

Natural killer (NK) cells display a prominent cytolytic activity against virus-infected cells and are indispensable in the innate antiviral response, particularly against herpesviruses. Despite their importance in the control of alphaherpesvirus infections, relatively little is known about the mechanisms that trigger NK cell cytotoxicity against alphaherpesvirus-infected cells. Here, using the porcine alphaherpesvirus pseudorabies virus (PRV), we found that the conserved alphaherpesvirus glycoprotein gB triggers NK cell-mediated cytotoxicity, both in virus-infected and in gB-transfected cells. In addition, we report that gB expression results in increased cell surface binding of porcine paired immunoglobulin-like type 2 receptor beta (PILRβ), an activating NK cell receptor. The interaction between PILRβ and viral gB may have consequences that stretch beyond the interaction with NK cells, including virus entry into host cells. The identification of gB as an NK cell-activating viral protein may be of importance in the construction of future vaccines and therapeutics requiring optimized interactions of alphaherpesviruses with NK cells.
KEYWORDS: NK cells, PILRβ, glycoprotein gB, herpes, natural killer cells, pseudorabies virus
ABSTRACT
Natural killer (NK) cells are components of the innate immunity and are key players in the defense against virus-infected and malignant cells. NK cells are particularly important in the innate defense against herpesviruses, including alphaherpesviruses. Aggravated and life-threatening alphaherpesvirus-induced disease has been reported in patients with NK cell deficiencies. NK cells are regulated by a diversity of activating and inhibitory cell surface receptors that recognize specific ligands on the plasma membrane of virus-infected or malignant target cells. Although alphaherpesviruses have developed several evasion strategies against NK cell-mediated attack, alphaherpesvirus-infected cells are still readily recognized and killed by NK cells. However, the (viral) factors that trigger NK cell activation against alphaherpesvirus-infected cells are largely unknown. In this study, we show that expression of the gB glycoprotein of the alphaherpesvirus pseudorabies virus (PRV) triggers NK cell-mediated cytotoxicity, both in PRV-infected and in gB-transfected cells. In addition, we report that, like their human and murine counterpart, porcine NK cells express the activating receptor paired immunoglobulin-like type 2 receptor beta (PILRβ), and we show that gB expression triggers increased binding of recombinant porcine PILRβ to the surfaces of PRV-infected cells and gB-transfected cells.
IMPORTANCE Natural killer (NK) cells display a prominent cytolytic activity against virus-infected cells and are indispensable in the innate antiviral response, particularly against herpesviruses. Despite their importance in the control of alphaherpesvirus infections, relatively little is known about the mechanisms that trigger NK cell cytotoxicity against alphaherpesvirus-infected cells. Here, using the porcine alphaherpesvirus pseudorabies virus (PRV), we found that the conserved alphaherpesvirus glycoprotein gB triggers NK cell-mediated cytotoxicity, both in virus-infected and in gB-transfected cells. In addition, we report that gB expression results in increased cell surface binding of porcine paired immunoglobulin-like type 2 receptor beta (PILRβ), an activating NK cell receptor. The interaction between PILRβ and viral gB may have consequences that stretch beyond the interaction with NK cells, including virus entry into host cells. The identification of gB as an NK cell-activating viral protein may be of importance in the construction of future vaccines and therapeutics requiring optimized interactions of alphaherpesviruses with NK cells.
INTRODUCTION
Natural killer (NK) cells can kill virus-infected and cancer cells and are of particular importance in the innate defense against herpesviruses, including alphaherpesviruses (1). In line with this, patients with deficiencies in NK cell activity may present with aggravated and life-threatening alphaherpesvirus disease, including herpes simplex encephalitis (2–4). NK cells are regulated by a diversity of activating and inhibitory cell surface receptors that recognize specific ligands variably expressed at the surfaces of virus-infected, malignant, and, also, normal cells. Specifically, the presence of activating receptor ligands at the cell surface is induced or increased upon viral infection or tumor transformation, while that of inhibitory receptor ligands is often reduced. The balance (or imbalance) of activating and inhibitory signals received by a NK cell upon encountering a potential target cell determines the initiation of the cytolytic response and its strength. As a result, NK cells can sense a wide array of alterations on the cell surfaces of target cells and respond immediately without the need to clonally expand (5, 6).
Research on pseudorabies virus (PRV), a porcine alphaherpesvirus displaying pathogenic and molecular similarities to human herpes simplex viruses (HSV), has recently resulted in the identification of two alphaherpesvirus NK cell evasion mechanisms. The expression of PRV proteins gD and US3 was found to suppress NK cell-mediated lysis of infected target cells, via reduced binding of the activating NK cell receptor DNAM-1 and increased binding of the inhibitory NK cell receptor CD300a, respectively (7, 8). Despite these NK cell evasion mechanisms, infection of cells with wild-type (WT) PRV (or HSV) still results in substantial NK cell-mediated lysis of the infected cells (7, 8).
Susceptibility to NK cell attack may depend in part on the reduced expression of major histocompatibility complex (MHC) class I molecules from the surfaces of infected cells. MHC class I molecules represent the most important ligands for inhibitory NK cell receptors and are downregulated by PRV and other herpesviruses in an attempt to lower recognition and elimination of infected cells by CD8+ cytotoxic T lymphocytes (9, 10). On the other hand, NK cell-mediated lysis of alphaherpesvirus-infected cells likely also depends on recognition of activating ligands on infected cells. However, (viral) factors that trigger NK cell reactivity are largely unknown. Older studies have suggested that viral envelope glycoproteins, which are expressed on the plasma membranes of infected cells, may increase NK cell reactivity. For both HSV-1 and PRV, viral glycoproteins, including the highly conserved glycoprotein gB, have been suggested to augment the activity of NK-like cells (11–13). However, these studies did not make use of purified NK cell populations and therefore do not allow discrimination of whether the activating effect of gB on NK cells is direct or indirect (e.g., cytokine driven), thus involving other white blood cells.
