

Human papillomaviruses affect an estimated 75% of the sexually active adult population in the United States, with 5.5 million new cases emerging every year. More than 200 HPV genotypes have been identified; a subset of them are linked to the development of cancers from these epithelial infections. Specific antiviral medical treatments for infected individuals are not available. This project examines the mechanisms that control viral genome replication and may allow the development of novel therapeutics.
KEYWORDS: DNA replication, E2, P300, papillomavirus, topoisomerase 1
ABSTRACT
Human papillomavirus (HPV) E2 proteins are integral for the transcription of viral genes and the replication and maintenance of viral genomes in host cells. E2 recruits the viral DNA helicase E1 to the origin. A lysine (K111), highly conserved among almost all papillomavirus (PV) E2 proteins, is a target for P300 (EP300) acetylation and is critical for viral DNA replication (E. J. Quinlan, S. P. Culleton, S. Y. Wu, C. M. Chiang, et al., J Virol 87:1497–1507, 2013, https://doi.org/10.1128/JVI.02771-12; Y. Thomas and E. J. Androphy, J Virol 92:e01912-17, 2018, https://doi.org/10.1128/JVI.01912-17). Since the viral genome exists as a covalently closed circle of double-stranded DNA, topoisomerase 1 (Topo1) is thought to be required for progression of the replication forks. Due to the specific effect of K111 mutations on DNA unwinding (Y. Thomas and E. J. Androphy, J Virol 92:e01912-17, 2018, https://doi.org/10.1128/JVI.01912-17), we demonstrate that the E2 protein targets Topo1 to the viral origin, and this depends on acetylation of K111. The effect was corroborated by functional replication assays, in which higher levels of P300, but not its homolog CBP, caused enhanced replication with wild-type E2 but not the acetylation-defective K111 arginine mutant. These data reveal a novel role for lysine acetylation during viral DNA replication by regulating topoisomerase recruitment to the replication origin.
IMPORTANCE Human papillomaviruses affect an estimated 75% of the sexually active adult population in the United States, with 5.5 million new cases emerging every year. More than 200 HPV genotypes have been identified; a subset of them are linked to the development of cancers from these epithelial infections. Specific antiviral medical treatments for infected individuals are not available. This project examines the mechanisms that control viral genome replication and may allow the development of novel therapeutics.
INTRODUCTION
Human papillomaviruses (HPVs) are a large family of double-stranded DNA viruses that are best known for their roles in cancers of the mucosal and cutaneous epithelia. The circular, 8-kb genome is comprised of a long control region (LCR) (1) and eight open reading frames (ORFs) (2–4) that encode two late structural proteins and eight multifunctional early proteins and splice variants. The E2 protein has integral roles in transcriptional regulation of viral genes and in viral replication. The papillomavirus (PV) replication origin contains E1 binding sites that are flanked by high-affinity E2 binding sites. Past in vitro studies have shown that E1 can initiate limited amounts of replication in the absence of E2 (5–7); however, E2 is essential for viral replication in vivo (8). Upon recruitment to the origin by E2, E1 assembles into double hexamers upon release from E2, and its helicase initiates strand separation. E1 is a DNA binding ATPase (9), and the structure of its helicase domain is similar to that of the SV40 large T protein (9–11). In eukaryotic cells, this helicase activity resides in the MCM complex. For eukaryotic cell studies, the E2 and E1 genes can be cotransfected with a minimal origin consisting of E2 and E1 binding sites flanked by a poly(A) tract that acts as a replication origin in transient assays.
We recently reported an intriguing acetylation site in bovine and HPV31 PV E2 at lysine 111 (K111), which is conserved in 99% of PVs. In both of these E2 proteins, the K111R arginine (R) mutant was defective for induction of E1-dependent viral ori replication. In contrast, the acetylation mimic glutamine at mutant 111 (Q111) was fully active. Importantly, both E2 mutant proteins coimmunoprecipitated with relevant factors, including E1, Brd4, TopBP1, and GPS2. While E2 K111R recruited E1 to the origin, replication protein A (RPA) was not detected, indicating the inability to form stable single-stranded DNA at the replication forks where detectable levels of RPA were deposited (12). To uncover the mechanism by which this acetylation regulates viral replication, we focused on topoisomerase 1 (Topo1), which is necessary for resolving the torsional stress resulting from unwinding double-stranded DNA as it is duplicated by polymerases (13).
