

Abstract

Hydroxylation and fluorination of proline alters the pyrrolidine ring pucker and the trans:cis amide bond ratio in a stereochemistry-dependent fashion, affecting molecular recognition of proline-containing molecules by biological systems. While hydroxyprolines and fluoroprolines are common motifs in medicinal and biological chemistry, the synthesis and molecular properties of prolines containing both modifications, i.e., fluoro-hydroxyprolines, have not been described. Here we present a practical and facile synthesis of all four diastereoisomers of 3-fluoro-4-hydroxyprolines (F-Hyps), starting from readily available 4-oxo-l-proline derivatives. Small-molecule X-ray crystallography, NMR spectroscopy, and quantum mechanical calculations are consistent with fluorination at C3 having negligible effects on the hydrogen bond donor capacity of the C4 hydroxyl, but inverting the natural preference of Hyp from C4-exo to C4-endo pucker. In spite of this, F-Hyps still bind to the von Hippel–Lindau (VHL) E3 ligase, which naturally recognizes C4-exo Hyp in a stereoselective fashion. Co-crystal structures and electrostatic potential calculations support and rationalize the observed preferential recognition for (3R,4S)-F-Hyp over the corresponding (3S,4S) epimer by VHL. We show that (3R,4S)-F-Hyp provides bioisosteric Hyp substitution in both hypoxia-inducible factor 1 alpha (HIF-1α) substrate peptides and peptidomimetic ligands that form part of PROTAC (proteolysis targeting chimera) conjugates for targeted protein degradation. Despite a weakened affinity, Hyp substitution with (3S,4S)-F-Hyp within the PROTAC MZ1 led to Brd4-selective cellular degradation at concentrations >100-fold lower than the binary Kd for VHL. We anticipate that the disclosed chemistry of 3-fluoro-4-hydroxyprolines and their application as VHL ligands for targeted protein degradation will be of wide interest to medicinal organic chemists, chemical biologists, and drug discoverers alike.
1. Introduction
(2S,4R)-4-Hydroxyproline (Hyp) is a nonessential amino acid, prevalently found in collagen and produced by the most common, irreversible, posttranslational modification in animals: proline hydroxylation.1−3 Introduction of the hydroxyl group on the five-membered ring of proline at the 4R position affects the pyrrolidine ring puckering, shifting the C4-endo pucker preference of proline to the C4-exo pucker of Hyp.4−6 The C4-exo conformer in Hyp is stabilized by the well-known gauche effect (Chart 1A).7 This particular conformer allows a gauche arrangement between the two electronegative substituents of two vicinal carbons, i.e., the prolyl amide group and the hydroxyl group (dihedral angle N–C5–C4–O of approximately 60 degrees).4,5,8 Notably, the C4-exo pucker conformation places the carbonyl of the i amino acid in the correct position to interact with the Hyp carbonyl (i + 1), enabling an n → π* interaction that in turn stabilizes the trans amide bond in Hyp-containing peptides and proteins.4,5 As a result of the gauche effect, conversely, the diastereoisomer (2S,4S)-4-hydroxyproline (hyp) adopts a C4-endo pucker conformation, which reduces the n → π* interaction and leads to a lower trans:cis amide ratio.9 Following similar considerations, the puckering system for the less common 3-hydroxyprolines can be explained and predicted.10,11
Chart 1. Chemical Structures of Hydroxyl- and Fluoro-prolines: (A) Conformational Preferences of 4-Substituted Hydroxyprolines and Fluoroprolines; (B) Examples of 4-Fluoro- and 4-Hydroxyprolines in Medicinal Chemistry; (C) 3-Fluoro-4-hydroxyprolines (F-Hyps), the Object of This Study.

The realization that the stereochemistry of electronegative atoms on proline rings can greatly affect the conformation of the pucker and the trans:cis amide bond ratio has prompted the design and synthesis of a diverse array of unnatural proline derivatives.12 Among these, monofluorinated13,14 and trifluoromethyl prolines15 have been extensively studied. Fluorination at C4 or C3 causes stabilization of the C4-endo/C4-exo ring pucker in a similar fashion to hydroxylation, according to the same gauche effect.13,14 To this end, 4-fluoro and 3-fluoro prolines have been incorporated in proteins such as collagen,16 ubiquitin,17 and GFP,18 used to probe prolyl isomerase enzyme activity by NMR19 and used as PET probes.20 Beyond proteins, fluorinated prolines have also found applications as building blocks for medicinal chemistry,21 reflecting the wide interest in fluorination as a strategy to finely tune conformational and physicochemical properties of biologically active small molecules and peptides.22−25 For instance, the incorporation of (4S)- or (4R)-4-fluoroprolines into inhibitors of fibroblast activation protein (FAP)26 and thrombin27 (Chart 1B) stabilizes a single conformational pucker of the pyrrolidine ring (the C4-endo in the case of FAP inhibitor and the C4-exo for thrombin inhibitor), which results in improved potency compared to the unsubstituted proline analogues. More recently, mono- and difluorinated prolines were discovered to enhance key CH/π interactions in peptide–protein complexes.28 Like fluorinated prolines, hydroxyprolines are common building blocks in medicinal chemistry: the Hyp core can be found in the structure of recently FDA approved inhibitors of the NS3-4A serine protease such as glecaprevir29 (Chart 1B). Curiously, despite the variety of fluorinated and hydroxylated proline derivatives reported in the literature, the simultaneous presence of these two modifications within the same pyrrolidine ring is unprecedented. Introduction of fluorine substituents can modulate the electronic and conformational properties of small molecules, thus impacting protein–ligand and protein–protein binding affinities, as well as metabolism and cell permeability.21 We therefore became interested in exploring the synthesis and stereoelectronic properties of fluorinated hydroxyprolines, which we call F-Hyps (Chart 1C). We hypothesized that the addition of the highly electronegative F atom adjacent to the hydroxyl group on the pyrrolidine ring could significantly alter the puckering preference of the ring and affect the cis:trans amide ratio. Moreover, it was anticipated that fluorination of hydroxyproline would fine-tune the acidity, nucleophilicity, and hydrogen-bond properties of the hydroxyl group, depending on the stereochemistry of the fluorine atom. To this end, previous reports have studied the influence of fluorine on the −OH group acidity in the context of 2-fluorocyclohexanols and have shown that fluorination can lead to a reduction of alcohol H-bond acidity as a result of intramolecular F···HO interactions.30
Beyond its structural role in collagen, Hyp has a central role in oxygen sensing, a biological process of fundamental physiological and therapeutic relevance.31 Specifically, hydroxylation of the hypoxia-inducible factor 1 alpha (HIF-1α) subunits at two conserved proline residues (Pro402 and Pro564 in HIF-1α), which is catalyzed primarily by the prolyl hydroxylase enzyme PHD2 (also known as EGLN1),32,33 triggers the binding of HIF-1α to the von Hippel–Lindau (VHL) Cullin RING E3 ubiquitin ligase (Figure 1A). This recognition event leads to HIF-1α polyubiquitylation by the VHL ligase complex and subsequent proteasomal degradation under normal oxygen levels, preventing the transcription of hypoxia response genes.34−36 Work from our laboratory and others leveraged the highly specific molecular recognition of Hyp by the VHL E3 ubiquitin ligase (Figure 1B) to design and develop potent small-molecule VHL ligands.37−41 In one application, VHL ligands work in their own right as disruptors of the VHL-HIF protein–protein interaction to block the ubiquitination of HIF-1α downstream of HIF hydroxylation. This blockade leads to HIF-1α stabilization in cells, triggering a hypoxic response under otherwise normoxic conditions (Figure 1A).39−41 In a distinct application, VHL ligands provide suitable drug-like starting points for conjugation into bivalent degrader molecules, also known as proteolysis-targeting chimeras (PROTACs), which hijack the VHL E3 ligase activity to trigger ubiquitination and degradation of specific target proteins.42−44 Many potent and selective Hyp-based PROTACs have been recently reported against a wide range of target proteins, including Brd4,45,46 RIPK2,47 c-Abl kinase,48 VHL itself,49 TBK1,50 and TRIM2451 among others. A notable example among the first to be disclosed is MZ1 (Figure 1C), a potent and selective degrader for the bromodomain and extra-terminal (BET) protein Brd4 with profound antiproliferative activity in acute myeloid leukemia cells.45,46 Our crystal structure of MZ1 bound in a ternary complex with VHL, elongin B, and elongin C (hereafter called VBC), and the bromodomain of the target protein Brd4 revealed a structural basis for the cooperative ternary complex recognition that underpins the mode of action of the degrader molecule.52 The design of bivalent degrader molecules is seeing increasing interest for drug development, due to their catalytic rather than occupancy-based mode of action and an extended duration of action at low concentrations in substoichiometric fashion.53 All current VHL-targeting degraders contain Hyp as the core building block. It would therefore be of interest and potentially beneficial to replace Hyp with novel scaffolds, for example by introducing substitutions that might fine-tune the molecular recognition and pharmacokinetic properties of the final compounds.
