

Kaposi’s sarcoma-associated herpesvirus (KSHV) is a pathogen causing cancer in the immune-deficient population. The reactivation of KSHV from latency is important for it to be carcinogenic. Our finding that SIRT6 has inhibitory effects on KSHV reactivation by interacting with the viral genome and suppressing viral gene expression is important because it might lead to a strategy of interfering with KSHV reactivation. Overexpression of SIRT6 repressed the activities of several KSHV promoters, leading to reduced gene expression and DNA replication by KSHV in a KSHV bacterial artificial chromosome-containing cell line. Depletion of SIRT6 favored reactivation of KSHV from SLK-iBACV-gfpK52 cells. More importantly, we reveal that SIRT6 interacts with KSHV DNA. Whether the interaction of SIRT6 with KSHV DNA occurs at a global level will be further studied in the future.
KEYWORDS: histone deacetylases (HDACs), Kaposi's sarcoma-associated herpesvirus (KSHV), latency, ori-Lyt, reactivation, replication and transcription activator (RTA), sirtuins (SIRTs)
ABSTRACT
Kaposi’s sarcoma-associated herpesvirus (KSHV; also called human herpesvirus 8 [HHV-8]), upon being reactivated, causes serious diseases in immunocompromised individuals. Its reactivation, especially how the cellular regulating mechanisms play roles in KSHV gene expression and viral DNA replication, is not fully understood. In searching for the cellular factors that regulate KSHV gene expression, we found that several histone deacetylases (HDACs) and sirtuins (SIRTs), including HDACs 2, 7, 8, and 11 and SIRTs 4 and 6, repress KSHV ori-Lyt promoter activity. Interestingly, the nuclear protein SIRT6 presents the greatest inhibitory effect on ori-Lyt promoter activity. A more detailed investigation revealed that SIRT6 exerts repressive effects on multiple promoters of KSHV. As a consequence of inhibiting the KSHV promoters, SIRT6 not only represses viral protein production but also inhibits viral DNA replication, as investigated in a KSHV-containing cell line, SLK-iBAC-gfpK52. Depletion of the SIRT6 protein using small interfering RNA could not directly reactivate KSHV from SLK-iBAC-gfpK52 cells but made the reactivation of KSHV by use of a small amount of the reactivator (doxycycline) more effective and enhanced viral DNA replication in the KSHV infection system. We performed DNA chromatin immunoprecipitation (ChIP) assays for SIRT6 in the SLK-iBAC-gfpK52 cell line to determine whether SIRT6 interacts with the KSHV genome in order to exhibit regulatory effects. Our results suggest that SIRT6 interacts with KSHV ori-Lyt and ORF50 promoters. Furthermore, the SIRT6-KSHV DNA interaction is significantly negated by reactivation. Therefore, we identified a cellular regulator, SIRT6, that represses KSHV replication by interacting with KSHV DNA and inhibiting viral gene expression.
IMPORTANCE Kaposi’s sarcoma-associated herpesvirus (KSHV) is a pathogen causing cancer in the immune-deficient population. The reactivation of KSHV from latency is important for it to be carcinogenic. Our finding that SIRT6 has inhibitory effects on KSHV reactivation by interacting with the viral genome and suppressing viral gene expression is important because it might lead to a strategy of interfering with KSHV reactivation. Overexpression of SIRT6 repressed the activities of several KSHV promoters, leading to reduced gene expression and DNA replication by KSHV in a KSHV bacterial artificial chromosome-containing cell line. Depletion of SIRT6 favored reactivation of KSHV from SLK-iBACV-gfpK52 cells. More importantly, we reveal that SIRT6 interacts with KSHV DNA. Whether the interaction of SIRT6 with KSHV DNA occurs at a global level will be further studied in the future.