RESULTS
Acetylation of K111 is necessary for Topo1 recruitment.
Chromatin immunoprecipitation (ChIP) assays were conducted under replication conditions to evaluate the presence of Topo1 at the replication origin. C33A cells were cotransfected with plasmids for the HPV31 replication reporter origin plasmid PFLORI31 along with E1, E2, or both viral genes together. The cells were processed 48 h posttransfection and analyzed by ChIP with FLAG antibodies using PCR primers flanking the origin. Occupation of the ori by wild-type (WT) E2 did not differ when E1 was cotransfected (Fig. 1A). The E2 K111R and K111Q mutants were detected at levels equivalent to those of the WT in the E1 cotransfected cells (replication conditions), demonstrating that these E2 mutant proteins are expressed and competent for accessing the ori. We then tested for the presence of Topo1 at the origin (Fig. 1B). K111Q expression resulted in levels of Topo1 severalfold higher than those of wild-type E2, while Topo1 was not detected in samples containing E2 K111R. These results support our hypothesis that the acetylation status of E2 K111 regulates the participation of Topo1 in an early stage of viral DNA replication. A second observation was that Topo1 was not present at the replication origin with expression of E1 alone. Topo1 has been previously reported to bind to both PVs E1 and E2 using bacterial protein expression and Southwestern blotting (14, 15). These reports suggested that the loading of Topo1 was facilitated by E1, and a replication model was proposed where E1 interacts directly with Topo1 after E2 dissociation (16). However, there have been no in-cell experiments to corroborate these protein-protein interactions or the localization of Topo1 to an active PV ori. To further investigate our observations, we performed a ChIP assay to determine if E2 in the absence of E1 is responsible for Topo1 recruitment in G1/S-phase synchronized cells (Fig. 1C). The amount of Topo1 present on the ori plasmid in this nonreplicating system was at baseline with E2 K111R and increased by 4.5-fold with wild-type E2. Interestingly, in the presence of the acetylation mimic K111Q, we observed a 6-fold increase in Topo1 at the origin, even in the absence of the amplification effect that we observed when cotransfected with the E1 DNA helicase.
FIG 1.

E2 K111R failed to recruit Topo1 to the replication origin. C33A cells were transfected with FLAG-E1, FLAG-E2, and HPV31 Ori. ChIP assays were conducted for E2 and E1 with anti-FLAG antibodies, and endogenous Topo1 with anti-Topo1 antibodies using primers at the HPV31 LCR. Values were normalized to inputs. (A) Equivalent amounts of E2 proteins were detected at the origin. (B) WT and K111Q show significantly higher Topo1 recruitment compared to that of the control transfected with only E1. (C) E2 recruitment of Topo1 in the absence of E1 shows K111R is not significantly different from the control samples, and K111Q is increased compared to the wild type. (D) ChIP for replication initiation factors at the LCR and E4 ORF in synchronized CIN612 cells relative to IgG antibody control. *, p < 0.05; **, p < 0.005; ***, p < 0.0001 by one-way analysis of variance (ANOVA).
We used the HPV31-positive CIN612 cell line derived from human cervical dysplasia to determine whether localization of Topo1 is specific to the origin or if it is associated with other regions of the PV genome during initiation of DNA replication. CIN612 cells were synchronized in G1/S using a double thymidine block, and the cells were harvested and processed for ChIP assays for the endogenously expressed E1, E2, RPA, and Topo1 proteins. We compared the localization of these replication-linked proteins at the LCR and at the E4 gene. In the context of the whole HPV31 genome, all were detected at the LCR; however, none of these proteins were found at the E4 gene (Fig. 1D). This implies that Topo1 concentrates at the LCR during replication.
Topo1 recruitment correlates with acetylation at K111 by P300.