Figure 1.

von Hippel-Lindau E3 ligase recognition of Hyp for VHL ligands and PROTACs. (A) Schematic representation of the VHL E3 ligase recognition and ubiquitylation of the endogenous substrate, inhibition of the catalytic activity of VHL by blocking the substrate recognition site, and PROTAC-induced targeted protein ubiquitylation by VHL hijacking. (B) Hyp-binding site with key polar interactions between VHL and Hyp in a C4-exo, trans amide conformation. (C) Examples of VHL inhibitors and VHL-based PROTACs built around the essential Hyp core.
Here we describe the synthesis of novel F-Hyps. We developed synthetic routes that allow efficient and robust synthesis for all four possible diastereoisomers and extensively studied their conformational and stereoelectronic properties using NMR spectroscopy, X-ray crystallography, and computational approaches. The simultaneous presence of F and OH on the proline ring was found to affect the conformational and electronic properties of the amino acid, which could be rationalized by means of small-molecule crystal structures and quantum mechanics calculations. Finally, to provide a proof-of-concept for the application of F-Hyps as building blocks to probe biology, we systematically incorporated F-Hyps in place of Hyp into a diverse set of HIF-1α peptides, VHL ligands, and VHL-targeting PROTACs and investigated the effect of fluorination of Hyp on VHL binding. We found that VHL exhibits a highly stereoselective recognition of the (3R,4S)-3-fluoro-4-hydroxyproline over the (3S,4S) epimer. We show that (3R,4S)-3-fluoro-4-hydroxyproline incorporation achieved ligands and PROTACs with affinities and cellular activities comparable to those of the parent Hyp-containing compounds, while incorporation of the (3S,4S) epimer led to Brd4-selective degradation at nanomolar concentration in spite of a loss of ∼20-fold binding affinity to VHL.
2. Results and Discussion
2.1. Chemistry
To prepare the desired 3-fluoro-4-hydroxy prolines, we envisaged a synthetic strategy based on electrophilic fluorination of an enolate or enolate equivalent of N-Boc-4-oxo-l-proline benzyl ester 1, which is known to be regioselectively enolized at the C3 under kinetic conditions.54 In a first set of experiments, the lithium, sodium, and potassium enolate of 1, generated by treatment of ketone 1 with the corresponding metal hexamethyldisylazides at −78 °C in tetrahydrofuran (THF), were reacted with N-fluorobenzenesulfonimide at −78 °C, and the reaction mixtures were allowed to slowly warm to room temperature overnight. In all cases, no fluorinated ketone was detected by 19F NMR, and complex reaction mixtures were obtained. We next attempted to prepare the trimethylsilyl enol ether 2 (Scheme 1) by quenching the lithium enolate of 1 with trimethylsilyl chloride, as described previously.54 Silyl enol ether 2 was isolated by pentane extraction and, after solvent removal, directly reacted with Selectfluor in acetonitrile at −30 °C. The reaction mixture was allowed to slowly warm to room temperature overnight, and we were pleased to observe a set of 19F NMR signals compatible with a diastereoisomeric mixture of fluorinated ketones 3a,b in a ∼1.5:1 ratio and 50% overall isolated yield. However, variability of yield and diastereoisomeric ratio was observed when scaling up this reaction, with considerable reduction of yield when the fluorination was performed in gram scale. As a reliable fluorination protocol was highly desirable, a flow chemistry approach was considered. No examples of fluorination of silyl enol ether in flow conditions were found in the literature, except for a closely related α-fluorination of activated carbonyl compounds with Selectfluor, reported by the Ley laboratory in 2009.55 Under optimized conditions, the silyl enol ether 2 and Selectfluor were dissolved in anhydrous acetonitrile (0.67 and 0.1 M, respectively) and pumped at a rate of 0.77 mL/min in a 10 mL flow reactor (residence time 6.5 min) heated at 50 °C. The mixing was provided by a T piece junction before the coiled reactor. The crude product was treated with a saturated solution of NH4Cl and filtered over a short silica column, providing the fluorinated ketones 3a,b in a ∼1:1 ratio and 55% isolated yield.
Scheme 1. Synthesis of F-Hyps N-Protected Amino Acids.

Because any attempt to separate the diastereoisomers by column chromatography failed, we decided to subject the diastereoisomeric mixture to sodium borohydride reduction. Three fluorohydrines were isolated, 4a in 58%, 4b in 30%, and 4c in 11% yield. Despite being complicated by the well-known conformational isomerism shown by N-Boc prolines, the 1H NMR spectra of 4a, 4b, and 4c in CDCl3 showed remarkable differences in the coupling constants between H2 and H3 (see Figure 2A for numbering of the proline ring): JH2–H3 = 5.9 Hz in 4a and close to zero in 4b and 4c. This observation suggested that in 4a the fluorine atom and the carboxylate may be present in a cis conformation, in which the angle between H2 and H3 is consistent with a relatively large JH2–H3 value. On the other hand, the JH2–H3 value observed for 4b and 4c suggests an angle between H2 and H3 around 90°, possible when the fluorine atom and the carboxylate arrange in a trans conformation. The stereochemistry of the main products 4a and 4b was unequivocally assigned by means of NOESY and 19F-detected 19F–1H HOESY NMR experiments (Figure 2A). In the case of 4a, the relative intensities of NOESY peaks for H2–H3 and H3–H4 were consistent with a cis arrangement of H2–H3–H4 (Figure 2A). The relative intensity of the 19F–1H HOESY peaks for H2–F and H4–F were equally low and compatible with a trans arrangement of the F atom with both H2 and H4. For 4b, a cis arrangement could be deduced only for H3–H4, as the intensity of the NOESY peak for H2–H3 was 40% lower than the one for H3–H4 (Figure 2A). Consistently, the relative intensity of the 19F–1H HOESY peaks for H2–F was 87% higher than the one for H4–F, confirming a cis arrangement between H2–F and a trans relationship between H4–F (Figure 2A). These observations reveal that the main products of the reduction, 4a,b, result from a hydride delivery anti to the fluorine atom. This same preference had previously been observed in the reduction of conformationally locked fluorocyclohexanones.30
Figure 2.

Structural assignment of F-Hyps. (A) Numbering of the F-Hyp ring and stereochemical assignment for 4a,b by NMR (NOESY values of cross-peaks in black, 19F–1H HOESY values of cross-peaks in blue). (B) Molecular structure of compounds 4a–c and 10 showing 50% displacement ellipsoids resulting from small-molecule X-ray crystallography.
Mitsunobu esterification of 4a and 4b with 4-nitrobenzoic acid was conducted, followed by deprotection by sodium azide in methanol.56 This allowed access to the fourth diastereoisomer (4d) when starting from 4a and to a product identical to 4c when starting from 4b. These results confirmed that the minor product of the borohydride reduction, 4c, is the epimer at the C4 of 4b. As expected, for the newly synthesized compound 4d, the coupling constant between H2 and H3 (5.2 Hz) is very similar to its C4 epimer 4a. The stereochemical assignment of 4a–c was also confirmed by X-ray diffraction of the isolated products (Figure 2B and Supp. Figures 3–5). Interestingly, 4a,b crystallized with a cis carbamate configuration (similarly to a previously reported N-Boc proline derivative9), whereas 4c crystallized with a trans carbamate configuration. For the (4R) derivatives 4a and 4c no transannular hydrogen bond between the carboxylate and the hydroxyl group was found. Finally, catalytic hydrogenation of the benzyl esters 4a–d afforded the Boc-protected amino acids 6a–d in good yield and sufficient purity to be used for further synthetic steps.
