

The role of IFN-induced genes (ISGs) in viral infection remains incompletely understood. While most ISGs are antiviral, some ISGs have been shown to promote viral infection, including HIV-1 infection. We previously showed that IFN-inducible LY6E protein promotes HIV-1 infection in human PMBCs and high-CD4-expressing SupT1 cells. Here we found that LY6E inhibits HIV-1 entry and replication in low-CD4-expressing MDMs and Jurkat cells. Mechanistically, we demonstrated that LY6E downregulates the cell surface receptor CD4, thus impairing the virus binding to target cells. This is in contrast to the situation of high-CD4-expressing cells, where LY6E predominantly promotes viral membrane fusion. The opposing role of IFN-inducible LY6E in modulating HIV-1 infection highlights the complex roles of ISGs in viral infection and viral pathogenesis.
KEYWORDS: CD4, LY6E, entry, human immunodeficiency virus
ABSTRACT
Lymphocyte antigen 6E (LY6E) is a GPI-anchored, interferon-inducible protein that has been shown to modulate viral infection in a cell type-dependent manner. Our recent work showed that LY6E promotes HIV-1 infection in some high-CD4-expressing cells, including human peripheral blood mononuclear cells (PBMCs) and the SupT1 cell line. In this work, we provide evidence that LY6E inhibits HIV-1 entry and spread in low-CD4-expressing Jurkat cells and human monocyte-derived macrophages (MDMs) through downregulation of the viral receptor CD4. We found that knockdown of LY6E in Jurkat cells and MDMs increases HIV-1 infection, yet overexpression of LY6E in Jurkat cells inhibits HIV-1 entry and replication. LY6E was found to be colocalized with CD4 on the plasma membrane of Jurkat cells and MDMs and enhances CD4 internalization. We artificially manipulated the CD4 level in Jurkat and SupT1 cells and found that overexpression of CD4 in Jurkat cells overcomes the inhibitory effect of LY6E; conversely, blocking the function of CD4 in SupT1 with a neutralizing antibody eliminates the enhancement of LY6E on HIV-1 entry. The CD4-dependent inhibitory phenotype of LY6E in low-CD4-expressing human MDMs can be recapitulated for a panel of transmitted founder viruses and laboratory-adapted HIV-1 strains. Given that HIV-1 can target low-CD4-expressing cells during acute infection yet replicates efficiently in high-CD4-expressing T cells at the late stage of disease, our observation that LY6E differentially modulates HIV-1 replication in a CD4-dependent manner has implications for understanding the complex roles of interferon (IFN)-induced proteins in AIDS pathogenesis.
IMPORTANCE The role of IFN-induced genes (ISGs) in viral infection remains incompletely understood. While most ISGs are antiviral, some ISGs have been shown to promote viral infection, including HIV-1 infection. We previously showed that IFN-inducible LY6E protein promotes HIV-1 infection in human PMBCs and high-CD4-expressing SupT1 cells. Here we found that LY6E inhibits HIV-1 entry and replication in low-CD4-expressing MDMs and Jurkat cells. Mechanistically, we demonstrated that LY6E downregulates the cell surface receptor CD4, thus impairing the virus binding to target cells. This is in contrast to the situation of high-CD4-expressing cells, where LY6E predominantly promotes viral membrane fusion. The opposing role of IFN-inducible LY6E in modulating HIV-1 infection highlights the complex roles of ISGs in viral infection and viral pathogenesis.
INTRODUCTION
Despite the success of current antiretroviral therapy, HIV/AIDS still remains one of the greatest threats to human public health, particularly in developing countries (1). The efficiency of HIV entry critically determines the capacity of the virus to spread and cause pathogenesis in patients (2). HIV entry into target cells proceeds with engagement of the viral envelope glycoprotein (Env) gp120 with the primary cellular receptor CD4, leading to Env conformational changes that further facilitate the interaction between the gp120 V3 loop and coreceptor CCR5 or CXCR4 (2). Coreceptor binding triggers the fusion machinery and subsequent formation of the six-helix bundle of Env that drives fusion between the viral and host cell membranes (2, 3). A functional block of receptor, coreceptors, or HIV Env can effectively impair HIV entry and spread.
As a typical type I transmembrane glycoprotein, CD4 and its expression patterns are pivotal in governing the HIV-1 permissiveness into target cells and AIDS pathogenesis (4). CD4 is primarily expressed in T lymphocytes, monocytes, macrophages, dendritic cells, and brain microglia, etc., which are the main targets of HIV infection (5). However, despite the essential role of CD4 during HIV entry, a fine regulation of the cell surface CD4 level is critical for viral replication and pathogenesis (6). Indeed, HIV actively modulates CD4 trafficking and expression by encoding some proteins, such as Nef, Vpu, and Env. In the early stage of infection, HIV-1 Nef accelerates the cell surface CD4 endocytosis and subsequent lysosomal degradation (7), whereas in the late stage, the translated Env precursor gp160 binds to newly synthesized CD4 and retains the latter in the endoplasmic reticulum (ER) (8). Vpu is involved in the degradation of newly synthesized CD4, thus releasing gp160 from the gp160-CD4 complex, leading to enhanced viral infectivity (9, 10). Interestingly, modulation of CD4 expression by cellular factors, especially those of so-called restriction and dependency factors, has been rarely reported. In contrast, several host proteins have recently been shown to block HIV entry and/or to impair HIV-1 infectivity through modulation of the viral Env function; these include IFITMs (11–13), SERINCs (14, 15), MARCH8 (16), and 90K (17). The currently proposed mechanisms include direct interactions between Env and host factors (11), an alteration of Env conformation (18), the inhibition of Env processing (11, 17), and negative imprinting of host factors (14, 15, 19, 20).
The lymphocyte antigen 6 complex, locus E gene (LY6E) is an interferon (IFN)-inducible glycosylphosphatidylinositol (GPI)-anchored protein that has been associated with some normal physiological functions of the cells, including T lymphocyte differentiation and development (21), T-cell receptor (TCR)-mediated cell signaling and T cell activation (22–24), cellular adhesion (25), and oncogenesis and cancer progression (26–28). As with many other IFN-induced genes (ISGs), the role of LY6E in modulating viral infections has been investigated but with mixed results. For instance, LY6E has been reported to inhibit replication of vesicular stomatitis virus (VSV) (29). However, for several other viruses, including Marek’s disease virus (MDV) (30), mouse adenovirus type 1 (MAV-1) (31), yellow fever virus (YFV), and West Nile virus (WNV) (32), LY6E appears to promote viral replication. Interestingly, overexpression of LY6E in STAT−/− fibroblasts and in Huh-7 hepatocytes has produced different outcomes upon YFV infection: in STAT−/− fibroblasts, LY6E significantly enhances YFV infection, while in contrast, in Huh-7 cells, LY6E has no apparent effect on YFV (32), revealing a cell type-dependent function of LY6E for YFV. One recent study reported that LY6E enhances the internalization of multiple flaviviruses, including WNV, dengue virus (DENV), and Zika virus (ZIKV), through a specific tubularization process (33). Another recent study showed a conserved enhancement of LY6E family proteins on viral infection by acting on the late step of viral entry (34). However, exactly how LY6E modulates viral infection in different cell types remains unknown.
For retroviruses, we have recently shown that LY6E promotes HIV-1 infection in high-CD4-expressing cell types, including primary peripheral blood mononuclear cells (PBMCs), SupT1 cells, and THP-1 cells (35), which is in accordance with earlier reports (36, 37). Our mechanistic studies revealed that LY6E promotes HIV-1 entry and gene expression, although detailed mechanisms still await to be elucidated (35). Another important question is whether or not the effect of LY6E in high-CD4-expressing PBMCs and SupT1 cells can also be demonstrated in low-CD4-expressing cells and, if not, what the possible underlying mechanisms are. In this study, we investigated the features of LY6E in modulating HIV-1 infection in low-CD4-expressing human monocyte-derived macrophages (MDMs) and Jurkat T cells, and we found that in contrast to its enhancing effect on HIV-1 infection in high-CD4-expressing cells, LY6E restricts HIV-1 in low-CD4-expressing MDMs and Jurkat cells, in part by downregulating the cell surface CD4, thus impairing the virus binding and uptake into target cells.
RESULTS
LY6E inhibits HIV-1 replication in Jurkat T cells.