We previously reported P300-mediated acetylation at K111 in bovine PV (BPV1) E2 in vitro (17). Through mutagenesis studies, we have identified several effects that result from the K111 acetylation; however, we have not directly addressed the involvement of P300. The functional assays were conducted in C33A cells, which have very low P300 levels that simulate the conditions in keratinocytes in the lower levels of the epithelium (18). To determine if P300 is the lysine acetyltransferase (KAT) specifically involved in K111 acetylation and consequently Topo1 recruitment, we performed ChIP assays after cells were cotransfected with plasmids expressing HPV31 E2 WT or either of the mutants, the PFLORI31 plasmid containing the replication origin, and P300. Topo1 ChIP assays comparing an environment with low P300 levels to an environment with P300 overexpressed demonstrated a 3-fold higher recruitment of Topo1 to the replication origin in the presence of wild-type E2 but caused no significant changes in samples transfected with the arginine or glutamine mutants (Fig. 2A). These results confirm that P300 acetylation at K111 increases Topo1 recruitment to the origin. However, considering that acetyltransferases can exhibit a level of redundancy that may allow these to be interchangeable, this result alone does not prove that P300 is the only acetyltransferase capable of inducing this E2 activity. We therefore performed an identical experiment with an overexpression of the CREB-binding protein (CBP) (Fig. 2B). Unlike with P300, there was no increase in the presence of Topo1 at the origin. K111R also exhibited no change under high CBP concentrations, but the acetylation mimic K111Q recruited slightly diminished levels of Topo1. These data corroborate that acetylation of K111 is P300 specific.
FIG 2.

Acetylation of K111 by P300 recruits Topo1 to the viral origin. C33A cells were transfected with HPV31 E2 and PFLORI31. This experiment was conducted under endogenous KAT levels and with overexpression of P300 (A) or CBP (B). ChIP for endogenous Topo1 at the HPV31 LCR with anti-Topo1 antibody and LCR3 primers. *, p < 0.05; **, p < 0.005 by one way ANOVA.
HPV replication is dependent on acetylation at K111 by P300.
Previous experiments suggested the regulatory role of E2 on PV replication by mutating lysine 111 to arginine, glutamine, and alanine (12). After confirming that Topo1 recruitment is P300 specific (Fig. 2), we investigated whether this observation is responsible for the phenotypes of the K111 mutants. We conducted luciferase replication assays where levels of P300 and CBP were manipulated. E2 along with the E1 helicase and the PV origin-containing luciferase reporter construct (PFLORI31) were cotransfected into C33A cells at basal or overexpressed levels of the histone acetyltransferases (HATs). Although P300 and CBP are often regarded as swappable, mounting evidence indicates that these acetyltransferases have unique functions (17, 19). We questioned whether elevated P300 levels that attracted Topo1 to the origin could govern viral genome replication or whether CBP could act similarly despite an inability to stimulate E2-mediated Topo1 recruitment. This was an important comparison since CBP levels do not vary between proliferating and differentiating keratinocytes (18). Luciferase results were normalized to internal renilla controls and compared to control samples that were not transfected with E1 (Fig. 3). We observed no significant variations in luciferase levels in the groups without E1 transfected. In the presence of E1, overexpressing P300 caused an ∼6-fold increase in DNA replication in the presence of E2 WT, while samples with CBP overexpression showed no increase (Fig. 3B). Notably, P300 did not increase luciferase in parallel experiments with E2 K111R or K111Q. These results demonstrate a difference in function between the two KATs that is linked to acetylation of a single lysine in the transactivation domain and its critical role in the recruitment of Topo1.
FIG 3.

PV replication is dependent on K111 acetylation by P300. C33A cells were cotransfected with HPV31 E2 K111 mutants, pFLORI31, and pRRL with overexpression of P300 (A) and CBP (B). The luminescence was normalized to renilla luciferase. Each of the samples was compared to their respective control without E1 to cancel out any background transcriptional effects. *, p < 0.05; **, p < 0.005 by paired t test.
Keratinocytes do not maintain HPV31 K111 mutant genomes.
The HPV31 E2 K111 mutants were transferred to the viral genome in an attempt to test their ability to support replication in keratinocytes. The HPV31 genomes and pBABE-puro were cotransfected into spontaneously immortalized keratinocyte (NIKS) cells using the X-tremeGENE HP transfection system, and cells were selected with puromycin. Following selection, the NIKS cells transfected with wild-type genomes produced many colonies and maintained episomal HPV31 genomes (Fig. 4A) by both PCR analysis (Fig. 4B) and Southern blotting (Fig. 4C). The puromycin-resistant cells containing the K111Q mutant genome proliferated noticeably slower than untransfected NIKS cells or the wild-type transfected cells, and after 10 days, the genomes were undetectable. The K111R cells were slow to develop colonies, and Southern blot analysis showed the presence of episomes as well as integrated genomes (Fig. 4C). With continued passage, these cells proliferated with a doubling time of less than 24 h, and the HPV31 episomes could no longer be detected (Fig. 4C).