With the aim to develop a stereoselective synthesis, and inspired by previous work on the synthesis of kainic acid analogues,57 the fluorination step was attempted on the 9-phenyl fluorenyl (Pf)-protected silyl enol ether 8, prepared from ketone 7, trimethylsilyl triflate, and triethylamine in dichloromethane (DCM) at −40 °C. Treatment of the silyl enol ether 8 with Selectfluor in acetonitrile at −20 °C afforded the fluorinated ketone 9 as a sole diastereoisomer in 50% yield. 1H NMR analysis revealed a coupling constant between H2 and H3 = 8.0 Hz, similar to that of previously reported, structurally related, cis derivatives.57−60 The stereochemical outcome of the fluorination was in contrast with the observations made on the alkylation of the same ketone by Zanato et al.,57 which mainly occurred from the less hindered face of the enolate 8. Subsequent reduction with sodium borohydride in ethanol/THF exclusively afforded the all-cis F-Hyp 10. The stereochemistry of compound 10 was confidently assigned by means of NMR experiments. The relative intensity of NOESY peaks for H2–H3 and H3–H4 were compatible with a cis arrangement of H2–H3–H4, and the relative intensity of the 19F–1H HOESY peaks for H2–F and H4–F (equally low) were compatible with a trans arrangement of the F atom with H2 and H4. Moreover, a coupling constant of JH2–H3 = 8.7 Hz in 10 was found, in accordance with the cis derivatives previously prepared.57−60 This assignment was subsequently confirmed by X-ray diffraction of 10, which crystallized with a transannular hydrogen bond between the carboxylate and the hydroxyl groups (Figure 2B). Mitsunobu esterification of 10 with 4-nitrobenzoic acid followed by treatment with lithium hydroxide led to the inverted alcohol, and no methyl ester hydrolysis was observed. Accordingly, the relative intensity of the 19F–1H HOESY peak for H2–F was compatible with a trans arrangement, whereas the intensity for H4–F was only compatible with a cis arrangement, being 150% higher than that of H2–F. Deprotection of the carboxylate could be achieved only after removal of the Pf group. To this end, 11 was first treated with 5% trifluoroacetic acid (TFA) in DCM in the presence of triisopropylsilane as carbocation scavenger, then with LiOH at 0 °C in water. To obtain F-Hyps building blocks, useful for further manipulations, the free F-Hyps were transformed into the Boc- or Fmoc-protected amino acids by reaction with Boc- or Fmoc-N-succinimidyl carbonate and sodium bicarbonate in aqueous dioxane, respectively.
2.2. Conformational Analysis and H-Bond Donor Capacity of F-Hyps
Recognition of Hyp by VHL is highly stereoselective and conformation-dependent: the C4-exo ring pucker of Hyp is essential to locate the hydroxyl group within a well-defined hydrogen-bond network in the Hyp-binding pocket. Additionally, binding of HIF-1α peptides and VHL ligands requires adoption of a trans conformation of the prolyl tertiary amide (Figure 1B). We therefore became interested in determining to what extent addition of the highly electronegative F atom adjacent to the hydroxyl group in Hyp would impact the C4-exo ring pucker of the pyrrolidine, affect the donor capacity of the adjacent hydroxyl group, and modulate the abundance of trans prolyl amide. We hypothesized that modulation of these conformational and molecular properties could significantly fine-tune the binding affinity to VHL and consequently be exploited for a novel class of VHL ligands.
We first studied both experimentally and computationally the conformational preferences of N-acetylproline methylesters 12a–d (Chart 2), extending the analysis to the (3R,4R)- and (3S,4R)-3-fluoro-4-hydroxyproline derivatives. Analogous methylesters have been previously used in the literature as reliable model compounds to study the conformational preferences of the (4R)-hydroxyproline (Hyp), (4S)-hydroxyproline (hyp), (3S)-fluoroproline (3-Fpro), and (3R)-fluoroproline (3-fpro) moieties.5,13 The compounds 12a–d were synthesized from 6a–d by (trimethylsilyl)diazomethane esterification, followed by Boc deprotection and N-acetylation. To predict molecular properties of compounds in solution, we performed density functional theory (DFT) calculations of trans C4-endo and exo puckers of 12a–d and the reference compounds Ac-Hyp-OMe, Ac-hyp-OMe, Ac-3-Fpro-OMe, Ac-3-fpro-OMe, and Ac-Pro-OMe (Chart 2). All calculations were carried out at the MN-15L/aug-cc-pVTZ(-F) level of theory in a water solvation continuum (see Computational Methods and Supp. Table 1 for details on the choice of density functional). The protocol correctly anticipates the ring pucker preferences of model compounds Ac-Pro-OMe, Ac-3-Fpro-OMe, and Ac-Hyp-OMe and predicts JH2–H3 couplings for Ac-Hyp-OMe, Ac-hyp-OMe, and F-Hyps 12a–d in excellent agreement with the corresponding NMR-observed shifts (Table 1 and Supp. Table 3).
Chart 2. Chemical Structure of F-Hyps 12a–d and the Corresponding Reference Compounds.

Table 1. Summary of Conformational Analysis, Second-Order Perturbation Energies, and H-Bond Donor Capacity of F-Hyps 12a–da.
| compound | ratio trans:cisb | JH2–H3 (Hz) | calculated J̅H2–H3 (Hz) | optimized ring pucker | rel. E of conformer (kcal/mol) | fraction of conformer (%)c |
 (kcal/mol)d (kcal/mol)d
|
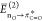 (kcal/mol)e (kcal/mol)e
|
V̅a(r) (a.u.)f |
|---|---|---|---|---|---|---|---|---|---|
| 12a | 89:11 | 4.6 | 5.7 | twisted C2-endo–C3-exo | 0.8 | 12 | 0.8 | 1.1 | 0.339 |
| C4-endo (with transannular H-Bond) | 0.1 | 40 | |||||||
| C4-exo | 0.0 | 47 | |||||||
| 12b | 85:15 | 2.4 | 1.2 | C4-endo | 0.0 | 92 | 1.0 | - | 0.378 |
| twisted C3-endo–C4-exo | 1.4 | 8 | |||||||
| 12c | 71:29 | <0.5 | 0.1 | C4-endo | 0.0 | 78 | 0.9 | - | 0.357 |
| C4-endo (with transannular H-Bond) | 0.8 | 21 | |||||||
| twisted C3-endo–C4-exo | 2.7 | 1 | |||||||
| 12d | 87:13 | 4.9 | 7.0 | C3-exo | 0.0 | 60 | 0.4 | 1.7 | 0.375 |
| C5-endo | 0.2 | 40 |
DFT calculations were carried out at the MN15-L/aug-cc-pVTZ(-F) level of theory in a water solvation continuum. See Supp. Table 3 for full results.
Measured as the ratio of proton-decoupled 19F NMR signals in D2O.
Boltzmann population obtained from relative E.
Charge transfer from acetyl O lone pair to π* orbital in the carbonyl in C2.
Charge transfer from F lone pair to π* orbital in the carbonyl in C2.
Analysis of the abundance of C4-endo and exo ring puckers in F-Hyps 12a–d revealed that conformational preferences upon 2,3-trans fluorination are driven by the stabilizing gauche conformation between the N and the F atoms. Indeed, 2,3-trans fluorination of Ac-hyp-OMe to yield 12c further stabilizes the C4-endo pucker of hyp (99% abundance), whereas 2,3-trans fluorination of Ac-Hyp-OMe to yield 12b shifts the natural preference of hyp from C4-exo to C4-endo (92% abundance). The case of 2,3-cis fluorination of hydroxyproline was found to be more complex. The steric congestion surrounding C3 in 3-fpro has been invoked to justify the reduced conformational preference of the ring for either pucker.13 Consistently, F-Hyps 12a and 12d showed very modest preference to adopt a C4-endo or -exo ring pucker, with ∼50% abundance of each (ΔEendo/exo < 0.25 kcal/mol). Noncovalent contacts in the molecules were studied by topological noncovalent interactions (NCI) analysis62 of the electron density distribution. In 12a, the NCI study showed a transannular H-bond between the hydroxyl group and the carbonyl O in C2, along with a consistent van der Waals contact between the hydroxyl proton and the ring in 12a. In contrast, in 12d, NCI analysis detected attractive F···C=O contacts (Figure 3A and B). The ring pucker of the predicted major components of F-Hyps 12a–c was captured in the related crystallized compounds 4a–c using small-molecule X-ray crystallography (Figure 2 and Supp. Figure 1), providing further support for the theoretical calculations. Taken together, the conformational analysis revealed that fluorination of Ac-Hyp-OMe to yield (3R,4S)- and (3S,4S)-3-fluoro-4-hydroxyprolines 12b and 12d stabilizes a C4-endo pucker and consequently would be expected to lower the population of the desired C4-exo ring pucker required for binding to VHL.
Figure 3.

Topological and conformational analysis and H-bond donating capacity of F-Hyps. NCI isosurface plots for 12a (A) and 12d (B).62 Reduced density gradient shown at an isovalue of 0.3, with interaction strength colored red (attractive), white (van der Waals contact), and blue (repulsive), ranging from −0.02 to 0.05 a.u. (C) Visualization of the H-bond donor capacity and major conformers of 12a–d, Ac-Hyp-OMe, and Ac-hyp-OMe. The Kenny electrostatic potential (Vα(r)) correlates with the H-bond donor capacity of the OH.30
To quantify the energetic extent of the potential nO → πC=O* charge transfer in F-Hyps 12a–d, we next performed natural bond order (NBO) analyses.63 This nO → πC=Ointeraction is known to stabilize the trans conformer of the amide,64 which is critical for binding of Hyp to VHL.65 We compared the nO → πC=O* charge transfer energies with the trans:cis ratio of F-Hyp derivatives 12a–d (observed by 19F NMR) and of 4-hydroxyprolines and 3-fluoroprolines reported in the literature13 (Table 1 and Supp. Table 3). The computational and experimental values for Ac-Hyp-OMe and Ac-hyp-OMe were in good agreement (higher charge transfers are indicative of higher abundance of the trans conformer). Instead, the trans:cis ratio for F-Hyps 12a–d did not correlate with nO → πC=O energies. This has been also reported in 3-fluoroproline,9 suggesting that a combination of other factors, including inductive and steric effects, might be involved. A higher amount of cis amide was observed by NMR for 12b,c, featuring the fluorine atom in the 3-(R) position, compared to 12a,d, bearing the fluorine atom in the 3-(S) position. This correlates well with the trans:cis ratio experimentally measured for 3-fluoroprolines.13 Based on the observed trans:cis ratio in 12b,d by NMR, we conclude that fluorination of Hyp does not stabilize the trans amide conformer, which would otherwise be expected to enhance the affinity of VHL ligands.
