smoothened (Smo) genes confer resistance to targeted cancer drugs such as imatinib (Kit D820Y and D716N) and vismodegib (Smo D473H/N/Y).23 These clinically identified mutations may all result from A3B-mediated lesions, which can be further processed by DNA repair mechanisms to give the observed amino acid change. Oligonucleotides (10-mers) containing mutation sites within the identified substrate sequences 5′-ATCA-3′ (Kit D820Y), 5′-ATCG-3′ (Kit D716N), and 5′-GTCG-3′ (Smo D473H/N/Y) were rapidly deaminated at a similar rate to the 5′-TTATTCATAT-3′ 10-mer substrate. Furthermore, the oligonucleotide representing the PIK3CA E542K mutation previously associated with A3B-activity24 was also deaminated. In contrast, no deamination of 5′-TACA-3′ (Kit T670I), which is not a predicted A3B substrate, was observed after 20 h. These findings provide a mechanistic rationale for A3B-mediated generation of mutations that sustain the emergence of resistant clones under the selective pressure of targeted cancer therapy.
Deamination of 5′-GTCG-3′ and 5′-ATCG-3′-containing substrates at rates comparable to the 5′-TTATTCATAT-3′ 10-mer suggests that their productive deamination is determined by both specific interactions of −1, −2, and +1 substrate nucleotides, as well as the modified C, with A3B-CTD. Furthermore, the secondary structure and conformation of the substrate may be important, consistent with the reported increased A3B binding of hairpin versus linear oligonucleotides.25
Previous A3B CTD NMR deamination studies of +1 base variations in the context of 40-mers are consistent with our results,9 thereby suggesting that the preference for bases immediately adjacent to the substrate C is conserved and independent from the composition or length of the overall oligonucleotide. This suggests that interactions with residues close to, but not within, the catalytic site are required for tight and specific binding of ssDNA, or for optimal positioning of the substrate C for productive deamination, or both. As discussed above, structural data on A3B ssDNA binding is fully consistent with the preference for T at position −1 and gives insights into possible induced-fit mechanisms for substrate binding. However, structural data to inform on potential interactions of −2 and +1 nucleotides is lacking because the reported A3B–ssDNA crystal structure includes alterations to A3B loop regions 1 and 3.13 Due to their proximity to the substrate-binding site, these loops are likely to be critical to binding of the −2 and +1 nucleobases (Figure S3).
Conflicting evidence is available regarding the substrate specificity of the full-length A3B protein compared to CTD alone: A study by Haché et al. showed that APOBEC substrate specificity is not affected by the NTD sequence and is determined by the CTD alone.26 By contrast, Byeon et al. reported a similar extent of deamination by full-length A3B in cell extracts at 5 h for 40-mer substrates with varying nucleobases at the +1 position; however, initial deamination rates were not reported.9 More detailed analyses of the impact of full-length protein on deamination rate and substrate specificity are required and will be the subject of future work.
Our studies investigate the impact of substrate length and composition on deamination rate by A3B. Direct comparison with other A3 isoforms is limited by the available data; however, a preference of A3A for a pyrimidine at position −2 has been reported.9,22 In contrast, we observe that A3B-mediated deamination is enhanced with purine bases at the −2 position consistent with the findings of Byeon et al.9 These results indicate a potential for A3A and A3B substrate specificity [XTCY (A3A)22 and YTCZ (A3B; this study), where X is a pyrimidine base, Y is a purine base, and Z can be any base (although T may be suboptimal)]. Importantly, this is consistent with differences observed in cancer patient samples, where A3A and A3B associated mutations can be distinguished based on the identity of the −2 base.11 Furthermore, the mutational pattern observed in patient samples with A3B gene deletion but A3A gene retention is consistent with an A3A-substrate preference.11 In the cellular environment, ssDNA is coated with replication protein A (RPA); however, a recent study showed that full-length A3B can compete with RPA binding to ssDNA.27 Together, these studies suggest that our findings are biologically and clinically relevant. Similar studies on related isoforms such as A3F and A3H would be highly desirable to dissect the contribution of each APOBEC family member to the mutational load observed in distinct disease states.
In summary, our studies reveal a broader A3B substrate sequence recognition than previously reported, which may widen the clinical context of A3B-mediated mutational signatures. TCG sequences are removed from bioinformatic characterization so that the events are not confused with CpG islands, where methylated C can be deaminated directly to T in a non-APOBEC-driven manner.24 However, our results suggest that G is equal or even preferred to A and the contribution of A3B to mutagenesis in the genome may therefore be underestimated. We observe equivalent deamination rates for optimized short tetranucleotide sequences compared to 10-mer nucleotides suggesting that induced-fit recognition of A3B CTD for its substrates can be triggered by an optimal tetranucleotide binding motif, which may facilitate the rational design of substrate-based A3B-selective inhibitors. The substrate preference identified in this study also translates to biologically relevant DNA sequences. We demonstrate that A3B CTD deaminates oligonucleotides derived from sequences associated with clinical resistance to cancer therapy, thereby supporting previous evidence that A3B activity contributes to the evolution of resistance in cancer.