FIG 4.
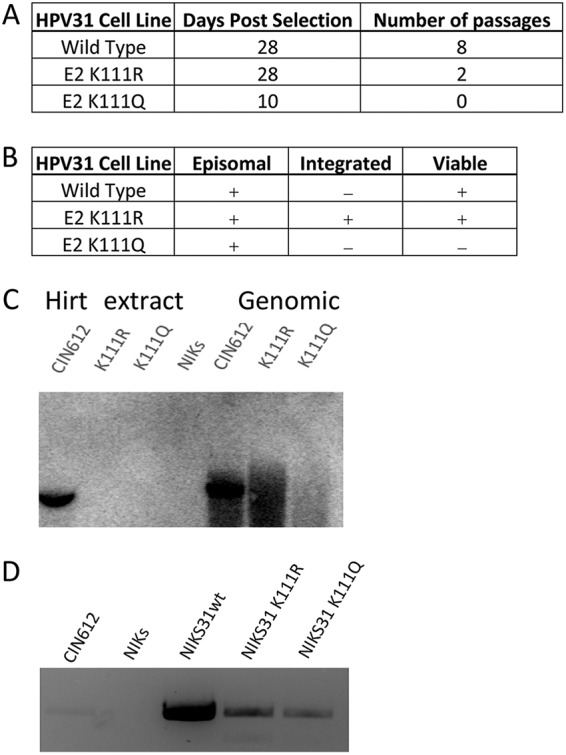
K111R does not maintain episomes in keratinocytes. Keratinocytes were transfected with HPV31 genomes and selected with puromycin. (A) Cells were maintained for 4 weeks post selection and proliferated at different rates with the E2 mutant genomes. (B) Episomal DNA was isolated, and the presence of HPV31 genomes was detected by PCR. (C) Episomal and genomic DNA isolates were visualized by Southern blotting; (D) differences in the maintenance of genomes and viability of cells were recorded.
DISCUSSION
The role of topoisomerase 1 in relieving torsional stress due to DNA unwinding is of particular importance for a small, closed, circular DNA like the PV episome. In such a system, the initial unwinding at the origin could create sufficient supercoiling to stall replication forks and require Topo1 at initiation. Another proposed role of Topo1 in mammalian replication is to aid in the unfolding of packaged chromosomes with subsequent origin recognition (20). The link between topoisomerase 1 and viral replication has been established for simian virus 40 (SV40). During SV40 replication, an initiation complex comprised of Topo1, replication protein A (RPA), and SV40 T-antigen assembles on the replication origin (21). Since increased levels of Topo1 protein did not increase SV40 DNA synthesis, it is thought that the role of Topo1 is linked to its chromatin-unfolding activity (22). Topo1 has been reported to directly interact with E1 and E2, both of which were proposed to stimulate its enzymatic activity (14, 23, 24). In addition, E2 and Topo1 were shown to independently enhance E1 binding at the origin (14). However, these reports used in vitro interaction designs and bacterially expressed proteins. HPV genomes resemble and associate with chromatin (25), which suggests that histone displacement might be a second function for Topo1 during PV replication.
We previously reported that the K111 E2 mutations did not alter E2 DNA binding or E1 recruitment to the replication origin. However, the K111R mutant was unable to induce origin unwinding and RPA accumulation (12). We suspected the requirement for Topo1 during replication because of the relevant functions and made two key discoveries. First, we found that E2 rather than E1 is responsible for recruiting Topo1 to the viral origin in live cells (Fig. 1C); intriguingly, less Topo1 was present at the origin when E1 was also introduced (Fig. 1B). These observations suggest sequential E2-mediated recruitment of Topo1, which does not remain on the origin at high levels when E1 is subsequently delivered by E2. This may occur concomitant with displacement of E2 from its complex with E1 as the latter assembles into double hexamers (26, 27). Formally, we cannot exclude a contribution of E1 in the context of its complex with E2; however, these data clearly demonstrate that E1 is not necessary or sufficient for Topo1 localization at the viral ori.
