

Although extensive studies of the transmembrane unit (gp41) of HIV-1 Env have led to a fusion inhibitor clinically used to block viral entry, the functions of different domains of gp41 in HIV-1 fusion and infectivity are not fully elucidated. The polar region (PR) of gp41 has been proposed to participate in HIV-1 membrane fusion in biochemical analyses, but its role in viral entry and infectivity remain unclear. In our effort to characterize three nucleotide mutations of an HIV-1 RNA element that partially overlaps the PR coding sequence, we identified a novel function of the PR that determines viral fusion and infectivity. We further demonstrated the structural and functional impact of six PR mutations on HIV-1 Env stability, viral fusion, and infectivity. Our findings reveal the previously unappreciated function of the PR and the underlying mechanisms, highlighting the important role of the PR in regulating HIV-1 fusion and infectivity.
KEYWORDS: HIV-1, entry, envelope glycoprotein, fusion, gp160, gp41, infectivity, polar region, trimer
ABSTRACT
HIV-1 enters cells through binding between viral envelope glycoprotein (Env) and cellular receptors to initiate virus and cell fusion. HIV-1 Env precursor (gp160) is cleaved into two units noncovalently bound to form a trimer on virions, including a surface unit (gp120) and a transmembrane unit (gp41) responsible for virus binding and membrane fusion, respectively. The polar region (PR) at the N terminus of gp41 comprises 17 residues, including 7 polar amino acids. Previous studies suggested that the PR contributes to HIV-1 membrane fusion and infectivity; however, the precise role of the PR in Env-mediated viral entry and the underlying mechanisms remain unknown. Here, we show that the PR is critical for HIV-1 fusion and infectivity by stabilizing Env trimers. Through analyzing the PR sequences of 57,645 HIV-1 isolates, we performed targeted mutagenesis and functional studies of three highly conserved polar residues in the PR (S532P, T534A, and T536A) which have not been characterized previously. We found that single or combined mutations of these three residues abolished or significantly decreased HIV-1 infectivity without affecting viral production. These PR mutations abolished or significantly reduced HIV-1 fusion with target cells and also Env-mediated cell-cell fusion. Three PR mutations containing S532P substantially reduced gp120 and gp41 association, Env trimer stability, and increased gp120 shedding. Furthermore, S532A mutation significantly reduced HIV-1 infectivity and fusogenicity but not Env expression and cleavage. Our findings suggest that the PR of gp41, particularly the key residue S532, is structurally essential for maintaining HIV-1 Env trimer, viral fusogenicity, and infectivity.
IMPORTANCE Although extensive studies of the transmembrane unit (gp41) of HIV-1 Env have led to a fusion inhibitor clinically used to block viral entry, the functions of different domains of gp41 in HIV-1 fusion and infectivity are not fully elucidated. The polar region (PR) of gp41 has been proposed to participate in HIV-1 membrane fusion in biochemical analyses, but its role in viral entry and infectivity remain unclear. In our effort to characterize three nucleotide mutations of an HIV-1 RNA element that partially overlaps the PR coding sequence, we identified a novel function of the PR that determines viral fusion and infectivity. We further demonstrated the structural and functional impact of six PR mutations on HIV-1 Env stability, viral fusion, and infectivity. Our findings reveal the previously unappreciated function of the PR and the underlying mechanisms, highlighting the important role of the PR in regulating HIV-1 fusion and infectivity.
INTRODUCTION
HIV-1 enters cells through binding between viral envelope glycoprotein (Env) and cellular receptors to initiate fusion of virus and cell membranes (1). The precursor polyprotein of HIV-1 Env (gp160) is cleaved by a cellular protease into a surface unit (gp120) and a transmembrane unit (gp41), which are noncovalently bound to form a trimer on virions. HIV-1 gp120 and gp41 are responsible for virus binding to target cells and membrane fusion, respectively. The binding of gp120 to the HIV-1 receptor CD4 and a coreceptor triggers refolding of gp41 into a trimer of hairpins with a six-helix bundle core, which leads to membrane fusion and viral entry and infection (2). As a class I fusion glycoprotein, HIV-1 gp41 contains an N-terminal ectodomain, a transmembrane domain, and a C-terminal cytoplasmic tail. The ectodomain of gp41 is comprised of an N-terminal fusion peptide (FP), connected through a flexible polar region (PR) to an N-terminal heptad repeat (NHR), a C-terminal heptad repeat (CHR), and a membrane-proximal external region (MPER). Despite extensive studies of HIV-1 Env, the functions of different domains of gp41 in HIV-1 fusion and immunogenicity are not fully elucidated (3).
The PR comprises 17 residues including 7 polar amino acids at the N terminus of gp41, which was initially proposed to participate in peptide-based membrane fusion in biochemical analyses (4, 5). However, the function of the PR in HIV-1 entry and infectivity remains unclear. In previous studies, the PR was also referred to as the FP-proximal region (FPPR) or the FP-proximal segment (6, 7). It has been suggested that the FPPR and MPER can synergistically contribute to initiation of the HIV-1 membrane fusion process and viral infection (6, 8). The structure of the gp41 ectodomain indicates helical refolding of the FPPR and part of the MPER as well as insertion of the MPER into the viral membrane within trimeric gp41, suggesting important roles of FPPR and MPER in viral fusion (7). However, these studies have not defined the mechanisms by which the PR modulates HIV-1 fusion and infectivity.
The HIV-1 Rev response element (RRE) is a highly structured cis-acting RNA element essential for viral replication, which is located in the env gene partially overlapping the gp120 and gp41 coding sequences (9). Binding of Rev to RRE is required for efficient nuclear export of viral mRNA and protein synthesis. The stem-loop secondary structure of the RRE is critical for Rev protein binding and its functions (9). To study the effect of HIV-1 RNA modification on viral gene expression, Lichinchi et al. examined single and combined mutations of three nucleotides in the HIV-1 RRE. They reported that N6-methyladenosine (m6A) at A7883 in the stem-loop of RRE is critical for Rev binding and enhanced viral mRNA nuclear export (10). However, they did not examine whether these RRE mutations that overlap gp41 coding sequences affect HIV-1 fusion and infectivity.
In this study, we found that the three RRE mutations reported by Lichinchi and colleagues (10) altered amino acid sequences of the PR and fusogenicity of gp41 but did not affect RRE function. We performed targeted mutagenesis and structural and functional analyses of three highly conserved residues in the PR that have not been studied previously. These point mutations abolish or substantially reduce HIV-1 fusion efficiency and viral infectivity by destabilizing the Env trimer. Our data reveal an undefined mechanism by which the PR determines HIV-1 fusion and infectivity. These findings indicate that the highly conserved PR may represent a new target for antiretroviral and vaccine development against HIV-1 entry and infection.
RESULTS
RRE mutations overlapping the gp41 coding sequence do not affect RRE function.
To study the effect of HIV-1 RNA modification on viral gene expression, Lichinchi et al. examined single and combined mutations of three nucleotides (U7871C, A7877G, and A7883G) in the RRE of the HIV-1LAI strain and suggested that m6A modification at A7883 is critical for Rev binding and enhanced viral mRNA nuclear export (10). We noticed that these three RRE mutations also changed 1 to 3 amino acids (aa) of gp41 compared with wild-type (WT) HIV-1 sequence (Fig. 1A). Our bioinformatic analyses of 63,612 RRE sequences in the HIV Sequence Database revealed that these three nucleotides are highly conserved, with a 0.2% to 7.3% mutation frequency (Fig. 1B). The three polar residues (S532, T534, and T536) are within the 17-aa PR of gp41 located between the fusion peptide (FP) and N-terminal heptad repeat (4) (Fig. 1C) and are highly conserved among 57,645 HIV-1 isolates (Fig. 1D). Of note, residues 533 and 541 in the PR show polymorphisms in different HIV-1 subtypes (Fig. 1D). These data suggest the importance of these residues of the PR in HIV-1 replication.
FIG 1.