In the current study, we show that the expression of PRV gB activates primary porcine NK cells directly. In addition, we demonstrate that porcine NK cells express the activating NK cell receptor paired immunoglobulin-like type 2 receptor beta (PILRβ) and that expression of PRV gB results in enhanced binding of recombinant porcine PILRβ to the surfaces of PRV-infected and gB-transfected cells.
RESULTS
Expression of PRV gB triggers NK cell-mediated killing of PRV-infected and gB-transfected cells.
To investigate whether the gB glycoprotein of PRV is involved in NK cell-mediated lysis of PRV-infected cells, swine kidney (SK) cells were mock infected or infected with wild-type (WT) PRV or isogenic gB-null virus (multiplicity of infection [MOI] of 10), coincubated with freshly isolated interleukin-2 (IL-2)-primed porcine NK cells, and subsequently assessed for their susceptibility to NK cell-mediated cytotoxicity by flow cytometry. In line with earlier reports (7, 8), PRV-infected SK cells showed significantly increased susceptibility to NK cell-mediated cytotoxicity compared to mock-infected cells (Fig. 1A). Importantly, increased killing of infected cells depended to a large extent on the expression of PRV gB, as cells infected with gB-null virus displayed a statistically significant reduction in their susceptibility to NK cell-mediated lysis. This difference was not due to a possible heterogeneity in virus replication efficiency or percentage of infected cells, since infection with either virus (WT or gB-null PRV) resulted in infection of virtually all cells and in similar viral protein expression levels (Fig. 1B and C).
FIG 1.

Expression of PRV gB contributes to NK cell-mediated killing of PRV-infected cells. (A) SK cells were mock infected or infected with WT PRV or isogenic gB-null PRV (MOI, 10), collected at 12 hpi, and subsequently incubated with IL-2-primed porcine primary NK cells at a target/effector cell ratio of 1:25 for 4 h at 37°C. Viability of target cells was assessed by propidium iodide staining and flow cytometric analysis, and the percent NK cell-mediated lysis was calculated. The dot plot shows the results of 8 independent repeats, and the mean value is marked by a horizontal line. Statistically significant differences are indicated with asterisks (**, P < 0.01; ***, P < 0.001). (B) Mock-infected SK cells and SK cells infected with WT PRV or isogenic gB-null PRV were collected at 12 hpi and subsequently analyzed by Western blotting for expression of gB, gD, gE, and tubulin. (C) SK cells were collected at 12 hpi and subsequently analyzed by flow cytometry for the expression of PRV gB (left upper panel), PRV gE (right upper panel), and MHC class I (left lower panel). An overlay of the fluorescence intensities of the different samples (open histograms) and isotype controls (shaded histogram) is shown (black, mock-infected SK cells, blue, WT PRV-infected SK cells, red, gB-null PRV-infected SK cells). The graph (right lower panel) shows the mean fluorescence intensity (MFI) of MHC class I expression on infected SK cells. The data shown in the graph were calculated based on the MFI minus that of the isotype control-labeled cells. The dot plot shows the results of three independent repeats, and the mean value is marked by a horizontal line. Statistically significant differences are indicated with asterisks (**, P < 0.01; ***, P < 0.001).
As mentioned above, herpesviruses, including PRV, often cause a reduced cell surface expression of MHC class I in an attempt to lower recognition and elimination of infected cells by CD8+ cytotoxic T lymphocytes, which at the same time may increase susceptibility of the infected cells to NK cell-mediated lysis (9, 10). To investigate whether the differences in NK cell cytotoxicity observed for cells infected with WT or gB-null PRV were due to a difference in MHC class I downregulation, both viruses were tested for their ability to downregulate surface expression of MHC class I upon infection. Figure 1C shows that cells infected with either virus showed equal downregulation of MHC class I.
The cytolytic assays on PRV-infected cells show that expression of gB is required for wild-type levels of NK cell-mediated killing, but they do not establish whether expression of gB per se is sufficient to trigger NK cell-mediated target cell lysis. To investigate whether expression of PRV gB alone, in the absence of other viral proteins, is sufficient to increase susceptibility of cells toward NK cell-mediated lysis, rabbit kidney (RK13) epithelial cells stably expressing PRV gB were used (14). Wild-type RK13 cells and PRV gB-expressing RN/008 cells were coincubated with primary IL-2-primed porcine NK cells and subsequently assessed for NK cell-mediated cytotoxicity by flow cytometry (Fig. 2A). As shown in Fig. 2A, RK13 cells expressing PRV gB (RN/008) were significantly more susceptible to NK cell-mediated lysis than parental RK13 cells. Flow cytometry and Western blot analysis confirmed expression of PRV gB in the stably transfected RN/008 cell line (Fig. 2B). In conclusion, these data show that expression of the gB protein of PRV, in the absence of other viral proteins, is sufficient to trigger NK cell-mediated lysis of target cells and that expression of gB contributes significantly to NK cell-mediated lysis of PRV-infected cells.
FIG 2.
