

A 2014 EV-D68 outbreak in the United States has been linked to the development of acute flaccid myelitis in children. Unfortunately, no treatment options against EV-D68 are currently available, and the development of effective therapeutics is urgently needed. Here, we characterize and validate a new EV-D68 drug target, the 2Apro, and identify telaprevir—an FDA-approved drug used to treat hepatitis C virus (HCV) infections—as a potent antiviral with a novel mechanism of action toward 2Apro. 2Apro functions as a viral protease that cleaves a peptide sequence corresponding to the VP1-2A polyprotein junction. The binding of telaprevir potently inhibits its enzymatic activity, and using drug resistance selection, we show that the potent antiviral activity of telaprevir was due to 2Apro inhibition. This is the first inhibitor to selectively target the 2Apro from EV-D68 and can be used as a starting point for the development of therapeutics with selective activity against EV-D68.
KEYWORDS: antiviral agents, antiviral pharmacology, enterovirus, protease inhibitors
ABSTRACT
Enterovirus D68 (EV-D68) is a viral pathogen that leads to severe respiratory illness and has been linked with the development of acute flaccid myelitis (AFM) in children. No vaccines or antivirals are currently available for EV-D68 infection, and treatment options for hospitalized patients are limited to supportive care. Here, we report the expression of the EV-D68 2A protease (2Apro) and characterization of its enzymatic activity. Furthermore, we discovered that telaprevir, an FDA-approved drug used for the treatment of hepatitis C virus (HCV) infections, is a potent antiviral against EV-D68 by targeting the 2Apro enzyme. Using a fluorescence resonance energy transfer-based substrate cleavage assay, we showed that the purified EV-D68 2Apro has proteolytic activity selective against a peptide sequence corresponding to the viral VP1-2A polyprotein junction. Telaprevir inhibits EV-D68 2Apro through a nearly irreversible, biphasic binding mechanism. In cell culture, telaprevir showed submicromolar-to-low-micromolar potency against several recently circulating neurotropic strains of EV-D68 in different human cell lines. To further confirm the antiviral drug target, serial viral passage experiments were performed to select for resistance against telaprevir. An N84T mutation near the active site of 2Apro was identified in resistant viruses, and this mutation reduced the potency of telaprevir in both the enzymatic and cellular antiviral assays. Collectively, we report for the first time the in vitro enzymatic activity of EV-D68 2Apro and the identification of telaprevir as a potent EV-D68 2Apro inhibitor. These findings implicate EV-D68 2Apro as an antiviral drug target and highlight the repurposing potential of telaprevir to treat EV-D68 infection.
IMPORTANCE A 2014 EV-D68 outbreak in the United States has been linked to the development of acute flaccid myelitis in children. Unfortunately, no treatment options against EV-D68 are currently available, and the development of effective therapeutics is urgently needed. Here, we characterize and validate a new EV-D68 drug target, the 2Apro, and identify telaprevir—an FDA-approved drug used to treat hepatitis C virus (HCV) infections—as a potent antiviral with a novel mechanism of action toward 2Apro. 2Apro functions as a viral protease that cleaves a peptide sequence corresponding to the VP1-2A polyprotein junction. The binding of telaprevir potently inhibits its enzymatic activity, and using drug resistance selection, we show that the potent antiviral activity of telaprevir was due to 2Apro inhibition. This is the first inhibitor to selectively target the 2Apro from EV-D68 and can be used as a starting point for the development of therapeutics with selective activity against EV-D68.
INTRODUCTION
Enterovirus D68 (EV-D68) is a highly contagious pathogen known for causing respiratory tract infections in humans (1). A 2014 EV-D68 outbreak in the United States caused severe respiratory illness for more than a thousand patients (2, 3). The viral outbreak coincided with several cases of acute flaccid myelitis (AFM), a debilitating neurological disease with symptomatic similarities to poliomyelitis, in pediatric patients (4). Recent studies implicate EV-D68 as the agent responsible for neurological disease (5–7). Several efforts to develop direct-acting antivirals against enteroviruses have been reported (8–13); unfortunately, many of the antivirals demonstrated to have activity against EV-D68 either are not effective against contemporary strains, have cell type-dependent antiviral efficacy, or have concerning side effects (11, 14). Despite the significant global health and socioeconomic impact posed by EV-D68, there is currently no vaccine or specific antiviral against EV-D68 infection, and treatment for severe cases of the disease is limited to supportive care.
Upon translation of the viral genome, the polyprotein of enteroviruses is cleaved by the 2A protease (2Apro), 3Cpro, and 3CDpro to produce several precursor proteins that eventually lead to the production of mature viral proteins. 2Apro and 3Cpro are between 140 and 150 and between 180 and 220 amino acids in length, respectively. Both proteases contain a catalytic cysteine nucleophile and chymotrypsin-like fold (15). The difference in substrate recognition sequences allows 2Apro and 3Cpro to perform different roles in viral polyprotein processing: EV 2Apro cleaves a conserved amino acid sequence at the VP1-2A junction, while 3Cpro cleaves at all other junctions, including at the 3C N and C termini (15). 3Cpro from poliovirus 1 (PV-1) (16), hepatitis A virus (HAV) (17), human rhinovirus 14 (HRV14) (18), EV-A71 (19), and EV-D68 (20) adopts two β-barrel subdomains containing 6 β-sheets each, whereas 2Apro from human rhinovirus 2 (HRV2) (21), coxsackievirus B4 (CVB4) (22), and EV-A71 (23) has an N-terminal β-barrel consisting of only 4 β-sheets. 2Apro also contains a noncatalytic zinc atom which is also found in the hepatitis C virus (HCV) NS3-4A serine protease (24) but which is not found in EV 3Cpro proteins. For EV-D68, only the 3Cpro has been characterized. Although the EV-D68 2Apro was predicted to have proteolytic activity, based on sequence alignment, the functional activity for EV-D68 2Apro has not been reported.
We hypothesized that the 2Apro from EV-D68 is a protease specific for the VP1-2A junction site and may represent a valid target for antiviral drugs. To date, up to 19 viral protease inhibitors have been approved by the FDA to treat HIV and HCV infections (25). The success of viral protease inhibitors has stimulated drug discovery and development efforts for other human-infecting viral pathogens. Here we characterize the in vitro protease activity for EV-D68 2Apro and demonstrate its substrate specificity for the peptide sequence encoded in the VP1-2A polyprotein junction. Screening of the Selleckchem protease inhibitor library using a fluorescence resonance energy transfer (FRET) assay allowed us to identify telaprevir (VX-950) (Fig. 1) as a potent inhibitor of EV-D68 by targeting the viral 2Apro. The close analog of telaprevir, boceprevir (Fig. 1), had no enzymatic inhibition against 2Apro or antiviral activity, suggesting that the mechanism of action of telaprevir is specific toward 2Apro.
FIG 1.

Chemical structures of telaprevir and boceprevir. The electrophilic ketones in telaprevir and boceprevir are in orange. Both compounds are peptidomimetic protease inhibitors that have been approved for the treatment of hepatitis C virus (HCV) infections.
RESULTS AND DISCUSSION
2Apro from EV-D68 is a protease with substrate specificity toward the VP1-2A junction.
The enzymatic activities of 2Apro have been reported for other picornaviruses, such as EV-A71 (26), PV (27), CVB3 (28), and rhinovirus (29), yet the protein function for EV-D68 2Apro has not been reported. We found that EV-D68 2Apro (US/KY/14-18953) and EV-A71 2Apro (E2004104-TW-CDC) had a sequence identity of 49.3% and a sequence similarity of 69.3% (Fig. 2), but crucially, the catalytic cysteine and histidine residues in the protease active site were conserved. Therefore, we hypothesized that the 2Apro protein from EV-D68 might function as a protease.
FIG 2.

The sequence of 2Apro from EV-D68 US/KY/14-18953 aligned to other picornaviral 2Apro sequences was used to compare amino acid conservation. Sequences were obtained from the Virus Pathogen Resource website (https://www.viprbrc.org) and aligned using the Clustal Omega (v1.2.4) multiple-sequence-alignment tool. EV-D68 US/KY/14-18953 was 55% identical with 69% positive substitutions to human poliovirus 1 Mahoney, 56% identical with 76% positive substitutions to coxsackievirus B4 JVB/Benschoten/NY/51, 50% identical with 70% positive substitutions to EV-A71 E2004104-TW-CDC, and 49% identical with 70% positive substitutions to coxsackievirus A16 Tainan/5079/98.