RRE mutations overlapping the gp41 coding sequence do not affect RRE function. (A) Alignment of nucleotide (nt) and amino acid (aa) sequences of overlapped HIV-1 RRE and Env in wild-type (WT) and mutants (M1 to M5) based on HIV-1NL4-3 (GenBank accession number M19921.2). (B) Secondary structure of the hairpin loop IIB of the RRE of HIV-1NL4-3. The mutation frequencies of the indicated nucleotides shown in parentheses are based on analysis of 63,612 RRE sequences in the HIV Sequence Database. Three nucleotides that were mutated are shown in red, which are equivalent to U7871, A7877, and A7883 in the HIV-1LAI strain reported by Lichinchi et al. (10). (C) HIV-1 gp41 domains (residue numbers are based on HIV-1NL4-3 strain). FP, fusion peptide; PR, polar region; NHR, N-terminal heptad repeat; CHR, C-terminal heptad repeat; MPER, membrane-proximal external region; TM, transmembrane domain; CT, cytoplasmic tail. (D) Mutation frequency of the PR amino acids in HIV-1 gp41. A total of 57,645 aligned HIV-1 Env sequences from the HIV database were analyzed. Frequency of each residue (total, 17 aa) in the PR was calculated to generate sequence logo using WebLogo. (E and F) Single-cycle HIV-1 pseudotyped with VSV-G was generated by transfecting pNL4-3 E-R-Rev-/YFP (WT or M1 to M5 separately) and plasmids encoding VSV-G or Rev fused with mApple (Rev-mApple). Plasmid encoding mApple alone was used as a negative control. Cell lysates were used for immunoblotting, and the results are shown below the bar figures (one representative from three independent experiments is shown); viruses, as indicated, were used to infect HeLa cells to quantify HIV-1 infectivity. HSP90 was used as a loading control. All experiments were performed with triplicate samples and repeated at least three times, and means ± standard errors of the means are shown. No statistical difference in levels of infectivity was observed between WT and each mutant.
To investigate the effect of these mutated RREs on Rev interaction during HIV-1 infection, we engineered rev- and env-deficient reporter HIV-1 strains containing the WT or each mutant RRE in the presence or absence of Rev. Because the nucleotide mutations in RRE resulted in amino acid changes in Env (Fig. 1A), the reporter HIV-1 defective for Env expression was pseudotyped with vesicular stomatitis virus glycoprotein G (VSV-G) to allow endocytosis-mediated viral entry (11). We found that HIV-1 bearing mutant RREs responded to trans-complemented Rev similarly to WT HIV-1, with comparable infectivity (Fig. 1E and F), indicating that these RRE mutations do not affect Rev-RRE interactions when Rev is expressed in trans.
PR mutations significantly decrease HIV-1 infectivity without affecting viral production.
To investigate the role of these nucleotides in HIV-1 replication, we generated five HIV-1 mutants (M1 to M5) with single or combined mutations at T7814C, A7820G, and A7826G in the RRE of the HIV-1NL4-3 strain (Fig. 1A). These mutations caused 1- to 3-aa changes in gp41, namely M1 (S532P and T534A), M2 (T536A), M3 (S532P, T534A, and T536A), M4 (S532P), and M5 (T534A) (Fig. 1A). Intriguingly, when equal amounts of HIV-1 were used to infect target cells, M1, M3, and M4 lost infectivity, while M2 and M5 reduced infectivity over 10-fold compared with the level of WT HIV-1 (Fig. 2A), suggesting that these three residues in gp41 are critical for HIV-1 infection.
FIG 2.

PR mutations significantly decrease HIV-1 infectivity without affecting viral production. (A) HIV-1 infectivity of equal amounts of p24 of WT and mutants was quantified with TZM-bl cells. (B) Lysates of transfected HEK293T cells were analyzed for HIV-1 Gag, CA, and gp160 expression by immunoblotting. GAPDH was a loading control. (C) HIV-1 p24 levels in supernatants of transfected HEK293T cells were quantified by ELISA. All experiments were performed with triplicate samples and repeated at least three times, and means ± standard errors of the means are shown. Dunnett's multiple-comparison test was used for statistical analysis. ***, P < 0.0001, for the comparison of the result with an individual mutant to that with WT HIV-1.
To examine the effect of these gp41 mutations on HIV-1 production, we compared mutant viruses with replication-competent WT HIV-1 generated from proviral DNA-transfected HEK293T cells. Relative to WT HIV-1 proteins expressed in virus-producing cells, mutants M1 to M5 showed comparable levels of HIV-1 Gag, capsid ([CA] p24), gp160, and gp41 proteins (Fig. 2B). HIV-1 gp160 is cleaved into gp120 and gp41 by furin or a related cellular protease primarily at a motif before the first residue of the FP of gp41 or at a secondary site located 8 aa N-terminal to the first site (12, 13). The first mutation (S532P) is 22 aa and 30 aa from the primary and secondary cleavage sites of gp160, respectively (2). These gp41 mutations did not alter the gp160 cleavage sites, and cleaved gp41 levels in virus-producing cells were comparable between WT and mutants M1 to M5 (Fig. 2B), suggesting that gp160 cleavage is not affected by these mutations. However, compared with WT HIV-1-producing cells, cleaved gp120 was undetectable in cells expressing the M1, M3, and M4 mutants and significantly decreased in cells expressing the M5 mutant (Fig. 2B), suggesting that these mutations may reduce gp120 stability or increase gp120 shedding. Furthermore, similar p24 levels of WT and mutant viruses were detected in the supernatants of transfected cells (no statistically significant difference) (Fig. 2C), indicating that these PR mutations did not affect HIV-1 production and release.
PR mutations decrease gp120 association and reduce viral fusion and infectivity.
To investigate whether the decreased infectivity of the mutants was attributed to impaired HIV-1 entry due to the gp41 mutations, we compared the expression and cleavage of gp160 of WT and mutant HIV-1 produced from HEK293T cells transfected separately with proviral DNA constructs. All mutants showed comparable levels of Gag and p24 in both cell lysates and purified virions relative to levels of WT HIV-1 (Fig. 3A and B, respectively). In cell lysates, mutants M1 to M5 expressed levels of gp160 and gp41 similar to those of WT HIV-1. While M2 had a level of gp120 similar to that of WT HIV-1, M1 and M3 to M5 had undetectable or significantly lower levels of gp120 (Fig. 3A, left panel). Compared with purified WT virions, virions of M1 to M5 showed increased gp160 levels and significantly decreased gp120 levels. Relative to WT virions, gp41 levels of M1, M3, and M4 virions were significantly reduced, while M2 and M5 showed comparable or slightly reduced gp41 levels (Fig. 3B, left panel). The reduced gp41 and increased gp160 in HIV-1 particles of M1, M3, and M4 relative to levels in WT HIV-1 (Fig. 3B, left panel) suggest underpackaging of furin-processed trimers in mutant virions.
FIG 3.

PR mutations decrease gp120 association and reduce HIV-1 fusion and infectivity. HEK293T cells were transfected with full-length WT or mutant (M1 to M5) proviral constructs without (control) or with (+Env) a WT gp160 expression plasmid. (A) Cell lysates were used for immunoblotting of the indicated HIV-1 proteins. GAPDH was a loading control of cell lysates. (B) Purified HIV-1 virions were used for immunoblotting of the indicated HIV-1 proteins. (C and D) Densitometry quantification of gp160, gp120, and gp41 bands in immunoblotting of cell lysates (control samples in panel A) and purified HIV-1 (control samples in panel B), respectively. Relative levels of gp120/gp160 or gp41/gp160 ratios were calculated from three independent experiments. The values of WT HIV-1 were set as 100%, and relative levels are shown as means ± standard errors of the means (n = 3). *, P < 0.05; **, P < 0.005; ***, P < 0.0001, for the comparison of the result with an individual mutant to that with WT HIV-1 (E) WT or mutant HIV-1 generated from transfected HEK293T cells without (control) or with Env overexpression (+Env) were quantified for viral infectivity using TZM-bl cells. (F) Virion-cell fusion was determined by flow cytometry-based BlaM-Vpr assays using TZM-bl cells (10 or 50 ng of p24 for HIV-1 with or without Env trans-supplementation, respectively). All experiments were performed with triplicate samples for panels E and F and repeated at least three times, and means ± standard errors of the means are shown. Dunnett's multiple-comparison test was used for statistical analysis. ***, P < 0.0001, for the comparison of the result with an individual mutant to that with WT HIV-1.
Based on quantification of three independent experiments and compared with WT HIV-1, the gp120/gp160 levels of M1, M3, M4, and M5 mutants were significantly reduced in cell lysates (Fig. 3C), while the gp120/gp160 levels of M1, M2, M3, and M4 mutants were significantly decreased in purified virions (Fig. 3D). Moreover, the gp41/gp160 levels of all mutant viruses were similar to those of WT HIV-1 in cell lysates (Fig. 3C) but were significantly lower than those of WT HIV-1 in virions (Fig. 3D). Together, these results suggest that reduced gp120-gp41 trimer stability in mutant HIV-1 (particularly M1, M3, and M4 that contain the S532P mutation) leads to significantly decreased viral infectivity.
Env overexpression rescues HIV-1 infectivity and fusion of the PR mutants.