We then became interested in assessing whether fluorination could lead to a strengthening of the H-bond network of Hyp in the recognition site of VHL through inductive effects. To address this question, we calculated the weighted electrostatic potential V̅α(r) of F-Hyps 12a–d, evaluated at a distance of 0.55 Å from the hydroxyl hydrogen atom along the O–H bond (Table 1 and Figure 3C).57 This descriptor correlates linearly with relative H-bond acidity (pKAHY), which is a measure of the H-bond donor capacity of the hydroxyl group.30,66 An increase in H-bond donor capacity in Hyp could be potentially exploited to strengthen its H-bond with His115 (Figure 1B). However, fluorination of Ac-Hyp-OMe to yield 12b and 12d induced only a modest increase in the H-bond acidity of the hydroxyl H atom, regardless of the stereochemistry in C3. This suggests that the inductive effect of the fluorine may compete with a transannular H-bond to the F atom in the compounds.30,67 Nonetheless, we observed relatively long F···H–O distances (2.5–3.3 Å) along with undetectable n → σ* charge transfer between the lone pairs in the F and the acceptor σO—H* orbital, as by NBO analysis. Topological analysis of electron densities using NCI did not reveal any F···H–C contact, either. Thus, capture of the proton in 12b and 12d may arise from a weak electrostatic stabilization rather than a typical H-bond.30 Together, these analyses indicate that C3 fluorination of Hyp may only modestly improve the H-bond donor capacity of the hydroxyl group, regardless of the stereochemistry in C3. For completeness, we also studied the proton acidity of Ac-hyp-OMe and its fluoro derivatives 12a and 12c. A substantial decrease in H-bond donor capacity of the hydroxyl group in Ac-hyp-OMe compared to Ac-Hyp-OMe was anticipated, since in Ac-hyp-OMe the hydroxyl group engages in a transannular H-bond with the carbonyl O in C2. Fluorination of Ac-hyp-OMe to form 12a and 12c induces a higher increase in H-bond donor capacity compared to fluorination of Ac-Hyp-OMe, suggesting that in those compounds the inductive effect of fluorine is not competing with an F···H–O contact. The increase in acidity is utmost notorious in 12c, in which the fluorine atom is antiperiplanar to the O. Indeed, it has been proposed that this arrangement increases the electronegativity of the fluorine.30 The increasing acidity of the protons (Ac-hyp-OMe < 12a < 12c) can be further rationalized by the decreasing fraction of transannular H-bond (Ac-hyp-OMe > 12a > 12c, Figure 3C), which at the same time is consistent with an increase in the H-bond distance. Additional topological analyses of electron densities and second-order perturbation energies were also consistent with a weakening of the transannular H-bond upon fluorination of Ac-hyp-OMe, particularly in 12c (Supp. Table 4). Thus, C3 fluorination of Hyp and hyp has a distinct impact on proton acidity. In Hyp the inductive effect of F is compensated by a weak electrostatic stabilization of the H atom. In contrast, in hyp the acidity increases in a stereoselective fashion assisted by a weakening of the transannular H-bond.
Taken together, the conformational analysis of F-Hyps 12b,d indicates that C3-fluorination of Ac-Hyp-OMe, irrespective of the relative stereochemistry, does not modulate conformational preferences and hydrogen-bonding potential of the Hyp core fragment in the directions that would be expected to strengthen the binding affinity of VHL ligands. First, we do not observe stabilization of the C4-exo ring pucker, which is critical for recognition (Figure 1B). Rather, we observe a preference for the C4-endo pucker in both 12b and 12d, albeit to varying extent. Second, we do not detect an increase in the abundance of the trans amide conformer by NMR, which is required for binding to VHL. And third, fluorination induces only a very minor increase in the H-bond donor capacity of the hydroxyl group, regardless of the stereochemistry in C3. Nevertheless, fluorine often participates in noncanonical ligand–protein interactions that cannot be inferred from studying the isolated ligand and that generally behave in a highly nonadditive fashion, making it difficult to predict them computationally. We therefore decided to proceed to interrogate the molecular recognition of F-Hyp-containing peptides and peptidomimetics by VHL experimentally.
2.3. Synthesis and Biophysical and Structural Binding Studies of F-Hyp-Containing Peptides of HIF-1α
Hyp-containing HIF-1α epitope peptides are excellent model substrates for studying the binding to VHL, as their binding mode and affinities have been extensively characterized.32,33,37 To assess to what extent VHL recognizes F-Hyps as an artificial post-translational modification of proline, modified peptides from the C-terminal oxygen-dependent degradation domain of HIF-1α (of sequence DEALA(Hyp)YIPMDDDFQLRSF) that contained (3S,4S)- or (3R,4S)-F-Hyps in place of Hyp were synthesized (Chart 3).
Chart 3. Structures of Model HIF-1α Peptides Biophysically Evaluated in This Work.

To this end, Fmoc-protected (3S,4S)- and (3R,4S)-F-Hyps were prepared from the corresponding Boc-protected amino acids 6b and 6d via acidic deprotection of the Boc group followed by Fmoc installation by reaction with Fmoc-OSu. The first 13 C-terminal amino acids (YIPMDDDFQLRSF) of the 19-mer peptides were coupled via conventional automated solid-phase peptide synthesis on a Rink amide resin before the N-Fmoc-protected F-Hyps were manually coupled to resin-bound peptide via HATU/HOAT coupling. After Fmoc deprotection of F-Hyps, peptide elongation was performed manually, the N-terminal Fmoc group was removed, and the peptides were cleaved from the resin using a TFA/triisopropylsilane/H2O (95:2.5:2.5) mixture. The crude peptides were purified by preparative HPLC to obtain the epimeric peptides 13a,b. The corresponding peptides containing the (3S,4R)- and (3R,4R)-F-hyp were not prepared because it is well-known that such stereochemical arrangement of the hydroxyl group is not tolerated in the VHL binding site.8 Binding of the modified F-Hyp-containing peptides 13a,b to VBC was assessed using isothermal titration calorimetry (ITC, Figure 4A). Both peptides 13a,b presented nanomolar binding affinity to VBC (Kd = 12 ± 2 nM and 52 ± 3 nM, respectively, Table 2). Remarkably, 13a showed comparable binding affinity to the native peptide 13c (prepared as previously reported),37 for which a Kd = 11 ± 1 nM was measured (Table 2). Among the fluorinated peptides, 13a, containing the (3R,4S)-F-Hyp isomer, showed a 4-fold higher affinity for VBC than 13b, suggesting that VHL can recognize F-Hyps in a diastereoselective fashion.
Figure 4.

Binding affinity and binding mode of F-Hyp-containing HIF-1α peptides. (A) Overlay of ITC titration curves obtained for F-Hyps-containing peptides 13a,b against VBC. (B) Crystal structure of peptide 13a (blue) in complex with VBC (green) overlaid with the structure of VBC in complex with the Hyp-containing peptide 13c (yellow). Key hydrogen bond interactions and distances in Å between F-Hyp and Ser111 and His115 of VHL are shown in red; the F0–Fc omit map contoured at 3.5 σ around the F-Hyp residue is shown in blue. (C) Overlay of the crystal structure of peptide 13a (blue) in complex with VBC (green) with the structure of F-Hyp in the C4-endo conformation (orange, from small-molecule X-ray crystallography of 4b). In black: distances in Å between the hydroxyl group of F-Hyp in the C4-endo conformation (in orange) and the residues Ser111 and His115 of VHL.
Table 2. Thermodynamic Binding Parameters of Peptides 13a–c and Ligands 14a,b to VBC.
| Kd (nM) | ΔH (kcal/mol) | –TΔS (kcal/mol) | ΔG (kcal/mol) | N.R.a | |
|---|---|---|---|---|---|
| 13a | 12 ± 2 | –10.1 ± 0.1 | –0.75 ± 0.05 | –10.8 ± 0.1 | 2 |
| 13b | 52 ± 3 | –9.7 ± 0.3 | 0.0 ± 0.2 | –9.94 ± 0.04 | 2 |
| 13c | 11 ± 1 | –12.8 ± 0.1 | 2.0 ± 0.2 | –10.9 ± 0.1 | 2 |
| 14a | 244 ± 6 | –7.2 ± 0.5 | –1.8 ± 0.5 | –9.02 ± 0.01 | 3 |
| 14b | 3,080 ± 10 | –3.9 ± 0.5 | –3.8 ± 0.4 | –7.55 ± 0.03 | 2 |
N.R. = number of replicates.