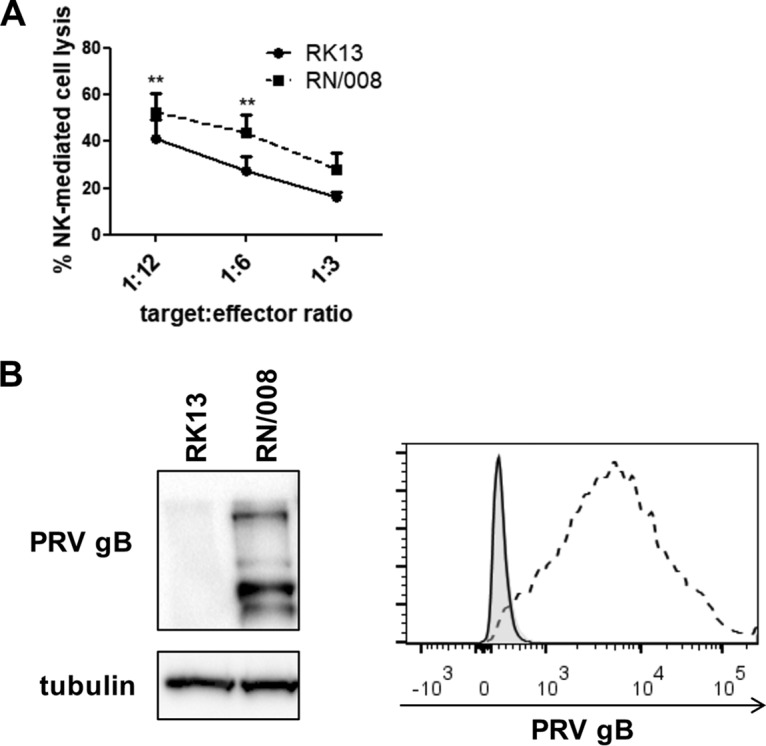
Expression of PRV gB alone, in the absence of other viral proteins, is sufficient to trigger NK cell-mediated lysis of target cells. (A) RK13 and RN/008 cells were incubated with IL-2-primed porcine primary NK cells at target/effector cell ratios of 1:12, 1:6, and 1:3 for 4 h at 37°C. Viability of target cells was assessed by propidium iodide staining and flow cytometric analysis, and the percent NK cell-mediated lysis was calculated. Data represent mean + standard error of the mean (SEM) of 4 independent repeats. Statistically significant differences are indicated with asterisks (**, P < 0.01). (B) RK13 and RN/008 cells were assessed by Western blotting for expression of PRV gB and tubulin (left panel) and by flow cytometry for surface expression of PRV gB (right panel) (black, RK13 cells; dashed line, RN/008 cells; shaded histogram, isotype control).
Porcine NK cells express PILRβ.
In infected cells, PRV gB is expressed in different host cell membranes, including the plasma membrane (15), where it may interact with receptors expressed on other cells, including immune cells. HSV-1 and PRV gBs have been reported to interact with the human and murine orthologs of paired immunoglobulin-like type 2 receptor α (PILRα) (16–20). PILRα is an inhibitory immune receptor that is typically not expressed on NK cells but is closely related to an activating member of the paired immunoglobulin-like type 2 receptors, PILRβ, which is expressed on both human and mouse NK cells (21–23). Although HSV-1 gB does not appear to interact with the human ortholog of PILRβ (18, 19), we wanted to address whether PILRβ is expressed in porcine NK cells and whether recombinant porcine PILRβ may show differential binding to WT or gB-null PRV-infected cells. To this end, we first investigated PILRβ expression in porcine NK cells by reverse transcription-PCR (RT-PCR). Figure 3A shows that, indeed, PILRβ transcript is expressed in primary porcine NK cells (Fig. 3A). As expected, negative controls (i.e., with no RNA template and inactivated reverse transcriptase activity) did not yield a PCR signal (Fig. 3A).
FIG 3.

Porcine NK cells express PILRβ, and expression of PRV gB increases binding of porcine recombinant PILRβ to the cell surfaces of PRV-infected and gB-transfected cells. (A) RT-PCR shows porcine PILRβ mRNA expression in primary porcine blood NK cells (NTC, control without template RNA; no RT, control reaction wherein reverse transcriptase activity is inhibited; PC, positive control). (B) Recombinant porcine PILRβ was produced, purified, and checked by SDS-PAGE followed by Coomassie blue staining (left panel) and by Western blotting via detection of the human Fc tag (right panel). (C) Swine kidney (SK) cells were mock infected or infected with wild-type PRV (WT) or gB-null PRV and at 14 hpi were incubated with recombinant Fc-tagged porcine PILRβ, followed by staining with fluorescently labeled anti-human-Fc antibodies and flow cytometric analysis. The histogram (left panel) shows PILRβ binding to mock-infected cells (black), WT PRV-infected cells (blue), and gB-null PRV-infected cells (red). The graph (right panel) shows MFI values of cells incubated with recombinant PILRβ minus that of cells that were stained only with secondary antibody. Dot plots show the result of four independent repeats, and the mean value is marked by a horizontal line. Statistically significant differences are indicated with asterisks (*, P < 0.05; **, P < 0.01). (D) RK13 cells and gB-expressing RK13 (RN/008) cells were incubated with recombinant Fc-tagged porcine PILRβ, followed by staining with fluorescently labeled anti-human-Fc antibodies and flow cytometric analysis. The histogram (left panel) shows PILRβ binding to RK13 cells (black) and RN/008 cells (blue). The graph (right panel) shows MFI values of cells incubated with recombinant PILRβ minus that of cells that were stained only with secondary antibody. Dot plots show the result of three independent repeats, and the mean value is marked by a horizontal line. Statistically significant differences are indicated with asterisks (*, P < 0.05).
Expression of PRV gB increases binding of recombinant porcine PILRβ to PRV-infected and gB-transfected cells.