We chose the EV-D68 US/KY/14-18953 strain 2Apro sequence since this virus demonstrated neurotropism and was associated with paralysis in children during the 2014 United States EV-D68 outbreak (30). We first attempted to express EV-D68 2Apro in an Escherichia coli bacterial expression system using the BL21(DE3), BL21(DE3)pLysS, or Rosetta strain and the pET28b(+) vector. However, several trials of expression with different IPTG (isopropyl-β-d-thiogalactopyranoside) concentrations, induction times, and temperatures failed to produce detectable amounts of 2Apro, precluding its characterization. Next, we switched to the SUMO tag expression strategy (31). His-SUMO-2Apro was successfully expressed in a satisfactory yield (5 mg/liter) under optimized conditions (0.5 mM IPTG, 18°C, and overnight) (Fig. 3A, lane 1). The His-SUMO tag was subsequently cleaved from the Ni-nitrilotriacetic acid (NTA) resin-bound His-SUMO-2Apro protein by incubating with the SUMO protease. Upon purification, tag-free EV-D68 2Apro was obtained in good purity, as confirmed using SDS-PAGE gels (Fig. 3A, lane 2). The identity of E. coli-expressed EV-D68 2Apro was confirmed by mass spectrometry (MS). The detected mass was 16,095 ± 0.6 Da, which matches the calculated mass of 16,097.6 Da. In addition, liquid chromatography (LC)-tandem mass spectrometry (MS/MS) sequencing showed 66% sequence coverage (Fig. 3A).
FIG 3.

EV-D68 2Apro is a viral protease that cleaves the peptide sequence at the VP1-2A junction. (A) SDS-PAGE of His-SUMO-2Apro and nontagged EV-D68 2Apro proteins. Lane M, protein ladder; lane 1, His-SUMO-2Apro; lane 2, EV-D68 2Apro. LC-MS/MS sequencing of EV-D68 2Apro was performed to confirm the identity of the expressed protein. Purified EV-D68 2Apro was loaded onto an SDS-PAGE gel (15%) and stained with Coomassie brilliant blue dye to visualize the bands. The band corresponding to 2Apro (∼16 kDa) was cut from the gel and stored in sterile H2O for LC-MS/MS. Sequencing was performed by the Taplin Biological Mass Spectrometry Facility (Harvard Medical School, Boston, MA). (B) Proteolytic reaction progress curves of 1 μM His-SUMO-2Apro and 1 μM EV-D68 2Apro proteins with 20 μM FRET-1 substrate at 30°C. (C) Michaelis-Menten plot of 1 μM His-SUMO-2Apro and 1 μM EV-D68 2Apro proteins with the FRET-1 substrate. The initial velocity (Vinitial) was calculated in the first 30 min of the reaction linear regression. (D) Proteolytic reaction progress curves of 1 μM His-SUMO-2Apro and 1 μM EV-D68 2Apro proteins with 20 μM the FRET-2 substrate at 30°C.
We next set out to determine its enzymatic activity and substrate specificity. For this purpose, we designed two FRET-based substrates: FRET-1, 4-((4-(dimethylamino)phenyl)azo)benzoic acid (Dabcyl)–KIRIVNT/GPGFGGE–5-[(2-aminoethyl)amino]naphthalene-1-sulfonic acid (Edans), corresponding to the VP1-2A junction from the EV-D68 polyprotein precursor, and FRET-2, Dabcyl-KEALFQ/GPPQFE-Edans, corresponding to the 2C-3A cleavage junction from the viral polyprotein, which is the substrate for the EV-D68 3Cpro (20). It was found that the FRET-1 substrate was efficiently cleaved by the EV-D68 2Apro (Fig. 3B, black) with a Vmax and a Km of 3.7 ± 0.1 relative fluorescence units (RFU)/s/μM and 49.8 ± 3.8 μM, respectively (Fig. 3C, black). In contrast, the FRET-2 substrate was cleaved significantly less efficiently than the FRET-1 substrate (Fig. 3D, black), suggesting a degree of substrate specificity of EV-D68 2Apro toward FRET-1. It was also noted that the His-SUMO-2Apro construct had lower enzymatic activity than the tag-free 2Apro (Fig. 3B and C, red), suggesting that the SUMO tag might interfere with the enzymatic activity to some extent. As such, all following experiments were carried out with the tag-free 2Apro.
Telaprevir is a potent protease inhibitor of the EV-D68 2Apro.
In light of the specific enzymatic activity of EV-D68 2Apro, we hypothesized that inhibiting EV-D68 2Apro by a protease inhibitor might be an effective antiviral strategy. We therefore screened the Selleckchem protease inhibitor library using the protease assay with the FRET-1 peptide. Specifically, after 1 h of incubation of the EV-D68 2Apro with screening compounds, the reaction was initiated by the addition of the fluorescent substrate FRET-1. The progress curves for substrate cleavage were monitored for an additional 2 h. One compound, telaprevir, dramatically reduced the 2Apro activity in a FRET-based assay in a concentration-dependent manner with a 50% inhibitory concentration (IC50) of 0.2 ± 0.1 μM (Fig. 4A). When telaprevir was tested against EV-D68 3Cpro, we did not observe a significant reduction in substrate cleavage (Fig. 4B). In addition, when telaprevir was tested against EV-A71 2Apro, we did not observe noticeable inhibition at 1 μM (Fig. 4C). Boceprevir (Fig. 1), the structural analog of telaprevir which similarly contains an α-ketoamide and which is an FDA-approved HCV NS3-4A protease inhibitor, did not show significant inhibition against EV-D68 2Apro in the enzymatic assay at 1 μM (Fig. 4D), although slight inhibition was observed at 30 μM. Therefore, the inhibition of EV-D68 2Apro by telaprevir appears to be through selective binding interactions. For the EV-D68 2Apro inhibition by telaprevir, the kinetic progress curves displayed a biphasic character in the presence but not in the absence of inhibitor (Fig. 5A), which is a hallmark of a slow/tight binding mechanism (32–34). In this mechanism, the inhibitor first associates with its target in an initial complex that then rearranges into a more stable form. In order to measure the reversibility of the EV-D68 2Apro–telaprevir complex, we performed a drug release dialysis experiment whereby preformed EV-D68 2Apro–telaprevir complex was dialyzed against a 100-fold volume of reaction buffer and monitored for activity every 24 h in order to monitor drug release. No recovery of enzymatic activity was observed even after 4 days in the dialysis buffer, indicating the irreversible binding of telaprevir to the EV-D68 2Apro (Fig. 5B).
FIG 4.

Telaprevir inhibits EV-D68 2Apro in a FRET-based functional assay. (A) IC50 of telaprevir against EV-D68 2Apro. The EV-D68 2Apro protein (1.0 μM) was incubated with DMSO or various concentrations of telaprevir for 1 h at 30°C. The reaction was initiated by addition of 20 μM FRET-1 substrate, and the reaction was monitored for 2 h. The initial velocity was calculated from the first 30 min of the reaction by linear regression. (B) Telaprevir has no effect against EV-D68 3Cpro. EV-D68 3Cpro (1 μM) was preincubated with DMSO, 1 μM or 30 μM telaprevir, or 1 μM rupintrivir for 1 h before the addition of 20 μM FRET-2 substrate. The reaction was monitored for 2 h. (C) Telaprevir has no effect on the enzymatic activity of EV-A71 2Apro protease. The EV-A71 2Apro protein (1 μM) was preincubated with DMSO and 1 μM telaprevir for 1 h before the addition of 20 μM FRET-3 substrate (Dabcyl-KSRTAITTL/GKFGQQSGE-Edans). The reaction was monitored for 2 h. (D) Boceprevir has weak inhibition of the EV-D68 2Apro protein. The EV-D68 2Apro protein (1 μM) was preincubated with DMSO, 1 μM telaprevir, or 1 μM or 30 μM boceprevir for 1 h before the addition of 20 μM the FRET-1 substrate. The reaction was monitored for 2 h.
FIG 5.

Telaprevir binds EV-D68 2Apro in a biphasic irreversible reaction. (A) Proteolytic progress curves in the absence or presence of various concentrations of telaprevir. The EV-D68 2Apro protein (0.2 μM) was added to 80 μM FRET-1 substrate with various concentrations of telaprevir in 200 μl of reaction buffer at 30°C to initiate the proteolytic reaction. The reaction was monitored for over 6 h. The details of the curve fitting are detailed in Materials and Methods. E, enzyme; I, inhibitor. (B) Drug release dialysis of the 2Apro-telaprevir complex. EV-D68 2Apro (1.0 μM) was treated with 2.0 μM telaprevir or DMSO for 1 h at 30°C before dialyzing at 4°C. Aliquots were taken every 24 h to measure activity (the values are the mean ± SE from two replicates). Red asterisks indicate that no protease activity was detected. (C) The best-fit Kobs values in panel A were replotted against the inhibitor concentration with the equation Kobs = k2[I]/(KI + [I]), where the best-fit k2 was equal to 0.0011 ± 0.0003 s−1, KI was equal to 0.225 ± 0.063 μM (n = 3), and the calculated k2/KI was 4,750 M−1 s−1.
Given the results that the formation of the EV-D68 2Apro–telaprevir complex was irreversible, we set the value of k-2 at 0 in the reaction scheme (Fig. 5A; equation 1) during data fitting. The equations used to fit the kinetic progress curves with the irreversible two-step model are shown in the Materials and Methods section. It was found that in the two-step reaction mechanism, telaprevir binds to EV-D68 2Apro rapidly with an equilibrium dissociation constant for the inhibitor (KI) of 225 nM in the first step. After the first complex formation, a slower inactivation step that forms a significantly tighter complex between telaprevir and EV-D68 2Apro occurs, with the second reaction rate constant (k2) being 0.00107 ± 0.00031 s−1 and the overall k2/KI value being 4,750 M−1 s−1 (Table 1 and Fig. 5C).