The second novel discovery derived from these experiments was that K111R failed to support while K111Q enhanced Topo1 recruitment in both a replicating (with E1) and nonreplicating (no E1) system (Fig. 1). These data imply that E2 acetylation at K111 is integral for the mobilization of Topo1 during PV replication. We show that K111R abrogates replication by restricting Topo1 recruitment to the viral origin, which effectively prevents DNA unwinding.
We endeavored to establish stable cell lines with the K111 mutants in the HPV31 genome. Our expectation was that the K111R mutant genome would not maintain episomes and would integrate into host chromosomes. The K111R genome behaved as expected, and HPV31 was present in genomic extracts and not in Hirt extracted samples (Fig. 4C). Interestingly, the K111Q genome arrested proliferation of the telomerase immortalized NIKS cells. We suspect that this acetylation mimic mutant caused overreplication of the viral genome that may have induced a DNA damage response. Further studies are planned to characterize the molecular phenotypes of the E2 K111R and K111Q genomes in primary human keratinocytes.
We reported that the lysine acetylation proteins p300, CBP, and pCAF have nonredundant activities necessary for full E2 transcriptional activity; however, we have not discerned the E2 lysines modified by these enzymes, as proteomic analyses indicated several acetylated residues (17). P300 is undetectable in basal epithelium and becomes elevated in upper strata. Furthermore, reduced levels stimulate proliferation while increased levels induce keratinocyte differentiation, thereby suggesting that P300 specifically coordinates epithelial cell growth and differentiation (18, 28). We propose that acetylation of E2 by P300 at K111 is involved in the switch between the maintenance and amplification modes of viral replication. Both the recruitment of Topo1 and viral replication were regulated by P300 in a K111-dependent manner. While CBP is similar to P300, it did not show the same pattern of protein levels between proliferating and differentiating keratinocytes. Moreover, CBP did not increase Topo1 at the viral origin or stimulate transient replication. This study infers that the molecular switch from maintenance replication to vegetative amplification hinges on P300-specific acetylation of a highly conserved lysine residue in the papillomavirus E2 protein, which coordinates recruitment of topoisomerase 1 to the viral origin.
MATERIALS AND METHODS
Cell culture, plasmids, and reagents.
C33A (D. Lowy) and J23T3 (H. Green) were maintained in Dulbecco’s modified Eagle medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (FBS) (Atlas Biologicals) and 100 U/ml penicillin-streptomycin (Life Technologies). Spontaneously immortalized keratinocyte (NIKS) cell lines were cocultured in F medium (29) with mitomycin C-treated J23T3 cells. C33A and NIKS cells were transfected with Lipofectamine 2000 at a ratio of 1:2 DNA to transfection reagent. CIN612 cells were cultured on J23T3 feeder cells in E medium to ∼70% confluence. The cells were treated with 2 mM thymidine in E medium overnight. The cells were then washed with phosphate-buffered saline (PBS) and cultured in E medium for 6 h. Thymidine was added again overnight at 2 mM. The cells were then washed with PBS and harvested. HPV31 E2 mutant plasmids were constructed by site-directed mutagenesis (QuikChange II; Agilent) and verified by sequencing of the entire E2 gene (12). HA-tagged HPV31 E2 was obtained from P. Howley (30), codon-optimized FLAG HPV31 E2 was supplied by A. McBride (31), and the HPV31 E1, pCI-RLuc, and PFLORI31 constructs were obtained from J. Archambault (32). Codon-optimized HA-31 E1 was cloned into the NheI and ApaI sites of pCDNA3.
Antibodies.
The antibodies used include mouse anti-FLAG M2 (Sigma-Aldrich) and mouse anti-HA (HA7; Sigma-Aldrich), anti-Topo1 (Cell Signaling), and glutathione- and M2-conjugated beads (Sigma-Aldrich).
Chromatin immunoprecipitation.
C33A cells were transfected with plasmids (1:1:1) expressing HPV31 E1, E2, and the HPV31 origin of replication (PFLORI31). Cells were harvested 48 h posttransfection and cross-linked in 1% paraformaldehyde for 10 min at room temperature. The cross-linking was stopped by adding 0.125 M glycine, and the cells were rinsed with cold PBS then collected by scraping. These samples were processed using the ChIP-It enzymatic shearing kit (Active Motif) using the anti-Topo1 antibody (Cell Signaling). The DNAs from the immunoprecipitations and their inputs were analyzed by quantitative PCR using the LCR4 primer pair designed against the HPV31 LCR (33).