To test whether expression of gB increases binding of PILRβ to the surfaces of PRV-infected target cells, a recombinant porcine PILRβ (pPILRβ-Fc) was produced. Purified protein was checked by SDS-PAGE followed by Coomassie blue staining and Western blotting (Fig. 3B). After purification, pPILRβ-Fc was used in a binding assay using mock-infected cells and cells infected with wild-type PRV or isogenic gB-null PRV. Figure 3C shows that binding of pPILRβ-Fc to the cell surface is significantly increased in cells infected with WT PRV but not in cells infected with gB-null PRV. Porcine PILRβ-Fc was also used in a binding assay on parental RK13 cells and on the PRV gB-expressing RN/008 cells. Remarkably, binding of pPILRβ-Fc to the cell surface was significantly increased in RN/008 cells (Fig. 3D). Addition of pPILRβ-Fc (80 μg/ml) to cytolytic assay mixtures, in an attempt to block the ability of gB to trigger NK cell cytotoxicity, did not result in a significant inhibition of NK cell-mediated killing of PRV-infected or gB-transfected cells (data not shown). In summary, our data show that recombinant porcine PILRβ shows increased binding to PRV gB-expressing cells (either PRV infected or gB transfected) but that addition of recombinant porcine PILRβ does not suppress the ability of gB to trigger NK cell cytotoxicity.
DISCUSSION
Our results show that expression of PRV gB triggers porcine NK cell-mediated cytotoxicity, in both infection and transfection assays. In addition, we found that, like their human and murine counterparts, porcine NK cells express the activating NK cell receptor PILRβ. We also observed a PRV gB-dependent increase in binding of recombinant porcine PILRβ to the surfaces of PRV-infected and gB-transfected cells.
Earlier results using total white blood cell populations already suggested an activating effect of PRV (and HSV-1) gB on NK cells (11–13), although it was unclear whether this represented a direct activating effect of gB on the NK cell population or, rather, an indirect effect mediated by other immune cell populations (e.g., via cytokine responses). Also, it was unclear whether cytolytic effects were mediated by NK cells or NK-like cells (11). Our results show that, in the absence of other immune cells, expression of PRV gB contributes substantially to the ability of PRV-infected cells to trigger NK cell cytotoxicity and that expression of PRV gB alone, in the absence of other viral proteins, is sufficient to trigger cytolytic activity in NK cells.
Although expression of PRV gB is sufficient to trigger NK cell cytotoxicity and contributes to NK cell-mediated lysis of PRV-infected cells, it is highly likely that additional factors contribute to NK cell-mediated killing of PRV-infected cells. In line with this, NK cell-mediated killing of cells infected with gB-null PRV, although significantly reduced compared to that of cells infected with WT PRV, was still significantly higher than killing of mock-infected cells (Fig. 1A). Although it is speculative at this point, such additional factors may include virus-induced upregulation of ligands for activating NK cell receptors such as natural cytotoxicity receptors (NCR), as has been described for HSV-1 (24).
Our binding assays using recombinant porcine PILRβ and (infected and transfected) PRV gB-expressing cells suggest that, in contrast to the case for HSV-1 gB and human PILRβ, PRV gB interacts with porcine PILRβ. The ability of HSV-1 gB to bind human PILRα but not PILRβ has been reported to be due to a tryptophan (W)-to-leucine (L) mutation in the amino acid sequence of PILRβ compared to that of PILRα (19). W108, together with tyrosine (Y2) and arginine (R95), is a critical residue in the sialic acid-binding domain of PILRα, and these residues are required for HSV-1 gB binding. In line with this, mutation of L108 in PILRβ to tryptophan restored the ability of this protein to bind HSV-1 gB (19). Interestingly, both porcine PILRα and PILRβ contain a tryptophan (W) residue at the location corresponding to W/L108 in the human orthologs, which could explain why PRV gB may interact with porcine PILRβ. Such interaction between PRV gB and porcine PILRβ may have important consequences for other aspects of PRV biology, including PRV entry in host cells. For example, it will be interesting to assess in future research whether PILRβ, like PILRα, may serve as a gB receptor during PRV fusion and entry in host cells (20).
Although it was not statistically significant, we observed a reproducibly reduced binding of PILRβ to the surfaces of gB-null PRV-infected cells compared to mock-infected cells (Fig. 3C). Although speculative at this point, it is possible that virus-induced shutoff of host protein synthesis and/or virus-induced alternations in host cell metabolism may result in reduced expression of natural host ligands for PILRβ.
We found that porcine NK cells express PILRβ transcripts. Since PRV has a wide host range, which include carnivores and ruminants, and almost invariably causes lethal infections in nonporcine susceptible hosts, it will be interesting in future research to address whether NK cells of these species express PILRβ and, if so, whether gB of PRV affects their binding to PRV-infected cells.
Since addition of recombinant porcine PILRβ to cytotoxicity assay mixtures did not lead to a reduced gB-mediated cell lysis by porcine NK cells, the NK cell-activating effect of PRV gB may not (solely) be due to PILRβ. However, a contribution of PILRβ in the NK cell-activating effect of PRV gB cannot be excluded at present. Indeed, for example, cells expressing PRV gB may show a higher affinity for native PILRβ that is expressed on NK cells than for recombinant PILRβ. This may prevent efficient inhibition of gB-mediated NK cell activation by recombinant PILRβ. In addition, it is possible that a relatively low threshold number of gB-PILRβ interactions is sufficient to activate NK cells during cell-to-cell contact.
Despite different attempts, we have currently been unable to design effective small interfering RNA (siRNA) strategies in primary porcine NK cells. Successful development of such assays will allow resolution of the contribution of PILRβ in the NK cell-activating effect of PRV gB.
Together, the results of this study show for the first time that, on the one hand, expression of PRV gB triggers NK cell cytotoxicity and, on the other hand, expression of PRV gB leads to an increased PILRβ binding.