To elucidate the binding mode of F-Hyp-containing HIF peptides, we solved the cocrystal structure of the ternary complex VBC with peptide 13a (to a resolution of 1.8 Å) and compared it to the cocrystal structure of the native peptide 13c.37 The VBC:13a complex crystallized in the same space group as VBC:13c (P43212), and the overall unit cell parameters and crystal packing were conserved. The F-Hyp-containing peptide 13a was found to adopt an identical binding mode when compared to the native peptide 13c (Figure 4B) and other HIF peptides,32,33 with all hydrogen bonds being conserved. After structural refinement, the pyrrolidine ring of the (3R,4S)-F-Hyp residue was modeled in a C3-endo conformation, as this satisfied better the extra electron density. However, the resolution was not sufficiently high to exclude the possibility of a C4-exo pucker, which was observed in the case of the cocrystal of VBC:13c, as the conformational difference between the two states (C3-endo and C4-exo) is very subtle. In the bound state, the hydroxyl group of the 13a F-Hyp forms two key hydrogen bonds to the side chains of His115 and Ser111, exactly as observed for Hyp in the native peptide 13c (Figure 4B). The newly introduced fluorine atom forms close contacts with an aromatic C–H of the indole ring of Trp117 (3.3 Å), the hydroxy group of Ser111 (3.2 Å), and the carbonyl of His110 (3.2 Å). An overlay of the VHL-cocrystallized peptide 13a with the small-molecule crystal structure of F-Hyp 4b (Figure 4C) reveals that, in order to preserve the key H-bond network of the Hyp core, the F-Hyp pyrrolidine needs to rearrange from its predominant C4-endo conformation to a C3-endo/C4-exo pucker that is only modestly populated in solution, according to DFT calculations (Table 1). This suggests that the pyrrolidine ring of the F-Hyp in 13a must undergo a significant change in the conformational equilibrium to bind to the protein, possibly at the expense of the ΔG of binding. Because only a very subtle difference in the ΔG of binding was found between the native peptide 13c and 13a (Table 2), presumably the energetic penalty required for the pucker rearrangement from free to bound state must be compensated for by significantly favorable contacts between the protein and the ligand environment around the newly introduced F atom.
2.4. Synthesis, Binding Affinity, and X-ray Crystal Structure of F-Hyp-Containing VHL Ligands
Encouraged by the positive results on the binding of F-Hyp-containing peptides to VHL, we proceeded replacing the Hyp core of the prototypical ligand VH032 (Figure 1C, ref (39)) with (3S,4S)- and (3R,4S)-F-Hyp to study the molecular recognition within the context of VHL ligands. Ligands 14a,b (Figure 5A) were prepared according to previously reported procedures,38−40 and no particular optimization of reaction conditions was required. Binding of the modified F-Hyp-containing VHL ligands 14a,b to VBC was assessed by ITC (Table 2, Figure 5B). Ligand 14a retained nanomolar binding affinity to VBC (Table 2), which is within 2-fold of the parent Hyp-containing ligand VH032 (Kd = 185 ± 7 nM).39 In contrast, ligand 14b showed a much lower affinity (Table 2), corresponding to a reduction in binding affinity of almost 20-fold compared to VH032. The ΔH of binding measured for the VHL ligands correlated well with the binding affinities.
Figure 5.

Binding affinity and binding mode of F-Hyp-containing VHL ligands. (A) Chemical structure of ligands 14a,b. (B) Overlay of ITC titration curves obtained for the F-Hyps-containing ligands 14a,b against VBC. (C) The F0–Fc omit map contoured at 4σ (shown in yellow) for ligands 14a,b in complex with VBC. (D) Crystal structure of ligand 14b in complex with VBC. Key H-bond interactions are shown in red.
These results confirmed the trend observed for the F-Hyp-containing peptides: the (3R,4S) isomer of F-Hyp binds to VBC with higher affinity than the (3S,4S) isomer, as a result of diastereoselective recognition of F-Hyps. As expected, in the case of small molecules, the difference in binding affinity between the two diastereoisomers is greater than in the case of peptides, as the binding of such ligands relies on fewer contacts and interactions beyond the most energetically favorable ones involving the core hydroxyproline moiety. To elucidate the binding mode of the two epimers of F-Hyp-containing ligands to VHL and to rationalize the difference in binding affinity, we soaked crystals of VBC into solutions of 14a,b and solved the crystal structures of the binary complexes. No difference in the binding mode between the two diastereoisomers was found (Figure 5C,D and Supp. Figure 2), and moreover the binding mode of the nonfluorinated analogue (VH032) was recapitulated by the fluorinated analogues 14a,b. Similarly to what was observed in the case of peptide 13a, the fluorine atom in 14a forms close contacts with an aromatic C–H of the indole ring of Trp117 (3.2 Å), the hydroxyl group of Ser111 (3.2 Å), and the carbonyl of His110 (3.1 Å). For 14b, close contacts between the fluorine atom and Trp117 (3.3 Å) and the phenolic group of Tyr98 (3.0 Å) were found. The resolution of the structures (2.7 Å for 14a and 2.3 Å for 14b) did not allow us to unambiguously determine the ring puckering of the F-Hyps of 14a,b, which was modeled as C4-exo, as for the nonfluorinated ligand VH032 (Figure 5C,D and Supp. Figure 2). We therefore conclude that, as in the case of F-Hyp-containing peptides, a comparably large energetic penalty must be overcome for the ligands to bind to VHL in a C4-exo conformation.
To provide insights into the preferential recognition of 14a by VHL, we carried out quantum mechanics/molecular mechanics (QM/MM) single-point energy (SPE) calculations in a water solvation continuum, using the ligand-bound crystal structures of VHL in complex with VH032,3914a and 14b. The obtained molecular electrostatic potential surfaces reveal a markedly distinct effect of fluorination in the charge density around the hydroxyproline core (Figure 6) and allow for the rationalization of the observed preferential binding. In 14a, the fluorine induces a substantial increase in the electrostatic potential surrounding the hydroxyl O compared to VH032 and 14b (note the yellow to red shift in Figure 6, bottom view). In contrast, in 14b the fluorine creates an electron-rich bump that contrasts with the smaller, electron-poor surface of the H in VH032 (note the blue to green shift in Figure 6, top view). These alterations are anticipated to affect the molecular recognition. An increase in the electron density of the hydroxyl group, as in 14a, would strengthen its H-bond acceptor interaction with the side chain of Ser111. Conversely, the bump and the inversion of the electrostatic potential induced by fluorine in 14b are potentially disruptive for binding because they face the electron-rich aromatic system of Tyr98, which is at a short distance of 3.0 Å (Figures 5D and 6) and known to be very sensitive toward binding of small molecules.65 These observations were consistent with MM/generalized-Born surface area (MM-GBSA) calculations performed using the same crystal structures (Figure 6). The MM-GBSA interaction energy (EMM-GBSA) of Ser111 was higher for VHL in complex with 14a than with VH032 and 14b (ΔEMM-GBSA = −0.6 and −1.4 kcal/mol, respectively), whereas the interaction of Tyr98 was weakened when 14b was bound, compared to VH032 and 14a (ΔEMM-GBSA = +0.9 and +0.6 kcal/mol, respectively).
Figure 6.

Structural basis of diasteroselective recognition of F-Hyps by VHL. Molecular electrostatic potential surface of VH032, 14a, and 14b in complex with VHL, obtained using QM/MM SPE calculations in a water solvation continuum at the MN15-L/aug-cc-pTVZ(-F) level of theory. Tyr98 and Ser111 from VHL are shown in gray, and their interaction energy as per MM-GBSA calculations is shown below.
We next investigated the impact of fluorination on the pharmacokinetic properties of hydroxyproline-containing compounds in the context of VHL ligands 14a,b (Table 3). Despite a small increase in LogD7.4 upon fluorination, microsomal stability of compound 14b was similar to that of the reference compound VH032, while ligand 14a proved to be even more metabolically stable. F-Hyp containing VHL ligands also retained high plasma stability. We also performed a parallel artificial membrane permeability assay (PAMPA) to measure the passive permeability of 14a,b. The compounds exhibit stereodependent permeability in spite of the same LogD7.4 value (Table 3), with 14a exhibiting higher permeability than the parent nonfluorinated ligand VH032.