INTRODUCTION
Kaposi’s sarcoma-associated herpesvirus (KSHV; also called human herpesvirus 8 [HHV-8]) causes serious diseases in immunocompromised individuals. The KSHV-related malignancies include lymphoproliferative cancers and several forms of Kaposi’s sarcoma (KS) that can involve the oral cavity, lymph nodes, and viscera (1–4). Like all other herpesviruses, KSHV infection exists in two different states: latency and lytic infection (5). KSHV evades host immunity by setting up a lifelong latent infection. In latency, viral infection does not result in the production of viral particles and is associated with genomic persistence, but the virus exhibits highly restricted gene expression patterns. Latency can be converted to lytic infection (5). In the lytic replication phase, virtually the entire viral genome is transcribed in a temporally regulated cascade of gene expression: early genes are regulatory and have catalytic functions that lead to viral DNA replication, followed by the expression of late genes encoding structural components. The effects of histone deacetylases (HDACs) on the reactivation of KSHV have been widely investigated. In a cell culture model, KSHV can be reactivated by HDAC inhibitors (HDACis), resulting in viral gene expression, viral DNA replication, and the release of mature virions (6–8).
HDACs are a group of enzymes that serve to remove acetyl groups from ε-N-acetyl lysine amino acids in histones and other proteins (9, 10). In humans, 18 HDACs have been identified and classified into four classes (11, 12). The class I HDACs are Rpd3-like proteins and include HDAC1, HDAC2, HDAC3, and HDAC8; the class II HDACs are Hda1-like proteins and contain HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, and HDAC10; the class III HDACs are Sir2-like proteins and include sirtuin 1 (SIRT1), SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, and SIRT7; and class IV has only HDAC11 (11). HDACs play critical regulatory functions in both nuclear and cytoplasmic processes, including transcriptional initiation and elongation, protein stability, and multiprotein complex formation (11). Together with histone acetyltransferases (HATs), histone methylases, and phosphorylation kinases, HDACs play roles in establishing, maintaining, and modifying the histone code (13, 14). During latency, KSHV genomic DNA resides in the nucleus and is connected to chromosomes, where it experiences the biological process of chromatinization. We hypothesize that HDACs participate in modifying the histone code of the chromatinized KSHV genome, resulting in dynamic changes to the KSHV chromatin structure.
Two elements, RTA (reactivation and transcription activator, encoded by ORF50) and ori-Lyt (origin of lytic DNA replication), are important for the reactivation of KSHV. RTA is the first viral protein generated after KSHV reactivation, and its binding with ori-Lyt is necessary for KSHV replication (15–18). Two almost identical ori-Lyts exist in the KSHV genome and are needed for KSHV DNA replication in the lytic phase (19, 20). Interestingly, the ori-Lyt sequence contains AP-1 binding elements, A+T-rich domains, and the TATA box, which are the elements of the gene promoter, although no open reading frame (ORF) has been identified to be under the control of ori-Lyt (19, 20). RTA activates the ori-Lyt promoter and initiates transcription across GC-rich tandem repeats, as determined using a luciferase assay (21). In addition, the ori-Lyt DNA recruits DNA replication factors to form a KSHV DNA replication compartment. Therefore, it is important to know whether ori-Lyt is affected by any viral or cellular regulatory factors. We have been interested in knowing whether HDACs interact with ori-Lyt DNA and affect the activities of ori-Lyt. In the present study, we used a KSHV-carrying cell line, SLK-iBAC-gfpk52, to investigate the effects of HDACs on the reactivation of KSHV and found that sirtuin 6 (SIRT6) plays an important regulatory function in KSHV gene expression and KSHV reactivation.
RESULTS
Biological effects of HDACs and SIRTs on ori-Lyt-directed promoter activity.