Our finding that expression of PRV gB triggers NK cell cytotoxicity increases our understanding of the interaction of alphaherpesviruses with this particularly important immune cell population and may lead to clinical applications. Indeed, activation of NK cells has been proposed as a valuable strategy in the design of alphaherpesvirus vaccines (25–31). In addition, NK cell-mediated elimination of virus-infected cells has been reported to interfere with efficient tumor cell lysis by HSV-based oncolytic vectors in a mouse model of glioblastoma (32). Our data, which demonstrate that a conserved alphaherpesvirus glycoprotein triggers NK cell cytotoxicity, may therefore have important consequences regarding the construction of future vaccines and oncolytic vectors with optimized NK cell-activating properties.
MATERIALS AND METHODS
Infections of SK cells.
As described earlier (7, 8), swine kidney (SK) cells were cultivated in minimal essential medium (MEM) (Life Technologies, Thermo Fisher Scientific) supplemented with 10% (vol/vol) fetal calf serum (FCS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.05 mg/ml gentamicin. SK cells were detached from cell culture flasks using trypsin, seeded in suspension culture flasks (Sarstedt, Nümbrecht, Germany) at 1.2 × 107 cells/8.5 ml, inoculated with PRV Kaplan wild-type virus (33) or the isogenic gB-null mutant strain (14) at a multiplicity of infection (MOI) of 10, and put on a rocking platform at 37°C. For cytolytic assays, cells were collected at 12 h postinoculation (hpi) and subjected to the cytolytic assay. For assays to determine binding of recombinant porcine PILRβ, cells were collected at 14 hpi.
RK13 cell lines.
Rabbit kidney (RK13) cells were cultivated with MEM supplemented with 10% FCS, l-glutamine, sodium pyruvate, and nonessential amino acids. RN/008 cells were cultivated with MEM supplemented with 10% FCS, l-glutamine, sodium pyruvate, nonessential amino acids, and Geneticin (500 μg/ml). RN/008 is a previously generated and characterized RK13 cell line that stably expresses PRV gB-008, which is a C-terminally truncated gB derivative constructed by PCR by insertion of stop codons before the second alpha-helical domain, thereby deleting the YQRL internalization motif (14, 34).
Primary porcine NK cell isolation and culture.
NK cell isolation was performed as previously described before (35). In brief, heparinized blood samples (50 U/ml blood; LEO Pharma) were obtained from the external jugular veins of pigs (12- to 27-week-old crossbred pigs derived from Rattlerow-Segher hybrid sows and Pietrain boars) that were kept as blood donors under standard conditions at the Faculty of Veterinary Medicine, Merelbeke, Belgium, and were from a commercial farm. The blood sampling procedure was approved by the Ethical Committee of the Faculty of Veterinary Medicine (EC2013/62 and EC2017/121). Primary porcine NK cells were isolated from porcine peripheral blood mononuclear cells (PBMC) by negative magnetically active cell sorting (MACS) depletion of CD3+ and CD172a+ cells followed by fluorescence-activated cell sorting (FACS) purification using antibodies against porcine CD172a (IgG1, clone 74-22-15a [36]), CD3 (IgG1, clone PPT3 [37]), and CD8a (IgG2a, clone 11/295/33 [38]) as previously described (35). Porcine NK cells were purity sorted based on CD3− CD172a− CD8a+ expression using a BD FACS Aria III cell sorter (BD Biosciences), resulting in a ≥98% pure porcine NK cell population.
Primary porcine NK cells were cultured in 96-well flat-bottom plates (Nunc, Thermo Fisher Scientific) at a density of 5 × 106 cells/ml in RPMI (Gibco) supplemented with 10% (vol/vol) fetal calf serum (FCS) (Thermo Fisher Scientific), 100 U/ml penicillin (Gibco), and 100 μg/ml streptomycin (Gibco) (referred to as porcine NK medium). For cytolytic assays, porcine NK cells were primed with recombinant human IL-2 (20 ng/ml) for 16 to 18 h before the assay.
Construction of recombinant PILRβ.
The sequence encoding the extracellular portion of the porcine PILRβ receptor (GenBank accession number JQ890108.2) was amplified starting from the pcDNA3.1+DYK-PILRβ plasmid (custom made; GenScript, USA) using the primers Fw 5′-ACGCGTCGACACCATGGGGCTGCCCCTGCT-3′ and Rev 5′-ACGCGTCGACTTCCGTACTCGGGGAATGCCTTTGG-3′. Amplification was performed with the Herculase II fusion DNA polymerase kit (Agilent Technologies, Santa Clara, CA, USA) for 2 min at 95°C, followed by 30 cycles of 20 s at 95°C, 20 s at 60°C, and 1 min at 72°C, followed by a 3-min elongation step at 72°C. The PCR product was digested with SalI restriction enzyme and subcloned in the SalI-digested pRB1-cc Fcmut vector in frame with the sequence coding for a dimeric human IgG1 portion, which was mutagenized to obtain a mutated Fc that does not bind to Fc receptors. The pRB1-cc PILRβ Fcmut construct was transfected into the HEK293T cell line (human embryonic fibroblasts) using JetPEI DNA transfection reagent (Polyplus transfection). Supernatants were collected from the transfected cells cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% Ultralow IgG fetal bovine serum (Life Technologies, UK), and the recombinant porcine PILRβ-Fcmut molecule was purified by affinity chromatography using a HiTrap protein G HP column (GE Healthcare, UK). The purified protein was checked by SDS-PAGE followed by Coomassie blue staining and by Western blotting using a horseradish peroxidase (HRP)-conjugated goat anti-human IgG antibody (Thermo Fisher Scientific) (see below).
Western blotting.
