

HCV represents a global health problem as more than 71 million people are chronically infected worldwide. Direct-acting antiviral agents can cure HCV infection in most patients, but due to the high cost of these agents and the emergence of resistant mutants, they do not represent a feasible and affordable strategy to eradicate the virus. Therefore, a vaccine is an urgent goal that requires efforts to understand the correlates of protection for HCV clearance. Here, we describe for the first time the generation of novel vaccines against HCV based on alphavirus DNA replicons expressing HCV antigens. We demonstrate that potent T cell immune responses, as well as humoral immune responses, against HCV can be achieved in mice by using a combined heterologous prime/boost immunization protocol consisting of the administration of alphavirus replicon DNA vectors as the priming immunization followed by a boost with a recombinant modified vaccinia virus Ankara vector expressing HCV antigens.
KEYWORDS: HCV, MVA, alphavirus replicon, immune response, mice, poxvirus, vaccine
ABSTRACT
Hepatitis C is a liver disease caused by the hepatitis C virus (HCV) affecting 71 million people worldwide with no licensed vaccines that prevent infection. Here, we have generated four novel alphavirus-based DNA-launched self-amplifying RNA replicon (DREP) vaccines expressing either structural core-E1-E2 or nonstructural p7-NS2-NS3 HCV proteins of genotype 1a placed under the control of an alphavirus promoter, with or without an alphaviral translational enhancer (grouped as DREP-HCV or DREP-e-HCV, respectively). DREP vectors are known to induce cross-priming and further stimulation of immune responses through apoptosis, and here we demonstrate that they efficiently trigger apoptosis-related proteins in transfected cells. Immunization of mice with the DREP vaccines as the priming immunization followed by a heterologous boost with a recombinant modified vaccinia virus Ankara (MVA) vector expressing the nearly full-length genome of HCV (MVA-HCV) induced potent and long-lasting HCV-specific CD4+ and CD8+ T cell immune responses that were significantly stronger than those of a homologous MVA-HCV prime/boost immunization, with the DREP-e-HCV/MVA-HCV combination the most immunogenic regimen. HCV-specific CD4+ and CD8+ T cell responses were highly polyfunctional, had an effector memory phenotype, and were mainly directed against E1-E2 and NS2-NS3, respectively. Additionally, DREP/MVA-HCV immunization regimens induced higher antibody levels against HCV E2 protein than homologous MVA-HCV immunization. Collectively, these results provided an immunization protocol against HCV by inducing high levels of HCV-specific T cell responses as well as humoral responses. These findings reinforce the combined use of DREP-based vectors and MVA-HCV as promising prophylactic and therapeutic vaccines against HCV.
IMPORTANCE HCV represents a global health problem as more than 71 million people are chronically infected worldwide. Direct-acting antiviral agents can cure HCV infection in most patients, but due to the high cost of these agents and the emergence of resistant mutants, they do not represent a feasible and affordable strategy to eradicate the virus. Therefore, a vaccine is an urgent goal that requires efforts to understand the correlates of protection for HCV clearance. Here, we describe for the first time the generation of novel vaccines against HCV based on alphavirus DNA replicons expressing HCV antigens. We demonstrate that potent T cell immune responses, as well as humoral immune responses, against HCV can be achieved in mice by using a combined heterologous prime/boost immunization protocol consisting of the administration of alphavirus replicon DNA vectors as the priming immunization followed by a boost with a recombinant modified vaccinia virus Ankara vector expressing HCV antigens.
INTRODUCTION
Hepatitis C virus (HCV) is an enveloped, positive-sense, single-stranded RNA virus that belongs to the family Flaviviridae. The open reading frame of HCV encodes a large polyprotein that is further processed into eight mature proteins, including structural (core, E1, and E2) and nonstructural (p7, NS2, NS3, NS4 and NS5) proteins (1). According to the World Health Organization (WHO), at least 71 million people were estimated to be infected with chronic HCV in 2015, and 400,000 deaths were due to liver cirrhosis and hepatocellular carcinoma, the two main HCV-derived complications caused by chronicity (http://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/). In the past few years, several direct-acting antivirals (DAAs) have been developed and approved for therapy and are able to eliminate HCV in more than 95% of treated patients (see the review in reference 2). However, DAAs do not represent a feasible solution to eradicate HCV for several reasons: (i) there is a large proportion of silent infections that are untreated while being highly contagious; (ii) the high cost of DAAs make them inaccessible to low-income countries; (iii) DAAs fail to protect against reinfection; (iv) DAAs do not prevent the emergence of drug-resistant variants (3). Therefore, only an effective prophylactic or therapeutic vaccine represents an achievable solution to control HCV, and efforts need to be undertaken to understand the immunogenicity of HCV and the correlates of protection in order to develop optimized vaccine candidates that fulfil the requirements of an effective worldwide solution against HCV.
Several vaccine candidates against HCV have been developed using different platforms, such as proteins, adenovirus, DNA, or modified vaccinia virus Ankara (MVA)-based vaccines (see reviews in references 4 to 7). DNA-based vaccines confer a number of advantages such as the simplicity of DNA manipulation, generation, transportation, and storage as well as low cost of production, high stability, and a good safety profile (see reviews in references 6 and 8). However, an important drawback of DNA vaccines is their low immunogenicity when they are administered alone, needing multiple booster doses or other agents to enhance the duration of their immune effect. Furthermore, the strength of the immune response elicited by DNA vaccines may be limited and may need to be boosted by another vector such as an MVA-based vaccine (9–11).
MVA is an attenuated orthopoxvirus that was generated after more than 570 passages in primary chicken embryo fibroblast (CEF) cells (12) and has been tested in several preclinical and clinical trials against numerous infectious diseases, including HCV (13–17). Additionally, the immune response generated by MVA-based vectors can be improved using a DNA prime/MVA boost regimen (18–21). In this regard, we have previously studied the immunogenicity of an MVA-based vaccine candidate against HCV (termed MVA-HCV), which expresses all HCV proteins of genotype 1a, using both heterologous and homologous prime/boost immunization regimens. We showed that the heterologous regimen (DNAs encoding core, E1, E2, and NS3 HCV proteins for priming followed by MVA-HCV as a boost) induced significantly higher adaptive and memory HCV-specific CD4+ and CD8+ T cell immune responses than two doses of MVA-HCV (homologous prime/boost regimen) (21), confirming the beneficial use of DNA/MVA immunization regimens. However, no antibodies were described for DNA-HCV/MVA-HCV (21), suggesting that immunization with a regular mammalian expression DNA plasmid should be replaced with improved DNA vectors aimed at generating higher immune responses (9, 22, 23).
Alphavirus-based DNA-launched replicons (DREPs) are naked DNA vectors that, upon delivery into the cells, express an RNA replicon that is a self-amplifying recombinant RNA molecule (24). They encode an alphavirus replicase that amplifies the RNA replicon, transcribes the mRNA encoding the heterologous antigens, and generates double-stranded RNA molecules that stimulate several pattern recognition receptors (PRR) (25). PRR stimulation leads to the induction of type I interferon (IFN) and triggers a potent apoptosis that serves as a vaccine adjuvant by cross-priming antigen epitopes on major histocompatibility complex (MHC) class I molecules (26, 27). Additionally, it has been reported that DREP vectors can be optimized with the addition of a translational enhancer upstream and in frame with the heterologous antigen that provides increased expression of the heterologous antigen and enhancement of antigen-specific humoral immune responses (28, 29). Thus, due to all of these features, DREP replicons express high levels of transient heterologous genes and induce higher antigen-specific immune responses in vivo than conventional DNA vaccines (9, 22–24, 30) and are thus attractive vectors for vaccine development (31). Moreover, several preclinical studies have shown that the use of the DREP platform as a priming immunization followed by an MVA boost induced potent antigen-specific immune responses against several infectious diseases, such as those caused by Chikungunya virus (10, 32, 33), Ebola virus (11), and HIV (9, 22). However, no DREP vectors have been developed for HCV, and due to the lack of an effective HCV vaccine, the study of the DREP/MVA approach can be a step forward in HCV vaccine development.
In this study, we have generated and characterized four novel DREP vectors encoding HCV antigens (either structural core-E1-E2 or nonstructural p7-NS2-NS3 proteins) placed in frame in the absence or presence of a translational enhancer (grouped in DREP-HCV or DREP-e-HCV, respectively). We have analyzed their immunogenicity in mice in a heterologous prime/boost immunization protocol using MVA-HCV as a boost and an MVA-HCV/MVA-HCV regimen as a control group for well-characterized HCV-specific humoral and cellular immune responses (34). The results showed that the heterologous DREP/MVA immunization elicited significantly higher HCV-specific CD4+ and CD8+ T cell responses than a homologous MVA-HCV prime/boost immunization regimen. The most immunogenic schedule was a DREP-e-HCV priming immunization followed by an MVA-HCV boost. Furthermore, DREP/MVA-HCV immunizations induced higher levels of antibodies against HCV E2 protein than two doses of MVA-HCV. Our findings reveal that the combination of novel DREP-based HCV vaccines with MVA-HCV is an effective way to enhance HCV-specific T cell and humoral immunogenicity, forming a promising vaccine strategy against hepatitis C.
RESULTS
Generation of DREP-based vaccine candidates expressing HCV antigens.
To develop new and more immunogenic vaccines against hepatitis C virus, we have generated four novel DREP vectors expressing HCV antigens. These new DREP vaccine candidates are divided in two groups: (i) DREP-HCV, consisting of a DREP vector encoding HCV core, E1, and E2 structural proteins (termed DREP-C-E1-E2), and another DREP vector encoding HCV p7, NS2, and NS3 nonstructural proteins (termed DREP-p7-NS2-NS3); (ii) DREP-e-HCV, consisting of two DREP vectors encoding the same HCV antigens as before (termed DREP-e-C-E1-E2 and DREP-e-p7-NS2-NS3) but containing a translational enhancer (e) placed in frame at the N-terminal open reading frame of the HCV antigens (Fig. 1A). It was previously reported that this translational enhancer increased translational expression by one order of magnitude and improved antigen-specific immune responses (28, 29).