Jurkat is one of the commonly used cell lines for HIV study, yet compared to many other T cell lines such as SupT1 cells, Jurkat expresses a very low level of CD4 (38). To determine if the phenotype of LY6E we observed for HIV-1 infection in SupT1 cells (35) can be observed in Jurkat cells, we generated a stable Jurkat cell line that overexpresses an N-terminally FLAG-tagged LY6E, the same construct as used in SupT1 cells (35). The N-terminal FLAG tag was preceded by a pre-protrypsin signal sequence so that the protein can be targeted to and traffic through the cellular secretory pathway. We confirmed the expression of the FLAG-tagged LY6E protein by Western blotting (Fig. 1A) and determined its effect on multiround HIV-1 replication. To our surprise, we found that ectopic expression of LY6E attenuated the replication of HIV-1 NL4.3, as evidenced by reduced reverse transcriptase (RT) activities over a period of 24 days (Fig. 1B); the peak of HIV-1 infection was also delayed, i.e., from day 12 in mock-infected control cells to day 15 in LY6E-expressing cells (Fig. 1B). To rule out a possible artificial effect of FLAG tag on LY6E suppression of HIV-1 replication, we generated another stable Jurkat cell line expressing the wild-type (WT) LY6E. The expression levels of WT and FLAG-tagged LY6E were comparable, with that of WT LY6E slightly higher than the FLAG-tagged version based on the flow cytometric determination (Fig. 1C). We then performed a short-term HIV-1 replication assay (for 48 h) and observed that both WT and FLAG-tagged LY6E proteins inhibited HIV-1 infection, as measured by RT activity (Fig. 1D) and viral titer (Fig. 1E). It was of note that the WT LY6E protein exhibited a relatively stronger inhibitory effect (Fig. 1D and E), consistent with its slightly higher level of expression (Fig. 1C).
FIG 1.

LY6E inhibits HIV-1 replication in low-CD4-expressing Jurkat T cells. (A) Expression of FLAG-tagged LY6E in Jurkat cells (FLAG-LY6E or F-LY6E here) as examined by Western blotting using an anti-FLAG antibody. (B) Replication of infectious NL4.3 in Jurkat and Jurkat-F-LY6E cells. Replication was monitored by measuring the RT activity of the supernatants containing the newly released virions every 2 days. (C) Expression of FLAG-tagged or WT LY6E proteins on the surface of Jurkat T cells as determined by flow cytometry using an anti-LY6E antibody. The mean fluorescence intensities (MFIs) of LY6E are as follows: unstained, 425; mock, 440; FLAG-LY6E, 6,015; LY6E, 6,277. (D and E) RT activity and viral titer (infectivity) of HIV-1 NL4.3 virions produced from vector control Jurkat cells, Jurkat-LY6E cells, and Jurkat-FLAG-LY6E cells. The viral infectivity or titer was determined by infecting HeLa-TZM indicator cells, followed by measuring the firefly luciferase activity of infected cells. (F) Expression of endogenous LY6E in Jurkat cells and shRNA knockdown efficiency. LY6E was knocked down by lentiviral shRNA transduction, and the knockdown efficiency was determined by flow cytometry by using an anti-LY6E antibody. The second antibody alone indicates cells being incubated with FITC-conjugated secondary antibody only, which serves as a negative control. The MFIs of LY6E are as follows: unstained, 1,077; second antibody alone, 1,110; shRNA LY6E, 1,279; shRNA control, 2,099. (G and H) Effect of LY6E knockdown on RT activity and viral titer in Jurkat cells in short-term replication assay. Experiments were performed the same as described for panels D and E, except that LY6E knockdown Jurkat cells were used for infection for 48 h. (I) Effect of LY6E knockdown on HIV-1 long-term replication. Approximately 2.5 ng p24 equivalent HIV-1NL4.3 was applied to 1 × 105 Jurkat shRNA control or shRNA LY6E cells for infection, and media were replaced with fresh RPMI 1640 12 h postinfection. Virus-containing supernatants were collected every 2 days, and cells were split every 4 days. Released HIV-1 virions in the supernatants were quantified by an RT assay. In all cases, data are means ± SD (standard deviation) of the results of at least 3 independent experiments. *, P < 0.05; **, P < 0.01. Unless otherwise specified, all data shown were from Jurkat cells expressing FLAG-LY6E (see details below).
We next utilized short hairpin RNA (shRNA) that targets the endogenous LY6E in Jurkat cells and determined its effect on HIV-1 replication. While the level of endogenous LY6E in Jurkat cells was not high, shRNA treatment led to its reduction to a level close to the background shown by flow cytometry (Fig. 1F). In accordance, we observed increased HIV-1 RT activity and viral titers in shRNA-treated cells compared to those of the shRNA control (Fig. 1G and H). A long-term replication assay revealed increased HIV-1 RT activity on day 10 in Jurkat cells expressing shRNA LY6E compared to that in shRNA control cells (Fig. 1I) All together, these results suggested that endogenous LY6E in Jurkat cells intrinsically inhibits HIV-1 replication.
Knockdown of endogenous LY6E in human MDMs increases replication of CCR5-tropic primary HIV-1 isolates, including TF viruses.
We next evaluated the effect of endogenous LY6E on HIV-1 replication in human primary MDMs, which are known to express low levels of CD4 (38). We pretransfected MDMs of three healthy donors with small interfering RNA (siRNA) targeting LY6E, infected these cells with a panel of CCR5-tropic HIV-1 isolates, i.e., AD8, YU2, or transmitted/founder (TF) viruses WITO, RHPA, and THRO, for 48 h, and measured their short-term replications. The siRNA knockdown efficiency of LY6E in these PBMCs was determined by reverse transcription-quantitative PCR (qRT-PCR) (∼50% to 60%), as shown in Fig. 2A, C, and E. In all cases, we observed increased viral replication in LY6E knockdown cells compared to that of the siRNA control, despite some donor-to-donor variations (Fig. 2B, D, and F). Overall, these results revealed that endogenous LY6E protein restricts HIV-1 infection in low-CD4-expressing human MDMs.
FIG 2.

Knockdown of LY6E in human MDMs increases HIV-1 infection. (A, C, and E) siRNA control or siRNAs targeting LY6E were transfected into primary human MDMs derived from three healthy donors prior to infection by different HIV-1 isolates. Knockdown efficiency was quantified by qRT-PCR. (B, D, and F) Infectious HIV-1 isolates AD8, YU2, and three transmitted/founder viruses were used to infect MDMs for ∼48 h. The viral infectivity was measured by infecting indicator HeLa-TZM cells and plotted as relative light units (RLU). Data for infectivity are means ± SD (standard deviation) of the results of triplicate experiments; no statistical analysis can be performed in this case. For siRNA knockdown efficiency, the values were obtained by averaging numbers from all siRNA-treated and infected cells in the same donor. *, P < 0.05, **, P < 0.01.
LY6E impedes HIV-1 entry in low-CD4-expressing Jurkat cells and MDMs.
We evaluated how LY6E might modulate HIV-1 infection in Jurkat cells. Given that LY6E has a GPI anchor, which normally targets the protein of interest to the lipid raft microdomain on the plasma membrane (39), we tested whether LY6E affected the early step of HIV-1 infection, i.e., at the viral entry level. We first carried out a BlaM-Vpr assay and examined entry of NL4.3 Env-mediated HIV-1 in Jurkat cells either overexpressing LY6E, expressing LY6E shRNA, or expressing shRNA control; VSV-G-mediated entry served as a control. As shown in Fig. 3A, NL4.3-mediated entry was reduced by ∼50% when LY6E was overexpressed but was enhanced by 40% when LY6E was knocked down. As was expected, VSV-G-mediated entry was insensitive to LY6E, regardless of LY6E overexpression or knockdown, which was consistent with our previous results (35).
FIG 3.

LY6E impairs HIV-1 entry in Jurkat cells and downregulates the cell surface CD4. (A) Entry mediated by NL4.3 Env or VSV-G was determined by infecting individual cell lines with an HIV-1 vector expressing BlaM-Vpr. Fusion efficiency was determined by measuring cleaved CCF2 (blue) from uncleaved CCF2 (green). Relative entry efficiency was calculated by setting the fusion of empty vector to 1.0 and plotted. (B) Single-round infection of HIV-1 inGLuc bearing Env of NL4.3, BH10, HXB2, JRFL, or three transmitted/founder viruses was determined by infecting Jurkat-CCR5 or Jurkat-CCR5-LY6E cells. Entry efficiency was determined by measuring the Gaussia luciferase activity of harvested supernatants. Relative entry was calculated and plotted by setting the entry of NL4.3 into Jurkat empty vector cell to 1.0. (C and D) MDMs derived from two healthy donors (donor 1 and donor 2) were transfected with siRNA targeting either LY6E or scramble control. Cells were then infected with HIV-1-inGLuc bearing the Env of CCR5-tropic NL(AD8). The LY6E knockdown efficiency was examined by qPCR (C). The single-round infection rate was determined by measuring the Gaussia luciferase activities at 48 h postinfection (D). VSV-G-mediated single-round infection served as a negative control. (E) Effect of LY6E on CXCR4 expression in Jurkat and Jurkat-LY6E cells. Jurkat cells transduced with empty pQCXIP vector control or pQCXIP encoding LY6E were stained with a primary anti-CXCR4 monoclonal antibody, followed by incubation with a secondary FITC-conjugated anti-mouse antibody; cells were fixed and analyzed by flow cytometry. The MFIs of CXCR4 are as follows: second antibody alone, 785; LY6E, 20,381; pQCXIP, 21,011. Ab, antibody. (F) Detection of LY6E and CD4 expression on the surface of Jurkat, Jurkat-LY6E, and Jurkat-shRNA LY6E cells. The MFIs of LY6E are as follows: second antibody alone, 910; shRNA LY6E, 1,111; mock, 2,214; LY6E, 9,913. The MFIs of CD4 are as follows: second antibody alone, 910; shRNA LY6E, 777; mock, 445; LY6E, 287. (G) Examination of the total and surface levels of CD4 in Jurkat cells. Cells were nonpermeabilized or permeabilized to determine the surface or total level of CD4. The MFIs of CD4 in nonpermeabilized Jurkat cells are as follow: second antibody alone, 401; mock, 1,777; LY6E, 1,287. The MFIs of CD4 in permeabilized Jurkat cells are as follows: second antibody alone, 2,101; mock, 12,098; LY6E, 10,117. The “2nd antibody alone” indicates cells being incubated with FITC-conjugated secondary antibody only, which served as a negative control. “Unstained” indicates cells stained with neither first nor second antibody. (H) Binding of infectious HIV-Gag-iGFP particles to Jurkat or Jurkat-LY6E cells. Cells were incubated with purified HIV-1 virions for 2 h on ice. Unbound virus was removed by an extensive wash with cold PBS. Binding was measured by detection of GFP-positive cells by flow cytometry. Results shown are representatives of a typical experiment. The experiment was repeated once with similar results. Unless otherwise specified, all data shown are means ± SD of the results of at least 3 independent experiments. *, P < 0.05; ***, P < 0.001.