Acknowledgments
This work was supported by Cancer Research UK Grant C309/A11566. T.K. is funded by Cancer Research UK Accelerator Award C1362/A20263. We thank O. Rossanese for critical review of the manuscript and helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.8b00639.
Experimental methods, Figures S1–S3, deamination rates of investigated oligonucleotides (Table S1), exemplar 1H NMR spectra (Figures S4–S6), and data analysis (Figure S7) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Harris R. S.; Dudley J. P. (2015) APOBECs and virus restriction. Virology 479–480, 131–145. 10.1016/j.virol.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter J. D.; Smith H. C. (2018) Modeling the embrace of a mutator: APOBEC selection of nucleic acid ligands. Trends Biochem. Sci. 43, 606–622. 10.1016/j.tibs.2018.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.; Ito F.; Zhang G.; Fernandez B.; Yang H.; Chen X. S. (2015) DNA cytosine and methylcytosine deamination by APOBEC3B: enhancing methylcytosine deamination by engineering APOBEC3B. Biochem. J. 471, 25–35. 10.1042/BJ20150382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X.; Yang H.; Arutiunian V.; Fang Y.; Besse G.; Morimoto C.; Zirkle B.; Chen X. S. (2017) Structural determinants of APOBEC3B non-catalytic domain for molecular assembly and catalytic regulation. Nucleic Acids Res. 45, 7494–7506. 10.1093/nar/gkx362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira V. C.; Soares M. A. (2013) The role of cytidine deaminases on innate immune responses against human viral infections. BioMed Res. Int. 2013, 683095. 10.1155/2013/683095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns M. B.; Temiz N. A.; Harris R. S. (2013) Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 45, 977–983. 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson S.; Fenton T. (2015) APOBEC3 genes: retroviral restriction factors to cancer drivers. Trends Mol. Med. 21, 274–284. 10.1016/j.molmed.2015.02.007. [DOI] [PubMed] [Google Scholar]
- Swanton C.; McGranahan N.; Starrett G. J.; Harris R. S. (2015) APOBEC enzymes: mutagenic fuel for cancer evolution and heterogeneity. Cancer Discovery 5, 704–712. 10.1158/2159-8290.CD-15-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byeon I.-J. L.; Byeon C.-H.; Wu T.; Mitra M.; Singer D.; Levin J. G.; Gronenborn A. M. (2016) Nuclear magnetic resonance structure of the APOBEC3B catalytic domain: structural basis for substrate binding and DNA deaminase activity. Biochemistry 55, 2944–2959. 10.1021/acs.biochem.6b00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns M. B.; Lackey L.; Carpenter M. A.; Rathore A.; Land A. M.; Leonard B.; Refsland E. W.; Kotandeniya D.; Tretyakova N.; Nikas J. B.; Yee D.; Temiz N. A.; Donohue D. E.; McDougle R. M.; Brown W. L.; Law E. K.; Harris R. S. (2013) APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370. 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K.; Roberts S. A.; Klimczak L. J.; Sterling J. F.; Saini N.; Malc E. P.; Kim J.; Kwiatkowski D. J.; Fargo D. C.; Mieczkowski P. A.; Getz G.; Gordenin D. A. (2015) An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat. Genet. 47, 1067–1072. 10.1038/ng.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard B.; Hart S. N.; Burns M. B.; Carpenter M. A.; Temiz N. A.; Rathore A.; Vogel R. I.; Nikas J. B.; Law E. K.; Brown W. L.; Li Y.; Zhang Y.; Maurer M. J.; Oberg A. L.; Cunningham J. M.; Shridhar V.; Bell D. A.; April C.; Bentley D.; Bibikova M.; Cheetham R. K.; Fan J.-B.; Grocock R.; Humphray S.; Kingsbury Z.; Peden J.; Chien J.; Swisher E. M.; Hartmann L. C.; Kalli K. R.; Goode E. L.; Sicotte H.; Kaufmann S. H.; Harris R. S. (2013) APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 73, 7222–7231. 10.1158/0008-5472.CAN-13-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K.; Carpenter M. A.; Banerjee S.; Shaban N. M.; Kurahashi K.; Salamango D. J.; McCann J. L.; Starrett G. J.; Duffy J. V.; Demir Ö.; Amaro R. E.; Harki D. A.; Harris R. S.; Aihara H. (2017) Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 24, 131–139. 