FIG 1.

Generation and analysis of HCV protein expression of the different DREP-based HCV vaccine candidates. (A) Scheme of the four novel DREP-based HCV vaccine candidates generated in this study, expressing either core-E1-E2 or p7-NS2-NS3 HCV genes. DREP-C-E1-E2 and DREP-p7-NS2-NS3 were grouped to form the DREP-HCV vaccine candidate, while DREP-e-C-E1-E2 and DREP-e-p7-NS2-NS3 were grouped to form the DREP-e-HCV vaccine candidate. The DREP replicons contain the alphavirus replicase placed under the control of the cytomegalovirus (CMV) promoter, and the HCV genes (either core-E1-E2 or p7-NS2-NS3) were placed under the control of the alphavirus subgenomic promoter (SP). In DREP-e-C-E1-E2 and DREP-e-p7-NS2-NS3, the translational enhancer (e) is placed in frame and upstream of the HCV genes, as indicated. (B) PCR analysis. Primers hybridizing in the DREP-vector regions flanking the place where the HCV genes were inserted were used to confirm their correct insertion. (C) Expression of HCV proteins in human HEK293T cells mock transfected or transfected with DREP-HCV (mixture of DREP-C-E1-E2 and DREP-p7-NS2-NS3), DREP-e-HCV (mixture of DREP-e-C-E1-E2 and DREP-e-p7-NS2-NS3), or empty DREP-Ø at 48 h posttransfection.
The correct generation of the four DREP vectors encoding HCV genes was confirmed by PCR (Fig. 1B) and by DNA sequencing. Furthermore, to confirm that DREP vectors successfully process and express the HCV antigens, 293T cells were transfected with DREP-HCV (DREP-C-E1-E2 and DREP-p7-NS2-NS3) or DREP-e-HCV (DREP-e-C-E1-E2 and DREP-e-p7-NS2-NS3), and at 48 h posttransfection the correct expression of HCV core, E1, E2, and NS3 proteins was analyzed by Western blotting (Fig. 1C). Results showed that both DREP-HCV and DREP-e-HCV vaccine candidates efficiently process and express the HCV antigens, with polyproteins core-E1-E2 and p7-NS2-NS3 properly processed into mature structural and nonstructural HCV proteins. Furthermore, the presence of the translational enhancer in frame with the HCV antigens in DREP-e-HCV promoted higher levels of HCV core, E1, E2, and NS3 proteins than DREP-HCV (Fig. 1C).
In vitro induction of apoptosis-related proteins by DREP vectors.
It has been reported that the enhanced capacity to stimulate the immune system by DREP-based vaccines in comparison to that of standard DNA vaccines is the inherent adjuvant effect of DREP vectors due to the stimulation of the apoptotic pathway of transfected cells as a result of the double-stranded RNA molecules produced and amplified by the encoded alphavirus replicase (35–37). Thus, to demonstrate that the induction of apoptosis triggered by the alphavirus replicase contained in the DREP vector is independent of the presence of the HCV antigens or the translational enhancer, 293T cells were transfected with empty DREP vector (DREP-Ø), DREP-p7-NS2-NS3, and DREP-e-p7-NS2-NS3. A plasmid expressing Neisseria meningitidis Cas (nmCas), similar in size to the DREP vectors but not containing the alphavirus replicase gene, was used as a negative control. At 4, 24, and 34 h posttransfection we analyzed cell lysates for the levels of phosphorylated protein kinase R (PKR) and eukaryotic initiation factor 2 alpha (eIF2α), two apoptosis-related proteins involved in IFN action (38, 39) (Fig. 2). The results showed that DREP vectors induced higher levels of phosphorylated PKR than cells transfected with control plasmid nmCas and mock-transfected cells (Fig. 2A). Furthermore, DREP vectors also induced higher levels of phosphorylated eIF2α than the corresponding controls (Fig. 2B and C), with a higher ratio of phosphorylated eIF2α/total eIF2α, as shown in the quantification graph of Fig. 2C.
FIG 2.

In vitro induction of apoptosis-related proteins by the DREP replicase. HEK293T cells were mock transfected or transfected with DREP-Ø, DREP-p7-NS2-NS3, or DREP-e-p7-NS2-NS3 vector. Plasmid nmCas, lacking the alphavirus replicase gene, was used as a negative control. At 4, 24, and 34 h posttransfection, cells were harvested, lysed with 1× Laemmli buffer–β-mercaptoethanol, and fractionated in 10% SDS-PAGE gels. The presence of total and phosphorylated (P-) PKR (A) and eIF2α (B) proteins was detected by Western blotting. Rabbit anti-β-actin and rabbit anti-histone H3 antibodies were used as loading controls. (C) A quantitative ratio of phosphorylated eIF2α/total eIF2α is shown and was determined by quantifying the Western blot bands represented in panel B using the Image Lab software. The dashed line indicates the threshold level for the controls. A.U., arbitrary units.
Heterologous prime/boost immunization in mice with a DREP-HCV or DREP-e-HCV prime followed by MVA-HCV boost induced potent, broad, and polyfunctional HCV-specific T cell adaptive immune responses.
It has been previously reported that a heterologous DREP/MVA prime/boost immunization protocol in animal models is highly immunogenic against several infectious diseases and superior to the homologous combinations of DREP/DREP or MVA/MVA (9–11, 22, 32). Thus, with the aim to enhance the HCV-specific immune responses elicited by the previously described MVA-HCV vaccine candidate (21, 34) and to discover whether the translational enhancer could increase HCV-specific immune responses, we immunized mice according to a heterologous prime/boost immunization regimen using DREP-HCV or DREP-e-HCV vaccine as a priming immunization followed by an MVA-HCV boost (DREP-HCV/MVA-HCV or DREP-e-HCV/MVA-HCV, respectively) and compared the results to those with the homologous MVA-HCV/MVA-HCV prime/boost regimen (Fig. 3). Ten C57BL/6 mice from each group (DREP-HCV/MVA-HCV, DREP-e-HCV/MVA-HCV, MVA-HCV/MVA-HCV, DREP-Ø/MVA-wild type [WT], and MVA-WT/MVA-WT) were immunized, and 10 days postboost half of them (n = 5) were sacrificed to measure the HCV-specific CD4+ and CD8+ T cell adaptive immune responses by intracellular cytokine staining (ICS), as previously described (21, 34).
FIG 3.
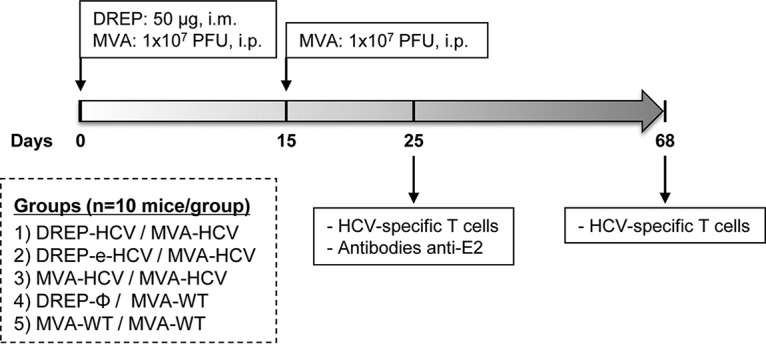
Immunization schedule. A prime/boost immunization protocol was performed in C57BL/6JOlaHsd mice to study the immunogenicity of DREP-HCV and DREP-e-HCV vaccine candidates, as described in Materials and Methods. Immunization groups are indicated, together with the time points at which animals were immunized (dose and route of administration are shown) and sacrificed to analyze the adaptive and memory HCV-specific T cell and humoral immune responses.
The magnitude of the total HCV-specific CD4+ and CD8+ T cell adaptive immune responses (consisting in the sum of cells expressing CD107a, a degranulation marker on the cell surface of activated T cells and secreting IFN-γ, tumor necrosis factor alpha [TNF-α], and/or interleukin-2 [IL-2] cytokines) against all HCV peptide pools of genotype 1a (consisting of core, E1, E2, p7, NS2, NS3, NS4, and NS5 proteins) was more potent and significantly higher in mice immunized with DREP-HCV/MVA-HCV and DREP-e-HCV/MVA-HCV than in mice receiving MVA-HCV/MVA-HCV. These results demonstrate the beneficial use of the DREP prime/MVA boost combination to enhance the HCV-specific CD4+ and CD8+ T cell responses (7.8- and 4-fold increase, respectively) compared to levels with MVA-HCV/MVA-HCV (Fig. 4A). The induced HCV-specific T cell immune responses were mediated by mainly CD8+ T cells in all the groups, reaching potent levels in mice immunized with DREP-HCV/MVA-HCV and DREP-e-HCV/MVA-HCV, with around 60% to 70% of the CD8+ T cells being HCV specific (Fig. 4A, right panel). Interestingly, the DREP-e-HCV/MVA-HCV regimen elicited significantly higher HCV-specific CD8+ T cell responses than DREP-HCV/MVA-HCV, confirming the positive effect of the translational enhancer in the enhancement of HCV-specific T cell immune responses (Fig. 4A, right panel).
FIG 4.