Cells were collected on ice, washed in phosphate-buffered saline (PBS), and lysed in lysis buffer, containing TNE (20 mM Tris-HCl, 150 mM NaCl, and 1 mM EDTA) with 10% NP-40 (Roche) and protease inhibitor cocktail (Sigma-Aldrich), on a shaker at 4°C for 1 h. Nuclei were removed by centrifugation (13,000 × g, 10 min, 4°C), and the protein content was measured using the bicinchoninic acid (BCA) protein assay kit (Thermo Scientific). Cell lysates were loaded on SDS-10% acrylamide gels and transferred to a P-Hybond polyvinylidene difluoride (PVDF) membrane (GE Healthcare), which was afterwards blocked using 5% milk powder (Nestlé) diluted in PBS-T (PBS supplemented with 0.1% Tween 20 [Sigma-Aldrich]) for 1 h at room temperature. Incubations with primary monoclonal antibodies (MAbs) (anti-PRV gB 1C11 [39]), anti-PRV gD 13D12 [39]), anti-PRV gE 18E8 [39]) or HRP-labeled anti-alpha-tubulin (Abcam, UK) or with HRP-labeled secondary antibodies (HRP-conjugated goat anti-mouse IgG or goat Fab anti-human IgG [Thermo Fisher Scientific]) were performed in 5% milk powder–PBS-T at 4°C overnight and for 1 h at room temperature, respectively. Blots were developed using chemiluminescence. Bands were detected using a ChemiDocMP imager (Bio-Rad) according to the manufacturer’s instructions.
Cytolytic assays.
In general, cytolytic assays were performed as described before (7, 8). In brief, mock-, PRV wild-type Kaplan-, or isogenic Kaplan gB null-infected SK cells or RK13 and RN/008 cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) proliferation dye (CellTrace CFSE cell proliferation kit; Invitrogen, Thermo Fisher Scientific) according to the manufacturer’s recommendations. Briefly, 1.0 × 106 target cells/ml were resuspended in porcine NK medium with 5 μM CFSE and incubated for 15 min at 37°C. The labeling reaction was stopped by addition of ice-cold porcine NK medium. Afterwards, cells were washed in porcine NK medium to remove excess CFSE and coincubated with IL-2-primed NK cells at different effector/target cell ratios (as indicated in the figure legends) for 4 h at 37°C. Thereafter, the viability of 5,000 target cells was assessed by propidium iodide staining and flow cytometry. The percentage of NK cell-mediated lysis was calculated using the formula (% dead targetNK − % dead targetspont)/(% dead targetmaximum − % dead targetspont).
Cell surface expression analysis via flow cytometry.
For flow cytometric analysis, cells were collected and washed in PBS. All incubation steps were performed in 96-well V-bottom plates for 40 min at 4°C. The different combinations of primary monoclonal antibodies (MAbs) and secondary reagents used for each assay are listed in Table 1 and were diluted in PBS. Sytox blue (Thermo Fisher Scientific) was used to discriminate live and dead cells. Flow cytometry was performed using a BD FACS Aria III (BD Biosciences), and samples were analyzed with FACSDiva software (BD Biosciences) and FlowJo software (doublet discrimination).
TABLE 1.
Primary and secondary antibodies used for cell surface expression analysis by flow cytometry
| Antigen (reference or source) | Clone | Isotype or recombinant | Labeling strategy |
|---|---|---|---|
| PILRβ ligands | Recombinant porcine PILRβ Fcmut | Secondary antibody R-PEa-conjugated goat Fab anti-human IgG | |
| PRV gB (39) | 1C11 | IgG2a | Secondary antibody R-PE-conjugated goat anti-mouse IgG |
| MHC I (VMRD, USA) | PT85A | IgG2a | Secondary antibody R-PE-conjugated goat anti-mouse IgG |
| PRV gE (39) | 18E8 | IgG1 | Secondary antibody R-PE-conjugated goat anti-mouse IgG |
R-PE, R-phycoerythrin.
One-step RT-PCR.
Total RNA was isolated from freshly isolated porcine NK cells using the RNeasy minikit (Qiagen) and was reverse transcribed into cDNA and immediately amplified by using one-step RT-PCR kit (Qiagen). The primers used for detection of PILRβ were Fw 5′-ATACGGTGAACAGGAGTGGC-3′ and Rev 5′-CGTACTCGGGGAATGCCTTT-3′. The cycle conditions were 30 min at 50°C, 1 cycle of 15 min at 95°C, 30 cycles of 30 s at 94°C, 30 s at 55°C, and 90 s at 72°C, and then 10 min at 72°C. A no-template control without template RNA was included. In addition, DNA contamination was excluded by using control reactions wherein reverse transcriptase activity is inhibited. Under these conditions, reverse transcriptase activity is inhibited by keeping control reaction mixtures on ice and placing them in the thermal cycler after it has reached 95°C for the HotStarTaq DNA polymerase activation step (before cycling). The pcDNA3.1+DYK-PILRβ plasmid (custom made; GenScript, USA) was used as a positive control.
Statistical analysis.
Statistical analysis was performed using GraphPad Prism 5 (GraphPad, CA, USA). Data were analyzed for statistical differences with a repeated-measures analysis of variance (ANOVA) at the 5% significance level. Post hoc comparisons between different conditions were performed using Tukey's range test. For comparing two groups, data were analyzed for statistical differences with a two-tailed (un)paired t test.
ACKNOWLEDGMENTS
Steffi De Pelsmaeker is supported by a Ph.D. grant from the Agency for Innovation by Science and Technology in Flanders (IWT-Vlaanderen). This research was supported by grants from the F.W.O.-Vlaanderen (grant number G.0176.15), the Special Research Fund of Ghent University (G.O.A. grant 01G01317), and the Belgian Science Policy (BELSPO-IAP phase VII BELVIR).
We thank Korneel Grauwet for performing experiments and collecting data on the topic that has resulted in the current paper during his Ph.D. thesis research at Ghent University. We also thank Cliff Van Waesberghe, Jiexiong Xie, and Isaura Christiaens for excellent technical assistance and Rudy Cooman and Jochen Lamote for animal caretaking.