TABLE 1.
Best-fit values of the curve fittings
| Protein | Kma (μM) | Vmaxa (RFU/s/μM) | IC50b (μM) | k2/KI (M−1 s−1) | k2c (s−1) | KIc (μM) |
|---|---|---|---|---|---|---|
| 2Apro | 49.8 ± 3.8 | 3.70 ± 0.12 | 0.24 ± 0.07 | 4,750 | 0.0011 ± 0.0003 | 0.225 ± 0.063 |
| 2Apro-N84T | 48.6 ± 3.3 | 0.99 ± 0.10 | 3.10 ± 0.85 | 17 ± 2d |
Km and Vmax were calculated as described in the legend to Fig. 3C, and the data are means ± SEs from 4 experiments.
IC50 is calculated as described in the legend to Fig. 9B, and the data are means ± SEs from 2 replicates.
k2 and KI were calculated as described in the legend to Fig. 5C, and the data are means ± SEs from 3 replicates.
The k2/KI of 2Apro-N84T was calculated as a slope via linear regression, as described in the text; the data are means ± SEs from 2 replicates.
Telaprevir inhibits EV-D68 replication in cell culture.
Given telaprevir’s potent inhibition of the enzymatic activity of EV-D68 2Apro, we set out to confirm its antiviral activity in cell culture. The antiviral activity of telaprevir was first measured using a cytopathic effect (CPE) assay in rhabdomyosarcoma (RD) cells infected with the EV-D68 US/KY/14-18953 strain. The half-maximal dose response (50% effective concentration [EC50]) for telaprevir against US/KY/14-18953 was found to be 0.6 ± 0.1 μM (Fig. 6A). Telaprevir was well tolerated in RD cells, having a 3-day half-maximal cellular cytotoxicity (50% cytotoxic concentration [CC50]) of 46.2 ± 14.3 μM (Fig. 6C), corresponding to a selectivity index of 77. As EV-D68 2Apro is highly conserved among different EV-D68 strains (data not shown), we hypothesized that telaprevir will display a similar antiviral potency against other contemporary EV-D68 strains. Indeed, telaprevir had similar efficacy against five 2014 strains, with EC50 values ranging from 0.4 ± 0.1 to 1.9 ± 0.2 μM (Fig. 6A). Additionally, the EC50 values of telaprevir against EV-D68 in A549, HeLa, and HEK293 cells were between 0.1 ± 0.0 and 1.0 ± 0.1 μM (Fig. 6B), with CC50 values ranging from 36.0 ± 8.7 to 95.1 ± 34.1 μM for these cell lines (Fig. 6C). In plaque assays, an EC50 value of 0.1 ± 0.0 μM was found against EV-D68 US/KY/14-18953, consistent with the CPE assay results (Fig. 6D). To test whether the antiviral efficacy of telaprevir is viral titer dependent, we performed a viral titer reduction assay. When tested at 10 μM, telaprevir inhibited viral replication when the multiplicity of infection (MOI) was as high as 0.2 (Fig. 6E). Finally, multicycle replication assays revealed at least a log unit reduction in viral titers at 48 and 72 h postinfection (hpi) after telaprevir treatment in all cell lines tested (Fig. 6F). Altogether, telaprevir inhibited a broad range of neurotropic EV-D68 strains, and the antiviral efficacy was independent of the cell type used, having notable potency in cell lines derived from human lung (A549 cells), muscle (RD cells), and neuronal (A172 cells) tissues.
FIG 6.

Telaprevir shows antiviral activity against multiple EV-D68 strains in different human cell lines. (A) The EC50 values of different strains of EV-D68 in RD cells were determined using a cytopathic effect (CPE) assay. (B) The EC50 of telaprevir against the US/KY/14-18953 strain in different cell lines was determined using a CPE assay. An MOI of 0.01 for RD cells and 0.3 for A172, A549, HeLa, and HEK293 cells was used for a 3-day infection (the values are the mean ± SE from three replicates). (C) The CC50 of telaprevir was determined by a CPE assay after 3 days of drug incubation (the values are the mean ± SE from three replicates). (D) A plaque reduction assay was used to obtain an orthogonal EC50 value using the US/KY/14-18953 strain in RD cells (the values are the mean ± SE from two replicates). (E) The titer reduction at different MOIs was determined, using the US/KY/14-18953 strain in RD cells, by a plaque reduction assay (the values are the mean ± SE from two replicates; red asterisks, no plaque formation). (F) Multicycle growth curves in different cell lines were determined with or without the addition of 3 μM telaprevir. Viral titers were quantified at different time points using a plaque assay (the values are the mean ± SE from two replicates). The same MOI titers were used for both multicycle growth curves and EC50 values.
Telaprevir blocks EV-D68 replication during the early stage(s) of the viral life cycle.
In order to gain insight into the cellular mechanism(s) that gives rise to the antiviral activity of telaprevir against EV-D68, we performed a time-of-addition experiment in RD cells using the US/KY/14-18953 strain. EVs complete a single replication cycle in approximately 9 to 14 h (35). It was found that maximum inhibition for postentry treatment occurred at 0, 2, and 4 hpi (Fig. 7), and after 4 h, a reduction in telaprevir efficacy was observed, suggesting that the antiviral mechanism occurs at an early or intermediate stage in the viral life cycle.
FIG 7.
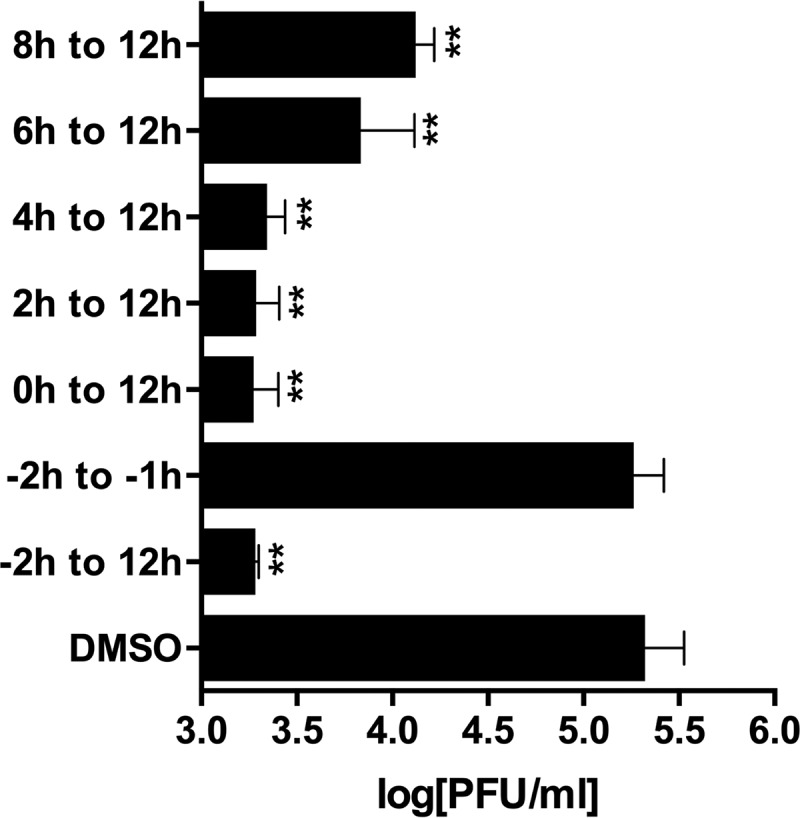
Telaprevir inhibits viral replication during the early to intermediate stage of viral replication. The US/KY/14-18953 strain was used to infect RD cells at an MOI of 1.0. Telaprevir (10 μM) was introduced before infection (−2 h) or after infection (0, 2, 4, 6, or 8 h). After 12 hpi, viruses were harvested, and the titer was determined in plaque assays (the values are the mean ± SE from two replicates). One-way ANOVA and Dunnett's multiple-comparison test were performed to identify statistically significant differences between the DMSO- and antiviral-treated groups (**, P < 0.01).
Next, the viral protein expression levels of the capsid VP1 were monitored by immunofluorescence imaging and Western blotting, and viral RNA levels were monitored by real-time quantitative PCR (RT-qPCR). Telaprevir significantly reduced the fluorescent signal of VP1 at 9 and 12 hpi (Fig. 8A, first to fourth rows), and a similar reduction was confirmed in the Western blot experiment (Fig. 8B and C). Telaprevir led to a dose-dependent reduction in protein VP1 levels at 9 hpi (Fig. 8C). Similarly, RT-qPCR quantification of viral RNAs showed that VP1 mRNA levels were significantly reduced upon telaprevir treatment (Fig. 8D). These results collectively confirmed the cellular antiviral activity of telaprevir and suggest that the mechanism of action may be through inhibition of the 2Apro enzyme.
FIG 8.