HCV-specific CD4+ and CD8+ T cell adaptive immune responses elicited in immunized mice. Five mice per group were sacrificed at 10 days postboost, and the splenic HCV-specific CD4+ and CD8+ T cell immune responses were analyzed by ICS, as described in Materials and Methods. P values indicate significantly differences between results for DREP-HCV/MVA-HCV, DREP-e-HCV/MVA-HCV, and MVA-HCV/MVA-HCV, as indicated (*, P < 0.05; ***, P < 0.001). (A) Magnitude of total HCV-specific CD4+ and CD8+ T cell adaptive immune responses directed against all HCV antigens. Percentages of CD4+ or CD8+ T cells expressing CD107a and/or producing IFN-γ and/or TNF-α and/or IL-2 against a mixture of core, E1, E2, p7, NS2, NS3, NS4, and NS5 genotype 1a HCV peptide pools are represented. (B) Breadth of HCV-specific CD4+ and CD8+ T cell adaptive immune responses. Percentages of core-, E1-, E2-, p7-, NS2-, NS3-, NS4-, or NS5-specific CD4+ and CD8+ T cells expressing CD107a and/or producing IFN-γ and/or TNF-α and/or IL-2 against each specific HCV peptide pool are represented. (C) Polyfunctionality of total HCV-specific CD4+ and CD8+ T cell adaptive immune responses directed against all HCV antigens. Responses are divided according to function in combined production of CD107a, IFN-γ, TNF-α, and/or IL-2 and grouped according to the color-coded pie charts, taking into consideration the number of functions (one, two, three, or four).
The profile of HCV-specific CD4+ and CD8+ T cell adaptive immune responses elicited by the different immunization groups showed that the responses were broad but significantly higher in magnitude in the two groups immunized with the heterologous DREP-HCV/MVA-HCV combinations than in those with homologous MVA-HCV/MVA-HCV immunizations (Fig. 4B). The majority of the HCV-specific CD4+ T cell responses were directed preferentially against E1 and E2 and to a lesser extent toward NS3 and NS5 in both DREP-HCV/MVA-HCV immunization groups while they were lower and directed mainly against NS5 in the MVA-HCV/MVA-HCV immunization group. Furthermore, DREP-e-HCV/MVA-HCV elicited higher E1-specific CD4+ T cell responses than DREP-HCV/MVA-HCV, while DREP-HCV/MVA-HCV induced higher E2-specific CD4+ T cell responses than DREP-e-HCV/MVA-HCV (Fig. 4B, left panel). Regarding the HCV-specific CD8+ T cell responses, in all the groups immunized with HCV vaccines, the responses were directed mainly against NS2, followed by NS3, with DREP-e-HCV/MVA-HCV eliciting a significantly higher magnitude of response against both antigens than DREP-HCV/MVA-HCV and with MVA-HCV/MVA-HCV inducing the lowest levels (Fig. 4B, right panel).
Last, since a dysfunction in T cell cytokine production and cytotoxic potential has been repeatedly associated with the failure to control HCV (40), the quality of the HCV-specific CD4+ and CD8+ T cell immune responses was measured by analyzing the profile of cytokine production (IFN-γ, TNF-α, and/or IL-2) and/or its cytotoxic potential (CD107a) against all HCV peptide pools (Fig. 4C). HCV-specific CD4+ T cell immune responses were similar and highly polyfunctional in animals immunized with DREP-HCV/MVA-HCV and DREP-e-HCV/MVA-HCV, with most of the CD4+ T cells having four functions (CD107a, IFN-γ, TNF-α, and IL-2) (Fig. 4C, left panel). MVA-HCV/MVA-HCV induced a similar polyfunctional profile, but the magnitudes for all the cell populations were significantly lower. On the other hand, HCV-specific CD8+ T cell immune responses were also polyfunctional and preferentially produced the following groups of cytokines in all of the groups immunized with HCV vaccines: CD107a, IFN-γ, TNF-α, and IL-2; CD107a, IFN-γ, and TNF-α; CD107a and IFN-γ or CD107a. In all cases the DREP-e-HCV/MVA-HCV immunization induced the highest magnitude of responses (Fig. 4C, right panel).
Heterologous prime/boost immunization in mice with a DREP-HCV or DREP-e-HCV prime followed by MVA-HCV boost induced potent, broad, and polyfunctional HCV-specific T cell memory immune responses.
Memory HCV-specific CD8+ T cells are important for protection against HCV infection (41). Thus, next we analyzed at 53 days postboost the HCV-specific CD4+ and CD8+ T cell memory immune responses induced in mice immunized with DREP-HCV/MVA-HCV, DREP-e-HCV/MVA-HCV, and MVA-HCV/MVA-HCV (n = 5 per group) (Fig. 3).
The magnitude of the total genotype 1a HCV-specific CD4+ and CD8+ T cell memory immune responses was again significantly higher in mice immunized with DREP-HCV/MVA-HCV and DREP-e-HCV/MVA-HCV than in mice immunized with MVA-HCV/MVA-HCV (8- and 5-fold increases, respectively); again, the DREP-e-HCV/MVA-HCV immunized group was the most immunogenic (Fig. 5A).
FIG 5.

HCV-specific CD4+ and CD8+ T cell memory immune responses elicited in immunized mice. Five mice per group were sacrificed at 53 days postboost, and the splenic HCV-specific CD4+ and CD8+ T cell immune responses were analyzed by ICS, as described in Materials and Methods. P values indicate significantly differences between results for DREP-HCV/MVA-HCV, DREP-e-HCV/MVA-HCV, and MVA-HCV/MVA-HCV, as indicated (***, P < 0.001). (A) Magnitude of total HCV-specific CD4+ and CD8+ T cell memory immune responses directed against all HCV antigens. Percentages of CD4+ or CD8+ T cells expressing CD107a and/or producing IFN-γ and/or TNF-α and/or IL-2 against a mixture of core, E1, E2, p7, NS2, NS3, NS4, and NS5 genotype 1a HCV peptide pools are represented. (B) Breadth of HCV-specific CD4+ and CD8+ T cell memory immune responses. Percentages of core-, E1-, E2-, p7-, NS2-, NS3-, NS4-, or NS5-specific CD4+ and CD8+ T cells expressing CD107a and/or producing IFN-γ and/or TNF-α and/or IL-2 against each specific HCV peptide pool are represented. (C) Polyfunctionality of total HCV-specific CD4+ and CD8+ T cell memory immune responses directed against all HCV antigens. Responses are divided according to function in combined production of CD107a, IFN-γ, TNF-α, and/or IL-2 and grouped according to the color-coded pie charts, taking into consideration the number of functions (one, two, three, or four).
The profile of HCV-specific CD4+ and CD8+ T cell memory immune responses elicited by the different immunization groups showed that most of the HCV-specific CD4+ T cell responses, similar to those in the adaptive phase, were directed against E2 and E1 and to a lesser extent toward NS3 and NS5 in both DREP-HCV/MVA-HCV immunization groups, while they were lower and directed preferentially against NS5 in the MVA-HCV/MVA-HCV immunization group (Fig. 5B, left panel). Furthermore, DREP-e-HCV/MVA-HCV elicited significantly higher E1-, E2-, and NS3-specific CD4+ T cell responses than DREP-HCV/MVA-HCV (Fig. 5B, left panel). Regarding the HCV-specific CD8+ T cell responses, similar to responses in the adaptive phase, all the groups immunized with HCV vaccines induced responses preferentially directed against NS2 and NS3, with DREP-e-HCV/MVA-HCV inducing a significantly higher magnitude against both antigens than DREP-HCV/MVA-HCV and with MVA-HCV/MVA-HCV eliciting the lowest levels (Fig. 5B, right panel).
Analysis of the quality of total HCV-specific CD4+ and CD8+ T cell memory immune responses (Fig. 5C), showed that, similar to results in the adaptive phase, HCV-specific CD4+ T cell immune responses were highly polyfunctional in animals immunized with DREP-HCV/MVA-HCV and DREP-e-HCV/MVA-HCV, with CD4+ T cells producing three cytokines and expressing the degranulation marker (IFN-γ, TNF-α, IL-2, and CD107a) and with DREP-e-HCV/MVA-HCV being the most immunogenic (Fig. 5C, left panel). MVA-HCV/MVA-HCV also induced a highly polyfunctional profile, but the magnitudes for all the cell populations were significantly lower. HCV-specific CD8+ T cell immune responses were also highly polyfunctional in all the immunization groups, and the majority of CD8+ T cells had three (CD107a, IFN-γ, and TNF-α) or four (CD107a, IFN-γ, TNF-α, and IL-2) functions, with the DREP-e-HCV/MVA-HCV immunization group inducing the highest magnitudes for all the cell populations (Fig. 5C, right panel).
Heterologous prime/boost immunization in mice with a DREP-HCV or DREP-e-HCV prime followed by MVA-HCV boost induced high levels of HCV-specific CD4+ and CD8+ TEM cells.
An important characteristic of a vaccine is the ability to generate functional and long-lasting memory cells, and it has been reported that higher expression of the memory surface marker CD127 on HCV-specific T cells is involved in HCV clearance after acute infection (42–44). Thus, next we analyzed the memory phenotype of the total HCV-specific CD4+ and CD8+ T cell populations induced by the different vaccination regimens by analyzing the expression levels of the memory surface markers CD127 and CD62L on CD4+ and CD8+ T cells and the secretion of IFN-γ, TNF-α, and IL-2 and expression of CD107a upon stimulation with all genotype 1a HCV peptide pools (Fig. 6). The results showed that, in all the immunization groups, the majority of the responses in the adaptive (Fig. 6A) and memory (Fig. 6B) phases were due to CD4+ and CD8+ T effector memory (TEM) cells (CD127+ CD62L−), followed by CD4+ and CD8+ T effector (TE) cells (CD127− CD62L−), with DREP/MVA-HCV immunization inducing higher responses than that with MVA-HCV/MVA-HCV. DREP-e-HCV/MVA-HCV elicited the highest magnitude in all of the populations in the adaptive and memory phases, except in adaptive CD4+ T cells, where DREP-HCV/MVA-HCV induced higher levels of CD4+ TE cells and levels of TEM cells similar to those induced by DREP-e-HCV/MVA-HCV.
FIG 6.