To examine whether HIV-1 Env expression in trans could rescue gp120 expression of the mutant viruses, we overexpressed WT Env of HIV-1NL4-3 in virus-producing cells and compared WT and mutant viruses (Fig. 3A and B, +Env). trans-Supplementation of WT Env significantly increased gp160 incorporation into virions (Fig. 3B, right panels) and rescued the infectivity of all mutants (Fig. 3E), confirming that low levels of gp120 and gp41 in M1 to M5 account for the loss or significantly reduced infectivity. Notably, Env trans-supplementation decreased WT HIV-1 infectivity by ∼40% and partially rescued infectivity of M1 to M5 relative to the level of WT HIV-1 (Fig. 3E), which was likely due to competitive binding of uncleaved gp160 on virion surfaces to cellular receptors, leading to decreased viral fusion of WT HIV-1.
To investigate whether the loss of infectivity of the mutant viruses was due to compromised viral entry, we measured virion-cell fusion efficiency of WT and mutant viruses using a beta-lactamase (BLaM)-Vpr assay (14). WT HIV-1 infection led to fusion with 72% of target cells, while M1, M3, and M4 mutants showed only background levels of virion-cell fusion (< 0.5%), and M2 and M5 showed 5% to 8% virion-cell fusion (Fig. 3F). We also tested virion-cell fusion for viruses generated with WT Env overexpression. The trans-supplementation of WT Env significantly increased virus-cell fusion of M1 to M5 to levels close to the level of WT HIV-1, indicating that WT Env overexpression rescued defective virion-cell fusion of the mutants (Fig. 3F). These data are consistent with the HIV-1 infectivity results (Fig. 3E), confirming that mutant viruses have significantly diminished virion-cell fusion and, thus, compromised HIV-1 entry into target cells.
PR mutations abolish or significantly decrease Env-mediated cell-cell fusion.
Because Env-mediated cell-cell fusion plays an important role in efficient HIV-1 transmission (2, 15), we sought to examine whether the gp41 mutations affected HIV-1 Env-mediated membrane fusion between cells. HEK293T cells that overexpressed Env (gp160) of WT HIV-1 and M1 to M5 were cocultured with TZM-bl cells to measure cell-cell fusion using a luciferase reporter assay (16). Compared with the level of WT Env (set as 1.0), similar levels of mutant Env (0.9 to 1.3) were detected in cell lysates of transfected HEK293T cells (Fig. 4A). The gp120 and gp41 products in cell lysates were less abundant than the precursor gp160 (Fig. 4A), likely due to overexpressed gp160 that could not be efficiently cleaved by endogenous furin in cells. Of note, the majority of unprocessed gp160 in cells could be a limitation of the cell-cell fusion assay relative to the performance of the virus-cell fusion assay.
FIG 4.
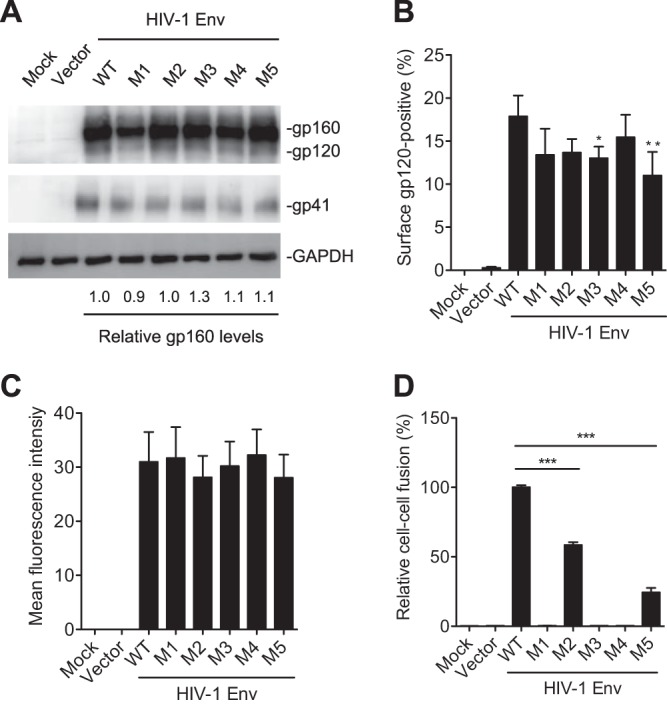
PR mutations abolish or significantly decrease Env-mediated cell-cell fusion. HEK293T cells were separately transfected with pRK empty vector, WT, or mutant Env (gp160) expression construct together with pCMV-Rev. HEK293T cells transfected without plasmid DNA were used as a mock control. (A) At 24 h posttransfection, HEK293T cells were collected and lysed for immunoblotting analysis with antibodies to gp160/gp120, gp41, or GAPDH. (B and C) At 24 h posttransfection, the cells were stained with gp120 monoclonal antibody (2G12) at 4°C to measure the cell surface gp120 expression using flow cytometry. Average percentages (B) and the mean fluorescence intensity (C) of gp120-positive cells from four independent experiments were determined. (D) Transfected HEK293T cells were cocultured with TZM-bl cells for 24 h and then lysed for firefly luciferase activity measurement of Env-mediated cell-cell fusion. Average percentages of HIV-1 Env-mediated cell-cell fusion from three independent experiments are shown, with the value for the WT set as 100%. All experiments were performed with triplicate samples and repeated at least three times, and means ± standard errors of the means are shown. Dunnett's multiple-comparison test was used for statistical analysis. *, P < 0.05; **, P < 0.01; ***, P < 0.0001, for the comparison of the result with an individual mutant Env to that with WT Env.
The level of cell surface gp120 and its trimer conformation are critical for Env-mediated cell-cell fusion (15). We performed flow cytometry to measure the level of cell surface gp120 expression in transfected HEK293T cells. The average percentages of gp120-positive cells were slightly lower in M3 and M5 mutant Env-expressing cells than in WT Env-expressing cells (Fig. 4B), while the mean fluorescence intensities of cell surface gp120 were comparable among mutants M1 to M5 and WT Env-expressing cells (Fig. 4C). Importantly, M1, M3, and M4 mutants containing S532P abolished Env-mediated cell-cell fusion, and M2 and M5 mutants significantly reduced cell-cell fusion compared with the level of WT Env (Fig. 4D). These data demonstrate that the PR mutations significantly decrease HIV-1 Env-mediated cell-cell fusion, suggesting that these PR mutations likely lead to conformation changes of Env trimers, thereby significantly reducing the fusogenicity of mutant Env.
PR mutations decrease gp120 association with gp41 and increase gp120 shedding.
The processed gp120 in cells that is not assembled into trimers can shed into the medium (17). To better understand the mechanisms of defective fusion of the mutants, we performed a gp120 shedding assay and compared gp120 processing and association indexes between WT and mutant Env proteins using a standard protocol and equations (18). Representative results of gp120 shedding of 35S-labeled Env of WT and mutants (M1 to M5) from one experiment are shown in Fig. 5A. The average results from four independent experiments demonstrate that gp120 shedding of M1, M3, and M4 mutants were significantly higher than those of WT Env, while gp120 processing levels were comparable (Fig. 5B). These results suggest that the common mutation S532P in M1, M3, and M4 significantly decreases gp120 and gp41 trimer association, thereby leading to reduced viral fusion and infectivity.
FIG 5.
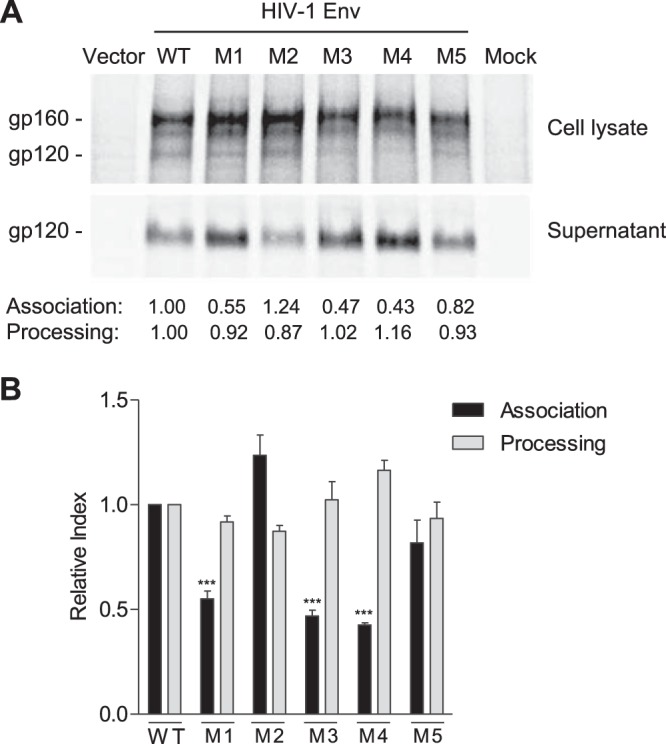
PR mutations decrease gp120 association and increase gp120 shedding. (A) Representative results of gp120 shedding of 35S-labeled WT and mutant Env (M1 to M5) from one of four separate experiments. HEK293T cells were transfected with WT or mutant Env expression constructs for the gp120 shedding assay. The gp120 processing and the association index were calculated as described in Materials and Methods and are shown below the shedding gel. Values are representative of the average of four separate experiments. (B) Summary results of four independent experiments of gp120 shedding. Dunnett's multiple-comparison test was used for statistical analysis. ***, P < 0.001, for a comparison of the association index of mutant M1, M3, or M4 with that of WT Env.