Table 3. Cellular Pharmacokinetics Parameters of VHL Ligands 14a,b.
| mouse microsomal stability (mL/min/g liver) | plasma stability T1/2 (min) | permeability (PAMPA) (nm s–1) | CHI LogD7.4 | |
|---|---|---|---|---|
| VH032 | 0.70a | >180a | 1.2a | 0.90a |
| 14a | <0.50 | >180 | 2.3 | 1.30 |
| 14b | 0.68 | >180 | 0.81 | 1.30 |
Values from Frost et al.40
Taken together, the crystal structures of VHL in complex with 14a,b reveal that diastereoselective recognition of 14a is mediated by strengthening of the H-bond acceptor interaction with Ser111, which stabilizes the less abundant C4-exo ring pucker of the compound (Table 1). This induced fit of the ligand is reflected in a slight loss in binding affinity of 14a compared to the Hyp-containing ligand VH032. Instead, steric and electronic repulsions of the fluorine with Tyr98 explain the significant loss of binding affinity in 14b. Additionally, we found that F-Hyps-containing compounds are metabolically stable and that fluorination of Hyp increases its lipophilicity. These positive data qualify F-Hyp VHL ligands 14a,b as suitable cellular probes and motivated us to pursue incorporation into VHL-targeting molecular degraders.
2.5. F-Hyp-Containing PROTACs: Effect of Fluorination on Targeted Protein Degradation
PROTACs are bivalent degrader molecules composed of a moiety that binds an E3 ubiquitin ligase (such as VHL) linked to another moiety that binds a target protein, such that the E3 and the target protein are brought into close spatial proximity. This triggers the polyubiquitylation and subsequent proteasomal degradation of the target protein, enabling post-translational elimination of a specific gene product inside the cell (Figure 1A).
Our crystal structure of the VHL:MZ1:Brd4 ternary complex and supporting biophysical studies have demonstrated that productive assembly of the ternary complex ligase:PROTAC:target depends on the de novo formation of PROTAC-mediated protein–protein interactions (PPIs) between the target protein and the ligase.45,52 These induced interactions in the ternary complex can result in preferential recognition of a given target protein even against members of highly conserved protein families, in spite of using nonselective target ligands. For instance, we have shown that a pan-selective inhibitor of the BET protein family, JQ1, can be derivatized using the VHL ligand VH032 to obtain a PROTAC (MZ1, Figure 1C) that is a preferential degrader for a single member of the BET family, Brd4. By changing the Hyp in PROTAC MZ1 with (3S,4S)- and (3R,4S)-F-Hyps we sought to generate two epimeric F-Hyp PROTACs (Figure 7A). These F-Hyp analogues are expected to have identical binding modes compared to MZ1, the same binary binding affinities for the target BET bromodomains, and extremely similar physicochemical properties, but considerably different binary binding affinity for the VHL ligase. We therefore hypothesized that F-Hyp analogues of MZ1 could help to understand whether and how changing the binary affinity for one component of the ternary complex can impact target degradation. Moreover, fluorination of the hydroxyproline core of a PROTAC may be useful to modulate physicochemical properties of the resulting PROTAC compound. We thus prepared the F-Hyp-containing PROTAC 15a,b, analogues of MZ1 (Figure 7A), which were synthesized according to the synthetic protocol reported previously by our group.45,46 First, we measured the binary binding affinity of 15a,b for VBC and observed that, consistently with what was found for the F-Hyps-containing peptides and VHL ligands (Table 2), PROTAC 15a, featuring the (3R,4S) isomer of F-Hyp, showed the highest binary binding affinity for VBC (Kd binary,VBC = 249 ± 6 nM, Supp. Table 5). PROTAC 15b was found to be a 2.5-fold weaker ligand (Kd binary,VBC = 603 ± 74 nM, Supp. Table 5). In previous work52 we have shown that MZ1 binds VHL and the bromodomain target proteins in a cooperative fashion, (Kd binary,VBC/Kd ternary = α > 1) as a result of favorable PROTAC-induced PPIs. Cooperative binding was found also for F-Hyp-containing PROTACs 15a,b, with α values of the system VHL:15a,b:Brd4 (α = 9.5 for 15a and α = 14.5 for 15b, Supp. Table 5) in line with the values previously measured for MZ1 (α = 9.5–17.645,52). Together these data are consistent with fluorination of Hyp maintaining the cooperative recognition and relative binding mode of MZ1 within the ternary complex.
Figure 7.

BET protein degradation induced by F-Hyp-containing PROTACs. (A) Chemical structure of PROTACs 15a,b. (B) HeLa cells were treated with 15a or 15b and vehicle control (0.01% DMSO) for 24 h. Abundance of individual BET protein was analyzed by Western blotting using corresponding specific antibodies after SDS-PAGE. (C) Antiproliferative activity of compounds 15a,b, MZ1, and inactive epimer cisMZ1. MV4;11 cells were treated with compounds for 72 h prior to quantitation of cell proliferation using the CellTiter-Glo luminescent cell viability assay. The relative signal reflects luminescence values normalized to DMSO control. (D) A549 cells stained with crystal violet dye, following a 7-day incubation with compounds 15a,b, MZ1, and inactive epimer cisMZ1. Dark background was added to improve legibility.
Encouraged by the biophysical validation, we next investigated to what extent F-Hyp-containing PROTACs could induce degradation of target proteins Brd2, Brd3, and Brd4 in cells. PROTAC 15a was able to induce potent degradation of Brd4 (both short and long isoforms) with a DC50 between 1 and 3 nM (Figure 7B). In contrast, degradation of Brd2 and Brd3 occurred only at higher concentrations (DC50 ≈ 10 nM), consistent with the preferential Brd4 degradation documented with MZ1 degrader.46,52 Compound 15b showed a reduced potency in terms of Brd4 degradation, with a DC50 between 10 and 30 nM. Crucially, however, degradation of cellular Brd2 and Brd3 could be observed only at the highest concentration of 15b tested (DC50 ≈ 3 μM, Figure 7B). To evaluate the impact of BET protein degradation on the antiproliferative activity of BET-sensitive cancer cell lines, compounds 15a,b were tested first in acute myeloid leukemia MV4;11 cells (Figure 7C) and compared with MZ1 and the inactive epimer cisMZ1.45,46 PROTAC 15a showed a marked antiproliferative effect (pEC50 = 6.5), albeit weaker than MZ1 (pEC50 = 7.3), with similar maximal response at the highest concentration used when compared to MZ1 (Emax = 92% and 98%, respectively). In contrast, PROTAC 15b showed a much weaker antiproliferative effect compared to MZ1 or 15a, and incomplete maximal response to baseline level was observed even at the highest concentration tested. A similar trend was observed in a colony-formation assay using BET-sensitive lung adenocarcinoma A549 cells treated with test compounds for 7 days (Figure 7D). In this assay, treatment with PROTACs 15a and MZ1 resulted in profound concentration-dependent cytotoxicity, whereas 15b affected cell growth only modestly even at the highest concentration tested. Taken together, the data support more potent degradation activity for degraders that incorporate the (3R,4S)-F-Hyp epimer, which is preferentially recognized by VHL. The markedly more profound growth inhibition activity observed between the two BET degraders investigated here is contributed at least in part by these differences in E3 ligase-binding affinities. At the same time, the data provide also strong evidence that pan-selective degradation of all three BET proteins Brd2, Brd3, and Brd4 leads to a much more profound antiproliferative impact in these cancer cell lines compared to selective Brd4 degradation, as was recently observed in other cancer cell lines too.68 Crucially nevertheless, we show that losing over 20-fold binding affinity at one of the two binding ends of a PROTAC still leads to effective degradation of the target protein at concentrations ∼100-fold lower than the binding Kd of the warhead ligand.
3. Conclusion
We devise a simple, efficient, and scalable synthetic protocol for the preparation of all four diastereoisomers of F-Hyps starting from readily available N-Boc-4-oxo-l-proline benzyl ester. Stereoselective fluorination of the proline ring is demonstrated when starting from N-phenylfluorenyl-protected 4-oxo-l-proline methyl ester. By means of computational studies on model compounds and small-molecule X-ray crystallography we have delineated the effect of fluorination on the conformational preferences and H-bond donor capacity of the Hyp core. Hydroxyproline has a crucial role in structural proteins, e.g., collagen and elastin, and cell signaling pathways such as hypoxic response. Conformational control in proline systems is thus a subject of intense investigation, with important biological and technical applications. Our discovery that fluorination of Hyp inverts the natural preference from the C4-exo pucker to the C4-endo pucker could have unforeseen applications in protein engineering, e.g., enabling design of new molecular switches based on F-Hyps.