KSHV sets up latency in infected endothelial cells and B lymphocytes and can be reactivated by several exogenous stimulators, such as gene-expressing activators, among which HDAC inhibitors have been mostly investigated. KSHV lytic DNA replication requires the origin and its activity. ori-Lyt is not only the origin of KSHV DNA replication but also has promoter activity. To know whether the HDACs and sirtuins (SIRTs) are the repressors of the ori-Lyt promoter, we cotransfected an ori-Lyt controlling a luciferase-expressing plasmid with each of the 18 HADC- or SIRT-expressing plasmids into HEK293T cells, as shown in Fig. 1. At 24 h after transfection, the luciferase activity of the cells was analyzed with an IVIS Spectrum imaging system, which recorded the photos (Fig. 1, top) and quantified the luciferase activity units (Fig. 1, middle). The cells were then collected for the Western blot assay to show the expression of HDACs or SIRTs using anti-FLAG antibody because each HDAC- or SIRT-expressing plasmid is tagged with a FLAG tag at its N terminus. As can be seen at the bottom of Fig. 1, all HDACs and SIRTs were expressed after transfection in HEK293T cells. Our results show that the luciferase activities were significantly affected by only some HDACs and SIRTs, specifically, HDACs 2, 7, 8, and 11 and SIRTs 4 and 6. SIRT6 appeared to have the most inhibitory effects on the ori-Lyt promoter. Therefore, SIRT6 was selected for further investigation.
FIG 1.

Luciferase assay to screen the effects of HDACs and SIRTs on the promoter activity of ori-Lyt. The ori-Lyt-driven luciferase plasmid was cotransfected with the vector (pcDNA3) or HDAC- or SIRT-expressing plasmid into HEK293T cells for 24 h. Then, the cells were applied to an IVIS imaging system to examine the luciferase activities. (A) (Top) The IVIS system was used to visualize the signal and take photos. The depth of the color correlates with the strength of luciferase activity. (Middle) The activities quantitated from three independent experiments were statistically averaged and compared to the activities of the pori-Lyt-luc and pcDNA3 groups. The fold change is shown. (Bottom) Whole-cell lysate samples were collected after luciferase assay, and Western blotting was performed to detect the expression of HDACs and SIRTs. (B) Luciferase assay to compare the suppressive effects on the promoter activity of ori-Lyt between SIRT6 and SIRT4. p/sec/cm2/sr, number of photons per second per square centimeter per steradian; MW, molecular weight.
The biological effects of SIRT6 on ori-Lyt are dose dependent.
We wondered whether the repressive effects of SIRT6 on the ori-Lyt promoter are dose dependent. To that end, we cotransfected an ori-Lyt-controlling luciferase-expressing plasmid with different amounts of an SIRT6-expressing plasmid into HEK293T cells, as shown in Fig. 2. At 24 h after transfection, the luciferase activity of the cells was analyzed with the IVIS Spectrum imaging system. The cells were then collected for the Western blot assay to show the expression of SIRT6. As can be seen, the ori-Lyt promoter activity was lower when more SIRT6-expressing plasmid was used for transfection into HEK293T cells. The inhibited activities of the ori-Lyt promoter were shown not only by the color change but also by the quantitative activity. Therefore, the repressive effects of SIRT6 on the ori-Lyt promoter are dose dependent.
FIG 2.

Luciferase assay to determine the effects of SIRT6 on the promoter activity of ori-Lyt. The ori-Lyt-driven luciferase plasmid was cotransfected with the vector (pcDNA3) or different amounts of the SIRT6-expressing plasmid into HEK293T cells for 24 h. The amount of total DNA used in the transfection system was adjusted so that the amount was the same in each group by adding pcDNA3. Then, the cells were applied to an IVIS imaging system to examine the luciferase activities. (Top) The IVIS system was used to visualize the signal and take photos. The depth of the color strands represents the strength of luciferase activity. (Middle) Activities quantitated from three independent experiments were statistically averaged, and the activity units are shown on the y axis. (Bottom) Whole-cell lysate samples were collected after the luciferase assay, and Western blotting was performed to detect the expression of SIRT6.
Biological effects of SIRT6 on other KSHV promoters: pORF50 and pORF59.