S532A mutant of the PR reduces HIV-1 infectivity and fusogenicity.
Our bioinformatic analyses revealed that, at residue 532 in the PR of 57,645 HIV-1 Env sequences, 96.6% and 3.3% sequences have serine and alanine, respectively (Table 1). These data suggest that the majority of circulating HIV-1 isolates contain residue S532 in the PR, while viruses containing A532 in the PR can also be infectious and transmitted in human patients. To compare the role of S532 and A532 in regulating HIV-1 Env incorporation, infectivity, and fusogenicity, we generated a single S532A mutant of the PR (referred to as M6) for related functional analyses (Fig. 6A). Transfection of HEK293T cells with a proviral DNA construct of M6 yielded levels of extracellular p24 release similar to those of the WT HIV-1 level (Fig. 6B), indicating comparable viral production and release. However, M6 infectivity decreased by 74% compared with that of WT HIV-1 (Fig. 6C, control samples). Similar expression levels of gp160, gp120, gp41, Gag, and p24 were observed in HEK293T cells producing M6 or WT HIV-1 and in purified virions (Fig. 6D, control samples), suggesting that S532A mutation does not significantly affect HIV-1 protein expression and Env incorporation in virions. To examine whether HIV-1 Env expression in trans could rescue M6 infectivity, we overexpressed WT Env of HIV-1NL4-3 in virus-producing cells and compared WT and M6 levels of expression (Fig. 6D, +Env). trans-Supplementation of WT Env significantly increased expression of gp160/120 in cells and incorporation of gp160/gp120 into virions (Fig. 6D) and increased M6 infectivity to the level of WT HIV-1 (Fig. 6C, +Env).
TABLE 1.
The frequency of residue 532 in the PR of 57,645 HIV-1 Env sequencesa
| Amino acid at position 532 | No. of Env sequences | Frequency (%) |
|---|---|---|
| S | 55,694 | 96.615 |
| A | 1,891 | 3.280 |
| P | 30 | 0.052 |
| T | 17 | 0.029 |
| G | 6 | 0.010 |
| Xb | 3 | 0.005 |
| L | 3 | 0.005 |
| —c | 1 | 0.002 |
The analysis was based on a total of 57,645 aligned HIV-1 Env sequences from the Los Alamos National Laboratory HIV Sequence Database. The amino acid position is based on the HIV-1NL4-3 Env sequence (GenBank accession number M19921.2).
Unknown amino acids due to ambiguous sequences.
Gap in sequence alignment.
FIG 6.

S532A mutant of the PR reduces HIV-1 infectivity and fusogenicity. (A) Alignment of nucleotide (nt) and amino acid (aa) sequences of overlapped HIV-1 RRE and Env in wild-type (WT) and S532A mutants (M6) based on HIV-1NL4-3 (GenBank accession number M19921.2). (B) HIV-1 p24 levels in supernatants of transfected HEK293T cells were quantified by ELISA (n = 6). (C to E) HEK293T cells were transfected with full-length WT or M6 mutant proviral constructs without (control) or with (+Env) a WT gp160 expression plasmid. HIV-1 infectivity of equal amounts of p24 of WT and mutants was quantified with TZM-bl cells (C). (D) Lysates of transfected HEK293T cells or purified HIV-1 were analyzed for the indicated HIV-1 proteins by immunoblotting. GAPDH was a loading control (D). Virion-cell fusion was determined by flow cytometry-based BlaM-Vpr assays using TZM-bl cells (10 ng p24 for HIV-1 with or without Env trans-supplementation) (E). (F to H) HEK293T cells were separately transfected with a pRK empty vector, WT, or M6 Env (gp160) expression construct together with pCMV-Rev. HEK293T cells transfected without plasmid DNA were used as a mock control. At 24 h posttransfection, HEK293T cells were collected and lysed for immunoblotting analysis with antibodies to gp160/gp120, gp41, or GAPDH (loading control). Relative gp160 levels were quantified and normalized to the level of GAPDH (F). At 24 h posttransfection, the cells were stained with gp120 monoclonal antibody (2G12) at 4°C to measure the cell surface gp120 expression using flow cytometry. Average percentages (G) and the mean fluorescence intensity (H) of gp120-positive cells from three independent experiments were determined. (I) Transfected HEK293T cells were cocultured with TZM-bl cells for 24 h and then lysed for firefly luciferase activity measurement of Env-mediated cell-cell fusion. Average percentages of HIV-1 Env-mediated cell-cell fusion from four independent experiments are shown, with the value for the WT set as 100%. All experiments were performed with triplicate samples and repeated at least three times, and means ± standard errors of the means are shown. Dunnett's multiple-comparison test was used for statistical analysis. *, P < 0.05; ***, P < 0.0001, for results with M6 mutant compared to those with WT HIV-1.
To investigate whether reduced infectivity of M6 was due to compromised viral entry, we measured virion-cell fusion efficiency of WT and M6 using the BlaM-Vpr assay. M6 showed 30% lower virion-cell fusion than the WT HIV-1 (Fig. 6E). Moreover, the trans-supplementation of WT Env significantly increased virus-cell fusion of M6 to a level similar to that of WT HIV-1 (Fig. 6E). These data indicated that M6 had reduced virion-cell fusion and, thus, compromised HIV-1 entry into target cells. We further examined whether M6 mutation affected HIV-1 Env-mediated cell-cell fusion. The expression levels of WT and M6 Env were comparable in cell lysates of transfected HEK293T cells (Fig. 6F). The average percentage of gp120-positive cells and the mean fluorescence intensity of cell surface gp120 were also similar in M6 mutant Env-expressing cells and WT Env-expressing cells (Fig. 6G and H, respectively). In contrast, M6 mutant Env-mediated cell-cell fusion was significantly reduced by 77% relative to the level of WT Env (Fig. 6I). These data suggest that S532A mutation significantly decreases Env-mediated cell-cell fusion, potentially through an alteration to the Env stability required for optimal fusogenicity.
Structural models of the PR in the Env trimer of the prefusion state.
To better understand the structural basis of the PR in determining HIV-1 Env trimer stability, we analyzed the published three-dimensional (3D) structures of HIV-1 Env trimer in the prefusion state (19, 20). The structural model shows that the PR interacts with the CHR element of a neighboring protomer, indicating the role of PR in stabilizing the gp120-gp41 trimer (Fig. 7). The S534/T536/T538 residues in HIV-1BG505 (equivalent to S532/T534/T536 residues in HIV-1NL4-3, respectively) are located right in the trimer junction of gp120 and gp41 (Fig. 7A and B). S534 and T538 in HIV-1BG505 are exposed to and interact with the neighboring CHR by forming hydrogen bonds with residues E647 and N656 (Fig. 7C). Interestingly, S534 (S532 in HIV-1NL4-3) interacts with a neighboring protomer through its main-chain carbonyl group (Fig. 7C), which may be affected by structural and stability deviations caused by a different amino acid that has a different α-helical propensity (21, 22). This would help explain abolished or significantly reduced HIV-1 infectivity and fusogenicity of mutants containing S532P (mutants M1, M3, and M4) or S532A (mutant M6) in the HIV-1NL4-3 sequence. Although residue T536 in HIV-1BG505 (equivalent to T534 in HIV-1NL4-3) does not make direct contact with protomer 2 (Fig. 7C), T534A (mutant M5) of HIV-1NL4-3 also had a significant impact on the infectivity (Fig. 2A and 3E) and fusogenicity (Fig. 3F and 4D), which may result from altered structure or stability of the region of protomer 1 proximal to the FP (Fig. 7C). Overall, our structural analysis suggests that these PR mutations lead to loss of this interaction and alter the helical structure of the PR, which would destabilize the Env trimer and affect the association of gp120 and gp41.
FIG 7.

Structural models of the PR in the Env trimer of prefusion state. (A) PR sequence alignment between HIV-1BG505 and HIV-1NL4-3. (B) PR location at the trimer junction of gp120 (tan) and gp41 (blue). One protomer is shown in ribbon representation. The neighboring protomers are in surface representation and are shown in gray and teal blue. (C) PR (blue) interaction with gp41 C-terminal heptad repeat (CHR) of a neighboring protomer (gray). Fusion peptide (FP) is shown in red. The hydrogen bonds between residues are shown in dashed lines. Residue numbers and the structure prediction in panels B and C are based on the HIV-1BG505 strain (19, 20). Of note, residue numbers of HIV-1BG505 shown in panels B and C are two numbers greater than those of HIV-1NL4-3.