10.1038/nsmb.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starrett G. J.; Luengas E. M.; McCann J. L.; Ebrahimi D.; Temiz N. A.; Love R. P.; Feng Y.; Adolph M. B.; Chelico L.; Law E. K.; Carpenter M. A.; Harris R. S. (2016) The DNA cytosine deaminase APOBEC3H haplotype I likely contributes to breast and lung cancer mutagenesis. Nat. Commun. 7, 12918. 10.1038/ncomms12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue E. C.; Bloch N.; Dhuey E.; Zhang R.; Cao P.; Herate C.; Chauveau L.; Hubbard S. R.; Landau N. R. (2014) A DNA sequence recognition loop on APOBEC3A controls substrate specificity. PLoS One 9, e97062. 10.1371/journal.pone.0097062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K.; Carpenter M. A.; Kurahashi K.; Harris R. S.; Aihara H. (2015) Crystal structure of the DNA deaminase APOBEC3B catalytic domain. J. Biol. Chem. 290, 28120–28130. 10.1074/jbc.M115.679951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byeon I.-J. L.; Ahn J.; Mitra M.; Byeon C.-H.; Hercík K.; Hritz J.; Charlton L. M.; Levin J. G.; Gronenborn A. M. (2013) NMR structure of human restriction factor APOBEC3A reveals substrate binding and enzyme specificity. Nat. Commun. 4, 1890. 10.1038/ncomms2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harjes S.; Solomon W. C.; Li M.; Chen K.-M.; Harjes E.; Harris R. S.; Matsuo H. (2013) Impact of H216 on the DNA binding and catalytic activities of the HIV restriction factor APOBEC3G. J. Virol. 87, 7008–7014. 10.1128/JVI.03173-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelico L.; Pham P.; Calabrese P.; Goodman M. F. (2006) APOBEC3G DNA deaminase acts processively 3′ → 5′ on single-stranded DNA. Nat. Struct. Mol. Biol. 13, 392–399. 10.1038/nsmb1086. [DOI] [PubMed] [Google Scholar]
- Wan L.; Nagata T.; Morishita R.; Takaori-Kondo A.; Katahira M. (2017) Observation by real-time NMR and interpretation of length- and location-dependent deamination activity of APOBEC3B. ACS Chem. Biol. 12, 2704–2708. 10.1021/acschembio.7b00662. [DOI] [PubMed] [Google Scholar]
- Bohn M.-F.; Shandilya S. M. D.; Silvas T. V.; Nalivaika E. A.; Kouno T.; Kelch B. A.; Ryder S. P.; Kurt-Yilmaz N.; Somasundaran M.; Schiffer C. A. (2015) The ssDNA mutator APOBEC3A is regulated by cooperative dimerization. Structure 23, 903–911. 10.1016/j.str.2015.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvas T. V.; Hou S.; Myint W.; Nalivaika E. A.; Somasundaran M.; Kelch B. A.; Matsuo H.; Yilmaz N. K.; Schiffer C. A. (2018) Substrate sequence selectivity of APOBEC3A implicates intra-DNA interactions. Sci. Rep. 8, 7511. 10.1038/s41598-018-25881-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes S. A.; Beare D.; Boutselakis H.; Bamford S.; Bindal N.; Tate J.; Cole C. G.; Ward S.; Dawson E.; Ponting L.; Stefancsik R.; Harsha B.; Kok C. Y.; Jia M.; Jubb H.; Sondka Z.; Thompson S.; De T.; Campbell P. J. (2017) COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45, D777–D783. 10.1093/nar/gkw1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson S.; Chakravarthy A.; Su X.; Boshoff C.; Fenton T. R. (2014) APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 7, 1833–1841. 10.1016/j.celrep.2014.05.012. [DOI] [PubMed] [Google Scholar]
- Hou S.; Silvas T. V.; Leidner F.; Nalivaika E. A.; Matsuo H.; Kurt Yilmaz N.; Schiffer C. A. (2018) Structural analysis of the active site and DNA binding of human cytidine deaminase APOBEC3B. bioRxiv 10.1101/304170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haché G.; Liddament M. T.; Harris R. S. (2005) The retroviral hypermutation specificity of APOBEC3F and APOBEC3G is governed by the C-terminal DNA cytosine deaminase domain. J. Biol. Chem. 280, 10920–10924. 10.1074/jbc.M500382200. [DOI] [PubMed] [Google Scholar]
- Adolph M. B.; Love R. P.; Feng Y.; Chelico L. (2017) Enzyme cycling contributes to efficient induction of genome mutagenesis by the cytidine deaminase APOBEC3B. Nucleic Acids Res. 45, 11925–11940. 10.1093/nar/gkx832. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.