We next examined whether SIRT6 could have any biological effects on other KSHV promoters, such as the pORF50 and pORF59 promoters. Both ORF50 (RTA) and ORF59 are essential for KSHV reactivation and viral DNA replication, with RTA being the immediate early (IE) gene and ORF59 being the late gene. To know whether SIRT6 has any effect on these promoters, we cotransfected a pORF50 or pORF59 promoter-controlling luciferase-expressing plasmid (pORF50_luc or pORF59_luc) with different amounts of a SIRT6-expressing plasmid into HEK293T cells, as shown in Fig. 3. At 24 h after transfection, the luciferase activity of the cells was analyzed with the IVIS Spectrum imaging system. As can be seen, SIRT6 repressed the activities of both the ORF50 (RTA) promoter and the ORF59 promoter, and the repressive effects of SIRT6 on pORF50 or pORF59 were dose dependent. Therefore, SIRT6 might have a broad effect on KSHV gene expression.
FIG 3.

Luciferase assay to determine the effects of SIRT6 on the promoter activity of ORF50 or ORF59. The ORF50- or ORF59-driven luciferase plasmid was cotransfected with the vector (pcDNA3) or different amounts of SIRT6-expressing plasmid into HEK293T cells for 24 h. The amount of total DNA used in the transfection system was adjusted so that it was the same in each group. Then, the cells were applied to an IVIS imaging system to examine the luciferase activities. (Top) The activities quantitated from three independent experiments were statistically averaged, and the activity units are shown on the y axis. (Bottom) Whole-cell lysate samples were collected after luciferase assay, and Western blotting was performed to detect the expression of SIRT6.
SIRT6 seemed to have inhibitory effects on all 3 KSHV promoters, which raised the question of the specificity of the assays. To know whether the suppressive effect of SIRT6 is specific, we cotransfected SIRT6 with ori-Lyt- or other promoter-controlling luciferase plasmids, which are listed in Fig. 4. As shown at the top of Fig. 4, SIRT6 exerted more repressive effects on the ori-Lyt promoter than on the human cytomegalovirus (HCMV) or the murine cytomegalovirus (MCMV) MIE promoter, herpes simplex virus 1 thymidine kinase (TK) promoter, or the respiratory syncytial virus (RSV) promoter. Although SIRT6 could significantly inhibit the RSV promoter, no significant effects of SIRT6 on the other promoters, such as TK, HCMV MIE, and MCMV MIE, were observed. The expression of SIRT6 is shown at the bottom of Fig. 4. These results demonstrate that SIRT6 has the most suppressive effects on the ori-Lyt of KSHV and that its inhibitory effects on KSHV promoters are specific.
FIG 4.
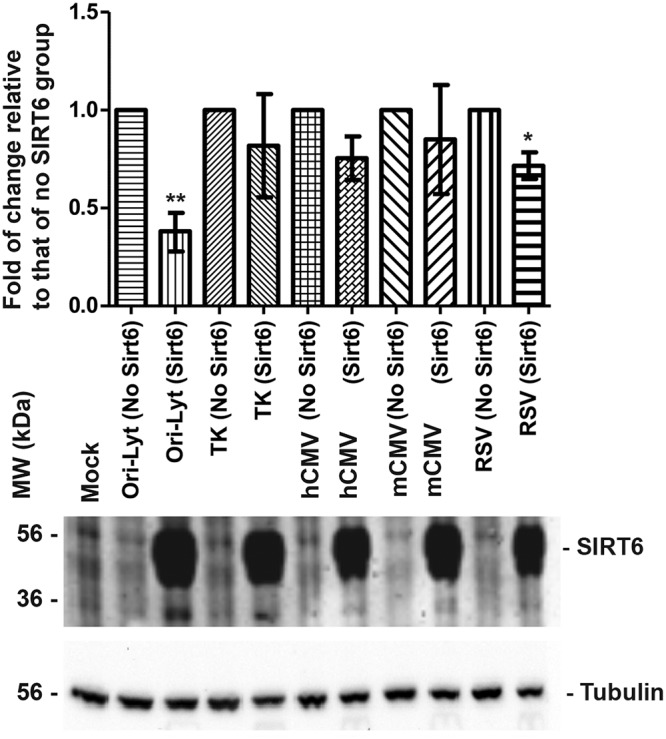
Luciferase assay to determine the effects of SIRT6 on different promoter activities. The indicated luciferase plasmid was cotransfected with the vector (pcDNA3) or SIRT6-expressing plasmid into HEK293T cells for 24 h. The amount of total DNA used in the transfection system was adjusted so that it was the same in each group. Then, the cells were applied to an IVIS imaging system to examine the luciferase activities. (Top) The activities quantitated from three independent experiments were statistically averaged, and the activity units are shown in the y axis. (Bottom) The whole-cell lysate samples were collected after luciferase assay, and Western blotting was performed to detect the expression of SIRT6.