DISCUSSION
Here, we report that the three RRE mutations (M1 to M3) tested by Lichinchi and colleagues (10) altered the coding sequence of the PR and abolished or significantly reduced fusogenicity of gp41 and viral infectivity. However, these mutations did not affect the RRE function in a single-cycle HIV-1 infection assay. Of note, Lichinchi et al. showed only reduced gp120 mRNA expression levels and nuclear export of HIV-1 RNA of M2 and M3 relative to levels of the WT in transfected HEK293T cells (10), but they did not investigate the effect of these RRE mutations on Env protein expression, viral fusion, and infectivity. Furthermore, in contrast to the report by Lichinchi et al. of m6A modifications in the stem-loop of HIV-1 RRE (10), two other independent studies did not identify any m6A peaks in the RRE region (23, 24). Although different experimental approaches in these three studies might partially account for the discrepancy of the m6A mapping results, our current study is important to clarify the effects of these RRE mutations on HIV-1 fusion and infectivity given their overlapping of the gp41 coding sequence.
Based on our results presented in Fig. 2 and 6, we summarized the effects of the PR mutations on HIV-1 biological activities, including viral infectivity, virion-cell and cell-cell fusion, and gp120 association and processing (Table 2). In particular, we found that three mutations (M1, M3, and M4) containing S532P abolished HIV-1 fusion and infectivity by destabilizing the gp120 and gp41 association. Furthermore, an S532A mutation (M6) that shows a 3.3% frequency in all analyzed HIV-1 Env sequences also significantly reduced viral infectivity and fusion. These data indicate lower replication fitness of S532A variant, which are consistent with results from a recent study using deep mutational analyses of HIV-1 Env (25). Taken together, these results also highlight the importance of residue S532 of gp41 in regulating HIV-1 fusion and infectivity, indicating that the PR is critical for HIV-1 fusion and infectivity. We did not perform the gp120 shedding experiment with the M6 mutant because this mutant showed cleavage and virion incorporation of gp120 and gp41 similar to that of WT HIV-1 (Fig. 6D) and higher infectivity and virus-cell fusion ability than mutants M1 to M5 (Table 2).
TABLE 2.
Summary of the effects of mutations in the PR on HIV-1 biological activitiesa
| Groupb | Mutation(s) in PR of gp41 | Viral infectivity (%) | Virion-cell fusion (%) | Cell-cell fusion (%) | gp120 association (%) | gp120 processing (%) |
|---|---|---|---|---|---|---|
| WT | None | 100 | 100 | 100 | 100 | 100 |
| M1 | S532P, T534A | 0 | 0.4 | 0 | 55 | 92 |
| M2 | T536A | 6.2 | 16.7 | 58.0 | 124 | 87 |
| M3 | S532P, T534A, T536A | 0.4 | 0.3 | 0 | 47 | 102 |
| M4 | S532P | 0.5 | 0.5 | 0 | 43 | 116 |
| M5 | T534A | 2.3 | 5.3 | 24.0 | 82 | 93 |
| M6 | S532A | 25.9 | 69.9 | 22.7 | ND | ND |
All percentages represent the average results of relative levels from at least 3 independent experiments. ND, not done.
WT, wild-type HIV-1NL4-3; M1 to M6, HIV-1NL4-3-derived PR mutations 1 to 6, respectively.
Analysis of the published structures of soluble disulfide-stabilized gp120 and gp41 complex (19, 20) suggests that the PR promotes gp120 and gp41 association by stabilizing the trimer via an interaction with the CHR. It has been shown that a six-helix bundle core is formed before fusion pore opening (26) and that the gp41 peptide consisting of residues 17 to 70, including the PR and NHR-derived N36 peptide, can interact with the CHR-derived C34 to form more stable six-helical bundles than those formed by N36 and C34 peptides (5). A previous study of the crystal structure of residues 528 to 683 of gp41 (gp41528–683) showed that both the PR and the MPER form helical extensions from the gp41 six-helical bundle core structure (7). These biophysical and structural studies further support our notion that the PR maintains the association of g120 and gp41 and Env trimer stability.
Our results are consistent with an earlier study by Freed et al. showing that different point mutations in the FP or PR of HIV-1 gp41 largely reduced or blocked Env-mediated syncytium formation but significantly increased the levels of gpl20 secretion into the extracellular medium (27). Poumbourios and colleagues have studied the effects of different and combined mutations in the PR and MPER of HIV-1AD8 on viral fusion and infection, including the double-residue mutations of L537A/W666A or I535A/V539G that are defective for cell-cell fusion, HIV-1 entry, and infection (6, 8). Their results suggested that the PR and MPER act synergistically in forming a fusion-competent gp120-gp41 complex and in stabilizing the trimer of gp41 hairpins and that prefusion Env complex can be adversely affected by MPER mutations. In contrast, our results highlight the importance of previously uncharacterized residues of the PR (S532, T534, and T536) in stabilizing Env trimers and in determining gp41 fusogenicity and HIV-1 infectivity. Together, these results indicate that both the PR and MPER could be conserved functional targets for the discovery of fusion inhibitors or neutralizing antibodies.
Although extensive studies of HIV-1 Env have led to a peptide-based fusion inhibitor (T20) clinically used to block viral entry, drug resistance of T20 and other anti-HIV drugs is a significant barrier to eradication of HIV-1 infection (1). Our findings indicate that the PR is structurally essential for maintaining HIV-1 Env trimer, suggesting that the PR might be targeted for design of more effective anti-HIV-1 peptides. Indeed, structure-based design of peptides targeting the CHR of gp41 significantly improves pharmacological properties of HIV-1 fusion inhibitors (28, 29). A top priority in HIV-1 vaccine development is to generate HIV-1 Env trimer-based immunogens to elicit broadly neutralizing antibodies (bNAbs), and bNAbs that bind to gp120 and gp41 have demonstrated broad and potent HIV-1 neutralization (3). HIV-1 neutralization by the bNAb (ACS202) that recognizes a trimer-specific epitope at the gp120-gp41 interface is highly sensitive to substitutions of multiple residues in the FP and the PR of gp41 (30), suggesting that both regions could be conserved targets for bNAb. The N terminus of the FP has been shown as a promising target of HIV-1 vaccine to elicit bNAbs (31). Monoclonal antibodies to the PR have been identified in HIV-1-infected patients (32) although it is unclear whether these antibodies could be potential bNAbs. One limitation of our study is the use of HEK293T cell-generated HIV-1NL4-3 that contains a tier 1A Env. It would be helpful to further examine the observed phenotypes in CD4+ T cells that may incorporate less unprocessed gp160 and to test a tier 2 Env that could be less prone to shedding (33).
In summary, our findings indicate that the PR of gp41, particularly the key residue S532, is structurally essential for maintaining HIV-1 Env trimer, viral fusion, and infection. Our results reveal the previously unappreciated function of the PR and the underlying mechanisms, highlighting the important role of the PR in determining HIV-1 fusogenicity and infectivity.
MATERIALS AND METHODS
Cell culture.
The HEK293T cell line (American Type Culture Collection [ATCC], CRL-3216), HeLa cells (ATCC, CCL-2), and GHOST/R5/X4 cells were kind gifts from Vineet KewalRamani (National Cancer Institute, USA) and maintained in complete Dulbecco’s modified Eagle’s medium (DMEM) as described previously (24, 34). TZM-bl cells (35) were obtained through the NIH AIDS Reagent Program (NIH-ARP; catalog number 8129). All cell lines utilized were maintained at 37°C in 5% CO2 and tested negative for mycoplasma contamination using a universal mycoplasma detection kit (ATCC 30-1012K).
Plasmids, cloning, and mutagenesis.
Mutations in pNL4-3 were introduced using an Agilent QuikChange Lightning Multi Site-directed mutagenesis kit (catalog number 210515-5) according to the manufacturer’s instructions. The sequences of the PCR primers used for mutagenesis are summarized in Table 3. To generate the yellow fluorescent protein (YFP) reporter virus constructs, plasmids encoding full-length pNL4-3 and the RRE (WT and M1 to M5) were digested with BamH I and NheI to generate a fragment that was inserted into pNL4-3 E-R-Rev-/YFP (11) using the same restriction sites. WT Env and mutant (M1 to M6) expression constructs were generated using a pRK vector (36) as described previously (17). All mutations were confirmed by DNA sequencing.
TABLE 3.