Telaprevir blocks EV-D68 translation and transcription. (A) An immunofluorescence experiment was performed using the US/KY/14-18953 strain at an MOI of 1.0 to infect RD cells. At 6, 9, and 12 hpi, the cells were fixed and stained with anti-VP1. The drug concentrations used for rupintrivir, boceprevir, and telaprevir were 0.1 μM, 10 μM, and 10 μM, respectively. (B and C) Western blot analysis of US/KY/14-18953 at an MOI of 1.0 at 6, 9, and 12 h (B) and with different telaprevir concentrations (C) was performed using anti-VP1 antibody. (D) Viral mRNA levels were quantified at 6, 9, and 12 hpi with US/KY/14-18953 at an MOI of 1.0 using RT-qPCR. The results of one representative experiment from two replicates are shown for both the Western blot and immunofluorescence assays. Asterisks indicate a statistically significant difference in comparison with the DMSO control (Student's t test, *, P < 0.05; ***, P < 0.001). The values are the means from three independent experiments ± standard deviation.
Selection of EV-D68 resistant to telaprevir.
We set out to independently confirm the drug target of telaprevir through the selection of drug-resistant viruses using a serial viral passage experiment (12, 36–38). As shown in Table 2, the efficacy of telaprevir was reduced at passage 03 (P3), and the EC50 value was found to be >30 μM at passage 05 (P5). In order to determine the mutations that give rise to a loss of drug efficacy, the 2Apro gene segment was sequenced, and only one mutation, N84T, was identified. Sequence analysis using all the EV-D68 strains in the Virus Pathogen Resource (ViPR) database showed no EV-D68 containing the T84 residue (data not shown).
TABLE 2.
Serial viral passage experiments with telaprevir against EV-D68
| Passage no. | Selection pressure (μM)a | EC50b (μM) | 2Apro mutation |
|---|---|---|---|
| 00 | 0.6 ± 0.1 | No mutation | |
| 01 | 0.5 | 1.1 ± 0.4 | NDc |
| 02 | 1 | ND | ND |
| 03 | 2 | 2.9 ± 0.2 | No mutation |
| 04 | 4 | ND | ND |
| 05 | 8 | >30 (resistant) | N84T |
US/KY/14-18953 (MOI, 0.1) was treated with telaprevirat the indicated concentrations.
EC50s were quantified using the CPE assay and are means ± SEs.
ND, not determined.
In order to determine whether N84T accounts for the reduction of drug sensitivity to 2Apro, we expressed the EV-D68 2Apro-N84T mutant protein and tested its enzymatic activity using the FRET-based assay. Vmax and Km values for the mutant enzyme were found to be 1.0 ± 0.1 RFU/s/μM and 48.6 ± 3.3 μM (Table 1), respectively. Fitting of the kinetic data with the two-step inhibition model revealed that k2/KI was only 17 M−1 s−1 (Table 1), whereas it was 4,750 M−1 s−1 for the wild-type (WT) EV-D68 2Apro. We could not get accurate individual KI and k2 values from curve fitting because we did not observe a saturation of the Kobs-versus-inhibitor concentration ([I]) plot in the telaprevir concentration range tested (data not shown). However, this indicates that the KI of telaprevir in inhibiting the EV-D68 2Apro-N84T protein is over 30 μM. The mutant protein had reduced enzymatic activity compared with that of WT EV-D68 2Apro (Vmax, 1.0 ± 0.1 RFU/s/μM for N84T versus 3.7 ± 0.1 RFU/s/μM for the WT). Similarly, the efficacy (k2/KI) of telaprevir in inhibiting the 2Apro-N84T mutant was reduced nearly 300-fold compared to that for the WT 2Apro (k2/KI, 4,750 M−1 s−1 for the WT versus 17 M−1 s−1 for the N84T mutant) (Fig. 9A; Table 1). Moreover, when we measured the telaprevir IC50 value against the EV-D68 2Apro-N84T protein as we did for WT 2Apro, the IC50 value increased from 0.2 ± 0.1 μM to 3.1 ± 0.9 μM (Fig. 9B; Table 1). Overall, it appears that the mutation of asparagine to threonine at residue 84 significantly impairs the enzymatic activity of 2Apro as well as the sensitivity of telaprevir to 2Apro.
FIG 9.

The 2Apro-N84T mutation reduces the inhibitory activity of telaprevir in functional and antiviral assays. (A) Proteolytic progress curves in the absence or presence of various concentrations of telaprevir for EV-D68 2Apro-N84T are shown. The EV-D68 2Apro-N84T protein (0.8 μM) was added to 80 μM FRET-1 substrate, and the mixture was monitored for over 6 h. (B) Normalized IC50 titrations for telaprevir against WT EV-D68 2Apro and 2Apro-N84T proteins in the enzymatic assay. (C) Telaprevir sensitivity of WT EV-D68 (EV-D68 P0), viral passage-selected resistant virus (EV-D68 P5), recombinant WT EV-D68 (rEV-D68), and recombinant mutant EV-D68 (rEV-D68 2Apro-N84T). The results shown were derived from a CPE assay (the values are the mean ± SE from three replicates).
To independently verify that a single N84T mutation in the EV-D68 2Apro sequence caused the observed drug resistance in cell culture, we constructed a recombinant EV-D68 (rEV-D68) generated from a pHH21 vector named rEV-D68 2Apro-N84T that contained a single 2Apro-N84T mutation in the viral genome (see Materials and Methods) (39). As shown in Fig. 9C, the antiviral efficacies of telaprevir against WT EV-D68 (EV-D68 P0, black) and recombinant virus rEV-D68 (blue) were very similar, with EC50 values of 0.6 ± 0.1 and 0.4 ± 0.0 μM, respectively. In contrast, both the passage 05 viruses (EV-D68 P5, red) and the recombinant mutant virus rEV-D68 2Apro-N84T (green) showed resistance to telaprevir, with EC50 values being greater than 20 μM (Fig. 9C). Interestingly, we observed that the recombinant virus containing the N84T mutation grew much slower than the WT virus (data not shown), suggesting that it might have compromised the fitness of replication.
Telaprevir binding affects the thermal stability of WT EV-D68 2Apro.
The binding of telaprevir to EV-D68 2Apro was confirmed in a differential scanning fluorimetry (DSF) assay (40, 41). DSF is typically applied to assess protein stability and drug binding. As the protein unfolds at increasing temperature, more hydrophobic residues are exposed, which gives an increasing fluorescence signal. If a small molecule is able to bind to a protein target and stabilize it, the melting temperature (Tm) should increase. Telaprevir was able to significantly stabilize WT EV-D68 2Apro at 30 μM and increase the Tm by 13.78°C (Fig. 10A). For the EV-D68 2Apro-N84T mutant, telaprevir had a minimal effect on protein stability and the Tm increased by only 4.26°C, when tested at 100 μM (Fig. 10B).
FIG 10.

Binding of telaprevir to EV-D68 2Apro and the N84T mutant using differential scanning fluorimetry (DSF). EV-D68 2Apro (A) or EV-D68 2Apro-N84T (B) (5 μM each) was incubated with DMSO (black), boceprevir (blue) or telaprevir (red), and the results were plotted to determine the Tm (right).
The α-ketoamide of telaprevir is required for antiviral activity and in inhibiting 2Apro in vitro.
In the scenario of HCV NS3-4A inhibition by telaprevir, telaprevir fits into an elongated, relatively hydrophobic active-site cavity, positioning the α-ketoamide in the vicinity of the catalytic serine (42), allowing for nucleophilic attack. In our case, given the observation that telaprevir inhibition of EV-D68 2Apro was nearly irreversible (Fig. 5B), we reasoned that the ketone electrophile in telaprevir might be essential for its enzymatic inhibition and antiviral activity, possibly by forming a covalent bond with the catalytic cysteine (C107) in the active site of EV-D68 2Apro. To test this hypothesis, we synthesized a telaprevir analog with reduced α-ketoamide (Fig. 11A). It was found that reduction of the α-ketoamide to the hydroxy-telaprevir (telaprevir-OH) significantly attenuated its inhibition of the enzymatic activity of 2Apro (Fig. 11B) as well as its antiviral activity against EV-D68 in cell culture (Fig. 11C). The lack of antiviral activity of telaprevir-OH was not due to its cellular cytotoxicity, as telaprevir-OH was not cytotoxic to RD cells up to 10 μM and the determined CC50 was 52.6 ± 2.5 μM (Fig. 11D). Overall, the results from the dialysis assay (Fig. 5B) and the telaprevir-OH inhibition assay collectively suggest that telaprevir might form a covalent complex with the EV-D68 2Apro by reacting with the catalytic cysteine at the active site.
FIG 11.