SIRT6 represses KSHV reactivation.
From the results shown above, we hypothesized that SIRT6 overexpression might be able to repress the reactivation of KSHV. To examine this, we used a KSHV-infected cell line, SLK-iBAC-gfpK52, that harbors a KSHV bacterial artificial chromosome (BAC) in which green fluorescent protein (GFP) was fused with a KSHV late protein, K52. The cells normally have no fluorescence and are maintained in culture medium with hygromycin but can be induced by doxycycline (Dox) to produce GFP. The presence of GFP indicates the reactivation of KSHV by doxycycline from the SLK-iBAC-gfpK52 cells. If SIRT6 can inhibit the reactivation of KSHV, the SIRT6-positive cells should show a negative staining of KSHV proteins and replicated DNA.
We transfected the SLK-iBAC-gfpK52 cells with SIRT6-expressing plasmid for 24 h, and then doxycycline (10 ng/ml) was added to the cells for another 48 h. First, we performed an immunofluorescent assay (IFA) to know whether SIRT6 represses KSHV RTA protein production. As shown in Fig. 5A, we observed that the RTA protein was present at a lower level in almost all the SIRT6-positive cells than in the SIRT6-negative cells. This is more clearly visualized in the ×40 magnification image in Fig. 5B. We counted the total number of cells per the number of RTA-positive cells, the number of SIRT6-positive cells per the number of RTA-positive cells and the number of SIRT6 positive cells per the number of RTA-negative cells and found that SIRT6 significantly reduced the level of RTA expression (Fig. 5D). RTA was present in 34% of the cells at 48 h after doxycycline treatment, while only 8% RTA-positive cells were seen among the SIRT6-positive ones. Then, we performed DNA fluorescent in situ hybridization (FISH) to determine whether viral DNA replication could be repressed by SIRT6 overexpression. As shown in the Fig. 5C, almost all the SIRT6-positive cells were negative for KSHV DNA replication. We counted the total number of cells, the number of SIRT6-positive cells, and the number of DNA replication-positive cells. As shown in Fig. 5E, 33% of the cells were positive for viral DNA replication, while only 5% of the SIRT6-positive cells were positive for viral DNA replication. Therefore, overexpression of SIRT6 significantly suppressed the reactivation of KSHV.
FIG 5.

Immunofluorescence assay (IFA) to determine KSHV protein expression and FISH to examine viral DNA replication. (A and B) Cells of the SLK-iBAC-gfpK52 cell line were transfected with a SIRT6-expressing plasmid; 24 h later, the cells were treated with 100 nM doxycycline. Twenty-four hours later, the cells were fixed for IFA to detect the KSHV protein RTA. The pictures were taken under a confocal microscope at magnifications of ×10 (A) and ×40 (B). (C) FISH was performed to examine DNA replication. (D) The proportions of RTA-positive cells counted among total cells, SIRT6-positive cells, and SIRT6-negative cells were determined. (E) The proportions of DNA replication-positive cells among total cells, SIRT6-positive cells, and SIRT6-negative cells were determined. A Student's t test was performed to calculate statistical significance. **, P < 0.001. DAPI, 4′,6-diamidino-2-phenylindole.