PCR primers used in generating HIV-1 gp41 mutants M1 to M6
| PCR primer | DNA sequence (5′–3′)a |
|---|---|
| M1 | TGGGCGCAGCGCCAATGGCGCTGACGGTACA |
| M2 | TGGGCGCAGCGTCAATGACGCTGGCGGTACA |
| M3 | TGGGCGCAGCGCCAATGGCGCTGGCGGTACA |
| M4 | TGGGCGCAGCGCCAATGACGCTGACGGTACA |
| M5 | TGGGCGCAGCGTCAATGGCGCTGACGGTACA |
| M6 | TGGGCGCAGCGGCAATGACGCTGACGGTACA |
Mutated nucleotides compared with HIVNL4-3 sequence (GenBank accession number M19921.2) are shown in boldface.
Mutation frequency analyses of residues in HIV-1 RRE and the PR.
The total 63,612 aligned RRE sequences were downloaded from the HIV Sequence Database of the Los Alamos National Laboratory (https://www.hiv.lanl.gov). An in-house Python script was used to calculate mutation frequencies of nucleotides at positions 7814, 7820, and 7826 (starting from the U3 region of the pNL4-3 sequence; GenBank accession number M19921.2). For PR sequence analysis, a total of 57,645 aligned HIV-1 Env sequences were downloaded from the HIV Sequence Database (https://www.hiv.lanl.gov). A sequence logo was generated using WebLogo (http://weblogo.threeplusone.com/) based on frequencies of each residue in the PR (17 aa) of the 57,645 HIV-1 Env sequences.
Single-cycle HIV-1 production and infection assays for RRE activity.
Replication-competent WT and mutant HIV-1 stocks were generated by transfection of HEK293T cells with pNL4-3 or mutant proviral DNA plasmids using calcium phosphate, as described previously (37), or Lipofectamine 2000 (Invitrogen) according to instructions. Viral infectivity assays and immunoblotting experiments shown in Fig. 1E and F were carried out as described previously (11). Briefly, HEK293T cells were transfected with the following plasmids: 1,000 ng of pE-R-Rev-/YFP, 200 ng of pRev-mApple or mApple control plasmid, and 100 ng of pVSV-G using polyethylenimine (PEI) (catalog no. 23966; Polysciences, Inc.). Culture medium was exchanged at 4 h posttransfection with supernatants, and producer cell lysates were harvested at 48 h. Supernatants were filtered (0.45-μm-pore-size filter) and used to infect HeLa cells in 12-well plates; target cells were fixed at 48 h postransduction using 4% paraformaldehyde, permeabilized using 0.2% Triton X-100, and stained with 4,6-diamidino-2-phenylindole (DAPI). YFP and DAPI fluorescence were measured using a Cytation 5 imaging reader (Biotek Instruments, Inc.) operated by Gen5 software (version 2.07) using the following excitation/emission monochromator ranges, respectively (wavelengths in nanometers): 490 to 510/520 to 550 (YFP) and 340 to 380/420 to 480 (DAPI). YFP fluorescence was normalized to cell number based on the relative DAPI signal.
Immunoblotting.
HIV-1 producer cell monolayers were washed with phosphate-buffered saline (PBS) and lysed using radioimmunoprecipitation assay (RIPA) buffer (10 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 0.1% sodium dodecyl sulfate [SDS], 1% Triton X-100, 1% sodium deoxycholate) containing complete protease inhibitor cocktail (Roche). Cell lysates were prepared for immunoblotting by a freeze-thaw step, centrifugation for 10 min at 1,000 × g, and boiling in 2× dissociation buffer (62.5 mM Tris-HCl [pH 6.8], 10% glycerol, 2% SDS, 10% β-mercaptoethanol) at a 1:1 ratio prior to SDS-PAGE and transfer to nitrocellulose membranes. Immunoblot analyses were performed as described previously (11) using mouse HIV-1 Rev antibodies (ab85529 [Abcam] or sc-69729 [Santa Cruz Biosciences]) and rabbit anti-HSP90 antibody (sc-7947; Santa Cruz Biosciences) and detected using anti-mouse or anti-rabbit secondary antibodies conjugated to either of two infrared fluorophores, IRDye 680 or IRDye 800 (LiCor Biosciences). Rabbit anti-human glyceraldehyde 3-phosphate dehydrogenase (GAPDH) polyclonal antibody was purchased from Bio-Rad (catalog no. AHP1628). The HIV-1 antibodies were obtained from the NIH-ARP: anti-gp120 (which recognizes both gp160 and gp120; catalog no. 288), anti-gp41 (catalog no. 13049), anti-gp160 (catalog no. 188), anti-HIV-1 gp120 monoclonal (2G12) (catalog no. 1476), and anti-p24 (catalog no. 6458). Relative protein expression levels were quantified using ImageJ software (version 1.51w) based on the densitometry of the protein bands.
HIV-1 p24 quantification and viral infectivity assays.
HIV-1 p24 levels in replication-competent viral stocks were quantified by an enzyme-linked immunosorbent assay (ELISA) using anti-p24-coated plates (AIDS and Cancer Virus Program, NCI-Frederick, MD) as described previously (24). To compare the infectivity of WT and mutant HIV-1, viruses with equal amounts of p24 (0.4 ng/sample) were used to infect TZM-bl cells in 24-well plates. At 48 h postinfection, TZM-bl cells were washed twice with PBS and lysed for luciferase assay (Promega) according to the manufacturer’s instructions. GHOST/R5/X4 cells were also used to validate the HIV-1 infectivity results derived from TZM-bl cells (37). Cell protein concentrations were quantified using a bicinchoninic acid assay (Pierce), and all luciferase results were normalized based on total protein input.
HIV-1 purification.
To study the association and incorporation of gp120 and gp41 to virions, WT and mutant viruses were purified as described previously (38). The cell culture media containing viruses were pelleted through 25% sucrose using an SW28 rotor at 141,000 × g for 90 min. After resuspension in PBS, the viruses were layered on top of 6 to 18% OptiPrep (D1556; Sigma-Aldrich) in an SW55 Ti rotor tube and ultracentrifuged at 250,000 × g for 90 min. Fractions from 14.4% to 18% containing virions were pooled, mixed with PBS, and ultracentrifuged at 250,000 × g for 1 h using the SW55 Ti rotor to pellet purified virions. The purified virions were resuspended in the lysis buffer (Cell Signaling) supplemented with protease inhibitor cocktail (Sigma-Aldrich) and used for immunoblotting as described previously (24, 38).
HIV-1 virion-cell fusion assay and flow cytometry.
A fusion assay using beta-lactamase-Vpr (BlaM-Vpr) was performed as previously described with modifications (14, 16). HEK293T cells were seeded onto 10-cm-diameter dishes and cotransfected with 10 μg of WT pNL4-3 or pNL4-3-derived mutants M1 to M6 and 5 μg of HIV-1 YU2 Vpr beta-lactamase expression vector (pMM310) (catalog no. 11444; NIH-ARP), together with pIII NL4-3env (39) (a gift from Eric Freed, NCI-Frederick, MD) or empty vector control plasmid. Supernatants containing virions were collected at 48 h posttransfection, and virions were concentrated by ultracentrifugation. TZM-bl cells were incubated with equal amounts of viruses and spinoculated at 1,680 × g at 4°C for 1 h to allow synchronized virus binding and then further incubated at 37°C for 3 h to allow virion fusion. TZM-bl cells were washed with 1× PBS twice to remove unbound viruses and further loaded with the coumarin cephalosporin fluorescein acetoxymethyl ester (CCF2-AM) dye for 1 h at room temperature, according to the manufacturer’s recommendations (catalog no. K1032; Invitrogen). Cells were washed and incubated in development medium (10% fetal bovine serum [FBS], 2.5 mM probenecid, phenol red-negative DMEM) for 16 h at room temperature. Samples were fixed in 3.7% formaldehyde, washed, and resuspended in 1× PBS. Uncleaved CCF2-AM dye was detected at excitation (409 nm)/emission (518 nm), and cleaved CCF2 was detected at excitation (409 nm)/emission (447 nm) using Attune NxT flow cytometer (Invitrogen). The flow cytometry data were analyzed with FlowJo software (version 10; Tree Star).
HIV-1 Env-mediated cell-cell fusion and detection of cell surface Env expression by flow cytometry.
HEK293T cells (1 × 106) were transfected with pRK-based WT and mutant Env expression constructs (2 μg pRK-M1–6) with an HIV-1 Rev expression construct (0.8 μg pCMV-Rev) and an HIV-1 Tat expression construct (pCMV-Tat; 0.8 μg). Cells at 24 h posttransfection were cocultured with TZM-bl cells at a 1:1 ratio for 24 h, lysed, and measured for firefly luciferase activity (16). Protein concentrations were quantified by Bradford assay and used to normalize the firefly luciferase activity. For the measurement of cell surface expression of Env, the above remainder transfected HEK293T cells (5 × 105) were collected and washed twice with staining buffer (2% FBS-containing PBS) and then stained with anti-HIV-1 gp120 monoclonal (2G12) from NIH-ARP (1:200) for 30 min. After cells were washed with staining buffer and stained with fluorescein isothiocyanate (FITC)-conjugated anti-human IgG (Fc specific) (F9512, 1:100; Sigma-Aldrich) for 30 min, the cells were washed and resuspended with staining buffer and then detected using a Guava EasyCyte flow cytometer (Guava Technologies). The flow cytometry data were analyzed with FlowJo software (version 10; Tree Star) as described previously (40).