We declare no conflict of interest.
S.D.P. and H.W.F. designed the research. S.D.P. and E.D. performed the experiments. B.K. T.C.M., C.C., and M.V. provided reagents. S.D.P. and H.W.F. analyzed the results. S.D.P., E.D., B.K., T.C.M., C.C., M.V., and H.W.F. interpreted the data. S.D.P. and H.W.F. made the figures and wrote the manuscript.
REFERENCES
- 1.Waggoner SN, Reighard SD, Gyurova IE, Cranert SA, Mahl SE, Karmele EP, McNally JP, Moran MT, Brooks TR, Yaqoob F, Rydyznski CE. 2016. Roles of natural killer cells in antiviral immunity. Curr Opin Virol 16:15–23. doi: 10.1016/j.coviro.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Etzioni A, Eidenschenk C, Katz R, Beck R, Casanova JL, Pollack S. 2005. Fatal varicella associated with selective natural killer cell deficiency. J Pediatr 146:423–425. doi: 10.1016/j.jpeds.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 3.Almerigogna F, Fassio F, Giudizi MG, Biagiotti R, Manuelli C, Chiappini E, Galli L, Romagnani S, De Martino M. 2011. Natural killer cell deficiencies in a consecutive series of children with herpetic encephalitis. Int J Immunopathol Pharmacol 24:231–238. doi: 10.1177/039463201102400128. [DOI] [PubMed] [Google Scholar]
- 4.Orange JS. 2013. Natural killer cell deficiency. J Allergy Clin Immunol 132:515–525. doi: 10.1016/j.jaci.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. 2008. Functions of natural killer cells. Nat Immunol 9:503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 6.Della Chiesa M, Marcenaro E, Sivori S, Carlomagno S, Pesce S, Moretta A. 2014. Human NK cell response to pathogens. Semin Immunol 26:152–160. doi: 10.1016/j.smim.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 7.Grauwet K, Cantoni C, Parodi M, De Maria A, Devriendt B, Pende D, Moretta L, Vitale M, Favoreel HW. 2014. Modulation of CD112 by the alphaherpesvirus gD protein suppresses DNAM-1-dependent NK cell-mediated lysis of infected cells. Proc Natl Acad Sci U S A 111:16118–16123. doi: 10.1073/pnas.1409485111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grauwet K, Vitale M, De Pelsmaeker S, Jacob T, Laval K, Moretta L, Parodi M, Parolini S, Cantoni C, Favoreel HW. 2016. Pseudorabies virus US3 protein kinase protects infected cells from NK cell-mediated lysis via increased binding of the inhibitory NK cell receptor CD300a. J Virol 90:1522–1533. doi: 10.1128/JVI.02902-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schuren ABC, Costa AI, Wiertz EJHJ. 2016. Recent advances in viral evasion of the MHC class I processing pathway. Curr Opin Immunol 40:43–50. doi: 10.1016/j.coi.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 10.Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. 2012. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol 12:239–252. doi: 10.1038/nri3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kimman TG, De Bruin TGM, Voermans JJM, Bianchi ATJ. 1996. Cell-mediated immunity to pseudorabies virus: cytolytic effector cells with characteristics of lymphokine-activated killer cells lyse virus-infected and glycoprotein gB- and gC-transfected L14 cells. J Gen Virol 77:987–990. doi: 10.1099/0022-1317-77-5-987. [DOI] [PubMed] [Google Scholar]
- 12.Bishop GA, Glorioso JC, Schwartz SA. 1983. Relationship between expression of herpes simplex virus glycoproteins and susceptibility of target cells to human natural killer activity. J Exp Med 157:1544–1561. doi: 10.1084/jem.157.5.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bishop GA, Marlin SD, Schwartz SA, Glorioso JC. 1984. Human natural killer cell recognition of herpes simplex virus type 1 glycoproteins: specificity analysis with the use of monoclonal antibodies and antigenic variants. J Immunol 133:2206–2214. [PubMed] [Google Scholar]
- 14.Nixdorf R, Klupp BG, Karger A, Mettenleiter TC. 2000. Effects of truncation of the carboxy terminus of pseudorabies virus glycoprotein B on infectivity. J Virol 74:7137–7145. doi: 10.1128/JVI.74.15.7137-7145.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Favoreel HW, Mettenleiter TC, Nauwynck HJ. 2004. Copatching and lipid raft association of different viral glycoproteins expressed on the surfaces of pseudorabies virus-infected cells. J Virol 78:5279–5287. doi: 10.1128/JVI.78.10.5279-5287.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Satoh T, Arii J, Suenaga T, Wang J, Kogure A, Uehori J, Arase N, Shiratori I, Tanaka S, Kawaguchi Y, Spear PG, Lanier LL, Arase H. 2008. PILRα is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132:935–944. doi: 10.1016/j.cell.2008.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J, Shiratori I, Satoh T, Lanier LL, Arase H. 2008. An essential role of sialylated O-linked sugar chains in the recognition of mouse CD99 by paired Ig-like type 2 receptor (PILR). J Immunol 180:1686–1693. doi: 10.4049/jimmunol.180.3.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan Q, Longnecker R. 2010. The Ig-like V-type domain of paired Ig-like type 2 receptor alpha is critical for herpes simplex virus type 1-mediated membrane fusion. J Virol 84:8664–8672. doi: 10.1128/JVI.01039-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu Q, Lu G, Qi J, Wang H, Xuan Y, Wang Q, Li Y, Zhang Y, Zheng C, Fan Z, Yan J, Gao GF. 2014. PILR and PILR have a siglec fold and provide the basis of binding to sialic acid. Proc Natl Acad Sci U S A 111:8221–8226. doi: 10.1073/pnas.1320716111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arii J, Uema M, Morimoto T, Sagara H, Akashi H, Ono E, Arase H, Kawaguchi Y. 2009. Entry of herpes simplex virus 1 and other alphaherpesviruses via the paired immunoglobulin-like type 2 receptor. J Virol 83:4520–4527. doi: 10.1128/JVI.02601-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson MD, Cheung J, Martindale DW, Scherer SW, Koop BF. 2006. Comparative analysis of the paired immunoglobulin-like receptor (PILR) locus in six mammalian genomes: duplication, conversion, and the birth of new genes. Physiol Genomics 27:201–218. doi: 10.1152/physiolgenomics.00284.2005. [DOI] [PubMed] [Google Scholar]