Phenotypic profile of adaptive and memory HCV-specific CD4+ and CD8+ T cells elicited in immunized mice. Five mice per group were sacrificed at 10 (A) and 53 (B) days postboost, and the memory phenotypic profile of splenic HCV-specific CD4+ and CD8+ T cells was analyzed by ICS, as described in Materials and Methods. (A) Graphs indicate the percentages of T central memory (TCM; CD127+ CD62L+), T effector memory (TEM; CD127+ CD62L−), and T effector (TE; CD127− CD62L−) adaptive HCV-specific CD4+ or CD8+ T cells expressing CD107a and/or producing IFN-γ and/or TNF-α and/or IL-2 against all genotype 1a HCV peptide pools. Below the graphs, representative fluorescence-activated cell sorting plots of the memory phenotypic profile of adaptive NS2- and NS3-specific CD8+ T cells are represented. CD8+ T cells expressing CD127 and/or CD62L are depicted in black as density plots, while the NS2- or NS3-specific CD8+ T cells expressing CD107a and/or producing IFN-γ and/or TNF-α and/or IL-2 are depicted in blue, with their percentages being indicated. (B) Percentages of memory HCV-specific CD4+ and CD8+ T cells with a T central memory (TCM), TEM, or TE phenotype and expressing CD107a and/or producing IFN-γ and/or TNF-α and/or IL-2 against all HCV peptide pools. **, P < 0.005; ***, P < 0.001.
Heterologous prime/boost immunization in mice with a DREP-HCV or DREP-e-HCV prime followed by MVA-HCV boost induced antibodies against HCV E2 protein.
There is increasing evidence for the importance of HCV-specific antibodies in HCV clearance, suggesting that if a vaccine can induce different ways of HCV control, i.e., not only cellular but also strong humoral responses, it will be more difficult for the virus to escape and cause chronicity (45). Thus, to know whether the different immunization groups induced antibodies against HCV E2 protein, which is considered the main target of antibodies (6), we analyzed by enzyme-linked immunosorbent assay (ELISA) the total IgG levels of antibodies against HCV E2 protein in pooled sera obtained from mice at the peak of the response (10 days postboost) (Fig. 7A). The results showed that all vaccinated groups elicited antibodies against E2, albeit at low levels, with heterologous DREP/MVA-HCV immunization inducing higher antibody levels than MVA-HCV/MVA-HCV immunization and with no significant differences between DREP-HCV/MVA-HCV and DREP-e-HCV/MVA-HCV immunizations (Fig. 7A). Moreover, the analysis of the isotypes of the anti-HCV E2 antibodies allowed us to determine levels of IgG1, IgG2c, and IgG3 binding antibodies in the vaccinated groups compared to those in the control groups; however, due to the low levels of antibodies present in mouse serum, we could not define a statistically significant Th1/Th2 ratio (Fig. 7B). Additionally, the antibodies induced by the different DREP/MVA-HCV and MVA-HCV/MVA-HCV immunization regimens were not able to neutralize HCV H77, using an HCV pseudoparticle assay, or cross-react against other HCV genotypes (data not shown), mainly due to the low levels of antibodies elicited.
FIG 7.

HCV-specific humoral immune responses elicited in immunized mice. (A) Levels of HCV E2-specific total IgG binding antibodies were measured by ELISA in serial 2-fold dilutions of pooled serum samples (n = 10 per group) obtained from immunized mice at 10 days postboost. Absorbance values were measured at 450 nm. The mean and standard deviations are indicated. Blue asterisks indicate significant differences between results with DREP-HCV/MVA-HCV and MVA-HCV/MVA-HCV, while red asterisks indicate significant differences between results with DREP-e-HCV/MVA-HCV and MVA-HCV/MVA-HCV (*, P < 0.05). (B) Levels of HCV E2-specific IgG1, IgG2c, and IgG3 isotype antibodies in diluted 1/50 individual serum samples (n = 10 per group) obtained from immunized mice at 10 days postboost. Absorbance (optical density [OD]) values were measured at 450 nm. The mean and standard deviations are indicated.
DISCUSSION
Novel antiviral treatments for HCV are still very expensive and do not allow rapid global treatment campaigns. Despite the fact that many high-income countries have announced their decision to provide treatment for hepatitis C to all persons infected with the virus, some estimations made using prices of the generic sofosbuvir and daclatasvir calculate that the treatment will still cost at least $200 per patient, making it a nonfeasible solution to eradicate the virus or to treat patients in low-income countries (46). The WHO has set 2030 as a deadline to reduce the rates of new HCV infections by 90% (https://www.who.int/hepatitis/publications/hep-elimination-by-2030-brief/en/), but all the efforts to eradicate HCV should contemplate better screening programs to reduce under-diagnosis and an enhanced prevention that could only be achieved via vaccination (3, 5). The observation that approximately 25% of individuals acutely infected with HCV are able to clear the virus spontaneously raises hope for the possibility that a vaccine can induce protective immunity against HCV.
The development of novel vaccines that can efficiently stimulate HCV-specific immune responses is a public health priority. On one hand, alphavirus-based vectors are considered an interesting approach to develop specific immune responses against heterologous antigens due to their high level of protein expression and their ability to induce apoptosis (47). In this regard Ip et al. described three recombinant Semliki Forest virus (SFV) vectors expressing HCV nonstructural proteins (NS2-NS3-NS4-NS5) in different combinations that induced robust and effective HCV-specific immune responses (48). However, while Ip et al. described a virus-based vaccine, here we describe for the first time an alphavirus-based DNA vaccine (DREP) against HCV that includes not only the nonstructural proteins (p7-NS2-NS3) but also structural proteins (core-E1-E2), increasing the breadth of the immune response.
On the other hand, MVA-based vectors are also promising vaccine candidates (49). We have previously described the generation of MVA-HCV, an MVA-based HCV vaccine that was designed to express all HCV antigens in order to obtain broader T and B cell responses. A priming immunization with regular mammalian expression DNA vectors encoding core, E1, E2, and NS3 HCV proteins followed by a boost with MVA-HCV induced higher CD4+ and CD8+ T cell responses than MVA-HCV/MVA-HCV immunization. However, the CD4+ T cell response was very low (21). Therefore, although the different vaccines generated are immunogenic, novel vaccine candidates and/or immunization approaches that could improve the magnitude, breadth, and durability of the HCV-specific immune responses are needed.
Here, we have reported the generation and characterization of novel DREP-based HCV vaccine candidates. When DREP-HCV and DREP-e-HCV were characterized in vitro, we confirmed that the presence of the translational enhancer in the DREP-e-HCV vaccine increased HCV protein expression compared to that with DREP-HCV, confirming its positive effect in the DREP-based HCV system. Moreover, both DREP-based HCV vaccines were able to promote apoptosis in transfected cells, an important adjuvant effect that is linked to the induction of cross-priming and further stimulation of the immune responses. Thus, our DREP-based HCV vaccine candidates contain ideal characteristics to be considered potential vaccines against HCV (26, 27, 37).
It has been widely reported that potent HCV-specific CD4+ and CD8+ T cell responses are associated with spontaneous viral clearance and play an important role in HCV infection (see reviews in references 50 and 51). Strong and sustained cytotoxic T lymphocytes (CTL) are key factors in a successful vaccine against HCV (52), but the induction and maintenance of proper CTL responses and the generation of CD8+ T cell memory depend on the presence of functional and efficient CD4+ T cells, making a CD4+ T cell response a key factor that contributes to control of the infection (53, 54).
In this study, we demonstrated in mice that a prime/boost immunization regimen consisting of a priming immunization with DREP-based HCV vaccines followed by a boost with MVA-HCV induced potent HCV-specific CD4+ and CD8+ T cell adaptive and memory immune responses that were significantly higher than those with homologous MVA-HCV/MVA-HCV immunization. DREP-e-HCV/MVA-HCV was the most immunogenic combination, eliciting the highest levels of CD4+ and CD8+ HCV-specific immune responses, with up to 70% of the total CD8+ T cells being HCV specific in the adaptive phase, and demonstrating for the first time the positive effect of the translational enhancer in the enhancement of antigen-specific T cell immune responses. In this study we did not use the homologous combination of DREP vectors for immunizations as we have previously described in the HIV, Chikungunya virus, and Ebola virus systems that heterologous DREP/MVA was superior to a DREP/DREP combination (9–11, 22, 32, 33).
Additionally, it is important that the early T cell response should be vigorous and broad, including CD4+ and CD8+ T cell responses targeting several HCV antigens, to avoid the emergence of viral escape mutations in targeted CD8+ T cell epitopes (5, 55–57). A study in the chimpanzee model suggested that the breath of CD8+ T cells was associated with resolution of HCV infection and a limited emergence of escape mutations (58).
The majority of vaccine candidates that have been tested in phase I/II clinical studies included only nonstructural proteins (TG4040, Ad6-NSmut- and ChAd3-NSmut, MVA-NSmut, and ChronVac-C [ClinicalTrials registration no. NCT00563173]), eliciting strong T cell responses that were only reactive against NS3-NS5 HCV proteins (13, 14, 59). Adenoviruses encoding structural proteins E1-E2 and p7 protein with a protein boost (E1-E2 with MF59 adjuvant) in mice and guinea pigs induced humoral and cellular responses (60), but this protocol has not been tested in humans. Other clinical studies have used subunit vaccines of HCV structural proteins (E1-E2/MF59, core-ISCOMATRIX, and core peptide C35-44) as immunogens that induced antibodies against them but showed low T cell responses and did not include nonstructural proteins (61–63). Moreover, the GI-5005 vaccine candidate (based on a recombinant whole-yeast immunogen) showed promising results but included only core and NS3 proteins (64). Here, we describe novel vaccine candidates that elicited potent T cell responses broadly distributed against several structural and nonstructural HCV antigens, with CD4+ T cells directed mainly against E1 and E2 and with CD8+ T cells directed against NS2 and NS3, thus providing a new vaccine strategy to increase the breadth and magnitude of HCV responses.