To confirm these results, we established a Jurkat/CCR5 cell line and expanded our analysis to some other CCR5- and CXCR4-tropic viruses using an HIV-intron Gaussia luciferase (HIV-inGLuc) system; this assay allowed us to measure a single-round HIV-1 infection, with higher sensitivity than that of the BlaM-Vpr assay (11). With this assay, we observed that LY6E inhibited infection of three X4-tropic viruses, i.e., NL4.3, HXB2, and BH10, two R5-tropic viruses, i.e., AD8 and JRFL, and three TF viruses, i.e., pBJOX2000, pX1632, and pCH119 (Fig. 3B).
We also knocked down the endogenous LY6E expression in human MDMs of two healthy donors by siRNA and examined its effect on HIV-1 infection using this sensitive HIV-inGLuc-based assay. Real-time PCR assays confirmed that LY6E mRNA was effectively depleted by siRNA in MDMs (up to 60%) (Fig. 3C). Subsequent infection of these MDMs by AD8 Env-bearing HIV-inGLuc showed that knockdown of LY6E enhanced, albeit modestly, the AD8 Env-mediated infection but had no significant effect on that of VSV-G (actually slightly decreased) (Fig. 3D). These results, along with the results of Jurkat/CCR5 cells overexpressing LY6E, collectively indicated a conserved inhibitory effect of LY6E on HIV-1 infection in low-CD4-expressing cells.
LY6E downregulates CD4 at the plasma membrane.
For T lymphocytes, HIV-1 entry requires an engagement of the viral Env glycoprotein with the cell surface receptor CD4 and coreceptor CXCR4 (2). We thus explored whether these two key molecules in target cells would be affected by LY6E. We found that despite a high-level expression of the FLAG-tagged LY6E in Jurkat cells, the CXCR4 level at the cell surface remained unaltered (Fig. 3E). In contrast, the CD4 level on the cell surface was downregulated in Jurkat cells overexpressing LY6E cells but was upregulated in Jurkat cells expressing shRNA targeting LY6E cells (Fig. 3F); these were based on the histogram plots of the flow cytometric analyses as well as the calculated mean fluorescence intensities (MFIs) measured by using a CD4-specific monoclonal antibody known as SIM.4 (Fig. 3F, see legend). Similar results were also obtained by using another anti-CD4 antibody known as SIM.2 (data not shown) that targeted a different epitope on the CD4 molecule (40), implying that the reduced signal revealed by SIM.4 was unlikely to be due to an epitope masking. Further experiments showed that only the cell surface expression level (nonpermeabilized) of CD4, but not the total level (permeabilized) of CD4, was reduced by LY6E (Fig. 3G).
We then examined whether HIV-1 binding to Jurkat-LY6E cells (Jurkat cells expressing LY6E) was altered by using purified HIV-Gag-internal GFP (iGFP) virions; the fluorescent HIV-1 particles allowed us to directly visualize and measure the HIV-1 binding to target cells by flow cytometry (59). In one representative experiment, shown in Fig. 3F, we observed an ∼9.6% HIV-Gag-iGFP-positive population in empty-vector-transduced Jurkat cells compared to an ∼5.5% population in Jurkat cells expressing LY6E (Fig. 3H). In another separate experiment, we observed 14.3% of iGFP binding to Jurkat control cells compared to 6.0% to the LY6E-expressing Jurkat cells (data not shown). Overall, these results demonstrated that LY6E in Jurkat cells inhibits HIV-1 entry and downregulates the cell surface CD4.
Manipulating the cell surface CD4 level in T cells can change the pattern of LY6E restriction or enhancement of HIV-1 infection.
We then directly compared the effects of LY6E on HIV-1 single-round infection in low-CD4-expressing Jurkat and high-CD4-expressing SupT1 cells using the HIV-inGluc assay. Consistent with the result of the BlaM-Vpr assay shown in Fig. 3A, LY6E overexpression decreased the HIV-inGLuc infection in Jurkat cells by ∼80% relative to the mock control (Fig. 4A). In contrast, HIV-1 infection in SupT1 cells overexpressing LY6E was increased by ∼30% (Fig. 4A), a modest but consistent result that was similar to what we had previously reported (35). The overexpression levels of LY6E in Jurkat and SupT1 cells were comparable, as examined by flow cytometry (Fig. 4B; see MFI values in the legend). Consistent with an earlier report (38), we found that the endogenous CD4 level on the surface of SupT1 cells was much higher than that in Jurkat cells (Fig. 4C). We therefore asked if an overexpression of CD4 in Jurkat cells would overcome the inhibitory effect of LY6E on HIV-1 entry and, vice versa, whether or not a neutralization of the functional CD4 molecule on the surface of SupT1 cells would reverse the enhancing effect of LY6E on HIV-1. To facilitate these comparisons, we first overexpressed CD4 molecules in both parental Jurkat cells and Jurkat-LY6E cells using retroviral transduction. As shown in Fig. 4D, the levels of overexpressed CD4 in parental Jurkat and Jurkat-LY6E cells were comparable. We found that in contrast to the marked suppression of HIV-1 infection by LY6E in parental Jurkat cells (Fig. 4E, compare column 2 with column 1), overexpression of CD4 in parental Jurkat cells and Jurkat cells overexpressing LY6E resulted in comparable levesl of HIV-1 infection (Fig. 4E, compare column 3 and column 4).
FIG 4.

Overexpression of CD4 in Jurkat cells overcomes the inhibitory effect of LY6E on HIV-1; conversely, neutralization of CD4 on the surface of SupT1 cells eliminates the enhancement of LY6E infection. (A) Comparison of HIV-1 single-round infection in Jurkat and SupT1 cells overexpressing LY6E. Cells were infected with HIV-1-NL4.3-inGLuc viral stocks; 24 h after infection, supernatants were collected for measurement of the Gaussia luciferase activity. (B and C) The expression levels of LY6E (B) and CD4 (C) on Jurkat and SupT1 cell surfaces were examined by flow cytometry. The “2nd antibody alone” indicates cells being incubated with FITC-conjugated secondary antibody only, which serves as a negative control. The MFIs of LY6E in Jurkat cells are as follows: second antibody alone, 355; LY6E, 9,981; mock, 1,011. The MFIs of LY6E in SupT1 cells are as follows: second antibody alone, 1,255; LY6E, 8,210; mock, 991. The MFI of CD4 in Jurkat cells is 3,033, and the MFI of CD4 in SupT1 cells is 42,131. (D) Examination of LY6E and CD4 expression levels in individual Jurkat cell lines by flow cytometry. The MFIs of CD4 are as follows: Jurkat-Mock, 598; Jurkat-CD4, 5,985; Jurkat-LY6E, 402; Jurkat-CD4-LY6E, 6,100. The MFIs of LY6E are as follows: Jurkat-Mock, 383; Jurkat-CD4, 385; Jurkat-LY6E, 1,402; Jurkat-CD4-LY6E, 1,300. (E) Single-round HIV-inGLuc infection in Jurkat, Jurkat-LY6E, Jurkat-CD4, or Jurkat-CD4-LY6E cells. Infectivity was determined by measuring the Gaussia luciferase activity of virions harvested at 24 h postinfection. Relative infection was plotted by setting the values of parental cells (column 1) to 1.0. (F and G) Single-round infection of HIV-inGLuc virions bearing HIV-1 NL4.3 Env (F) or VSV-G (G) into SupT1 or SupT1-LY6E cells was determined by pretreating cells with 0, 5, or 50 ng/ml anti-CD4 neutralizing antibodies for 1 h, followed by infection of cells for 36 h. Supernatants were harvested at 48 h postinfection and measured for Gaussia luciferase activity. Relative infection was shown by setting the values of luciferase activity of supernatants harvested from parental SupT1 cells without anti-CD4 treatment to 1.0. Data are means ± SD of the results of at least 3 independent experiments. NS, not significant; *, P < 0.05; **, P < 0.01.