Detection of HIV-1 Env expression, processing, and shedding.
HEK293T cells were cultured in six-well plates (5 × 105 cells/well) overnight and cotransfected with pRK plasmids expressing HIV-1 WT or M1 to M5 Env (3 μg) and Rev (1 μg) using PEI. One day after transfection, the medium was replaced with DMEM lacking cysteine and methionine and supplemented with dialyzed FBS, and metabolically labeled 35S protein-labeling mix (NEG772002MC PerkinElmer). After cells were cultured for 18 h, the medium was harvested, and the cells were lysed using a mild NP-40-based lysis buffer. The two fractions were treated, first with normal human serum (lot 6031174; Community Blood Bank) and protein A-Sepharose beads to clear background proteins and then with HIV-1 (subtype B)-positive patient sera (Community Blood Bank) and fresh beads to immunoprecipitate the radiolabeled Env proteins as described previously (41). The beads were mixed with SDS loading buffer, and then the protein was separated by SDS-PAGE. After vacuum fixation, protein expression was quantified using Quantity One software (Bio-Rad) (41). The association index, which indicates the ability of mutant gp120 to remain associated with the trimer complex on the cell membrane relative to WT gp120, was calculated using the following equation: association index = (lysate gp120mutant/supernatant gp120mutant) × (supernatant gp120wild-type/lysate gp120wild-type).
The processing index, a measure of how much mutant gp160 is converted to gp120 relative to the amount of WT gp120, was calculated as follows (18): processing index = (total gp120mutant/gp160mutant) × (gp160wild-type/total gp120wild-type).
Statistical analysis.
Dunnett’s multiple-comparison test was used for statistical analysis (GraphPad Prism, version 5) of all data, as indicated in figure legends. A P value of <0.05 was considered significant.
ACKNOWLEDGMENTS
We thank Eric Freed and Vineet KewalRamani for reagents, Dane Bowder for assistance in the gp120 shedding experiment, Serena Bonifati, Corine St. Gelais, and Jacob Yount for critical readings of the manuscript, and members of the Wu lab for helpful discussions. The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HIV-1 p24 Gag monoclonal (number 24-3) from Michael Malim; TZM-bl from John C. Kappes, Xiaoyun Wu, and Tranzyme, Inc.; anti-gp120 (number 288) from Michael Phelan; anti-HIV-1 gp41 monoclonal (Chessie 8, catalog number 13049) from George Lewis; anti-gp160 (number 188) and anti-HIV-1 gp120 monoclonal (2G12) from Polymun Scientific; and HIV-1 YU2 Vpr β-lactamase expression vector (pMM310) from Michael Miller.
This work was supported by NIH grants R01AI120209 and R01GM128212 (L.W.). S.-L.L. was supported by NIH grants R01AI112381 and R01GM132069.
W.L. and S.C. performed all experiments unless otherwise stated and contributed to manuscript preparation. J.Y. and W.L. performed HIV-1 and cell fusion experiments. R.B. and N.S. generated single-cycle HIV-1 with RRE mutants and performed viral infection assays. J.W. performed gp120-shedding experiments. Y.X. provided structural prediction of HIV-1 Env and data interpretation. S.-L.L., N.S., Y.X., S.-H. X., and L.W. analyzed data and contributed to experiment design. L.W. conceived the study, supervised the work, and wrote the paper. All authors contributed to manuscript editing and revision.
We declare that we have no competing interests.
REFERENCES
- 1.Melikyan GB. 2017. How entry inhibitors synergize to fight HIV. J Biol Chem 292:16511–16512. doi: 10.1074/jbc.H117.791731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Checkley MA, Luttge BG, Freed EO. 2011. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J Mol Biol 410:582–608. doi: 10.1016/j.jmb.2011.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwong PD, Mascola JR. 2018. HIV-1 vaccines based on antibody identification, B cell ontogeny, and epitope structure. Immunity 48:855–871. doi: 10.1016/j.immuni.2018.04.029. [DOI] [PubMed] [Google Scholar]
- 4.Peisajovich SG, Epand RF, Pritsker M, Shai Y, Epand RM. 2000. The polar region consecutive to the HIV fusion peptide participates in membrane fusion. Biochemistry 39:1826–1833. [DOI] [PubMed] [Google Scholar]
- 5.Sackett K, Wexler-Cohen Y, Shai Y. 2006. Characterization of the HIV N-terminal fusion peptide-containing region in context of key gp41 fusion conformations. J Biol Chem 281:21755–21762. doi: 10.1074/jbc.M603135200. [DOI] [PubMed] [Google Scholar]
- 6.Bellamy-McIntyre AK, Lay CS, Baar S, Maerz AL, Talbo GH, Drummer HE, Poumbourios P. 2007. Functional links between the fusion peptide-proximal polar segment and membrane-proximal region of human immunodeficiency virus gp41 in distinct phases of membrane fusion. J Biol Chem 282:23104–23116. doi: 10.1074/jbc.M703485200. [DOI] [PubMed] [Google Scholar]
- 7.Buzon V, Natrajan G, Schibli D, Campelo F, Kozlov MM, Weissenhorn W. 2010. Crystal structure of HIV-1 gp41 including both fusion peptide and membrane proximal external regions. PLoS Pathog 6:e1000880. doi: 10.1371/journal.ppat.1000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lay CS, Ludlow LE, Stapleton D, Bellamy-McIntyre AK, Ramsland PA, Drummer HE, Poumbourios P. 2011. Role for the terminal clasp of HIV-1 gp41 glycoprotein in the initiation of membrane fusion. J Biol Chem 286:41331–41343. doi: 10.1074/jbc.M111.299826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandes J, Jayaraman B, Frankel A. 2012. The HIV-1 Rev response element: an RNA scaffold that directs the cooperative assembly of a homo-oligomeric ribonucleoprotein complex. RNA Biol 9:6–11. doi: 10.4161/rna.9.1.18178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lichinchi G, Gao S, Saletore Y, Gonzalez GM, Bansal V, Wang Y, Mason CE, Rana TM. 2016. Dynamics of the human and viral m(6)A RNA methylomes during HIV-1 infection of T cells. Nat Microbiol 1:16011. doi: 10.1038/nmicrobiol.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Behrens RT, Aligeti M, Pocock GM, Higgins CA, Sherer NM. 2017. Nuclear export signal masking regulates HIV-1 Rev trafficking and viral RNA nuclear export. J Virol 91:e02107-16. doi: 10.1128/JVI.02107-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fenouillet E, Gluckman JC. 1992. Immunological analysis of human immunodeficiency virus type 1 envelope glycoprotein proteolytic cleavage. Virology 187:825–828. [DOI] [PubMed] [Google Scholar]
- 13.Hallenberger S, Bosch V, Angliker H, Shaw E, Klenk HD, Garten W. 1992. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature 360:358–361. doi: 10.1038/360358a0. [DOI] [PubMed] [Google Scholar]
- 14.Cavrois M, De Noronha C, Greene WC. 2002. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat Biotechnol 20:1151–1154. doi: 10.1038/nbt745. [DOI] [PubMed] [Google Scholar]