A cross-reactive response against different HCV genotypes is an important characteristic for a universal HCV vaccine (65) and also one of the most challenging goals due to high viral diversity (45, 66). Here, we have demonstrated that the combination of novel DREP-based vaccines and MVA-HCV (both expressing HCV antigens from genotype 1a, strain H77) in prime/boost immunization regimens elicited potent HCV-specific T cell immune responses against homologous HCV genotype 1a, a member of genotype 1, the most prevalent HCV genotype worldwide (49.1%) (67). Furthermore, to define whether T cells from vaccinated mice also recognize epitopes from genotype 1b, we performed an experiment similar to that shown in Fig. 4 but with homologous MVA-HCV immunization; mice elicited HCV-specific CD8+ T cell immune responses that cross-reacted well against pooled peptides from genotype 1b (data not shown). This is consistent with our previously described findings that a homologous genotype 1a MVA-HCV/MVA-HCV prime/boost immunization regimen in mice could also induce HCV-specific T cell immune responses against HCV genotype 1b (21) that were similar in magnitude, breadth, and polyfunctionality to the responses against genotype 1a described in the manuscript and other recent reports (21, 34), which suggests that the stronger DREP/MVA-HCV immunization regimen should behave similarly. Additionally, to detect antibody cross-reactivity against other HCV genotypes, mouse sera from the different immunization groups was reacted with ELISA plates coated with cell extracts of 293T cells transfected with DNA plasmid vectors expressing E1-E2 from genotypes 1 to 6. The results obtained did not have sufficient statistical power to conclude whether there is cross-reactivity (data not shown). It is likely that to achieve this goal, high levels of HCV antibodies are needed, which could probably be obtained if in the immunization protocol of DREP/MVA booster doses with purified E1-E2 proteins are included during vaccination.
In all human infectious viruses, but especially in those which cause chronicity, the capacity of T cells to produce more than one cytokine, together with their cytotoxic potential, is believed to correlate with protection (68, 69). In the case of HCV, polyfunctional HCV-specific CD8+ T memory cells are important to control HCV infection (50, 68). HCV-specific CD4+ and CD8+ T cell responses induced by DREP/MVA-HCV combinations were highly polyfunctional, secreting up to three cytokines at the same time (IFN-γ, TNF-α, and IL-2) together with the degranulation marker (CD107a).
Furthermore, after the contraction of the adaptive immune response, memory T cells are homeostatically maintained in order to respond rapidly to secondary infection (70), playing an important role in controlling infection (41, 69). Two relevant studies have shown that reinfected chimpanzees could not clear the virus after antibody-mediated depletion of CD4+ or CD8+ memory T cells but cleared the virus only when both lymphocyte subsets were present, demonstrating the essential role for memory CD8+ and CD4+ T cells in long-term protection (41, 71). In our study, DREP-HCV/MVA-HCV and DREP-e-HCV/MVA-HCV regimens induced higher levels of HCV-specific memory T cells of an effector memory phenotype (CD127+ CD62L−) than MVA-HCV/MVA-HCV, and the DREP-e-HCV/MVA-HCV immunization group again had the highest memory T cell response. Moreover, one of the parameters involved in HCV clearance after acute infection is the higher expression on the surface of HCV-specific T cells of the memory marker CD127 (42–44), a molecule that is part of the IL-7 receptor and that can be useful to predict the number of memory T cells generated after vaccination (42). In this study, we observed that DREP-HCV/MVA-HCV regimens induced high levels of HCV-specific memory T cells expressing CD127. This effect could reinforce the use of DREP/MVA as a potential immunization regimen able to elicit strong effector memory T cell responses against HCV after vaccination.
Thus, in this study we were able to enhance the magnitude, breadth, polyfunctionality, and durability of HCV-specific CD4+ and CD8+ T cell immune responses using novel DREP vaccines combined with MVA-HCV.
More recently there has been evidence of the important protective effect of the antibody response against E1 and E2 HCV glycoproteins (5, 72, 73). Furthermore, the presence of the translational enhancer upstream and in frame with the heterologous antigens inserted in a DREP vector enhanced antigen-specific humoral immune responses (28, 29). In our study, both DREP/MVA-HCV immunization regimens elicited higher levels of antibodies against E2 than MVA-HCV/MVA-HCV immunization although the levels were relatively low in all groups. However, no significant differences were found between immunization with DREP-HCV and that with DREP-e-HCV in the elicited antibodies against HCV E2 protein. Furthermore, the analysis of the IgG isotypes showed that IgG1, IgG2c, and IgG3 antibodies were produced in some of the mice of the vaccinated groups and were absent in the control groups, but due to the low levels induced and the variability between mice we could not conclude whether there is a trend toward a Th1 or Th2 response. Moreover, we did not detect neutralizing antibodies against homologous HCV H77 or cross-reactive antibodies against other HCV genotypes (data not shown), which could reflect the low levels of antibodies induced by the DREP/MVA-HCV and MVA-HCV/MVA-HCV immunization regimens. Thus, our results showed that although the DREP/MVA-HCV vaccination regimen induced highly potent HCV-specific T cell responses, it is not a strong regimen to elicit high levels of humoral responses. Combination of DREP-based vaccines and MVA-HCV with a protein HCV E2 component should be the approach to further enhance antibody responses.
Overall, here we report a novel study of the combination of the first described DREP-based HCV vaccines and MVA-HCV in a highly immunogenic regimen against HCV that is able to activate both arms of the immune system, cellular and humoral, with a preference for T cell activation. These results highlight the future use of DREP-based vectors and MVA-HCV as potential HCV vaccines to be further tested in clinical trials.
MATERIALS AND METHODS
Construction of DREP-HCV and DREP-e-HCV vaccine candidates.
In this study, we have generated four novel Semliki Forest virus (SFV)-based DREP vaccines against HCV: (i) DREP-C-E1-E2, expressing core, E1, and E2 proteins; (ii) DREP-p7-NS2-NS3 expressing p7, NS2, and NS3 proteins; (iii) DREP-e-C-E1-E2, expressing core, E1, and E2 proteins in frame with a translational enhancer; and (iv) DREP-e-p7-NS2-NS3 expressing p7, NS2, and NS3 proteins in frame with a translational enhancer. DREP-C-E1-E2 and DREP-p7-NS2-NS3 were combined to produce the DREP-HCV vaccine candidate, and DREP-e-C-E1-E2 and DREP-e-p7-NS2-NS3 were combined to produce the DREP-e-HCV vaccine candidate. The generation of the different DREP-based hepatitis C vaccine candidates was performed using a Gibson assembly (New England Biolabs) system according to the manufacturer’s recommendations. HCV genes (structural core, E1, and E2 genes or nonstructural p7, NS2 and NS3 genes; genotype 1a, strain H77; GenBank accession number NC_038882.1) were cloned into the empty DREP-Ø or DREP-e (containing a translational enhancer: NH2-NYIPTQTFYGRRWRPRPAARPWPLQATPVAPVVNFDLLKLAGDVESNPG-COOH) plasmid vector. Escherichia coli DH5α strain bacterial cultures transformed with the DREP vectors were cultured in Terrific Broth medium (VWR, Radnor, PA, USA), in the presence of ampicillin (Sigma-Aldrich, St. Louis, MO, USA) for 24 h, and then DREP plasmid DNA was purified using an EndoFree Plasmid Mega kit (Qiagen, Hilden, Germany). Their correct generation was checked by PCR and DNA sequencing (Macrogen, Seoul, South Korea). PCR was performed using primers annealing in the flanking regions where the heterologous HCV antigens were inserted, and the amplifications were performed with Phusion High-Fidelity DNA Polymerase (England Biolabs, Ipswich, MA, USA) according to the manufacturer’s recommendations.
Generation of a recombinant MVA-HCV vaccine candidate.
An MVA-HCV vaccine candidate was previously generated and contains the nearly full-length HCV genome of genotype 1a (strain H77). MVA-HCV expresses HCV core, E1, E2, p7, NS2, NS3, NS4A, NS4B, and NS5A proteins and a part of NS5B under the transcriptional control of a synthetic early/late promoter (21). We have also included in this study the attenuated parental wild-type MVA (MVA-WT) as a control. Viruses were grown in primary chicken embryo fibroblast (CEF) cells and purified by two sucrose cushions, as previously described (74).
Expression of HCV proteins by Western blot analysis.
To verify the correct expression of HCV proteins by the different DREP hepatitis C vaccine candidates, human embryonic kidney 293T (HEK293T) cells were mock transfected or transfected with 5 μg of DREP-HCV (2.5 μg of DREP-C-E1-E2 and 2.5 μg of DREP-p7-NS2-NS3), 5 μg of DREP-e-HCV (2.5 μg of DREP-e-C-E1-E2 and 2.5 μg of DREP-e-p7-NS2-NS3), or 5 μg of empty DREP-Ø vectors using PEImax (Polysciences, Warrington, PA, USA), according to the manufacturer’s recommendations. At 48 h posttransfection, cell lysates were harvested, pelleted, and resuspended in 1× Laemmli buffer plus β-mercaptoethanol. Next, cell extracts were fractionated in 10% polyacrylamide gels, and the expression of the different HCV proteins was analyzed by Western blotting using mouse polyclonal antibody against core protein (C7-50, diluted 1:5,000; ThermoFisher, Waltham, MA, USA), mouse monoclonal antibody against E1 (1:1,000; Acris Antibodies, Herford, Germany), goat polyclonal antibody against E2 (1:1,000; Abcam, Cambridge, UK), and mouse monoclonal antibody against NS3 (1:500; Santa Cruz, Dallas, TX, USA). Rabbit anti-β-actin antibody (diluted 1:1,000; Cell Signaling, Leiden, The Netherlands) was used as a loading control. Anti-mouse horseradish peroxidase (HRP)-conjugated (diluted 1:2,000; Sigma-Aldrich) or anti-goat HRP-conjugated (1:10,000; Sigma-Aldrich) antibodies were used as secondary antibodies. The immunocomplexes were detected by an enhanced chemiluminescence (ECL Plus) system (GE Healthcare, Chicago, IL, USA).