We then neutralized the CD4 molecules on the surface of parental SupT1 and SupT1-LY6E cells by using a monoclonal antibody against human CD4 (clone SK3, catalog number 344602; BioLegend). We observed that this treatment gradually abolished the LY6E-mediated enhancement of HIV-1 infection in SupT1-LY6E cells in an anti-CD4 antibody dose-dependent manner (Fig. 4F) yet had no apparent effect on the entry mediated by VSV-G (Fig. 4G). We attempted to knock down the total CD4 level in SupT1 cells by using shRNA lentiviral transduction; however, the knockdown efficiency was poor, and the results were not informative (data not shown). Nevertheless, the data shown here support the notion that the cell surface CD4 level is a critical determinant of the differential effects of LY6E on HIV-1 entry in Jurkat and SupT1 cells.
LY6E is associated with CD4 on the plasma membrane and promotes its internalization.
To investigate how the cell surface CD4 is downregulated by LY6E, we performed an internalization assay to determine the rate of CD4 endocytosis from the plasma membrane in Jurkat cells expressing or not expressing LY6E; this was achieved by using a phycoerythrin (PE)-Texas Red-conjugated anti-CD4 antibody, with the rationale that antibody binding would trigger endocytosis of CD4 coupled with the fluorophores. Using this assay, we observed that the half-life of CD4 on the plasma membrane of the parental Jurkat cells was approximately 30 min; in comparison, the half-life of CD4 in Jurkat-LY6E cells was shortened to less than 5 min (Fig. 5A), indicating that expression of LY6E greatly accelerated the CD4 internalization in Jurkat cells. We further examined the possible routes of CD4 internalization by using some pharmacological inhibitors: Dynasore blocks dynamin-2-mediated endocytic processes (41), whereas chlorpromazine (CPZ) and methyl-β-cyclodextrin (MBCD) block clathrin- and caveolin-dependent endocytosis pathways (42), respectively. We found that LY6E reduced the cell surface CD4 level up to 40% in dimethyl sulfoxide (DMSO)-treated cells, yet treatment of cells with Dynasore and MBCD, but not CPZ, virtually rescued the CD4 downregulation by LY6E (Fig. 5B), suggesting that LY6E enhances CD4 endocytosis likely through a cholesterol-related, lipid raft-dependent pathway.
FIG 5.

LY6E is associated with CD4 on the plasma membrane and promotes its internalization. (A) Kinetics of CD4 internalization in the presence or absence of LY6E. CD4 internalization was measured by incubating Jurkat or Jurkat-LY6E cells with a PE-Texas Red-conjugated anti-CD4 antibody (clone S3.5; Invitrogen) on ice for 2 h, followed by switching the temperature to 37°C at different periods of time. At each time point, one half of the harvested cells was treated with pH 3.0 in order to remove the surface CD4, the signals thus reflecting the internalized fraction of CD4. The other half was treated with pH 7.0 to measure the total CD4 level. The CD4 internalized rate was calculated as the ratio of pH 3.0-resistant PE-Texas Red fluorescence intensity versus the total cell-associated fluorescence intensity (pH 7.0). Relative rates of internalization were shown by setting the values of parental Jurkat cells at 60 min without acid wash to 1.0. (B) Effect of different chemical inhibitors on the steady-state level of cell surface CD4 expression. DMSO, 80 μM Dynasore, 10 μM CPZ, or 10 mM MBCD was applied to Jurkat or Jurkat-LY6E cells; 8 h after treatment, cells were collected to determine the cell surface expression of CD4 by flow cytometry. The MFI (geometric mean) of each sample was obtained and compared to the value of DMSO-treated parental cells, which was set to 1.0. (C) Lipid raft flotation assay. Jurkat or Jurkat-LY6E cells were lysed and homogenized in 1% Triton X-100 lysis buffer at 4°C, and harvested lysates were subjected to sucrose cushion ultracentrifugation to determine the water-soluble and detergent-resistant membrane fractions. Samples were collected from the top fraction to the bottom fraction upon completion of ultracentrifugation and subjected to SDS-PAGE; CD4/LY6E expression and distribution was determined by immunoblotting using specific antibodies. (D) Analysis of colocalization between LY6E and CD4. Jurkat-TAg cells (Jurkat cells expressing simian virus 40 [SV40] T antigen but with high transfection efficiency) were cotransfected with plasmids encoding CD4, GFP-GPI, or TIM-1. Thirty-six hours after transfection, cells were fixed and costained with specific antibodies. White arrows indicate colocalized areas of the plasma membrane that were positive for both LY6E and CD4. (E) Randomly selected cells (5 fields) were used for colocalization analysis using NIH ImageJ software, and the Pearson’s correlation coefficients were determined and plotted. (F) Analysis of colocalization between LY6E and CD4 in MDMs. MDMs were permeabilized with a permeabilization buffer (Fermentas) and intracellularly stained with Texas Red-conjugated anti-CD4 and anti-LY6E antibodies, followed by incubating cells with a FITC-conjugated anti-rabbit antibody. The nucleus was stained with DAPI. White arrows indicate colocalized LY6E and CD4. Images were collected under 100× magnification and processed through deconvolution software. *, P < 0.05; ***, P < 0.001.
To further explore how LY6E modulates CD4 from the cell surface, we examined a possible interaction between LY6E and CD4 by performing a coimmunoprecipitation (co-IP) assay. However, despite much effort, we observed only a weak CD4 signal in a co-IP complex immunoprecipitated by an anti-FLAG (LY6E) antibody in Triton X-100 (1%, vol/vol) lysis buffer, but not in radioimmunoprecipitation (RIPA) buffer (data not shown). This raised the possibility that the possible interaction between LY6E and CD4 was weak or that LY6E might have depleted CD4 from the lipid raft microdomain. Previously, CD4 has been shown to be present in lipid rafts and that CD4 in this microdomain is associated with the functional entry of HIV-1 (43–45). Our raft flotation assay revealed that, indeed, a portion of CD4 was present in the lipid raft microdomain of parental Jurkat cells (Fig. 5C, fractions 3, 4, and 5); however, overexpression of LY6E, which was predominantly present in the lipid rafts, led to the disappearance of CD4 from the lipid raft to some of the soluble fractions (Fig. 5C).
We next analyzed the possible subcellular colocalizations of LY6E and CD4 in Jurkat cells by immunostaining and microscopic imaging. We found that despite the significant downregulation of CD4 by LY6E, CD4 and LY6E were still partly colocalized, with a Pearson’s correlation coefficient of ∼0.6 (Fig. 5D and E). As would be expected, LY6E was strongly colocalized with a GPI-anchored green fluorescent protein (GFP) known to be in lipid rafts (46, 47), with a Pearson’s correlation coefficient of ∼0.7 (Fig. 5D and E). As a control, LY6E did not significantly colocalize with TIM-1, a cell surface molecule that is not present in lipid rafts (unpublished data), as reflected by a Pearson’s correlation coefficient of 0.3 (Fig. 5D and E). We also performed immunostaining of LY6E and CD4 in MDMs, and we observed that LY6E and CD4 colocalized in these cells (Fig. 5F). Collectively, these data showed that LY6E is associated with CD4, likely in the lipid raft microdomain of the plasma membrane; this would accelerate the internalization of CD4 via lipid raft-dependent pathways, resulting in impaired virus binding and infection.
DISCUSSION
LY6E has been well characterized in mediating the development and maturation of thymocytes (21). However, its exact role in viral infection remains debatable (30, 48). We recently reported that LY6E promotes HIV-1 entry in PBMCs and SupT1 cells, which express a high level of CD4, likely by enhancing viral membrane hemifusion and entry (35). Interestingly, in this work, we found that LY6E inhibits HIV-1 entry in Jurkat cells and MDMs, which express a low level of CD4. So, what are the factors that determine the differential effect of LY6E on HIV-1 infection in these two T cell lines? Among the multiple variables, we have focused on the possible influence of LY6E on the HIV-1 primary receptor CD4 and coreceptor CXCR4, primarily because of the predominant localization of these molecules on the plasma membrane. We demonstrated that LY6E does not affect CXCR4 yet specifically downregulates CD4 on the plasma membrane of Jurkat cells. We also showed that the downregulation of CD4 correlates with impaired binding and entry of HIV-1 into target cells, likely accounting for the diminished viral replication in this cell type. Interestingly, we found that only the cell surface, and not the total, level of CD4 in Jurkat cells was affected by LY6E and that this downregulation was likely due to an accelerated rate of CD4 internalization from the plasma membrane. By using a panel of chemical inhibitors, we observed that a dynamin-dependent, lipid raft-associated endocytic pathway is likely involved in the downregulation of CD4 by LY6E, which was in line with the colocalization of CD4 and LY6E in lipid rafts of the plasma membrane. These findings are interesting, considering that HIV-1 accessory protein Nef can downregulate CD4 from the cell surface yet mainly through clathrin-mediated endocytic pathways (35). At this time, we cannot rule out the possibility that CD4 and LY6E may also be associated with nonraft microdomains, thus contributing to the differential effect of LY6E on HIV-1. In addition, other unknown cellular proteins may function together with LY6E to modulate the cell surface CD4 molecule, therefore impairing HIV entry and replication.