The α-ketoamide of telaprevir is required for its inhibition of EV-D68 2Apro enzymatic activity as well as antiviral activity against the EV-D68 US/KY/14-18953 strain. (A) Synthesis of hydroxy-telaprevir (telaprevir-OH). (B) Proteolytic reaction progress curves with telaprevir and telaprevir-OH. In this assay, 1.0 μM EV-D68 2Apro protein was preincubated with either DMSO, 1 μM telaprevir, or 1 μM or 10 μM telaprevir-OH for 1 h at 30°C before the addition of 20 μM FRET-1 substrate in a 100-μl reaction buffer at 30°C to initiate the reaction. Proteolytic cleavage was monitored for 2 h. (C) Antiviral activity of telaprevir-OH in inhibiting the EV-D68 US/KY/14-18953 strain in RD cells using a CPE assay compared with that of telaprevir. (D) Cytotoxicity of telaprevir-OH against RD cells.
Structural model of telaprevir in complex with EV-D68 2Apro.
To provide a structural rationale of the inhibition mechanism of telaprevir as well as the importance of N84 in telaprevir binding, we set to build a structural model of EV-D68 2Apro. As EV-A71 2Apro and EV-D68 2Apro share 69.3% sequence similarities (Fig. 2), we built a homology model of EV-D68 2Apro in SWISS-MODEL using the X-ray crystal structure of EV-A71 2Apro (PDB accession number 4FVB) (23) as a template. The model showed that EV-D68 2Apro adopts a fold similar to that of EV-A71 2Apro (Fig. 12A and B): the N-terminal domain consists of a four-stranded antiparallel β-sheet with a short α-helix, and the C-terminal domain contains a six-stranded antiparallel β-barrel with a zinc finger motif. The catalytic triad consists of His18, Asp36, and Cys107, and these form a cavity between the C-terminal β-barrels and the bII2-cII loop (Fig. 12A). The N84 residue, which confers telaprevir resistance when mutated to threonine, is located at the bII2-cII loop with the side chain facing the substrate/drug binding pocket. In the docking model of EV-D68 2Apro in complex with telaprevir (Fig. 12C), there are several notable features that could explain the results from the functional assays. (i) The α-ketoamide from telaprevir is in close proximity to the catalytic C107 residue; therefore, it is possible for telaprevir to form a covalent complex with EV-D68 2Apro. (ii) The N84 side chain amide carbonyl from EV-D68 2Apro forms a hydrogen bond with the telaprevir norvaline amide NH, and the N84T mutation likely abolishes this hydrogen bond, leading to reduced efficacy. (iii) Telaprevir interacts extensively with EV-D68 2Apro: the carbonyl from telaprevir’s cyclohexylglycine residue forms an additional hydrogen bond with the E82 main chain amide NH; the cyclopropyl side chain from telaprevir forms hydrophobic interactions with residues A104 and Y86 at the S1 pocket; and the tert-butyl side chain from telaprevir forms hydrophobic interactions with residues I128, I81, and A123 in the S5 pocket. Overall, the modeling indicates that telaprevir binds to the active site of EV-D68 2Apro and the docking pose of telaprevir is consistent with the enzymatic assay results.
FIG 12.

Homology model and docking model of EV-D68 2Apro. (A) Homology model of EV-D68 2Apro generated in SWISS-MODEL using EV-A71 2Apro (PDB accession number 4FVB) as a template. (B) Alignment of the EV-D68 2Apro model with the X-ray crystal structure of EV-A71 2Apro (PDB accession number 4FVB). (C) Docking pose of telaprevir in the EV-D68 2Apro model. Docking was performed using Schrödinger Glide standard precision.
Conclusion.
In conclusion, herein we demonstrate the enzymatic activity of EV-D68 2Apro and validate that EV-D68 2Apro is a drug target. We observed that EV-D68 2Apro recognizes and cleaves a peptide substrate with the sequence specific for the VP1-2A junction of the EV-D68 polyprotein. Telaprevir was identified to be a potent inhibitor of EV-D68 2Apro activity. Telaprevir was found to have a k2/KI of 4,750 M−1 s−1 against EV-D68 2Apro, demonstrating remarkable potency for inhibition with a nearly irreversible off rate. The potency of telaprevir against EV-D68 2Apro was confirmed in multiple cell lines using several strains isolated from the 2014 EV-D68 outbreak, in which we observed a submicromolar-to-low-micromolar half-maximal effective concentration (EC50). As telaprevir has been approved by the FDA for the treatment of HCV infections and has a well-documented safety and pharmacokinetic profile for long-term treatment courses, it is promising to repurpose telaprevir directly for the treatment of EV-D68 infection.
The mechanism of action of telaprevir was independently unveiled by the serial passage experiment. Selection for drug resistance identified the N84T mutation near the active site of the EV-D68 2Apro, which attenuated the activity of telaprevir against EV-D68 2Apro in the enzymatic assay. In the docking model, the α-ketoamide from telaprevir is in close proximity to the catalytic C107 residue, suggesting that it is possible for covalent complex formation. The N84 side chain carbonyl forms a hydrogen bond with the norvaline main chain NH from telaprevir, and the N84T mutation likely abolishes this interaction. High-resolution X-ray crystal structures of EV-D68 2Apro and its complex with telaprevir are ultimately needed to corroborate our findings from this study. It is worth noting that although the N84T mutation confers resistance to telaprevir, the N84T mutation has not been observed among circulating EV-D68 strains. This might due to either the lack of selection pressure or the compromised fitness of replication of the N84T mutant virus. Interestingly, we observed that the recombinant virus containing the N84T mutation grows slower than the WT virus (data not shown), suggesting that replication fitness might be compromised. Together, these observations may alleviate the concern of potential drug resistance.
Overall, we have shown that EV-D68 2Apro is a viable antiviral drug target and that it is feasible to develop 2Apro inhibitors, such as telaprevir, as potent anti-EV-D68 antivirals. The next step is to test the in vivo antiviral efficacy of telaprevir using EV-D68-infected mouse models.
MATERIALS AND METHODS
Cell lines and viruses.
Human rhabdomyosarcoma (RD; ATCC CCL-136), HeLa (ATCC CCL-2), A549 (ATCC CCL-185), HEK293 (ATCC CRL-1573), and human brain glioblastoma A172 (ATCC CRL-1620) cells were maintained at 37°C in a 5% CO2 atmosphere and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin antibiotics. EV-D68 strains US/KY/14-18953 (ATCC NE-49132), US/MO/14-18947 (ATCC NR-49129), US/MO/14-18949 (ATCC NR-49130), US/IL/14-18952 (ATCC NR-49131), and US/IL/14-18956 (ATCC NR-49133) were purchased from ATCC and amplified in RD cells prior to infection assays.
Antiviral assays.
Cells for antiviral assays were seeded and grown overnight at 37°C in a 5% CO2 atmosphere to ∼90% confluence on the next day. For all infections, cells were washed with phosphate-buffered saline containing magnesium and calcium and infected with virus diluted in DMEM with 2% FBS and 30 mM MgCl2. Viruses were incubated for at least 1 h at 33°C in a 5% CO2 atmosphere, followed by addition of compound as well as 1% penicillin-streptomycin. For CC50 measurements, the experiment was performed similarly but excluded viral infection. For cytopathic effect (CPE) assays, cells were stained with 66 μg/ml neutral red for 2 h, and neutral red uptake was measured at an absorbance at 540 nm using a Multiskan FC microplate photometer (Thermo Fisher Scientific). The EC50 and CC50 values were calculated from best-fit dose-response curves using GraphPad Prism software. For plaque reduction assays, a 1.2% Avicel microcrystalline cellulose (FMC BioPolymer, Philadelphia, PA) overlay was used, and the cells were stained after 3 days as previously described (36).
Serial passage experiments and sequencing were performed using an approach similar to that described previously (36). Briefly, serial passages were initiated by allowing wild-type virus to replicate in the presence of telaprevir at the EC50. The virus was harvested after approximately 3 days, and the viral titers were determined by a plaque assay. The harvested virus was used to infect healthy cells in the presence of 2 times the concentration of telaprevir from the previous passage. 2Apro sequences were determined by first purifying viral RNA using a QIAamp viral RNA minikit (Qiagen, Hilden, Germany). Viral RNA was reverse transcribed using a SuperScript III first-strand reverse transcriptase (Invitrogen) with an oligo(dT) primer and amplified by PCR. Sequencing was done by Eton Bioscience, Inc.
For time-of-addition experiments, RD cells were infected with US/KY/14-18953 at an MOI of 1.0. After 12 h, the viruses in the infectious medium were harvested and the titers were determined using a plaque assay. One-way analysis of variance (ANOVA) and Dunnett’s multiple-comparison test were performed to identify statistically significant differences between the dimethyl sulfoxide (DMSO)- and antiviral-treated groups.
Protein expression and purification.