- 15.Wilen CB, Tilton JC, Doms RW. 2012. HIV: cell binding and entry. Cold Spring Harb Perspect Med 2:a006866. doi: 10.1101/cshperspect.a006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu J, Li M, Wilkins J, Ding S, Swartz TH, Esposito AM, Zheng YM, Freed EO, Liang C, Chen BK, Liu SL. 2015. IFITM proteins restrict HIV-1 infection by antagonizing the envelope glycoprotein. Cell Rep 13:145–156. doi: 10.1016/j.celrep.2015.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiang SH, Finzi A, Pacheco B, Alexander K, Yuan W, Rizzuto C, Huang CC, Kwong PD, Sodroski J. 2010. A V3 loop-dependent gp120 element disrupted by CD4 binding stabilizes the human immunodeficiency virus envelope glycoprotein trimer. J Virol 84:3147–3161. doi: 10.1128/JVI.02587-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finzi A, Xiang SH, Pacheco B, Wang L, Haight J, Kassa A, Danek B, Pancera M, Kwong PD, Sodroski J. 2010. Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol Cell 37:656–667. doi: 10.1016/j.molcel.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pancera M, Zhou T, Druz A, Georgiev IS, Soto C, Gorman J, Huang J, Acharya P, Chuang GY, Ofek G, Stewart-Jones GB, Stuckey J, Bailer RT, Joyce MG, Louder MK, Tumba N, Yang Y, Zhang B, Cohen MS, Haynes BF, Mascola JR, Morris L, Munro JB, Blanchard SC, Mothes W, Connors M, Kwong PD. 2014. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature 514:455–461. doi: 10.1038/nature13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwon YD, Pancera M, Acharya P, Georgiev IS, Crooks ET, Gorman J, Joyce MG, Guttman M, Ma X, Narpala S, Soto C, Terry DS, Yang Y, Zhou T, Ahlsen G, Bailer RT, Chambers M, Chuang GY, Doria-Rose NA, Druz A, Hallen MA, Harned A, Kirys T, Louder MK, O'Dell S, Ofek G, Osawa K, Prabhakaran M, Sastry M, Stewart-Jones GB, Stuckey J, Thomas PV, Tittley T, Williams C, Zhang B, Zhao H, Zhou Z, Donald BR, Lee LK, Zolla-Pazner S, Baxa U, Schon A, Freire E, Shapiro L, Lee KK, Arthos J, Munro JB, Blanchard SC, Mothes W, Binley JM, McDermott AB, Mascola JR, Kwong PD. 2015. Crystal structure, conformational fixation and entry-related interactions of mature ligand-free HIV-1 Env. Nat Struct Mol Biol 22:522–531. doi: 10.1038/nsmb.3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Padmanabhan S, Marqusee S, Ridgeway T, Laue TM, Baldwin RL. 1990. Relative helix-forming tendencies of nonpolar amino acids. Nature 344:268–270. doi: 10.1038/344268a0. [DOI] [PubMed] [Google Scholar]
- 22.Costantini S, Colonna G, Facchiano AM. 2006. Amino acid propensities for secondary structures are influenced by the protein structural class. Biochem Biophys Res Commun 342:441–451. doi: 10.1016/j.bbrc.2006.01.159. [DOI] [PubMed] [Google Scholar]
- 23.Kennedy EM, Bogerd HP, Kornepati AV, Kang D, Ghoshal D, Marshall JB, Poling BC, Tsai K, Gokhale NS, Horner SM, Cullen BR. 2016. Posttranscriptional m(6)A editing of HIV-1 mRNAs enhances viral gene expression. Cell Host Microbe 19:675–685. doi: 10.1016/j.chom.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tirumuru N, Zhao BS, Lu W, Lu Z, He C, Wu L. 2016. N6-Methyladenosine of HIV-1 RNA regulates viral infection and HIV-1 Gag protein expression. Elife 5:e15528. doi: 10.7554/eLife.15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haddox HK, Dingens AS, Hilton SK, Overbaugh J, Bloom JD. 2018. Mapping mutational effects along the evolutionary landscape of HIV envelope. Elife 7:e34420. doi: 10.7554/eLife.34420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Markosyan RM, Cohen FS, Melikyan GB. 2003. HIV-1 envelope proteins complete their folding into six-helix bundles immediately after fusion pore formation. Mol Biol Cell 14:926–938. doi: 10.1091/mbc.e02-09-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freed EO, Myers DJ, Risser R. 1990. Characterization of the fusion domain of the human immunodeficiency virus type 1 envelope glycoprotein gp41. Proc Natl Acad Sci U S A 87:4650–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu X, Zhu Y, Ye S, Wang Q, Xu W, Su S, Sun Z, Yu F, Liu Q, Wang C, Zhang T, Zhang Z, Zhang X, Xu J, Du L, Liu K, Lu L, Zhang R, Jiang S. 2015. Improved pharmacological and structural properties of HIV fusion inhibitor AP3 over enfuvirtide: highlighting advantages of artificial peptide strategy. Sci Rep 5:13028. doi: 10.1038/srep13028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Su S, Wang Q, Xu W, Yu F, Hua C, Zhu Y, Jiang S, Lu L. 2017. A novel HIV-1 gp41 tripartite model for rational design of HIV-1 fusion inhibitors with improved antiviral activity. AIDS 31:885–894. doi: 10.1097/QAD.0000000000001415. [DOI] [PubMed] [Google Scholar]
- 30.van Gils MJ, van den Kerkhof TL, Ozorowski G, Cottrell CA, Sok D, Pauthner M, Pallesen J, de Val N, Yasmeen A, de Taeye SW, Schorcht A, Gumbs S, Johanna I, Saye-Francisco K, Liang CH, Landais E, Nie X, Pritchard LK, Crispin M, Kelsoe G, Wilson IA, Schuitemaker H, Klasse PJ, Moore JP, Burton DR, Ward AB, Sanders RW. 2016. An HIV-1 antibody from an elite neutralizer implicates the fusion peptide as a site of vulnerability. Nat Microbiol 2:16199. doi: 10.1038/nmicrobiol.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu K, Acharya P, Kong R, Cheng C, Chuang G-Y, Liu K, Louder MK, O’Dell S, Rawi R, Sastry M, Shen C-H, Zhang B, Zhou T, Asokan M, Bailer RT, Chambers M, Chen X, Choi CW, Dandey VP, Doria-Rose NA, Druz A, Eng ET, Farney SK, Foulds KE, Geng H, Georgiev IS, Gorman J, Hill KR, Jafari AJ, Kwon YD, Lai Y-T, Lemmin T, McKee K, Ohr TY, Ou L, Peng D, Rowshan AP, Sheng Z, Todd J-P, Tsybovsky Y, Viox EG, Wang Y, Wei H, Yang Y, Zhou AF, Chen R, Yang L, Scorpio DG, McDermott AB, Shapiro L, Carragher B, Potter CS, Mascola JR, Kwong PD. 2018. Epitope-based vaccine design yields fusion peptide-directed antibodies that neutralize diverse strains of HIV-1. Nat Med 24:857–867. doi: 10.1038/s41591-018-0042-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buchacher A, Predl R, Strutzenberger K, Steinfellner W, Trkola A, Purtscher M, Gruber G, Tauer C, Steindl F, Jungbauer A. 1994. Generation of human monoclonal antibodies against HIV-1 proteins; electrofusion and Epstein-Barr virus transformation for peripheral blood lymphocyte immortalization. AIDS Res Hum Retroviruses 10:359–369. doi: 10.1089/aid.1994.10.359. [DOI] [PubMed] [Google Scholar]
- 33.Montefiori DC, Roederer M, Morris L, Seaman MS. 2018. Neutralization tiers of HIV-1. Curr Opin HIV AIDS 13:128–136. doi: 10.1097/COH.0000000000000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong C, Kwas C, Wu L. 2009. Transcriptional restriction of human immunodeficiency virus type 1 gene expression in undifferentiated primary monocytes. J Virol 83:3518–3527. doi: 10.1128/JVI.02665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O'Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E. 2000. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J Virol 74:8358–8367. doi: 10.1128/JVI.74.18.8358-8367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen S, Bonifati S, Qin Z, St Gelais C, Kodigepalli KM, Barrett BS, Kim SH, Antonucci JM, Ladner KJ, Buzovetsky O, Knecht KM, Xiong Y, Yount JS, Guttridge DC, Santiago ML, Wu L. 2018. SAMHD1 suppresses innate immune responses to viral infections and inflammatory stimuli by inhibiting the NF-kappaB and interferon pathways. Proc Natl Acad Sci U S A 115:E3798–E3807. doi: 10.1073/pnas.1801213115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.St Gelais C, de Silva S, Hach JC, White TE, Diaz-Griffero F, Yount JS, Wu L. 2014. Identification of cellular proteins interacting with the retroviral restriction factor SAMHD1. J Virol 88:5834–5844. doi: 10.1128/JVI.00155-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu W, Tirumuru N, St Gelais C, Koneru PC, Liu C, Kvaratskhelia M, He C, Wu L. 2018. N(6)-Methyladenosine-binding proteins suppress HIV-1 infectivity and viral production. J Biol Chem 293:12992–13005. doi: 10.1074/jbc.RA118.004215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murakami T, Freed EO. 2000. The long cytoplasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc Natl Acad Sci U S A 97:343–348. doi: 10.1073/pnas.97.1.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antonucci JM, Kim SH, St Gelais C, Bonifati S, Li TW, Buzovetsky O, Knecht KM, Duchon AA, Xiong Y, Musier-Forsyth K, Wu L. 2018. SAMHD1 impairs HIV-1 gene expression and negatively modulates reactivation of viral latency in CD4+ T cells. J Virol 92:e00292-18. doi: 10.1128/JVI.00292-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bowder D, Thompson J, Durst K, Hollingsead H, Hu D, Wei W, Xiang SH. 2018. Characterization of twin-cysteine motif in the V2-loop region of gp120 in primate lentiviruses. Virology 519:180–189. doi: 10.1016/j.virol.2018.04.013. [DOI] [PubMed] [Google Scholar]