Analysis of apoptosis-related proteins by Western blotting.
To analyze the induction of apoptosis-related proteins by the alphavirus replicase encoded by the DREP plasmid vectors, HEK293T cells were mock transfected or transfected with DREP-p7-NS2-NS3, DREP-e-p7-NS2-NdS3, and empty DREP-Ø, as described above. A plasmid containing bacterial N. meningitidis Cas9 (nmCas), similar in size to the DREP vector but not containing the alphavirus replicase gene, was used as a negative control. Cells were then harvested at 4, 24, and 34 h posttransfection, and cell extracts were prepared for Western blot analysis, as described above. Detection of total and phospho-PKR and total and phospho-eIF2α was performed using anti-PKR, anti-phospho-PKR, anti-eIF2α, and anti-phospho-eIF2α antibodies, respectively (diluted 1:1,000; Cell Signaling). Rabbit anti-β-actin (1:2,000; Cell Signaling) and rabbit anti-histone H3 (1:1,000; Cell Signaling) antibodies were used as loading controls in Western blotting of anti-PKR/anti-phospho-PKR and anti-eIF2α/anti-phospho-eIF2α, respectively. Densitometry quantification of the ratio between phospho-eIF2α and total eIF2α was determined by Image Lab.
C57BL/6 mice and immunizations.
Female 6- to 8-week-old C57BL/6JOlaHsd mice were acquired from Envigo and maintained at the Centro Nacional de Biotecnología (CNB) in a pathogen-free animal facility, according to the Federation of European Laboratory Animal Science Associations recommendations. Mouse experiments were approved by the Ethical Committee of Animal Experimentation (CEEA) of Centro Nacional de Biotecnología (Madrid, Spain) according to international guidelines and Spanish law under Royal Decree (RD) 53/2013 (permit number PROEX 331/14).
A prime/boost immunization regimen was performed to study the immunogenicity of DREP-HCV and DREP-e-HCV vaccine candidates (Fig. 3). Mice (n = 10 per group) were first immunized with 50 μg of DREP-HCV (25 μg of DREP-C-E1-E2 and 25 μg of DREP-p7-NS2-NS3), 50 μg of DREP-e-HCV (25 μg of DREP-e-C-E1-E2 and 25 μg of DREP-e-p7-NS2-NS3), 50 μg of DREP-Ø, or 1 × 107 PFU of MVA-HCV or MVA-WT. DREP immunizations were performed intramuscularly (i.m.) in both legs of mice in 50 μl of phosphate-buffered saline (PBS), while MVA immunizations were performed intraperitoneally (i.p.) in 200 μl of PBS. Two weeks after the prime immunization, mice received a second i.p. dose of 1 × 107 PFU/mouse of MVA-HCV or MVA-WT. At 10 and 53 days postboost, five mice in each group were sacrificed, and spleens were collected and processed to measure by intracellular cytokine staining (ICS) the adaptive and memory HCV-specific T cell immune responses, respectively. Additionally, at 10 days postboost, serum samples were obtained from mice (n = 10) to further analyze the presence of antibodies against HCV.
Peptides.
Purified HCV peptide pools of the HCV virus H77 strain (genotype 1a) were obtained through the Biodefense and Emerging Infectious Research Resources Repository (BEI Resources, National Institute of Allergy and Infectious Disease, National Institutes of Health, Bethesda, MD, USA) and were previously described (21, 34). Peptides that cover the entire HCV H77 genome as consecutive 13- to 19-mers overlapping by 11 or 12 amino acids were resuspended to a final concentration of 1 mg/ml and grouped into seven pools: core pool (28 peptides), E1 pool (29 peptides), E2 pool (56 peptides), p7 pool (8 peptides), NS2 pool (32 peptides), NS3 pool (98 peptides), NS4 pool comprising NS4A (7 peptides) plus NS4B (40 peptides), and NS5 pool comprising NS5A (71 peptides) plus NS5B (91 peptides). Peptides were used for ex vivo stimulation of splenocytes from immunized mice.
ICS assay.
Using an ICS assay, we analyzed the magnitude, breadth, polyfunctionality, and memory phenotype of the adaptive and memory HCV-specific T cell immune responses, as previously described (21, 34). Briefly, spleens were processed, and the splenocytes were stimulated for 6 h with 1 μg/ml of different HCV peptide pools of the HCV genotype 1a, H77 strain. Then, cells were stained for the different surface markers (CD4, CD8, CD62L, CD127, and CD107a), fixed, permeabilized (Cytofix/Cytoperm kit; BD Biosciences, Franklin Lakes, NJ, USA), and stained intracellularly with the appropriate fluorochrome-conjugated antibodies (IFN-γ, TNF-α, and IL-2), as previously described (21, 34).
ELISA.
Serum samples from immunized mice were used to analyze by ELISA total IgG binding antibody levels against HCV E2 protein (genotype 1a, isolate H77; Sino Biological Wayne, PA, USA) and isotypes IgG1, IgG2c, and IgG3, as previously described (34). ELISAs were performed with pooled or individual serum samples (n = 10) obtained at 10 days postboost from each immunization group. Absorbance was read at 450 nm.
Statistical analysis.
Statistical analysis of the ICS data was done as previously described (75, 76). Statistical significance in the ELISA was determined using the Holm-Sidak method, with an alpha of 5%. Statistically significant differences are indicated in the figure legends.
ACKNOWLEDGMENTS
This research was supported by MINECO Spanish grants SAF-2013-45232-R and SAF-2017-88089-R to M.E. and by Swedish Research Council SRC grant 2017-03086 to P.L. M.Q.M. received a Formación del Profesorado Universitario PhD fellowship from the Spanish Ministry of Education and a short-term EMBO fellowship.
We thank Cristina Sánchez Corzo and Victoria Jiménez for their technical assistance.
REFERENCES
- 1.Lohmann V. 8 October 2018. Hepatitis C virus cell culture models: an encomium on basic research paving the road to therapy development. Med Microbiol Immunol doi: 10.1007/s00430-018-0566-x. [DOI] [PubMed] [Google Scholar]
- 2.Pawlotsky J-M. 2016. Hepatitis C virus resistance to direct-acting antiviral drugs in interferon-free regimens. Gastroenterology 151:70–86. doi: 10.1053/j.gastro.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Bartenschlager R, Baumert TF, Bukh J, Houghton M, Lemon SM, Lindenbach BD, Lohmann V, Moradpour D, Pietschmann T, Rice CM, Thimme R, Wakita T. 2018. Critical challenges and emerging opportunities in hepatitis C virus research in an era of potent antiviral therapy: considerations for scientists and funding agencies. Virus Res 248:53–62. doi: 10.1016/j.virusres.2018.02.016. [DOI] [PubMed] [Google Scholar]
- 4.Ghasemi F, Rostami S, Meshkat Z. 2015. Progress in the development of vaccines for hepatitis C virus infection. World J Gastroenterol 21:11984–12002. doi: 10.3748/wjg.v21.i42.11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shoukry NH. 2018. Hepatitis C vaccines, antibodies, and T cells. Front Immunol 9:1480. doi: 10.3389/fimmu.2018.01480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo X, Zhong J-Y, Li J-W. 2018. Hepatitis C virus infection and vaccine development. J Clin Exp Hepatol 8:195–204. doi: 10.1016/j.jceh.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Man John Law L, Landi A, Magee WC, Lorne Tyrrell D, Houghton M. 2013. Progress towards a hepatitis C virus vaccine. Emerg Microbes Infect 2:e79. doi: 10.1038/emi.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee J, Arun Kumar S, Jhan YY, Bishop CJ. 2018. Engineering DNA vaccines against infectious diseases. Acta Biomater 80:31–47. doi: 10.1016/j.actbio.2018.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knudsen ML, Ljungberg K, Tatoud R, Weber J, Esteban M, Liljeström P. 2015. Alphavirus replicon DNA expressing HIV antigens is an excellent prime for boosting with recombinant modified vaccinia Ankara (MVA) or with HIV gp140 protein antigen. PLoS One 10:e0117042. doi: 10.1371/journal.pone.0117042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hallengard D, Lum F-M, Kummerer BM, Lulla A, Lulla V, Garcia-Arriaza J, Fazakerley JK, Roques P, Le Grand R, Merits A, Ng LFP, Esteban M, Liljestrom P. 2014. Prime-boost immunization strategies against Chikungunya virus. J Virol 88:13333–13343. doi: 10.1128/JVI.01926-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Öhlund P, García-Arriaza J, Zusinaite E, Szurgot I, Männik A, Kraus A, Ustav M, Merits A, Esteban M, Liljeström P, Ljungberg K. 2018. DNA-launched RNA replicon vaccines induce potent anti-Ebolavirus immune responses that can be further improved by a recombinant MVA boost. Sci Rep 8:12459. doi: 10.1038/s41598-018-31003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayr A, Stickl H, Müller HK, Danner K, Singer H. 1978. The smallpox vaccination strain MVA: marker, genetic structure, experience gained with the parenteral vaccination and behavior in organisms with a debilitated defence mechanism (author’s transl). Zentralbl Bakteriol B 167:375–390. [PubMed] [Google Scholar]