The EV-D68 2Apro gene from strain US/KY/14-18953 was ordered from GenScript (Piscataway, NJ) in the pET28b(+) vector with E. coli codon optimization. The 2Apro gene was subcloned into the pE-SUMOstar vector according to the manufacturer’s protocol. A plasmid encoding the 2Apro-N84T mutation was generated in the pE-SUMOstar vector using a QuikChange site-directed mutagenesis kit (Agilent, Santa Clara, CA). Plasmids encoding WT-2Apro and 2Apro-N84T were transformed into competent E. coli BL21(DE3) cells, and a single colony was picked and used to inoculate 10 ml of LB supplemented with 50 μg/ml kanamycin at 37°C and 250 rpm. The 10-ml inoculum was added to 1 liter of LB with 50 μg/ml kanamycin and grown to an optical density at 600 nm of 0.8 to 1.0, cooled to 18°C, and then induced using 0.5 mM IPTG. Induced cultures were incubated at 18°C for an additional 24 h and then harvested, resuspended in lysis buffer (25 mM Tris [pH 7.5], 750 mM NaCl, 2 mM dithiothreitol [DTT] with 0.5 mg/ml lysozyme, 0.5 mM phenylmethylsulfonyl fluoride [PMSF], 0.02 mg/ml DNase I), and lysed with alternating sonication and French press cycles. The cell debris were removed by centrifugation at 12,000 × g for 45 min (20% amplitude, 1 s on/1 s off). The supernatant was incubated with Ni-NTA resin for over 2 h at 4°C on a rotator. The Ni-NTA resin was thoroughly washed with 50 mM imidazole in wash buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 2 mM DTT); for the SUMO-tagged protein, the protein was directly eluted with 300 mM imidazole and for the nontagged 2Apro, the Ni-NTA resin was treated with SUMO protease 1 to remove the SUMO tag, and then 2Apro was washed off from the column with 10 mM imidazole in wash buffer. The imidazole was removed via dialysis or on a 10,000-molecular-weight-cutoff centrifugal concentrator spin column. The purity of the protein was confirmed with SDS-PAGE. The protein concentration was determined by the classic bicinchoninic acid assay using bovine serum albumin as a standard. EV-D68 3Cpro was expressed in the pET28b(+) vector as previously described (20).
Peptide synthesis.
The FRET-1 (Dabcyl- KIRIVNT/GPGFGGE-Edans), FRET-2 (Dabcyl- KEALFQ/GPPQFE-Edans), and FRET-3 (Dabcyl- KSRTAITTL/GKFGQQSGE-Edans) peptides were synthesized using the 9-fluorenylmethoxy carbonyl (Fmoc) solid-phase synthesis strategy (43). Briefly, Chemmatrix rink amide resin (loading concentration, 0.4 mmol/g) was used for the synthesis. For the coupling step, 5 eq of 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU), 5 eq Fmoc-AA-OH (where AA is amino acid), and 10 eq of N,N-diisopropylethylamine (DIEA) were used, and the reaction proceeded for 5 min under heating at 80°C with N2 bubbling. For the deprotection step, 5% piperazine plus 0.1 M 1-hydroxybenzotriazole (HOBt) was used and the reaction proceeded for 5 min under heating at 80°C with N2 bubbling. The final step of Dabcyl coupling used the same conditions as the coupling step. Peptide was cleaved from the resin using 95% trifluoracetic acid, 2.5% triisopropylsilane (TIS), and 2.5% H2O, and the reaction proceeded for 2 h at ambient temperature. The final peptide was precipitated from cold ether, dried, and purified by C18 reverse-phase preparative high-performance liquid chromatography (HPLC). The purity and identity of the two peptides were determined by HPLC and matrix-assisted laser desorption ionization mass spectrometry, respectively. The calculated molecular weight for FRET-1 was 1,944.26 (+1), and the detected molecular weight was 1,944.30 (+1). The calculated molecular weight for FRET-2 was 1,890.17 (+1), and the detected molecular weight was 1,890.88 (+1). The calculated molecular weight for FRET-3 was 1,765.04 (+1), and the detected molecular weight was 1,765.99 (+1).
Enzymatic assays.
For activity measurements for Km/Vmax as well as telaprevir IC50 measurements, a reaction volume of 100 μl contained 1.0 μM SUMO-2Apro, 2Apro, or the 2Apro-N84T mutant. The reaction buffer contained 50 mM Tris (pH 7.0), 150 mM NaCl, 10% glycerol, and 2 mM DTT, and the reaction was carried out at 30°C in a Cytation 5 imaging reader (Thermo Fisher Scientific) with filters for excitation at 360/40 nm and emission at 460/40 nm. Reactions were monitored every 90 s. For Km/Vmax measurements, a FRET-1 substrate concentration ranging from 0 to 200 μM was applied. The initial velocity of the proteolytic activity was calculated by linear regression for the first 30 min of the kinetic progress curves. The initial velocity was plotted against the FRET-1 concentration with the classic Michaelis-Menten equation in Prism (v5.0) software. For telaprevir IC50 measurements, 1.0 μM 2Apro or 2Apro-N84T was incubated with various concentrations of telaprevir at 30°C for 1 h in reaction buffer, and then the reaction was initiated by adding 20 μM FRET-1 substrate, the reaction was monitored for 2 h, and the initial velocity was calculated for the first 30 min by linear regression. The IC50 was calculated by plotting the initial velocity against various concentrations of telaprevir by use of a dose-response curve in Prism (v5.0) software. Dialysis assays were performed by using 10 ml 1.0 μM EV-D68 2Apro, which was incubated with 4.0 μl DMSO or 4.0 μl 5 mM telaprevir (final concentration, 2.0 μM) in reaction buffer at 30°C for 1 h. The mix was loaded into a 10,000-molecular-weight-cutoff dialysis cassette (G2; Thermo Fisher Scientific) and dialyzed in 2 liters of reaction buffer separately at 4°C. Every 24 h, 100-μl samples were taken to measure the enzymatic activity with 20 μM FRET-1 substrate. Progress curve kinetics measurements with telaprevir, used for curve fitting, were carried out as follows: 0.2 μM EV-D68 2Apro protein or 0.8 μM 2Apro-N84T protein was added to 80 μM FRET-1 substrate with various concentrations of telaprevir in 200 μl of reaction buffer at 30°C to initiate the proteolytic reaction. The reaction was monitored for over 6 h. The progress curves were fit to a standard exponential equation (equation 1) as described previously (44):
| (1) |
where P(t) is the fluorescence signal at time t, P0 is the background signal at time zero, A is the possible maximal signal increase, and Kobs is the observed rate constant for inhibition.
The best-fit Kobs values were replotted against the inhibitor concentration with equation 2:
| (2) |
where KI is the equilibrium dissociation constant of the initial binding step, and k2 is the rate of the 2nd tightly binding inactivation step.
For EV D68 2Apro-N84T, saturation for the Kobs-versus-[I] plot was not observed. k2/KI was derived from the slope of the plot instead.
Western blotting.
Total proteins were extracted from cells infected with EV-D68 strain US/KY/14-18953 using RAPI lysis buffer (50 mM Tris [pH 8.0], 1% NP-40, 0.1% SDS, 150 mM NaCl, 0.5% sodium deoxycholate, 5 mM EDTA, 10 mM NaF, 10 mM NaPPi, 2 mM phenylmethylsulfonyl, 1 mM PMSF). Equal amounts of extracted total proteins were separated by electrophoresis and transferred to a polyvinylidene difluoride (PVDF) membrane. Viral protein VP1 or host GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was detected using rabbit anti-VP1 (1:3,000 dilution; catalog number GTX132313; GeneTex) or mouse anti-GAPDH antibody (1:3,000 dilution; antibody MAB374; EMD Millipore), respectively. Horseradish peroxidase (HRP)-conjugated secondary antibody (1:3,000 dilutions; catalog number 32430 or 656120; Thermo Fisher Scientific) and Supersignal West Femto substrate (Thermo Fisher Scientific) were used to produce chemiluminescence for detection.
RNA extraction and RT-qPCR.
Total RNA was extracted from EV-D68 strain US/KY/14-18953-infected cells using TRIzol reagents (Thermo Fisher Scientific). After removing genomic DNA by RQ1 RNase-free DNase (Promega), 1.2 μg of total RNA was used to synthesize the first strand of cDNA of viral RNA and host mRNA using SuperScript III reverse transcriptase (Thermo Fisher Scientific) and oligo(dT)18. Viral RNA was amplified on a StepOne Plus real-time PCR system (Thermo Fisher Scientific) using FastStart Universal SYBR Green Master mix (carboxy-X-rhodamine; Roche) and virus-specific primers, and GAPDH was amplified to serve as a control using GAPDH-specific primers. The amplification conditions were 95°C for 10 min and 40 cycles of 15 s at 95°C and 60 s at 60°C. Melting curve analysis was performed to verify the specificity of each amplification.
Immunostaining.
EV-D68 strain US/KY/14-18953-infected cells were fixed with 4% formaldehyde for 10 min, followed by permeabilization with 0.2% Triton X-100 for another 10 min. After blocking with 10% bovine serum, cells were stained with rabbit anti-VP1 antibody, followed by staining with anti-rabbit immunoglobulin secondary antibody conjugated to Alexa Fluor 488 (Thermo Fisher Scientific). The nuclei were stained with 300 nM DAPI (4′,6-diamidino-2-phenylindole; Thermo Fisher Scientific) after secondary antibody incubation. Fluorescent images were acquired using a Leica SP5-II spectral confocal microscope (Leica).
DSF.