How can we explain the distinct phenotypes of LY6E in two different T lymphocyte cell lines, Jurkat and SupT1? Jurkat cells were originally derived from a male acute leukemia patient and express a low level of CD4, whereas SupT1 cells were derived from a human T cell lymphoblastic lymphoma patient and express a high level of CD4 (38, 49, 50). Here we hypothesize that LY6E modulates HIV-1 entry and replication through two different mechanisms in these cells (Fig. 6). In the first mechanism, where the cellular CD4 level is high, LY6E intrinsically promotes HIV-1 entry postbinding, especially at the step of HIV-1 fusion with cell membrane, as we have reported in SupT1 cells (35). In this case, even though LY6E can still downregulate CD4 (data not shown for SupT1 cells), there are sufficient amounts of CD4 to allow the efficient binding and infection of the virus. In the second mechanism, where the endogenous CD4 level is low in target cells, the downregulation of CD4 by LY6E becomes prominent, thus significantly affecting the virus binding and infection. Because Jurkat cells express much less CD4 on the plasma membrane (38), they are therefore more sensitive to changes in the cell surface level of CD4. In contrast, SupT1 cells express a high level of CD4, which makes them less sensitive to change in the cellular CD4 level, unless a minimal level of CD4 required for HIV-1 infection is not met. In addition to direct comparisons of LY6E phenotypes between Jurkat and SupT1 cells, we have also artificially manipulated the CD4 level in these two cell lines; we found that, indeed, overexpression of CD4 in Jurkat cells overcame the inhibitory effect of LY6E on HIV-1 entry and, conversely, that a functional block of CD4 by a neutralizing CD4 antibody in SupT1 cells alleviated the enhancement of LY6E on HIV-1 entry. These results collectively strengthen the notion that CD4 is a critical factor that determines the differential effect of LY6E on HIV-1 infection in different cell types.
FIG 6.
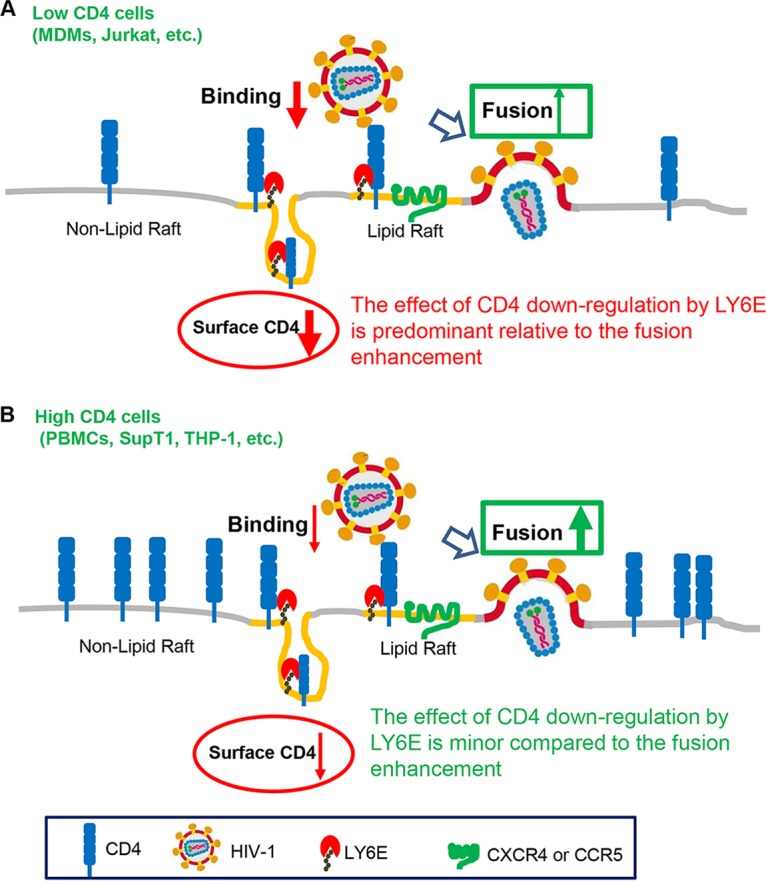
A working model for the differential roles of LY6E in HIV-1 infection, a CD4-dependent effect. (A) In low-CD4-expressing cells (such as Jurkat T cells, macrophages, and others), LY6E is associated with CD4 within the lipid raft microdomain, thus promoting its internalization from the plasma membrane; this would result in a decreased CD4 level on the cell surface and therefore impairs virus binding and entry. In this case, the effect of CD4 downregulation by LY6E is predominant relative to its effect on fusion enhancement; therefore, LY6E acts as a negative factor for HIV infection. (B) In high-CD4-expressing cells (PBMCs, SupT1 cells, and others), the effect of LY6E on the downregulating CD4 is minor compared to the LY6E-mediated enhancement of viral fusion. In this case, LY6E functions as a positive factor for HIV-1 infection.
One noticeable observation in this study is that CD4 overexpression in Jurkat cells did not fully convert the LY6E phenotype to that of SupT1 cells nor did a functional block of CD4 in SupT1 cells completely rescue the inhibitory effect of LY6E on HIV-1 entry, suggesting that additional factors in Jurkat and SupT1 cells are likely to be involved. Jurkat cells have been shown to express a greater amount of T cell receptor CD3 but fewer interleukin-7 receptor alpha (IL-7Rα) molecules on the surface than SupT1 cells (51). Since the ζ chain of CD3 is known to interact with LY6E, thus modulating CD3-mediated signaling (22, 23), it is possible that LY6E can differentially modulate HIV-1 infection through a mechanism involving LY6E-CD3 signaling cascades. Our observation that LY6E can relocate CD4 from the lipid raft microdomain, which is known to offer an effective platform for cell signaling (52), to nonraft fractions of the plasma membrane in Jurkat cells seems to support this possibility. In this respect, an RNA deep-sequencing analysis, together with functional proteomics characterization, of Jurkat and SupT1 cells should help further define the molecular pathways underlying the LY6E-medidated differential modulation of HIV-1 infection.
Low-CD4-expressing cells, such as dendritic cells and macrophages, are important initial targets of HIV infection and play essential roles in mediating host innate immunity in vivo (53, 54). As disease progresses, HIV-1 evolves and switches coreceptor usage for entry and more efficiently replicates in high-CD4-expressing T cells (55). Therefore, at the early stage of HIV-1 infection, the effect of LY6E on CD4 downregulation may be predominant, due to the intrinsic low level of CD4 in target cells; in this case, LY6E would function as an inhibitor of viral infection and transmission. However, at the advanced stage of infection, HIV-1 replicates in high-CD4 T cells, where the effect of LY6E on CD4 downregulation would become less effective, resulting in generally enhanced viral fusion and infection. While this model (Fig. 6) is supported by our previous data showing that LY6E enhances HIV-1 infection in high-CD4-expressing PBMCs, as well as by this current study showing that LY6E restricts HIV-1 entry in low-CD4-expressing MDMs, the specific and biological significance of LY6E in HIV-1 infection and AIDS progression in vivo requires further investigation. Because LY6E is an IFN-inducible protein, our work highlights a more complex role of IFN and its induced ISGs in virus-host interactions and possibly viral pathogenesis.
MATERIALS AND METHODS
Plasmids and cells.
The LY6E coding sequence was either FLAG tagged at the N terminus or untagged and then subsequently cloned into a pQCXIP (with puromycin selection marker) or PQCXIN (with neomycin selection marker) retroviral vector as reported previously (35). Short hairpin RNAs targeting LY6E were purchased from Sigma (TRCN0000155033). siRNA against LY6E was purchased from Dharmacon (SMARTpool: siGENOME LY6E siRNA). HIV-1 molecular clone pNL(AD8) and pAD8-Env were kindly provided by Eric Freed (NCI, MD). HIV-1 molecular clone pNL4.3 WT (catalog no. 114), transmitted/founder proviral constructs, WITO (catalog no. 11739), RHPA (catalog no. 11744), and THRO (catalog no. 11745), and Env plasmids pBJOX2000 Env (catalog no. 12655), pX1632 Env (catalog no. 12655), and pCH119 Env (catalog no. 12659) were obtained from NIH AIDS Reagent Program. HIV-inGLuc vector was kindly provided by the David Derse laboratory (56). HIV-Gag-iGFP was provided by Benjamin Chen (Icahn School of Medicine at Mount Sinai, NY). The pMX-CCR5 construct was provided by Li Wu’s laboratory (The Ohio State University, OH). The pCI-Neo-FLAG-TIM-1 encoding TIM-1 was described previously (57).