- 13.Habersetzer F, Honnet G, Bain C, Maynard-Muet M, Leroy V, Zarski J-P, Feray C, Baumert TF, Bronowicki J-P, Doffoël M, Trépo C, Agathon D, Toh M-L, Baudin M, Bonnefoy J-Y, Limacher J-M, Inchauspé G. 2011. A poxvirus vaccine is safe, induces T-cell responses, and decreases viral load in patients with chronic hepatitis C. Gastroenterology 141:890–899.e4. doi: 10.1053/j.gastro.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 14.Swadling L, Capone S, Antrobus RD, Brown A, Richardson R, Newell EW, Halliday J, Kelly C, Bowen D, Fergusson J, Kurioka A, Ammendola V, Del Sorbo M, Grazioli F, Esposito ML, Siani L, Traboni C, Hill A, Colloca S, Davis M, Nicosia A, Cortese R, Folgori A, Klenerman P, Barnes E. 2014. A human vaccine strategy based on chimpanzee adenoviral and MVA vectors that primes, boosts, and sustains functional HCV-specific T cell memory. Sci Transl Med 6:261ra153. doi: 10.1126/scitranslmed.3009185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan WG, Zubkova I, Kachko A, Wells F, Adler H, Sutter G, Major ME. 2017. Qualitative differences in cellular immunogenicity elicited by hepatitis C virus T-cell vaccines employing prime-boost regimens. PLoS One 12:e0181578. doi: 10.1371/journal.pone.0181578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Volz A, Sutter G. 2017. Modified vaccinia virus Ankara: history, value in basic research, and current perspectives for vaccine development. Adv Virus Res 97:187–243. doi: 10.1016/bs.aivir.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gómez CE, Perdiguero B, García-Arriaza J, Esteban M. 2013. Clinical applications of attenuated MVA poxvirus strain. Expert Rev Vaccines 12:1395–1416. doi: 10.1586/14760584.2013.845531. [DOI] [PubMed] [Google Scholar]
- 18.Abaitua F, Rodríguez JR, Garzón A, Rodríguez D, Esteban M. 2006. Improving recombinant MVA immune responses: potentiation of the immune responses to HIV-1 with MVA and DNA vectors expressing Env and the cytokines IL-12 and IFN-gamma. Virus Res 116:11–20. doi: 10.1016/j.virusres.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 19.Chea LS, Amara RR. 2017. Immunogenicity and efficacy of DNA/MVA HIV vaccines in rhesus macaque models. Expert Rev Vaccines 16:973–985. doi: 10.1080/14760584.2017.1371594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fournillier A, Frelin L, Jacquier E, Ahlén G, Brass A, Gerossier E, Holmström F, Broderick KE, Sardesai NY, Bonnefoy J-Y, Inchauspé G, Sällberg M. 2013. A heterologous prime/boost vaccination strategy enhances the immunogenicity of therapeutic vaccines for hepatitis C virus. J Infect Dis 208:1008–1019. doi: 10.1093/infdis/jit267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gómez CE, Perdiguero B, Cepeda MV, Mingorance L, García-Arriaza J, Vandermeeren A, Sorzano CÓS, Esteban M. 2013. High, broad, polyfunctional, and durable T cell immune responses induced in mice by a novel hepatitis C virus (HCV) vaccine candidate (MVA-HCV) based on modified vaccinia virus Ankara expressing the nearly full-length HCV genome. J Virol 87:7282–7300. doi: 10.1128/JVI.03246-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knudsen ML, Mbewe-Mvula A, Rosario M, Johansson DX, Kakoulidou M, Bridgeman A, Reyes-Sandoval A, Nicosia A, Ljungberg K, Hanke T, Liljeström P. 2012. Superior induction of T cell responses to conserved HIV-1 regions by electroporated alphavirus replicon DNA compared to that with conventional plasmid DNA vaccine. J Virol 86:4082–4090. doi: 10.1128/JVI.06535-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nordström EKL, Forsell MNE, Barnfield C, Bonin E, Hanke T, Sundström M, Karlsson GB, Liljeström P. 2005. Enhanced immunogenicity using an alphavirus replicon DNA vaccine against human immunodeficiency virus type 1. J Gen Virol 86:349–354. [DOI] [PubMed] [Google Scholar]
- 24.Ljungberg K, Liljeström P. 2015. Self-replicating alphavirus RNA vaccines. Expert Rev Vaccines 14:177–194. doi: 10.1586/14760584.2015.965690. [DOI] [PubMed] [Google Scholar]
- 25.Näslund TI, Kostic L, Nordström EK, Chen M, Liljeström P. 2011. Role of innate signalling pathways in the immunogenicity of alphaviral replicon-based vaccines. Virol J 8:36. doi: 10.1186/1743-422X-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nikonov A, Mölder T, Sikut R, Kiiver K, Männik A, Toots U, Lulla A, Lulla V, Utt A, Merits A, Ustav M. 2013. RIG-I and MDA-5 detection of viral RNA-dependent RNA polymerase activity restricts positive-strand RNA virus replication. PLoS Pathog 9:e1003610. doi: 10.1371/journal.ppat.1003610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schulz O, Diebold SS, Chen M, Näslund TI, Nolte MA, Alexopoulou L, Azuma Y-T, Flavell RA, Liljeström P, Reis e Sousa C. 2005. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 433:887–892. doi: 10.1038/nature03326. [DOI] [PubMed] [Google Scholar]
- 28.Forsell MNE, McInerney GM, Dosenovic P, Hidmark AS, Eriksson C, Liljeström P, Grundner C, Karlsson Hedestam GB. 2007. Increased human immunodeficiency virus type 1 Env expression and antibody induction using an enhanced alphavirus vector. J Gen Virol 88:2774–2779. doi: 10.1099/vir.0.83060-0. [DOI] [PubMed] [Google Scholar]
- 29.Sjöberg EM, Suomalainen M, Garoff H. 1994. A significantly improved Semliki Forest virus expression system based on translation enhancer segments from the viral capsid gene. Nat Biotechnol 12:1127–1131. doi: 10.1038/nbt1194-1127. [DOI] [PubMed] [Google Scholar]
- 30.Berglund P, Smerdou C, Fleeton MN, Tubulekas I, Liljeström P. 1998. Enhancing immune responses using suicidal DNA vaccines. Nat Biotechnol 16:562–565. doi: 10.1038/nbt0698-562. [DOI] [PubMed] [Google Scholar]
- 31.Lundstrom K. 2014. Alphavirus-based vaccines. Viruses 6:2392–2415. doi: 10.3390/v6062392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roques P, Ljungberg K, Kümmerer BM, Gosse L, Dereuddre-Bosquet N, Tchitchek N, Hallengärd D, García-Arriaza J, Meinke A, Esteban M, Merits A, Le Grand R, Liljeström P. 2017. Attenuated and vectored vaccines protect nonhuman primates against Chikungunya virus. JCI Insight 2:e83527. doi: 10.1172/jci.insight.83527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knudsen ML, Ljungberg K, Kakoulidou M, Kostic L, Hallengärd D, García-Arriaza J, Merits A, Esteban M, Liljeström P. 2014. Kinetic and phenotypic analysis of CD8+ T cell responses after priming with alphavirus replicons and homologous or heterologous booster immunizations. J Virol 88:12438–12451. doi: 10.1128/JVI.02223-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marín MQ, Pérez P, Gómez CE, Sorzano CÓS, Esteban M, García-Arriaza J. 2018. Removal of the C6 vaccinia virus interferon-β inhibitor in the hepatitis C vaccine candidate MVA-HCV elicited in mice high immunogenicity in spite of reduced host gene expression. Viruses 10:E414. doi: 10.3390/v10080414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barry G, Fragkoudis R, Ferguson MC, Lulla A, Merits A, Kohl A, Fazakerley JK. 2010. Semliki forest virus-induced endoplasmic reticulum stress accelerates apoptotic death of mammalian cells. J Virol 84:7369–7377. doi: 10.1128/JVI.02310-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Glasgow GM, McGee MM, Sheahan BJ, Atkins GJ. 1997. Death mechanisms in cultured cells infected by Semliki Forest virus. J Gen Virol 78:1559–1563. doi: 10.1099/0022-1317-78-7-1559. [DOI] [PubMed] [Google Scholar]
- 37.Li M-L, Stollar V. 2004. Alphaviruses and apoptosis. Int Rev Immunol 23:7–24. [DOI] [PubMed] [Google Scholar]
- 38.Lee SB, Esteban M. 1994. The interferon-induced double-stranded RNA-activated protein kinase induces apoptosis. Virology 199:491–496. doi: 10.1006/viro.1994.1151. [DOI] [PubMed] [Google Scholar]
- 39.Gil J, Alcamí J, Esteban M. 1999. Induction of apoptosis by double-stranded-RNA-dependent protein kinase (PKR) involves the alpha subunit of eukaryotic translation initiation factor 2 and NF-κB. Mol Cell Biol 19:4653–4663. doi: 10.1128/MCB.19.7.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swadling L, Klenerman P, Barnes E. 2013. Ever closer to a prophylactic vaccine for HCV. Expert Opin Biol Ther 13:1109–1124. doi: 10.1517/14712598.2013.791277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shoukry NH, Grakoui A, Houghton M, Chien DY, Ghrayeb J, Reimann KA, Walker CM. 2003. Memory CD8+ T cells are required for protection from persistent hepatitis C virus infection. J Exp Med 197:1645–1655. doi: 10.1084/jem.20030239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. 2003. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol 4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 43.Golden-Mason L, Burton JR, Castelblanco N, Klarquist J, Benlloch S, Wang C, Rosen HR. 2006. Loss of IL-7 receptor alpha-chain (CD127) expression in acute HCV infection associated with viral persistence. Hepatology 44:1098–1109. doi: 10.1002/hep.21365. [DOI] [PubMed] [Google Scholar]