The study of telaprevir binding to EV-D68-2Apro was performed by differential scanning fluorimetry (DSF) using 3.75 μM EV-D68-2Apro as previously described (40). Raw DSF data were exported and processed using StepOne (v2.3) software, and curve fitting and melting temperature (Tm) calculation were performed using the Boltzmann sigmoidal equation in Prism (v5) software.
Synthesis of telaprevir-OH.
Telaprevir (1 mmol) was dissolved in CH3OH, and the solution was cooled to 0°C. NaBH4 (4 mmol) was added portion wise, and the mixture was stirred at ambient temperature overnight. The reaction was quenched with 1 M HCl, and extraction with dichloromethane was performed (3 times). The final product was purified by flash column chromatography with a gradient of 2% to 10% CH3OH in CH2Cl2. The purity and identity of telaprevir-OH were confirmed by LC-MS. The calculated molecular weight was 682.85 (+1), and the detected molecular weight was 682.59 (+1).
Recombinant EV-D68.
A plasmid-based reverse genetic system for EV-D68 from US/MO/14-18947 was generated in a pHH21 vector, based on the study by Pan et al. (39).
Mass spectrometry.
LC-MS/MS sequencing of EV-D68 2Apro was performed at the Taplin Biological Mass Spectrometry Facility (Harvard Medical School, Boston, MA) according to a previously published procedure (45). Mass spectrometry was performed on an ultra-high-mass-range (UHMR) research option Q-Exactive HF mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) with the following parameters: a 1-kV capillary voltage, a 50-V source offset, a 200°C capillary temperature, and a −50 V desolvation voltage. The collision cell was set to 0 V, with the gas pressure set to 3. The resolution was set at 15,000 with 10 microscans per second. The maximum injection time was set at 200 ms, and the in-source trapping time was 4 ms. The instrument was optimized for low-mass detection. Deconvolution was performed with the UniDec (v2.7.3) program, using high-resolution native presets to determine the monomer mass. Uncertainty is presented as the standard deviation of the peak mass for three replicate measurements (46, 47).
Molecular modeling and docking.
The structural model of EV-D68 2Apro was generated by homology modeling using the SWISS-MODEL server (https://swissmodel.expasy.org/). The EV-A71 2Apro structure (PDB accession number 4FVB) was used as a template. The docking model of telaprevir in complex with the EV-D68 2Apro was generated using Schrödinger Glide standard precision docking. The coordinates of the docking site were set as follows: x, 7.5; y, 11.5; and z, 27.5.
ACKNOWLEDGMENTS
This research is supported by start-up funding from the University of Arizona and NIH grant AI119187 to J.W. M.T.M. and J.M.D. were funded by a Bisgrove Scholar Award from the Science Foundation Arizona and the National Institute of General Medical Sciences, National Institutes of Health, under award number R35 GM128624.
We thank Ivan V. Korendovych at Syracuse University for sharing the pE-SUMOstar plasmid.
REFERENCES
- 1.Holm-Hansen CC, Midgley SE, Fischer TK. 2016. Global emergence of enterovirus D68: a systematic review. Lancet Infect Dis 16:e64–e75. doi: 10.1016/S1473-3099(15)00543-5. [DOI] [PubMed] [Google Scholar]
- 2.Midgley CM, Watson JT, Nix WA, Curns AT, Rogers SL, Brown BA, Conover C, Dominguez SR, Feikin DR, Gray S, Hassan F, Hoferka S, Jackson MA, Johnson D, Leshem E, Miller L, Nichols JB, Nyquist AC, Obringer E, Patel A, Patel M, Rha B, Schneider E, Schuster JE, Selvarangan R, Seward JF, Turabelidze G, Oberste MS, Pallansch MA, Gerber SI, EV-D68 Working Group . 2015. Severe respiratory illness associated with a nationwide outbreak of enterovirus D68 in the USA (2014): a descriptive epidemiological investigation. Lancet Respir Med 3:879–887. doi: 10.1016/S2213-2600(15)00335-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oermann CM, Schuster JE, Conners GP, Newland JG, Selvarangan R, Jackson MA. 2015. Enterovirus d68. A focused review and clinical highlights from the 2014 U.S. outbreak. Ann Am Thorac Soc 12:775–781. doi: 10.1513/AnnalsATS.201412-592FR. [DOI] [PubMed] [Google Scholar]
- 4.Messacar K, Schreiner TL, Maloney JA, Wallace A, Ludke J, Oberste MS, Nix WA, Robinson CC, Glode MP, Abzug MJ, Dominguez SR. 2015. A cluster of acute flaccid paralysis and cranial nerve dysfunction temporally associated with an outbreak of enterovirus D68 in children in Colorado, USA. Lancet 385:1662–1671. doi: 10.1016/S0140-6736(14)62457-0. [DOI] [PubMed] [Google Scholar]
- 5.Messacar K, Asturias EJ, Hixon AM, Van Leer-Buter C, Niesters HGM, Tyler KL, Abzug MJ, Dominguez SR. 2018. Enterovirus D68 and acute flaccid myelitis—evaluating the evidence for causality. Lancet Infect Dis 18:e239–e247. doi: 10.1016/S1473-3099(18)30094-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greninger AL, Naccache SN, Messacar K, Clayton A, Yu G, Somasekar S, Federman S, Stryke D, Anderson C, Yagi S, Messenger S, Wadford D, Xia D, Watt JP, Van Haren K, Dominguez SR, Glaser C, Aldrovandi G, Chiu CY. 2015. A novel outbreak enterovirus D68 strain associated with acute flaccid myelitis cases in the USA (2012-14): a retrospective cohort study. Lancet Infect Dis 15:671–682. doi: 10.1016/S1473-3099(15)70093-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morrey JD, Wang H, Hurst BL, Zukor K, Siddharthan V, Van Wettere AJ, Sinex DG, Tarbet EB. 2018. Causation of acute flaccid paralysis by myelitis and myositis in enterovirus-D68 infected mice deficient in interferon alphabeta/gamma receptor deficient mice. Viruses 10:E33. doi: 10.3390/v10010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baggen J, Thibaut HJ, Strating J, van Kuppeveld FJM. 2018. The life cycle of non-polio enteroviruses and how to target it. Nat Rev Microbiol 16:368–381. doi: 10.1038/s41579-018-0005-4. [DOI] [PubMed] [Google Scholar]
- 9.van der Linden L, Wolthers KC, van Kuppeveld FJ. 2015. Replication and inhibitors of enteroviruses and parechoviruses. Viruses 7:4529–4562. doi: 10.3390/v7082832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ulferts R, van der Linden L, Thibaut HJ, Lanke KH, Leyssen P, Coutard B, De Palma AM, Canard B, Neyts J, van Kuppeveld FJ. 2013. Selective serotonin reuptake inhibitor fluoxetine inhibits replication of human enteroviruses B and D by targeting viral protein 2C. Antimicrob Agents Chemother 57:1952–1956. doi: 10.1128/AAC.02084-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rhoden E, Zhang M, Nix WA, Oberste MS. 2015. In vitro efficacy of antiviral compounds against enterovirus D68. Antimicrob Agents Chemother 59:7779–7781. doi: 10.1128/AAC.00766-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ulferts R, de Boer SM, van der Linden L, Bauer L, Lyoo HR, Mate MJ, Lichiere J, Canard B, Lelieveld D, Omta W, Egan D, Coutard B, van Kuppeveld FJ. 2016. Screening of a library of FDA-approved drugs identifies several enterovirus replication inhibitors that target viral protein 2C. Antimicrob Agents Chemother 60:2627–2638. doi: 10.1128/AAC.02182-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao Q, Yuan S, Zhang C, Wang Y, Wang Y, He G, Zhang S, Altmeyer R, Zou G. 2015. Discovery of itraconazole with broad-spectrum in vitro antienterovirus activity that targets nonstructural protein 3A. Antimicrob Agents Chemother 59:2654–2665. doi: 10.1128/AAC.05108-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Messacar K, Sillau S, Hopkins SE, Otten C, Wilson-Murphy M, Wong B, Santoro JD, Treister A, Bains HK, Torres A, Zabrocki L, Glanternik JR, Hurst AL, Martin JA, Schreiner T, Makhani N, DeBiasi RL, Kruer MC, Tremoulet AH, Van Haren K, Desai J, Benson LA, Gorman MP, Abzug MJ, Tyler KL, Dominguez SR. 9 November 2018. Safety, tolerability, and efficacy of fluoxetine as an antiviral for acute flaccid myelitis. Neurology doi: 10.1212/WNL.0000000000006670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skern T, Hampölz B, Guarne A, Fita I, Bergmann E, Petersen J, James MNG. 2002. Structure and function of picornavirus proteinases, p 199–212. In Semler BL, Wimmer E (ed), Molecular biology of picornaviruses. ASM Press, Washington, DC. [Google Scholar]
- 16.Mosimann SC, Cherney MM, Sia S, Plotch S, James MN. 1997. Refined X-ray crystallographic structure of the poliovirus 3C gene product. J Mol Biol 273:1032–1047. doi: 10.1006/jmbi.1997.1306. [DOI] [PubMed] [Google Scholar]