Jurkat E6.1 (ATCC) and SupT1 (NIH AIDS Research and Reagent Program; catalog no. 100) cell lines were maintained in RPMI 1640 (Gibco), supplemented with 100 U/ml penicillin/streptomycin (Gibco) and 10% fetal bovine serum (FBS) (Gibco). HEK293T cells and HeLa-TZM indicator cells were maintained in Dulbecco modified Eagle medium (DMEM), supplemented with 100 U/ml penicillin/streptomycin (Corning) and 10% FBS (Sigma). Jurkat-TAg cells were a kind gift of Heinrich Gottlinger (University of Massachusetts Medical School, MA). Anti-CD4 neutralizing antibody (clone SK3; catalog no. 344602) was purchased from Biolegend, CA. Anti-CD4 (clone 34915) antibody for immunoblotting was ordered from R&D, MN. Anti-CD4 (SIM.4) and anti-CD4 (SIM.2) antibodies for flow cytometry were produced from hybridoma (SIM.4 [catalog no. 512] and SIM.2 [catalog no. 511], respectively), which were ordered from AIDS Reagent Program, NIH. Anti-CXCR4 monoclonal antibody (catalog no. 4083) was obtained from the NIH AIDS Reagent Program. The anti-CD4 PE-Texas Red-S3.5 (Invitrogen; catalog no. MHCD0417) for the internalization assay was purchased from Invitrogen. Anti-LY6E (LS-C155101) antibody used for flow cytometry was purchased from LifeSpan BioSciences, WA.
MDM isolation and culture.
PBMCs from HIV-negative healthy donors were used to isolate MDMs. Briefly, PBMCs were plated in Teflon wells for 5 days to allow differentiation into MDMs. On day 5, cells were removed from the Teflon wells, washed, and counted. The cells were then plated in plastic tissue culture plates (24 wells) at 180,000 MDMs/well in 10% FBS–RPMI 1640 and used for viral infection.
Virus production.
Retroviral pseudotypes or infectious HIV-1 stocks were generated in HEK293T cells. Briefly, LY6E-expressing retroviral vectors, MLV Gag-pol and pMDG (encoding VSV-G), were cotransfected into HEK293T cells at ratio of 2:2:1. Similarly, shRNA LY6E-encoding lentiviral vectors, HIV-1 Gag-pol and pMDG, were cotransfected into HEK293T cells at ratio of 2:2:1. Virions released into supernatants were collected 24 and 36 h after transfection. For infectious HIV-1 production, proviral constructs were directly transfected into HEK293T cells by the calcium phosphate method. In some cases, pMDG was cotransfected to generate VSV-G-bearing infectious viral stocks. Cell debris was removed by centrifugation at 3,000 × g for 10 min, and supernatants were filtered through a 0.22-μm filter and stored at −80°C until use.
Generation of stable cell lines.
The collected virions were used to transduce Jurkat or SupT1 cells. Approximately 400 μl of viral stocks, supplemented with 5 μg/ml Polybrene, was mixed with 1 × 106 T cells, followed by spinoculation at 1,680 × g at 4°C for 1 h. Twenty-four hours after transduction, the cells were replenished with fresh RPMI 1640 containing 1 μg/ml puromycin or 800 μg/ml G418 sulfate for selection.
HIV-1 infection and replication.
For short-term replication in Jurkat cells, about 50 μl infectious HIV-1 virions (equivalent to about 25 ng p24) was spinoculated with 5 × 105 cells for 1 h at 1,680 × g at 4°C. For MDM infection, cells were transfected with 20 nM siRNA control or siRNA-LY6E by Lipofectamine 2000 (Invitrogen), followed by infection in a 24-well plate. In all cases, at 6 h postinfection, the cells were pelleted and resuspended in fresh RPMI 1640. Forty-eight hours after infection, the cells were collected for flow cytometric analyses. Virus-containing supernatants were harvested and used to infect HeLa-TZM cells for measuring viral infectivity.
For long-term replication, about 5 μl infectious HIV-1 (equivalent to 2.5 ng p24) was spinoculated with 1 × 105 Jurkat T cells for 1 h at 1,680 × g at 4°C. Six hours postinfection, the cells were pelleted and refed in fresh RPMI 1640 every 2 days. The cells were split once every 4 days, and virus-containing supernatants were harvested for reverse transcriptase (RT) assay and/or for determining viral infectivity.
RT assay.
HIV-1 production was determined by measuring the viral RT activity. Briefly, 10 μl of cell supernatants was mixed with 40-μl reaction cocktails {50 mM Tris-HCl (pH 7.9), 5 mM MgCl2, 0.5 mM EGTA, 0.05% Triton X-100, 2% (vol/vol) ethylene glycol, 150 mM KCl, 5 mM dithiothreitol (DTT), 0.3 mM GSH (reduced glutathione), 0.5 U/ml poly(rA) oligo(dT), and 0.1 μCi/μl [3H]dTTP (Perkin-Elmer)} and incubated at 37°C for 3 h. Samples were then placed on ice for 30 min with 150 μl prechilled 10% (mass/vol) trichloroacetic acid (TCA) and filtered onto an ELISA plate (Merck Millipore, Ireland). RT activity was measured in a Microbeta counter (Beckman Coulter).
Infectivity assay.
HIV-1 infectivity was determined by infecting indicator HeLa-TZM cells as previously described (11). Briefly, 36 to 48 h after infection, cells were washed once with phosphate-buffered saline (PBS) and lysed with lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100). Approximately 40 μl of cell lysates was incubated with 40 μl firefly luciferase substrates to determine HIV-1 infectivity in a microplate reader (FilterMax F5; Molecular Devices).
Virus binding and virion-cell fusion assay.
For the virion binding assay, Jurkat cells were incubated with HIV-iGFP particles at 4°C for 2 h. After three washes with PBS to remove unbound virus, cells were fixed with 3.7% formaldehyde for 10 min and subjected to flow cytometric analysis. A BlaM-Vpr-based HIV-1 entry assay was performed as previously described (35). Briefly, Jurkat cells were spinoculated with HIV-1 at 1,680 × g at 4°C. The CCF2 substrate was loaded into cells 1 h after infection, and then cells were washed three times with PBS. The cells were maintained in CO2-independent medium (plus 0.5 mM probenecid) overnight before being fixed with 3.7% formaldehyde and analyzed by flow cytometry.
Coimmunoprecipitation and Western blotting.
Cells were lysed in prechilled RIPA buffer (1% NP-40, 50 mM Tris-HCl, 150 mM NaCl, 0.1% SDS, and protease inhibitor mixture) or 1% Triton X-100 buffer (1% Triton X-100 in 1× PBS) for 20 min. Coimmunoprecipitation was performed by using anti-FLAG M2 affinity gel/beads (Sigma) to pull down FLAG-tagged LY6E in Jurkat or Jurkat-LY6E cell lysates (either in 1% Triton X-100 or RIPA buffer) overnight at 4°C. Unbound proteins were washed off three times with cold PBS or RIPA buffer. Samples were boiled with 5× SDS-PAGE sample buffer before being subjected to 10% SDS-PAGE. The coimmunoprecipitated CD4 was detected by using CD4 antibody (catalog no. MAB3791; R&D, MN). After the proteins were transferred to a polyvinylidene difluoride (PVDF) membrane, primary antibodies of interest were applied, and protein signals were detected using an Amersham Imager 600 (GE Healthcare), as previously described (11).
Lipid raft flotation assay.
Jurkat or Jurkat-LY6E cells (3 × 107 to 5 × 107) were washed 3 times with ice-cold PBS and lysed on ice for 20 min in 2 ml of 1% Triton X-100 lysis buffer supplemented with protease inhibitor cocktails (Sigma). Cell lysates were homogenized with 15 strokes of a Dounce homogenizer and further passed 5 times through 22G1 needles. Cells were then centrifuged for 5 min at 1,000 × g at 4°C to remove insoluble materials and nuclei. The supernatant was mixed with 2 ml of 80% sucrose in lysis buffer, placed at the bottom of ultracentrifuge tubes, and overlaid with 4 ml of 30% and 3.5 ml of 5% sucrose in Triton X-100 lysis buffer. Lysates were subjected to ultracentrifugation at 4°C in an SW41 rotor (Beckman) for 19 h at 39,000 rpm. Fractions of 1-ml volume were collected from top to the bottom and analyzed by Western blotting.
Detection of cell surface or intracellular protein expression by flow cytometry.
Cells were nonpermeabilized or permeabilized with permeabilization IC buffer (ThermoFisher Scientific). Cells were washed twice with cold PBS plus 2% FBS, detached with 1× PBS containing 5 mM EDTA, and incubated on ice with the appropriate primary antibodies for 1 h. After three washes with PBS plus 2% FBS, cells were further incubated with FITC (fluorescein isothiocyanate)- or TRITC tetramethyl rhodamine isothiocyanate)-conjugated secondary antibodies for 45 min. After two washes, the cells were fixed with 3.7% formaldehyde for 10 min and analyzed by flow cytometry (AttuneNxT flow cytometer; Invitrogen).
CD4 internalization assay.
CD4 internalization assays were adapted from the method of Aiken et al. (58). Briefly, Jurkat or Jurkat-LY6E cells were incubated with anti-CD4 PE-Texas Red-S3.5 (Invitrogen; catalog no. MHCD0417) in PBS plus 2% FBS on ice for 2 h, washed twice, and suspended in PBS–2% FBS buffer. The cells were then placed in a 37°C water bath for 0, 5, 15, 30, and 60 min, before being placed on ice for 5 min to stop the internalization process. Cells collected at different time points were aliquoted into 2 portions: one portion was incubated with cold low-pH PBS buffer (pH 3.0), while the other portion was incubated with cold normal PBS (pH 7.5) for 5 min. After removal of the supernatants, the PE-Texas Red signal in each sample was determined by flow cytometry. The CD4 internalized fraction was calculated as the ratio of pH 3.0-resistant PE-Texas Red signal to the total cell-associated signal.