- 44.Shin E-C, Park S-H, Nascimbeni M, Major M, Caggiari L, de Re V, Feinstone SM, Rice CM, Rehermann B. 2013. The frequency of CD127+ hepatitis C virus (HCV)-specific T cells but not the expression of exhaustion markers predicts the outcome of acute HCV infection. J Virol 87:4772–4777. doi: 10.1128/JVI.03122-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Torres-Cornejo A, Lauer GM. 2017. Hurdles to the development of effective HBV immunotherapies and HCV vaccines. Pathog Immun 2:102–125. doi: 10.20411/pai.v2i1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hill A, Simmons B, Gotham D, Fortunak J. 2016. Rapid reductions in prices for generic sofosbuvir and daclatasvir to treat hepatitis C. J Virus Erad 2:28–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Riezebos-Brilman A, de Mare A, Bungener L, Huckriede A, Wilschut J, Daemen T. 2006. Recombinant alphaviruses as vectors for anti-tumour and anti-microbial immunotherapy. J Clin Virol 35:233–243. doi: 10.1016/j.jcv.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 48.Ip PP, Boerma A, Regts J, Meijerhof T, Wilschut J, Nijman HW, Daemen T. 2014. Alphavirus-based vaccines encoding nonstructural proteins of hepatitis C virus induce robust and protective T-cell responses. Mol Ther 22:881–890. doi: 10.1038/mt.2013.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Esteban M. 2009. Attenuated poxvirus vectors MVA and NYVAC as promising vaccine candidates against HIV/AIDS. Hum Vaccin 5:867–871. [DOI] [PubMed] [Google Scholar]
- 50.Holz L, Rehermann B. 2015. T cell responses in hepatitis C virus infection: historical overview and goals for future research. Antiviral Res 114:96–105. doi: 10.1016/j.antiviral.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klenerman P, Thimme R. 2012. T cell responses in hepatitis C: the good, the bad and the unconventional. Gut 61:1226–1234. doi: 10.1136/gutjnl-2011-300620. [DOI] [PubMed] [Google Scholar]
- 52.Neumann-Haefelin C, Thimme R. 2013. Adaptive immune responses in hepatitis C virus infection. Curr Top Microbiol Immunol 369:243–262. doi: 10.1007/978-3-642-27340-7_10. [DOI] [PubMed] [Google Scholar]
- 53.Urbani S, Amadei B, Fisicaro P, Tola D, Orlandini A, Sacchelli L, Mori C, Missale G, Ferrari C. 2006. Outcome of acute hepatitis C is related to virus-specific CD4 function and maturation of antiviral memory CD8 responses. Hepatology 44:126–139. doi: 10.1002/hep.21242. [DOI] [PubMed] [Google Scholar]
- 54.Bourgeois C, Tanchot C. 2003. Mini-review CD4 T cells are required for CD8 T cell memory generation. Eur J Immunol 33:3225–3231. doi: 10.1002/eji.200324576. [DOI] [PubMed] [Google Scholar]
- 55.Bowen DG, Walker CM. 2005. Mutational escape from CD8+ T cell immunity: HCV evolution, from chimpanzees to man. J Exp Med 201:1709–1714. doi: 10.1084/jem.20050808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schulze Zur Wiesch J, Ciuffreda D, Lewis-Ximenez L, Kasprowicz V, Nolan BE, Streeck H, Aneja J, Reyor LL, Allen TM, Lohse AW, McGovern B, Chung RT, Kwok WW, Kim AY, Lauer GM. 2012. Broadly directed virus-specific CD4+ T cell responses are primed during acute hepatitis C infection, but rapidly disappear from human blood with viral persistence. J Exp Med 209:61–75. doi: 10.1084/jem.20100388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Semmo N, Klenerman P. 2007. CD4+ T cell responses in hepatitis C virus infection. World J Gastroenterol 13:4831–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meyer-Olson D, Shoukry NH, Brady KW, Kim H, Olson DP, Hartman K, Shintani AK, Walker CM, Kalams SA. 2004. Limited T cell receptor diversity of HCV-specific T cell responses is associated with CTL escape. J Exp Med 200:307–319. doi: 10.1084/jem.20040638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barnes E, Folgori A, Capone S, Swadling L, Aston S, Kurioka A, Meyer J, Huddart R, Smith K, Townsend R, Brown A, Antrobus R, Ammendola V, Naddeo M, O'Hara G, Willberg C, Harrison A, Grazioli F, Esposito ML, Siani L, Traboni C, Oo Y, Adams D, Hill A, Colloca S, Nicosia A, Cortese R, Klenerman P. 2012. Novel adenovirus-based vaccines induce broad and sustained T cell responses to HCV in man. Sci Transl Med 4:115ra1. doi: 10.1126/scitranslmed.3003155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chmielewska AM, Naddeo M, Capone S, Ammendola V, Hu K, Meredith L, Verhoye L, Rychlowska M, Rappuoli R, Ulmer JB, Colloca S, Nicosia A, Cortese R, Leroux-Roels G, Balfe P, Bienkowska-Szewczyk K, Meuleman P, McKeating JA, Folgori A. 2014. Combined adenovirus vector and hepatitis C virus envelope protein prime-boost regime elicits T cell and neutralizing antibody immune responses. J Virol 88:5502–5510. doi: 10.1128/JVI.03574-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Frey SE, Houghton M, Coates S, Abrignani S, Chien D, Rosa D, Pileri P, Ray R, Di Bisceglie AM, Rinella P, Hill H, Wolff MC, Schultze V, Han JH, Scharschmidt B, Belshe RB. 2010. Safety and immunogenicity of HCV E1E2 vaccine adjuvanted with MF59 administered to healthy adults. Vaccine 28:6367–6373. doi: 10.1016/j.vaccine.2010.06.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Drane D, Maraskovsky E, Gibson R, Mitchell S, Barnden M, Moskwa A, Shaw D, Gervase B, Coates S, Houghton M, Basser R. 2009. Priming of CD4+ and CD8+ T cell responses using a HCV core ISCOMATRIX vaccine: a phase I study in healthy volunteers. Hum Vaccin 5:151–157. [DOI] [PubMed] [Google Scholar]
- 63.Niu Y, Komatsu N, Komohara Y, Matsueda S, Yutani S, Ishihara Y, Itou M, Yamada A, Itoh K, Shichijo S. 2009. A peptide derived from hepatitis C virus (HCV) core protein inducing cellular responses in patients with HCV with various HLA class IA alleles. J Med Virol 81:1232–1240. doi: 10.1002/jmv.21518. [DOI] [PubMed] [Google Scholar]
- 64.Habersetzer F, Baumert TF, Stoll-Keller F. 2009. GI-5005, a yeast vector vaccine expressing an NS3-core fusion protein for chronic HCV infection. Curr Opin Mol Ther 11:456–462. [PubMed] [Google Scholar]
- 65.Wijesundara DK, Gummow J, Li Y, Yu W, Quah BJ, Ranasinghe C, Torresi J, Gowans EJ, Grubor-Bauk B. 2018. Induction of genotype cross-reactive, hepatitis C virus-specific, cell-mediated immunity in DNA-vaccinated mice. J Virol 92:e02133-17. doi: 10.1128/JVI.02133-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kelly C, Swadling L, Brown A, Capone S, Folgori A, Salio M, Klenerman P, Barnes E. 2015. Cross-reactivity of hepatitis C virus specific vaccine-induced T cells at immunodominant epitopes. Eur J Immunol 45:309–316. doi: 10.1002/eji.201444686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Petruzziello A, Marigliano S, Loquercio G, Cozzolino A, Cacciapuoti C. 2016. Global epidemiology of hepatitis C virus infection: an up-date of the distribution and circulation of hepatitis C virus genotypes. World J Gastroenterol 22:7824. doi: 10.3748/wjg.v22.i34.7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Badr G, Bédard N, Abdel-Hakeem MS, Trautmann L, Willems B, Villeneuve J-P, Haddad EK, Sékaly RP, Bruneau J, Shoukry NH. 2008. Early interferon therapy for hepatitis C virus infection rescues polyfunctional, long-lived CD8+ memory T cells. J Virol 82:10017–10031. doi: 10.1128/JVI.01083-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harari A, Dutoit V, Cellerai C, Bart P-A, Du Pasquier RA, Pantaleo G. 2006. Functional signatures of protective antiviral T-cell immunity in human virus infections. Immunol Rev 211:236–254. doi: 10.1111/j.0105-2896.2006.00395.x. [DOI] [PubMed] [Google Scholar]
- 70.Ahlers JD, Belyakov IM. 2010. Memories that last forever: strategies for optimizing vaccine T-cell memory. Blood 115:1678–1689. doi: 10.1182/blood-2009-06-227546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grakoui A, Shoukry NH, Woollard DJ, Han J-H, Hanson HL, Ghrayeb J, Murthy KK, Rice CM, Walker CM. 2003. HCV persistence and immune evasion in the absence of memory T cell help. Science 302:659–662. doi: 10.1126/science.1088774. [DOI] [PubMed] [Google Scholar]
- 72.Cashman SB, Marsden BD, Dustin LB. 2014. The humoral immune response to HCV: understanding is key to vaccine development. Front Immunol 5:550. doi: 10.3389/fimmu.2014.00550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ball JK, Tarr AW, McKeating JA. 2014. The past, present and future of neutralizing antibodies for hepatitis C virus. Antiviral Res 105:100–111. doi: 10.1016/j.antiviral.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ramírez JC, Gherardi MM, Esteban M. 2000. Biology of attenuated modified vaccinia virus Ankara recombinant vector in mice: virus fate and activation of B- and T-cell immune responses in comparison with the Western Reserve strain and advantages as a vaccine. J Virol 74:923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.García-Arriaza J, Nájera JL, Gómez CE, Sorzano COS, Esteban M. 2010. Immunogenic profiling in mice of a HIV/AIDS vaccine candidate (MVA-B) expressing four HIV-1 antigens and potentiation by specific gene deletions. PLoS One 5:e12395. doi: 10.1371/journal.pone.0012395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nájera JL, Gómez CE, García-Arriaza J, Sorzano CO, Esteban M. 2010. Insertion of vaccinia virus C7L host range gene into NYVAC-B genome potentiates immune responses against HIV-1 antigens. PLoS One 5:e11406. doi: 10.1371/journal.pone.0011406. [DOI] [PMC free article] [PubMed] [Google Scholar]