Luciferase replication assay.
C33A cells were seeded at a density of 2.5 × 104 cells/well 24 h prior to transfection into 96-well clear bottom plates (Corning). A luciferase-based replication assay was performed as previously reported (32). Ten nanograms each of HPV31 E1 and E2 and 2.5-ng pFLORI31/pFLORIBPV-1 constructs were cotransfected into C33A cells in a 96-well plate, 0.5 ng of the pCI-RLuc construct was transfected per well to normalize for transcriptional activation of the firefly luciferase gene, and the total DNA quantity was adjusted to 100 ng with pCI. Cells were incubated in the transfection medium overnight, and then the medium was replaced with normal growth medium (DMEM). Cells were lysed in Dual-Glo luciferase reagent (Promega) 72 h posttransfection, and both firefly and renilla luciferase activities were determined using the PHERAStar FS (BMG Labtech). Firefly luciferase activity was normalized to renilla luciferase activity and the E2 control for each sample.
ACKNOWLEDGMENTS
The research reported in this publication was supported by the National Cancer Institute under grant R01CA058376.
This content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health.
REFERENCES
- 1.Hou SY, Wu SY, Zhou T, Thomas MC, Chiang CM. 2000. Alleviation of human papillomavirus E2-mediated transcriptional repression via formation of a TATA binding protein (or TFIID)-TFIIB-RNA polymerase II-TFIIF preinitiation complex. Mol Cell Biol 20:113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kadaja M, Silla T, Ustav E, Ustav M. 2009. Papillomavirus DNA replication—from initiation to genomic instability. Virology 384:360–368. doi: 10.1016/j.virol.2008.11.032. [DOI] [PubMed] [Google Scholar]
- 3.Hebner CM, Laimins LA. 2006. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity. Rev Med Virol 16:83–97. doi: 10.1002/rmv.488. [DOI] [PubMed] [Google Scholar]
- 4.Mohr IJ, Clark R, Sun S, Androphy EJ, MacPherson P, Botchan MR. 1990. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science 250:1694–1699. [DOI] [PubMed] [Google Scholar]
- 5.Bonne-Andrea C, Tillier F, McShan GD, Wilson VG, Clertant P. 1997. Bovine papillomavirus type 1 DNA replication: the transcriptional activator E2 acts in vitro as a specificity factor. J Virol 71:6805–6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sedman J, Stenlund A. 1995. Co-operative interaction between the initiator E1 and the transcriptional activator E2 is required for replicator specific DNA replication of bovine papillomavirus in vivo and in vitro. EMBO J 14:6218–6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grossel MJ, Sverdrup F, Breiding DE, Androphy EJ. 1996. Transcriptional activation function is not required for stimulation of DNA replication by bovine papillomavirus type 1 E2. J Virol 70:7264–7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ustav M, Stenlund A. 1991. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. EMBO J 10:449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Enemark EJ, Joshua-Tor L. 2006. Mechanism of DNA translocation in a replicative hexameric helicase. Nature 442:270–275. doi: 10.1038/nature04943. [DOI] [PubMed] [Google Scholar]
- 10.Enemark EJ, Chen G, Vaughn DE, Stenlund A, Joshua-Tor L. 2000. Crystal structure of the DNA binding domain of the replication initiation protein E1 from papillomavirus. Mol Cell 6:149–158. [PubMed] [Google Scholar]
- 11.Hickman AB, Dyda F. 2005. Binding and unwinding: SF3 viral helicases. Curr Opin Struct Biol 15:77–85. doi: 10.1016/j.sbi.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Thomas Y, Androphy EJ. 2018. Human papillomavirus replication regulation by acetylation of a conserved lysine in the E2 protein. J Virol 92:e01912-17. doi: 10.1128/JVI.01912-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Champoux JJ. 2001. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem 70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 14.Hu Y, Clower RV, Melendy T. 2006. Cellular topoisomerase I modulates origin binding by bovine papillomavirus type 1 E1. J Virol 80:4363–4371. doi: 10.1128/JVI.80.9.4363-4371.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loo YM, Melendy T. 2004. Recruitment of replication protein A by the papillomavirus E1 protein and modulation by single-stranded DNA. J Virol 78:1605–1615. doi: 10.1128/JVI.78.4.1605-1615.