- 17.Allaire M, Chernaia MM, Malcolm BA, James MN. 1994. Picornaviral 3C cysteine proteinases have a fold similar to chymotrypsin-like serine proteinases. Nature 369:72–76. doi: 10.1038/369072a0. [DOI] [PubMed] [Google Scholar]
- 18.Matthews DA, Smith WW, Ferre RA, Condon B, Budahazi G, Sisson W, Villafranca JE, Janson CA, McElroy HE, Gribskov CL. 1994. Structure of human rhinovirus 3C protease reveals a trypsin-like polypeptide fold, RNA-binding site, and means for cleaving precursor polyprotein. Cell 77:761–771. [DOI] [PubMed] [Google Scholar]
- 19.Cui S, Wang J, Fan T, Qin B, Guo L, Lei X, Wang J, Wang M, Jin Q. 2011. Crystal structure of human enterovirus 71 3C protease. J Mol Biol 408:449–461. doi: 10.1016/j.jmb.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan J, George S, Kusov Y, Perbandt M, Anemuller S, Mesters JR, Norder H, Coutard B, Lacroix C, Leyssen P, Neyts J, Hilgenfeld R. 2013. 3C protease of enterovirus 68: structure-based design of Michael acceptor inhibitors and their broad-spectrum antiviral effects against picornaviruses. J Virol 87:4339–4351. doi: 10.1128/JVI.01123-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee W, Watters KE, Troupis AT, Reinen NM, Suchy FP, Moyer KL, Frederick RO, Tonelli M, Aceti DJ, Palmenberg AC, Markley JL. 2014. Solution structure of the 2A protease from a common cold agent, human rhinovirus C2, strain W12. PLoS One 9:e97198. doi: 10.1371/journal.pone.0097198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baxter NJ, Roetzer A, Liebig HD, Sedelnikova SE, Hounslow AM, Skern T, Waltho JP. 2006. Structure and dynamics of coxsackievirus B4 2A proteinase, an enzyme involved in the etiology of heart disease. J Virol 80:1451–1462. doi: 10.1128/JVI.80.3.1451-1462.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cai Q, Yameen M, Liu W, Gao Z, Li Y, Peng X, Cai Y, Wu C, Zheng Q, Li J, Lin T. 2013. Conformational plasticity of the 2A proteinase from enterovirus 71. J Virol 87:7348–7356. doi: 10.1128/JVI.03541-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stempniak M, Hostomska Z, Nodes BR, Hostomsky Z. 1997. The NS3 proteinase domain of hepatitis C virus is a zinc-containing enzyme. J Virol 71:2881–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Clercq E, Li G. 2016. Approved antiviral drugs over the past 50 years. Clin Microbiol Rev 29:695–747. doi: 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang CY, Huang AC, Hour MJ, Huang SH, Kung SH, Chen CH, Chen IC, Chang YS, Lien JC, Lin CW. 2015. Antiviral potential of a novel compound CW-33 against enterovirus A71 via inhibition of viral 2A protease. Viruses 7:3155–3171. doi: 10.3390/v7062764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toyoda H, Nicklin MJ, Murray MG, Anderson CW, Dunn JJ, Studier FW, Wimmer E. 1986. A second virus-encoded proteinase involved in proteolytic processing of poliovirus polyprotein. Cell 45:761–770. [DOI] [PubMed] [Google Scholar]
- 28.Maghsoudi N, Khodagholi F, Sadjadi M, Zeinodini M, Sabbaghian M. 2008. Purification and partial characterization of coxsackievirus B3 2A protease expressed in Escherichia coli. Int J Biol Macromol 43:238–244. doi: 10.1016/j.ijbiomac.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 29.Sommergruber W, Zorn M, Blaas D, Fessl F, Volkmann P, Maurer-Fogy I, Pallai P, Merluzzi V, Matteo M, Skern T. 1989. Polypeptide 2A of human rhinovirus type 2: identification as a protease and characterization by mutational analysis. Virology 169:68–77. [DOI] [PubMed] [Google Scholar]
- 30.Brown DM, Hixon AM, Oldfield LM, Zhang Y, Novotny M, Wang W, Das SR, Shabman RS, Tyler KL, Scheuermann RH. 2018. Contemporary circulating enterovirus D68 strains have acquired the capacity for viral entry and replication in human neuronal cells. mBio 9:e01954-18. doi: 10.1128/mBio.01954-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peroutka RJ III, Orcutt SJ, Strickler JE, Butt TR. 2011. SUMO fusion technology for enhanced protein expression and purification in prokaryotes and eukaryotes. Methods Mol Biol 705:15–30. doi: 10.1007/978-1-61737-967-3_2. [DOI] [PubMed] [Google Scholar]
- 32.Flores MV, Strawbridge J, Ciaramella G, Corbau R. 2009. HCV-NS3 inhibitors: determination of their kinetic parameters and mechanism. Biochim Biophys Acta 1794:1441–1448. doi: 10.1016/j.bbapap.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 33.Becker D, Kaczmarska Z, Arkona C, Schulz R, Tauber C, Wolber G, Hilgenfeld R, Coll M, Rademann J. 2016. Irreversible inhibitors of the 3C protease of coxsackie virus through templated assembly of protein-binding fragments. Nat Commun 7:12761. doi: 10.1038/ncomms12761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schulz R, Atef A, Becker D, Gottschalk F, Tauber C, Wagner S, Arkona C, Abdel-Hafez AA, Farag HH, Rademann J, Wolber G. 2018. Phenylthiomethyl ketone-based fragments show selective and irreversible inhibition of enteroviral 3C proteases. J Med Chem 61:1218–1230. doi: 10.1021/acs.jmedchem.7b01440. [DOI] [PubMed] [Google Scholar]
- 35.Chen TC, Chang HY, Lin PF, Chern JH, Hsu JT, Chang CY, Shih SR. 2009. Novel antiviral agent DTriP-22 targets RNA-dependent RNA polymerase of enterovirus 71. Antimicrob Agents Chemother 53:2740–2747. doi: 10.1128/AAC.00101-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Musharrafieh R, Ma C, Wang J. 2018. Profiling the in vitro drug-resistance mechanism of influenza A viruses towards the AM2-S31N proton channel blockers. Antiviral Res 153:10–22. doi: 10.1016/j.antiviral.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma C, Zhang J, Wang J. 2016. Pharmacological characterization of the spectrum of antiviral activity and genetic barrier to drug resistance of M2-S31N channel blockers. Mol Pharmacol 90:188–198. doi: 10.1124/mol.116.105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zuo J, Kye S, Quinn KK, Cooper P, Damoiseaux R, Krogstad P. 2015. Discovery of structurally diverse small-molecule compounds with broad antiviral activity against enteroviruses. Antimicrob Agents Chemother 60:1615–1626. doi: 10.1128/AAC.02646-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pan M, Gao S, Zhou Z, Zhang K, Liu S, Wang Z, Wang T. 2018. A reverse genetics system for enterovirus D68 using human RNA polymerase I. Virus Genes 54:484–492. doi: 10.1007/s11262-018-1570-3. [DOI] [PubMed] [Google Scholar]
- 40.Niesen FH, Berglund H, Vedadi M. 2007. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc 2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 41.Senisterra G, Chau I, Vedadi M. 2012. Thermal denaturation assays in chemical biology. Assay Drug Dev Technol 10:128–136. doi: 10.1089/adt.2011.0390. [DOI] [PubMed] [Google Scholar]
- 42.Romano KP, Ali A, Aydin C, Soumana D, Ozen A, Deveau LM, Silver C, Cao H, Newton A, Petropoulos CJ, Huang W, Schiffer CA. 2012. The molecular basis of drug resistance against hepatitis C virus NS3/4A protease inhibitors. PLoS Pathog 8:e1002832. doi: 10.1371/journal.ppat.1002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cady SD, Wang J, Wu Y, DeGrado WF, Hong M. 2011. Specific binding of adamantane drugs and direction of their polar amines in the pore of the influenza M2 transmembrane domain in lipid bilayers and dodecylphosphocholine micelles determined by NMR spectroscopy. J Am Chem Soc 133:4274–4284. doi: 10.1021/ja102581n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh J, Petter RC, Baillie TA, Whitty A. 2011. The resurgence of covalent drugs. Nat Rev Drug Discov 10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 45.Wang L, Fu B, Li W, Patil G, Liu L, Dorf ME, Li S. 2017. Comparative influenza protein interactomes identify the role of plakophilin 2 in virus restriction. Nat Commun 8:13876. doi: 10.1038/ncomms13876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reid DJ, Diesing JM, Miller MA, Perry SM, Wales JA, Montfort WR, Marty MT. 2018. MetaUniDec: high-throughput deconvolution of native mass spectra. J Am Soc Mass Spectrom 30:118–127. doi: 10.1007/s13361-018-1951-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marty MT, Baldwin AJ, Marklund EG, Hochberg GK, Benesch JL, Robinson CV. 2015. Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal Chem 87:4370–4376. doi: 10.1021/acs.analchem.5b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]