Immunostaining and imaging.
For CD4+ T cells, 5 × 105 Jurkat-TAg cells were transfected with 500 ng pQCXIP-FLAG-LY6E together with 500 ng of either pQCXIP-CD4 (Gregory Melikian, Emory University), pGFP-GPI (Harvey MaMahon, MRC Laboratory of Molecular Biology, United Kingdom), or pCIneo-TIM-1 (57) by Lipofectamine 2000 (Thermo Fisher Scientific) in combination with spinoculation at 1,640 × g for 1 h. Thirty-six hours after transfection, the cells were collected and washed once with 1× PBS. Cells were stained with anti-LY6E primary antibody together with another primary antibody: anti-CD4 (SIM.4), anti-GFP, or anti-TIM1 on ice for 1 h. Cells were washed with wash buffer (2% FBS in 1× PBS buffer) 3 times and stained with either FITC- or TRITC-conjugated anti-rabbit (for LY6E), anti-mouse (for GFP and CD4), or anti-goat (for TIM-1) secondary antibodies. After two additional washes, the cells were fixed with 4% paraformaldehyde for 10 min at room temperature and mounted. For MDMs, cells were seeded on a coverslip; 24 h after seeding, the cells were fixed with 4% paraformaldehyde at room temperature for 10 min and then permeabilized with 1× permeabilization buffer (1× IC buffer; Thermo Fisher Scientific) for 10 min. Cells were then blocked with 5% bovine serum albumin (BSA) and stained with anti-CD4 and anti-LY6E antibodies overnight at 4°C. After three washes with PBS, cells were incubated with anti-mouse–TRITC and anti-rabbit–FITC for 1 h, followed by DAPI (4′,6-diamidino-2-phenylindole) staining.
For fluorescence microscope, Z-stack images were collected using a Leica DMi8 inverted deconvolution microscope with a 100× oil immersion lens. Five fields that had been randomly selected were chosen, and individual cells were cropped for colocalization analyses using ImageJ software (NIH).
Statistical analyses.
All statistical analyses were carried out in GraphPad Prism6, with Student’s t tests or one-way analysis of variance (ANOVA) unless otherwise noted. Typically, data from at least 3 to 5 independent experiments were used for the analysis.
ACKNOWLEDGMENTS
This work was supported by NIH grants R01 AI112381 and R01 GM132069 to S.-L.L.
We thank Eric Freed for provision of PBMCs, MDMs, and other reagents for this study. We thank Emily Bowman and Nicholas Funderburg for proving some of the MDMs used in this work. We acknowledge the NIH AIDS Reagents Program for supplying other essential materials that made this work possible.
The authors declare that they have no competing interests.
REFERENCES
- 1.Deeks SG, Lewin SR, Havlir DV. 2013. The end of AIDS: HIV infection as a chronic disease. Lancet 382:1525–1533. doi: 10.1016/S0140-6736(13)61809-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilen CB, Tilton JC, Doms RW. 2012. HIV: cell binding and entry. Cold Spring Harb Perspect Med 2:a006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Melikyan GB. 2008. Common principles and intermediates of viral protein-mediated fusion: the HIV-1 paradigm. Retrovirology 5:111. doi: 10.1186/1742-4690-5-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalgleish AG, Beverley PCL, Clapham PR, Crawford DH, Greaves MF, Weiss RA. 1984. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 312:763–767. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- 5.Wyatt R, Sodroski J. 1998. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 280:1884–1888. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- 6.Levesque K, Finzi A, Binette J, Cohen EA. 2004. Role of CD4 receptor down-regulation during HIV-1 infection. Curr HIV Res 2:51–59. doi: 10.2174/1570162043485086. [DOI] [PubMed] [Google Scholar]
- 7.Garcia JV, Miller AD. 1991. Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature 350:508–511. doi: 10.1038/350508a0. [DOI] [PubMed] [Google Scholar]
- 8.Buonocore L, Rose JK. 1990. Prevention of HIV-1 glycoprotein transport by soluble CD4 retained in the endoplasmic reticulum. Nature 345:625–628. doi: 10.1038/345625a0. [DOI] [PubMed] [Google Scholar]
- 9.Willey RL, Maldarelli F, Martin MA, Strebel K. 1992. Human immunodeficiency virus type 1 Vpu protein regulates the formation of intracellular gp160-CD4 complexes. J Virol 66:226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vincent MJ, Abdul Jabbar M. 1995. The human immunodeficiency virus type 1 Vpu protein: a potential regulator of proteolysis and protein transport in the mammalian secretory pathway. Virology 213:639–649. doi: 10.1006/viro.1995.0035. [DOI] [PubMed] [Google Scholar]
- 11.Yu J, Li M, Wilkins J, Ding S, Swartz TH, Esposito AM, Zheng YM, Freed EO, Liang C, Chen BK, Liu SL. 2015. IFITM proteins restrict HIV-1 infection by antagonizing the envelope glycoprotein. Cell Rep 13:145–156. doi: 10.1016/j.celrep.2015.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilkins J, Zheng YM, Yu JY, Liang C, Liu SL. 2016. Nonhuman primate IFITM proteins are potent inhibitors of HIV and SIV. PLoS One 11:e0156739. doi: 10.1371/journal.pone.0156739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu J, Pan Q, Rong L, He W, Liu SL, Liang C. 2011. The IFITM proteins inhibit HIV-1 infection. J Virol 85:2126–2137. doi: 10.1128/JVI.01531-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosa A, Chande A, Ziglio S, De Sanctis V, Bertorelli R, Goh SL, McCauley SM, Nowosielska A, Antonarakis SE, Luban J, Santoni FA, Pizzato M. 2015. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 526:212–217. doi: 10.1038/nature15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Usami Y, Wu YF, Gottlinger HG. 2015. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 526:218–223. doi: 10.1038/nature15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tada T, Zhang YZ, Koyama T, Tobiume M, Tsunetsugu-Yokota Y, Yamaoka S, Fujita H, Tokunaga K. 2015. MARCH8 inhibits HIV-1 infection by reducing virion incorporation of envelope glycoproteins. Nat Med 21:1502–1507. doi: 10.1038/nm.3956. [DOI] [PubMed] [Google Scholar]
- 17.Lodermeyer V, Suhr K, Schrott N, Kolbe C, Sturzel CM, Krnavek D, Munch J, Dietz C, Waldmann T, Kirchhoff F, Goffinet C. 2013. 90K, an interferon-stimulated gene product, reduces the infectivity of HIV-1. Retrovirology 10:111. doi: 10.1186/1742-4690-10-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sood C, Marin M, Chande A, Pizzato M, Melikyan GB. 2017. SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J Biol Chem 292:6014–6026. doi: 10.1074/jbc.M117.777714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Compton AA, Bruel T, Porrot F, Mallet A, Sachse M, Euvrard M, Liang C, Casartelli N, Schwartz O. 2014. IFITM proteins incorporated into HIV-1 virions impair viral fusion and spread. Cell Host Microbe 16:736–747. doi: 10.1016/j.chom.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tartour K, Appourchaux R, Gaillard J, Nguyen XN, Durand S, Turpin J, Beaumont E, Roch E, Berger G, Mahieux R, Brand D, Roingeard P, Cimarelli A. 2014. IFITM proteins are incorporated onto HIV-1 virion particles and negatively imprint their infectivity. Retrovirology 11:103. doi: 10.1186/s12977-014-0103-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Godfrey DI, Masciantonio M, Tucek CL, Malin MA, Boyd RL, Hugo P. 1992. Thymic shared antigen-1. A novel thymocyte marker discriminating immature from mature thymocyte subsets. J Immunol 148:2006–2011. [PubMed] [Google Scholar]
- 22.Kosugi A, Saitoh S, Noda S, Miyake K, Yamashita Y, Kimoto M, Ogata M, Hamaoka T. 1998. Physical and functional association between thymic shared antigen-1 stem cell antigen-2 and the T cell receptor complex. J Biol Chem 273:12301–12306. doi: 10.1074/jbc.273.20.12301. [DOI] [PubMed] [Google Scholar]
- 23.Saitoh SI, Kosugi A, Noda S, Yamamoto N, Ogata M, Minami Y, Miyake K, Hamaoka T. 1995. Modulation of Tcr-mediated signaling pathway by thymic shared antigen-1 (Tsa-1) stem-cell antigen-2 (Sca-2). J Immunol 155:5574–5581. [PubMed] [Google Scholar]