Hydroxyproline-containing VHL-hijacking PROTACs are being considered as clinical candidates. Because the hydroxyproline moiety is absolutely critical for molecular recognition, all VHL ligands known to date contain the natural amino acid. The discovery that VHL can also recognize stereoselectively the (3R,4S) epimer of F-Hyp represents an important step forward by expanding the chemical space of chemical degraders. Incorporation of the weak-affinity (3S,4S) epimer of F-Hyp induced target-selective degradation at concentrations over 2 orders of magnitude lower than the binary Kd for VHL. This realization has important implications for drug design of degraders where only weak-affinity binding ligands may be available, for example, against poorly ligandable targets. It is expected that the structure–activity relationships extensively delineated for Hyp-containing VHL ligands38−40 should directly translate upon F-Hyp replacement, thereby allowing expansion of chemical space while fine-tuning molecular and pharmacokinetic properties of VHL inhibitors and VHL-targeting PROTACs. The structural, binding, and cellular PK characterization of F-Hyps and proof-of-concept application as chemical tools disclosed in this work together qualify F-Hyps as useful building blocks for incorporation in future small molecules of medicinal relevance and to probe cell biology. They also provide a blueprint to pursue other fluorohydrin-containing molecules that could inspire new applications to small-molecule discovery.
Acknowledgments
This project has received funding from the European Research Council (ERC) under the European Union’s Seventh Framework Programme (FP7/2007–2013) as a Starting Grant to A.C. (grant agreement no. ERC-2012-StG-311460 DrugE3CRLs). X.L. and K.-H.C. were supported by Marie Skłodowska-Curie Actions Individual Fellowships from the European Commission (H2020-MSCA-IF-2015-806323 and H2020-MSCA-IF-2014-655516, respectively). G.V.C. was supported by a Ph.D. Studentship from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, No. 7148-14-3). A.C.R. was supported by a BBSRC EASTBIO Doctoral Training Partnership award (BB/J01446X/1). Biophysics and drug discovery activities were supported by Wellcome Trust strategic awards to Dundee (100476/Z/12/Z and 094090/Z/10/Z, respectively). We thank Kevin Read and the DMPK team of the Dundee Drug Discovery Unit for cellular PK data, Professor B. Linclau for helpful discussions, P. Fyfe for support with in-house X-ray facilities, and the Diamond Light Source for beamtime (BAG proposals MX10071 and MX14980) and beamline support at beamlines I03, I04, and I04-1.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b05807.
Supplementary results (Tables S1–S6 and Figures S1–S5); materials and methods section; supplementary references; crystal structures have been deposited in the PDB with accession codes 6GFX, 6GFY, and 6GFZ for VBC in complex with compounds 13a, 14a, and 14b, respectively; data collection and refinement statistics; representative ITC titrations; NMR spectra of key intermediates and final compounds (PDF)
Crystal structures of compound 4a (CIF)
Crystal structure of compound 4b (CIF)
Crystal structure of compound 4c (CIF)
Crystal structure of compound 10 (CIF)
Author Present Address
⊥ M.S.G.: Division of Biomedical and Life Sciences, Faculty of Health and Medicine, Lancaster University, Lancaster LA1 4YQ, U.K.
The authors declare no competing financial interest.
Supplementary Material
References
- Gorres K. L.; Raines R. T. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 106. 10.3109/10409231003627991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. T.; Garneau-Tsodikova S.; Gatto G. J. Jr. Angew. Chem., Int. Ed. 2005, 44, 7342. 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- Loenarz C.; Coleman M. L.; Boleininger A.; Schierwater B.; Holland P. W.; Ratcliffe P. J.; Schofield C. J. EMBO Rep. 2011, 12, 63. 10.1038/embor.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRider M. L.; Wilkens S. J.; Waddell M. J.; Bretscher L. E.; Weinhold F.; Raines R. T.; Markley J. L. J. Am. Chem. Soc. 2002, 124, 2497. 10.1021/ja0166904. [DOI] [PubMed] [Google Scholar]
- Bretscher L. E.; Jenkins C. L.; Taylor K. M.; DeRider M. L.; Raines R. T. J. Am. Chem. Soc. 2001, 123, 777. 10.1021/ja005542v. [DOI] [PubMed] [Google Scholar]
- Hodges J. A.; Raines R. T. J. Am. Chem. Soc. 2005, 127, 15923. 10.1021/ja054674r. [DOI] [PubMed] [Google Scholar]
- Wolfe S. Acc. Chem. Res. 1972, 5, 102. 10.1021/ar50051a003. [DOI] [Google Scholar]
- Loenarz C.; Mecinovic J.; Chowdhury R.; McNeill L. A.; Flashman E.; Schofield C. J. Angew. Chem., Int. Ed. 2009, 48, 1784. 10.1002/anie.200805427. [DOI] [PubMed] [Google Scholar]
- Shoulders M. D.; Kotch F. W.; Choudhary A.; Guzei I. A.; Raines R. T. J. Am. Chem. Soc. 2010, 132, 10857. 10.1021/ja103082y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins C. L.; Bretscher L. E.; Guzei I. A.; Raines R. T. J. Am. Chem. Soc. 2003, 125, 6422. 10.1021/ja034015j. [DOI] [PubMed] [Google Scholar]
- Taylor C. M.; Hardré R.; Edwards P. J. B. J. Org. Chem. 2005, 70, 1306. 10.1021/jo0490043. [DOI] [PubMed] [Google Scholar]
- Pandey A. K.; Naduthambi D.; Thomas K. M.; Zondlo N. J. J. Am. Chem. Soc. 2013, 135, 4333. 10.1021/ja3109664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W.; Hardcastle K. I.; Conticello V. P. Angew. Chem., Int. Ed. 2006, 45, 8141. 10.1002/anie.200603227. [DOI] [PubMed] [Google Scholar]
- Newberry R. W.; Raines R. T. In Peptidomimetics I; Lubell W. D., Ed.; Springer International Publishing: Cham, 2017; p 1. [Google Scholar]
- Kubyshkin V.; Afonin S.; Kara S.; Budisa N.; Mykhailiuk P. K.; Ulrich A. S. Org. Biomol. Chem. 2015, 13, 3171. 10.1039/C5OB00034C. [DOI] [PubMed] [Google Scholar]
- Nishi Y.; Uchiyama S.; Doi M.; Nishiuchi Y.; Nakazawa T.; Ohkubo T.; Kobayashi Y. Biochemistry 2005, 44, 6034. 10.1021/bi047887m. [DOI] [PubMed] [Google Scholar]
- Crespo M. D.; Rubini M. PLoS One 2011, 6, e19425. 10.1371/journal.pone.0019425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner T.; Hess P.; Bae J. H.; Wiltschi B.; Moroder L.; Budisa N. PLoS One 2008, 3, e1680. 10.1371/journal.pone.0001680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C. A.; Talaty E. R.; Bann J. G. Chem. Commun. 2009, 3366. 10.1039/b821952d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skovgaard D.; Kjaer A.; Heinemeier K. M.; Brandt-Larsen M.; Madsen J.; Kjaer M. PLoS One 2011, 6, e16678. 10.1371/journal.pone.0016678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meanwell N. A.J. Med. Chem. 2018, 10.1021/acs.jmedchem.7b01788. [DOI] [Google Scholar]
- Chia P. W.; Livesey M. R.; Slawin A. M.; van Mourik T.; Wyllie D. J.; O’Hagan D. Chem. - Eur. J. 2012, 18, 8813. 10.1002/chem.201200071. [DOI] [PubMed] [Google Scholar]
- Gillis E. P.; Eastman K. J.; Hill M. D.; Donnelly D. J.; Meanwell N. A. J. Med. Chem. 2015, 58, 8315. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- O’Hagan D. Future Med. Chem. 2011, 3, 189. 10.4155/fmc.10.295. [DOI] [PubMed] [Google Scholar]
- Purser S.; Moore P. R.; Swallow S.; Gouverneur V. Chem. Soc. Rev. 2008, 37, 320. 10.1039/B610213C. [DOI] [PubMed] [Google Scholar]
- Jansen K.; Heirbaut L.; Verkerk R.; Cheng J. D.; Joossens J.; Cos P.; Maes L.; Lambeir A. M.; De Meester I.; Augustyns K.; Van der Veken P. J. Med. Chem. 2014, 57, 3053. 10.1021/jm500031w. [DOI] [PubMed] [Google Scholar]