- 22.Fournier N, Chalus L, Durand I, Garcia E, Pin J-J, Churakova T, Patel S, Zlot C, Gorman D, Zurawski S, Abrams J, Bates EEM, Garrone P. 2000. FDF03, a novel inhibitory receptor of the immunoglobulin superfamily, is expressed by human dendritic and myeloid cells. J Immunol 165:1197–1209. doi: 10.4049/jimmunol.165.3.1197. [DOI] [PubMed] [Google Scholar]
- 23.Shiratori I, Ogasawara K, Saito T, Lanier LL, Arase H. 2004. Activation of natural killer cells and dendritic cells upon recognition of a novel CD99-like ligand by paired immunoglobulin-like type 2 receptor. J Exp Med 199:525–533. doi: 10.1084/jem.20031885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chisholm SE, Howard K, Gómez MV, Reyburn HT. 2007. Expression of ICP0 is sufficient to trigger natural killer cell recognition of herpes simplex virus-infected cells by natural cytotoxicity receptors. J Infect Dis 195:1160–1168. doi: 10.1086/512862. [DOI] [PubMed] [Google Scholar]
- 25.Hanna J, Mandelboim O. 2007. When killers become helpers. Trends Immunol 28:201–206. doi: 10.1016/j.it.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 26.Wagstaffe HR, Mooney JP, Riley EM, Goodier MR. 2018. Vaccinating for natural killer cell effector functions. Clin Transl Immunol 7:e1010. doi: 10.1002/cti2.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goodier MR, Jonjić S, Riley EM, Juranić Lisnić V. 2018. CMV and natural killer cells: shaping the response to vaccination. Eur J Immunol 48:50–65. doi: 10.1002/eji.201646762. [DOI] [PubMed] [Google Scholar]
- 28.Sun JC, Lanier LL. 2018. Is there natural killer cell memory and can it be harnessed by vaccination? NK cell memory and immunization strategies against infectious diseases and cancer. Cold Spring Harb Perspect Biol 10:a029538. doi: 10.1101/cshperspect.a029538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neely HR, Mazo IB, Gerlach C, von Andrian UH. 2018. Is there natural killer cell memory and can it be harnessed by vaccination? Natural killer cells in vaccination. Cold Spring Harb Perspect Biol 10:a029488. doi: 10.1101/cshperspect.a029488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cooper MA, Fehniger TA, Colonna M. 2018. Is there natural killer cell memory and can it be harnessed by vaccination? Vaccination strategies based on NK cell and ILC memory. Cold Spring Harb Perspect Biol 10:a029512. doi: 10.1101/cshperspect.a029512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rydyznski CE, Waggoner SN. 2015. Boosting vaccine efficacy the natural (killer) way. Trends Immunol 36:536–546. doi: 10.1016/j.it.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alvarez-Breckenridge CA, Yu J, Price R, Wojton J, Pradarelli J, Mao H, Wei M, Wang Y, He S, Hardcastle J, Fernandez SA, Kaur B, Lawler SE, Vivier E, Mandelboim O, Moretta A, Caligiuri MA, Chiocca EA. 2012. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat Med 18:1827–1834. doi: 10.1038/nm.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaplan AS, Vatter AE. 1959. A comparison of herpes simplex and pseudorabies viruses. Virology 7:394–407. doi: 10.1016/0042-6822(59)90068-6. [DOI] [PubMed] [Google Scholar]
- 34.Favoreel HW, Van Minnebruggen G, Nauwynck HJ, Enquist LW, Pensaert MB. 2002. A tyrosine-based motif in the cytoplasmic tail of pseudorabies virus glycoprotein B is important for both antibody-induced internalization of viral glycoproteins and efficient cell-to-cell spread. J Virol 76:6845–6851. doi: 10.1128/JVI.76.13.6845-6851.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Pelsmaeker S, Devriendt B, Leclercq G, Favoreel HW. 2018. Porcine NK cells display features associated with antigen-presenting cells. J Leukoc Biol 103:129–140. doi: 10.1002/JLB.4A0417-163RR. [DOI] [PubMed] [Google Scholar]
- 36.Pescovitz MD, Lunney JK, Sachs DH. 1984. Preparation and characterization of monoclonal antibodies reactive with porcine PBL. J Immunol 133:368–375. [PubMed] [Google Scholar]
- 37.Yang H, Oura CAL, Kirkham PA, Parkhouse RME. 1996. Preparation of monoclonal anti-porcine CD3 antibodies and preliminary characterization of porcine T lymphocytes. Immunology 88:577–585. doi: 10.1046/j.1365-2567.1996.d01-682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jonjić S, Koszinowski UH. 1984. Monoclonal antibodies reactive with swine lymphocytes. I. Antibodies to membrane structures that define the cytolytic T lymphocyte subset in the swine. J Immunol 133:647–652. [PubMed] [Google Scholar]
- 39.Nauwynck HJ, Pensaert MB. 1995. Effect of specific antibodies on the cell-associated spread of pseudorabies virus in monolayers of different cell types. Arch Virol 140:1137–1146. doi: 10.1007/BF01315422. [DOI] [PubMed] [Google Scholar]