- 24.Su B, Waneck GL, Flavell RA, Bothwell ALM. 1991. The glycosyl phosphatidylinositol anchor is critical for Ly-6a/E-mediated T-cell activation. J Cell Biol 112:377–384. doi: 10.1083/jcb.112.3.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Classon BJ, Boyd RL. 1998. Thymic-shared antigen-1 (TSA-1). A lymphostromal cell membrane Ly-6 superfamily molecule with a putative role in cellular adhesion. Dev Immunol 6:149–156. doi: 10.1155/1998/53157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.AlHossiny M, Luo LL, Frazier WR, Steiner N, Gusev Y, Kallakury B, Glasgow E, Creswell K, Madhavan S, Kumar R, Upadhyay G. 2016. Ly6E/K signaling to TGF beta promotes breast cancer progression, immune escape, and drug resistance. Cancer Res 76:3376–3386. doi: 10.1158/0008-5472.CAN-15-2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yeom CJ, Zeng L, Goto Y, Morinibu A, Zhu Y, Shinomiya K, Kobayashi M, Itasaka S, Yoshimura M, Hur CG, Kakeya H, Hammond EM, Hiraoka M, Harada H. 2016. LY6E: a conductor of malignant tumor growth through modulation of the PTEN/PI3K/Akt/HIF-1 axis. Oncotarget 7:65837–65848. doi: 10.18632/oncotarget.11670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo L, McGarvey P, Madhavan S, Kumar R, Gusev Y, Upadhyay G. 2016. Distinct lymphocyte antigens 6 (Ly6) family members Ly6D, Ly6E, Ly6K and Ly6H drive tumorigenesis and clinical outcome. Oncotarget 7:11165–11193. doi: 10.18632/oncotarget.7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu SY, Sanchez DJ, Aliyari R, Lu S, Cheng G. 2012. Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci U S A 109:4239–4244. doi: 10.1073/pnas.1114981109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu HC, Niikura M, Fulton JE, Cheng HH. 2003. Identification of chicken lymphocyte antigen 6 complex, locus E (LY6E, alias SCA2) as a putative Marek's disease resistance gene via a virus-host protein interaction screen. Cytogenet Genome Res 102:304–308. doi: 10.1159/000075767. [DOI] [PubMed] [Google Scholar]
- 31.Stier MT, Spindler KR. 2012. Polymorphisms in Ly6 genes in Msq1 encoding susceptibility to mouse adenovirus type 1. Mamm Genome 23:250–258. doi: 10.1007/s00335-011-9368-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hackett BA, Cherry S. 2018. Flavivirus internalization is regulated by a size-dependent endocytic pathway. Proc Natl Acad Sci U S A doi: 10.1073/pnas.1720032115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mar KB, Rinkenberger NR, Boys IN, Eitson JL, McDougal MB, Richardson RB, Schoggins JW. 2018. LY6E mediates an evolutionarily conserved enhancement of virus infection by targeting a late entry step. Nat Commun 9:3603. doi: 10.1038/s41467-018-06000-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu J, Liang C, Liu SL. 2017. Interferon-inducible LY6E protein promotes HIV-1 infection. J Biol Chem 292:4674–4685. doi: 10.1074/jbc.M116.755819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu X, Qiu C, Zhu L, Huang J, Li L, Fu W, Zhang L, Wei J, Wang Y, Geng Y, Zhang X, Qiao W, Xu J. 2014. IFN-stimulated gene LY6E in monocytes regulates the CD14/TLR4 pathway but inadequately restrains the hyperactivation of monocytes during chronic HIV-1 infection. J Immunol 193:4125–4136. doi: 10.4049/jimmunol.1401249. [DOI] [PubMed] [Google Scholar]
- 37.Loeuillet C, Deutsch S, Ciuffi A, Robyr D, Taffe P, Munoz M, Beckmann JS, Antonarakis SE, Telenti A. 2008. In vitro whole-genome analysis identifies a susceptibility locus for HIV-1. PLoS Biol 6:319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee B, Sharron M, Montaner LJ, Weissman D, Doms RW. 1999. Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc Natl Acad Sci U S A 96:5215–5220. doi: 10.1073/pnas.96.9.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Varma R, Mayor S. 1998. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature 394:798–801. doi: 10.1038/29563. [DOI] [PubMed] [Google Scholar]
- 40.McCallus DE, Ugen KE, Sato AI, Williams WV, Weiner DB. 1992. Construction of a recombinant bacterial human CD4 expression system producing a bioactive CD4 molecule. Viral Immunol 5:163–172. doi: 10.1089/vim.1992.5.163. [DOI] [PubMed] [Google Scholar]
- 41.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. 2006. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell 10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 42.Dutta D, Donaldson JG. 2012. Search for inhibitors of endocytosis: intended specificity and unintended consequences. Cell Logist 2:203–208. doi: 10.4161/cl.23967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Popik W, Alce TM, Au WC. 2002. Human immunodeficiency virus type 1 uses lipid raft-colocalized CD4 and chemokine receptors for productive entry into CD4(+) T cells. J Virol 76:4709–4722. doi: 10.1128/JVI.76.10.4709-4722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Del Real G, Jiménez-Baranda S, Lacalle RA, Mira E, Lucas P, Gómez-Moutón C, Carrera AC, Martínez-A C, Mañes S. 2002. Blocking of HIV-1 infection by targeting CD4 to nonraft membrane domains. J Exp Med 196:293–301. doi: 10.1084/jem.20020308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Wilgenburg B, Moore MD, James WS, Cowley SA. 2014. The productive entry pathway of HIV-1 in macrophages is dependent on endocytosis through lipid rafts containing CD4. PLoS One 9:e86071. doi: 10.1371/journal.pone.0086071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keller P, Toomre D, Diaz E, White J, Simons K. 2001. Multicolour imaging of post-Golgi sorting and trafficking in live cells. Nat Cell Biol 3:140–149. doi: 10.1038/35055042. [DOI] [PubMed] [Google Scholar]
- 47.Hogue IB, Grover JR, Soheilian F, Nagashima K, Ono A. 2011. Gag induces the coalescence of clustered lipid rafts and tetraspanin-enriched microdomains at HIV-1 assembly sites on the plasma membrane. J Virol 85:9749–9766. doi: 10.1128/JVI.00743-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spindler KR, Welton AR, Lim ES, Duvvuru S, Althaus IW, Imperiale JE, Daoud AI, Chesler EJ. 2010. The major locus for mouse adenovirus susceptibility maps to genes of the hematopoietic cell surface-expressed LY6 family. J Immunol 184:3055–3062. doi: 10.4049/jimmunol.0903363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith SD, Shatsky M, Cohen PS, Warnke R, Link MP, Glader BE. 1984. Monoclonal antibody and enzymatic profiles of human malignant T-lymphoid cells and derived cell lines. Cancer Res 44:5657–5660. [PubMed] [Google Scholar]
- 50.Schneider U, Schwenk HU, Bornkamm G. 1977. Characterization of EBV-genome negative “null” and “T” cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. Int J Cancer 19:621–626. doi: 10.1002/ijc.2910190505. [DOI] [PubMed] [Google Scholar]
- 51.Patel ES, Chang LJ. 2012. Synergistic effects of interleukin-7 and pre-T cell receptor signaling in human T cell development. J Biol Chem 287:33826–33835. doi: 10.1074/jbc.M112.380113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mollinedo F, Gajate C. 2015. Lipid rafts as major platforms for signaling regulation in cancer. Adv Biol Regul 57:130–146. doi: 10.1016/j.jbior.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 53.Wu L, KewalRamani VN. 2006. Dendritic-cell interactions with HIV: infection and viral dissemination. Nat Rev Immunol 6:859–868. doi: 10.1038/nri1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shattock RJ, Moore JP. 2003. Inhibiting sexual transmission of HIV-1 infection. Nat Rev Microbiol 1:25–34. doi: 10.1038/nrmicro729. [DOI] [PubMed] [Google Scholar]
- 55.Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR. 1997. Change in coreceptor use correlates with disease progression in HIV-1-infected individuals. J Exp Med 185:621–628. doi: 10.1084/jem.185.4.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mazurov D, Ilinskaya A, Heidecker G, Lloyd P, Derse D. 2010. Quantitative comparison of HTLV-1 and HIV-1 cell-to-cell infection with new replication dependent vectors. PLoS Pathog 6:e1000788. doi: 10.1371/journal.ppat.1000788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li M, Ablan SD, Miao C, Zheng YM, Fuller MS, Rennert PD, Maury W, Johnson MC, Freed EO, Liu SL. 2014. TIM-family proteins inhibit HIV-1 release. Proc Natl Acad Sci U S A 111:E3699–E3707. doi: 10.1073/pnas.1404851111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aiken C, Konner J, Landau NR, Lenburg ME, Trono D. 1994. Nef induces Cd4 endocytosis–requirement for a critical dileucine motif in the membrane-proximal Cd4 cytoplasmic domain. Cell 76:853–864. doi: 10.1016/0092-8674(94)90360-3. [DOI] [PubMed] [Google Scholar]
- 59.Hubner W, McNerney GP, Chen P, Dale BM, Gordon RE, Chuang FYS, Li X-D, Asmuth DM, Huser T, Chen BK. 2009. Quantitative 3D video microscopy of HIV transfer across T cell virological synapses. Science 323:1743–1747. doi: 10.1126/science.1167525. [DOI] [PMC free article] [PubMed] [Google Scholar]