- Staas D. D.; Savage K. L.; Sherman V. L.; Shimp H. L.; Lyle T. A.; Tran L. O.; Wiscount C. M.; McMasters D. R.; Sanderson P. E.; Williams P. D.; Lucas B. J. Jr.; Krueger J. A.; Lewis S. D.; White R. B.; Yu S.; Wong B. K.; Kochansky C. J.; Anari M. R.; Yan Y.; Vacca J. P. Bioorg. Med. Chem. 2006, 14, 6900. 10.1016/j.bmc.2006.06.040. [DOI] [PubMed] [Google Scholar]
- Somovilla V. J.; Bermejo I. A.; Albuquerque I. S.; Martínez-Sáez N.; Castro-López J.; García-Martín F.; Compañón I.; Hinou H.; Nishimura S.-I.; Jiménez-Barbero J.; Asensio J. L.; Avenoza A.; Busto J. H.; Hurtado-Guerrero R.; Peregrina J. M.; Bernardes G. J. L.; Corzana F. J. Am. Chem. Soc. 2017, 139, 18255. 10.1021/jacs.7b09447. [DOI] [PubMed] [Google Scholar]
- Mullard A. Nat. Rev. Drug Discovery 2018, 17, 81. 10.1038/nrd.2018.4. [DOI] [PubMed] [Google Scholar]
- Graton J.; Wang Z.; Brossard A. M.; Monteiro D. G.; Le Questel J. Y.; Linclau B. Angew. Chem., Int. Ed. 2012, 51, 6176. 10.1002/anie.201202059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield C. J.; Ratcliffe P. J. Nat. Rev. Mol. Cell Biol. 2004, 5, 343. 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- Hon W. C.; Wilson M. I.; Harlos K.; Claridge T. D.; Schofield C. J.; Pugh C. W.; Maxwell P. H.; Ratcliffe P. J.; Stuart D. I.; Jones E. Y. Nature 2002, 417, 975. 10.1038/nature00767. [DOI] [PubMed] [Google Scholar]
- Min J. H.; Yang H.; Ivan M.; Gertler F.; Kaelin W. G. Jr.; Pavletich N. P. Science 2002, 296, 1886. 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- Kamura T.; Koepp D. M.; Conrad M. N.; Skowyra D.; Moreland R. J.; Iliopoulos O.; Lane W. S.; Kaelin W. G. Jr.; Elledge S. J.; Conaway R. C.; Harper J. W.; Conaway J. W. Science 1999, 284, 657. 10.1126/science.284.5414.657. [DOI] [PubMed] [Google Scholar]
- Stebbins C. E.; Kaelin W. G. Jr.; Pavletich N. P. Science 1999, 284, 455. 10.1126/science.284.5413.455. [DOI] [PubMed] [Google Scholar]
- Maxwell P. H.; Wiesener M. S.; Chang G. W.; Clifford S. C.; Vaux E. C.; Cockman M. E.; Wykoff C. C.; Pugh C. W.; Maher E. R.; Ratcliffe P. J. Nature 1999, 399, 271. 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- Van Molle I.; Thomann A.; Buckley D. L.; So E. C.; Lang S.; Crews C. M.; Ciulli A. Chem. Biol. 2012, 19, 1300. 10.1016/j.chembiol.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares P.; Gadd M. S.; Frost J.; Galdeano C.; Ellis L.; Epemolu O.; Rocha S.; Read K. D.; Ciulli A. J. Med. Chem. 2018, 61, 599. 10.1021/acs.jmedchem.7b00675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdeano C.; Gadd M. S.; Soares P.; Scaffidi S.; Van Molle I.; Birced I.; Hewitt S.; Dias D. M.; Ciulli A. J. Med. Chem. 2014, 57, 8657. 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost J.; Galdeano C.; Soares P.; Gadd M. S.; Grzes K. M.; Ellis L.; Epemolu O.; Shimamura S.; Bantscheff M.; Grandi P.; Read K. D.; Cantrell D. A.; Rocha S.; Ciulli A. Nat. Commun. 2016, 7, 13312. 10.1038/ncomms13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley D. L.; Van Molle I.; Gareiss P. C.; Tae H. S.; Michel J.; Noblin D. J.; Jorgensen W. L.; Ciulli A.; Crews C. M. J. Am. Chem. Soc. 2012, 134, 4465. 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Crews C. M. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 107. 10.1146/annurev-pharmtox-010715-103507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley D. L.; Crews C. M. Angew. Chem., Int. Ed. 2014, 53, 2312. 10.1002/anie.201307761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toure M.; Crews C. M. Angew. Chem., Int. Ed. 2016, 55, 1966. 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]
- Chan K. H.; Zengerle M.; Testa A.; Ciulli A. J. Med. Chem. 2018, 61, 504. 10.1021/acs.jmedchem.6b01912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M.; Chan K. H.; Ciulli A. ACS Chem. Biol. 2015, 10, 1770. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E. D.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Nat. Chem. Biol. 2015, 11, 611. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A. C.; Toure M.; Hellerschmied D.; Salami J.; Jaime-Figueroa S.; Ko E.; Hines J.; Crews C. M. Angew. Chem., Int. Ed. 2016, 55, 807. 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniaci C.; Hughes S. J.; Testa A.; Chen W.; Lamont D. J.; Rocha S.; Alessi D. R.; Romeo R.; Ciulli A. Nat. Commun. 2017, 8, 830. 10.1038/s41467-017-00954-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crew A. P.; Raina K.; Dong H. Q.; Qian Y. M.; Wang J.; Vigil D.; Serebrenik Y. V.; Hamman B. D.; Morgan A.; Ferraro C.; Siu K.; Neklesa T. K.; Winkler J. D.; Coleman K. G.; Crews C. M. J. Med. Chem. 2018, 61, 583. 10.1021/acs.jmedchem.7b00635. [DOI] [PubMed] [Google Scholar]
- Gechijian L. N.; Buckley D. L.; Lawlor M. A.; Reyes J. M.; Paulk J.; Ott C. J.; Winter G. E.; Erb M. A.; Scott T. G.; Xu M.; Seo H. S.; Dhe-Paganon S.; Kwiatkowski N. P.; Perry J. A.; Qi J.; Gray N. S.; Bradner J. E. Nat. Chem. Biol. 2018, 14, 405. 10.1038/s41589-018-0010-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd M. S.; Testa A.; Lucas X.; Chan K. H.; Chen W. Z.; Lamont D. J.; Zengerle M.; Ciulli A. Nat. Chem. Biol. 2017, 13, 514. 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes S. J.; Ciulli A. Essays Biochem. 2017, 61, 505. 10.1042/EBC20170041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barraclough P.; Hudhomme P.; Spray C. A.; Young D. W. Tetrahedron 1995, 51, 4195. 10.1016/0040-4020(95)00135-U. [DOI] [Google Scholar]
- Baumann M.; Baxendale I. R.; Martin L. J.; Ley S. V. Tetrahedron 2009, 65, 6611. 10.1016/j.tet.2009.05.083. [DOI] [Google Scholar]
- Gómez-Vidal J. A.; Silverman R. B. Org. Lett. 2001, 3, 2481. 10.1021/ol0161054. [DOI] [PubMed] [Google Scholar]
- Zanato C.; Watson S.; Bewick G. S.; Harrison W. T. A.; Zanda M. Org. Biomol. Chem. 2014, 12, 9638. 10.1039/C4OB02002B. [DOI] [PubMed] [Google Scholar]
- Gupta S.; Schafmeister C. E. J. Org. Chem. 2009, 74, 3652. 10.1021/jo802679n. [DOI] [PubMed] [Google Scholar]
- Sharma R.; Lubell W. D. J. Org. Chem. 1996, 61, 202. 10.1021/jo9514984. [DOI] [Google Scholar]
- Blanco M.-J.; Paleo M. R.; Penide C.; Sardina F. J. J. Org. Chem. 1999, 64, 8786. 10.1021/jo990283h. [DOI] [PubMed] [Google Scholar]
- Kenny P. W. J. Chem. Inf. Model. 2009, 49, 1234. 10.1021/ci9000234. [DOI] [PubMed] [Google Scholar]
- Johnson E. R.; Keinan S.; Mori-Sánchez P.; Contreras-García J.; Cohen A. J.; Yang W. J. Am. Chem. Soc. 2010, 132, 6498. 10.1021/ja100936w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glendening E. D.; Badenhoop J. K.; Reed A. E.; Carpenter J. E.; Bohmann J. A.; Morales C. M.; Landis C. R.; Weinhold F.. NBO 6.0.; Theoretical Chemistry Institute, University of Wisconsin: Madison, 2013.
- Newberry R. W.; Raines R. T. Acc. Chem. Res. 2017, 50, 1838. 10.1021/acs.accounts.7b00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares P.; Lucas X.; Ciulli A.. Bioorg. Med. Chem. 2018, 26, 2992. 10.1016/j.bmc.2018.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan E.; Compain G.; Mtashobya L.; Le Questel J. Y.; Besseau F.; Galland N.; Linclau B.; Graton J. Chem. - Eur. J. 2015, 21, 11462. 10.1002/chem.201501171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffredi G. T.; Gouverneur V.; Bernet B. Angew. Chem., Int. Ed. 2013, 52, 10524. 10.1002/anie.201303766. [DOI] [PubMed] [Google Scholar]
- Nabet B.; Roberts J. M.; Buckley D. L.; Paulk J.; Dastjerdi S.; Yang A.; Leggett A. L.; Erb M. A.; Lawlor M. A.; Souza A.; Scott T. G.; Vittori S.; Perry J. A.; Qi J.; Winter G. E.; Wong K.-K.; Gray N. S.; Bradner J. E. Nat. Chem. Biol. 2018, 14, 431. 10.1038/s41589-018-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.