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Archambault J, Melendy T. 2013. Targeting human papillomavirus genome replication for antiviral drug discovery. Antivir Ther 18:271–283. doi: 10.3851/IMP2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quinlan EJ, Culleton SP, Wu SY, Chiang CM, Androphy EJ. 2013. Acetylation of conserved lysines in bovine papillomavirus E2 by p300. J Virol 87:1497–1507. doi: 10.1128/JVI.02771-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong PP, Pickard A, McCance DJ. 2010. p300 alters keratinocyte cell growth and differentiation through regulation of p21(Waf1/CIP1). PLoS One 5:e8369. doi: 10.1371/journal.pone.0008369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henry RA, Kuo YM, Andrews AJ. 2013. Differences in specificity and selectivity between CBP and p300 acetylation of histone H3 and H3/H4. Biochemistry 52:5746–5759. doi: 10.1021/bi400684q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Falaschi A, Abdurashidova G, Sandoval O, Radulescu S, Biamonti G, Riva S. 2007. Molecular and structural transactions at human DNA replication origins. Cell Cycle 6:1705–1712. doi: 10.4161/cc.6.14.4495. [DOI] [PubMed] [Google Scholar]
- 21.Fairman MP, Prelich G, Tsurimoto T, Stillman B. 1989. Replication of SV40 in vitro using proteins derived from a human cell extract. J Cell Sci Suppl 12:161–169. [DOI] [PubMed] [Google Scholar]
- 22.Halmer L, Vestner B, Gruss C. 1998. Involvement of topoisomerases in the initiation of simian virus 40 minichromosome replication. J Biol Chem 273:34792–34798. [DOI] [PubMed] [Google Scholar]
- 23.Clower RV, Hu Y, Melendy T. 2006. Papillomavirus E2 protein interacts with and stimulates human topoisomerase I. Virology 348:13–18. doi: 10.1016/j.virol.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 24.Clower RV, Fisk JC, Melendy T. 2006. Papillomavirus E1 protein binds to and stimulates human topoisomerase I. J Virol 80:1584–1587. doi: 10.1128/JVI.80.3.1584-1587.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.You J. 2010. Papillomavirus interaction with cellular chromatin. Biochim Biophys Acta 1799:192–199. doi: 10.1016/j.bbagrm.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sedman J, Stenlund A. 1998. The papillomavirus E1 protein forms a DNA-dependent hexameric complex with ATPase and DNA helicase activities. J Virol 72:6893–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen G, Stenlund A. 2001. The E1 initiator recognizes multiple overlapping sites in the papillomavirus origin of DNA replication. J Virol 75:292–302. doi: 10.1128/JVI.75.1.292-302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muller A, Ritzkowsky A, Steger G. 2002. Cooperative activation of human papillomavirus type 8 gene expression by the E2 protein and the cellular coactivator p300. J Virol 76:11042–11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allen-Hoffmann BL, Schlosser SJ, Ivarie CAR, Meisner LF, O’Connor SL, Sattler CA. 2000. Normal growth and differentiation in a spontaneously immortalized near-diploid human keratinocyte cell line, NIKS. J Invest Dermatol 114:444–455. doi: 10.1046/j.1523-1747.2000.00869.x. [DOI] [PubMed] [Google Scholar]
- 30.Powell ML, Smith JA, Sowa ME, Harper JW, Iftner T, Stubenrauch F, Howley PM. 2010. NCoR1 mediates papillomavirus E8̂E2C transcriptional repression. J Virol 84:4451–4460. doi: 10.1128/JVI.02390-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oliveira JG, Colf LA, McBride AA. 2006. Variations in the association of papillomavirus E2 proteins with mitotic chromosomes. Proc Natl Acad Sci U S A 103:1047–1052. doi: 10.1073/pnas.0507624103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fradet-Turcotte A, Morin G, Lehoux M, Bullock PA, Archambault J. 2010. Development of quantitative and high-throughput assays of polyomavirus and papillomavirus DNA replication. Virology 399:65–76. doi: 10.1016/j.virol.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeSmet M, Kanginakudru S, Rietz A, Wu WH, Roden R, Androphy EJ. 2016. The replicative consequences of papillomavirus E2 protein binding to the origin replication factor ORC2. PLoS Pathog 12:e1005934. doi: 10.1371/journal.ppat.1005934. [DOI] [PMC free article] [PubMed] [Google Scholar]