We then performed a Western blot assay to detect KSHV protein production and PCR to detect viral DNA production, as shown in Fig. 6. A diagram of the experimental procedure is shown in Fig. 6A. We transfected cells of the SLK-iBAC-gfpK52 cell line with different amounts of pFLAG-SIRT6 for 24 h and stimulated the cells with doxycycline for 48 h, and the cells were collected for either protein detection or determination of DNA levels. As shown in Fig. 6B, the KSHV protein levels were reduced by the transfected pFLAG-SIRT6. The KSHV proteins detected included an immediate early (IE) protein (RTA), an early protein (K8), and a late one (K52). The viral DNA level was also seen to be repressed by SIRT6, as shown in Fig. 6C. These results were consistent with those shown in Fig. 5. Therefore, overexpression of SIRT6 inhibited KSHV reactivation.
FIG 6.

Western blot and PCR assays to determine the effects of SIRT6 on KSHV reactivation. (A) Diagram of the experiments performed with the SLK-iBCgfpK52 cell line. (B) Cells of the SLK-iBAC-gfpK52 cell line were treated as shown in panel A with different amounts of pFLA-SIRT6, and whole-cell lysates were used for the Western blot assay to detect KSHV proteins (RTA, K8, and K52), cellular protein (tubulin), and SIRT6. (C) The SLK-iBAC-gfpK52 cell line was treated as shown in panel A with different amounts of pFLA-SIRT6. The viral DNA and mitochondrial DNA (Mt DNA) were isolated from cells using the Hirt method at 48 h after transfection. Regular PCR (bottom) or real-time PCR (top) was performed to examine the viral DNA. Mitochondrial DNA was used as a loading control. **, P < 0.001.
Depletion of SIRT6 enhanced the reactivation of KSHV and viral replication.
The effects of SIRT6 on KSHV have been tested under conditions of overexpression, so we wondered whether the depletion of SIRT6 could favor KSHV reactivation and infection. To that end, we performed experiments to deplete SIRT6, using small interfering RNA (siRNA), from SLK-iBAC-gfpK52 cells, which were tested by IFA, as shown in Fig. 7A, and from HEK293T cells, which were tested by Western blotting, as seen in Fig. 7B. A small scrambled RNA designed for luciferase was used as the control (the group with no siRNA [siRNA−]). The results of IFA and Western blotting showed that SIRT6 was depleted (Fig. 7A and B). The decrease in the level of SIRT6 was caused by the siRNA for 24 h. First, we wondered if the siRNA could enhance KSHV infection. As shown in Fig. 7C, the HEK293T cells were treated with siRNA or scrambled small RNA (no siRNA) for 24 h, washed with minimal essential medium (MEM), and infected with KSHV for another 48 h. The cells were lysed to isolate KSHV DNA, which was used for real-time PCR. We found that siRNA enhanced the level of KSHV DNA replication by ∼10 times.
FIG 7.

Effects of depletion of SIRT6 on reactivation of KSHV and viral replication. (A) siRNA (0.4 nM/ml) or scrambled small RNA (no siRNA) was transfected into SLK-iBAC-gfpK52 cells for 24 h, the medium was changed, and the cells were incubated for another 24 h to perform IFA to examine for SIRT6 (red) and DAPI (4′,6-diamidino-2-phenylindole; blue) staining. (B) HEK293T cells were transfected with siRNA or scrambled small RNA (no siRNA) for 24 h to examine SIRT6 by Western blotting. (C) HEK293T cells were treated with siRNA or scrambled small RNA (siRNA−) for 24 h, washed with MEM, and infected with KSHV for another 48 h. The cells were lysed to isolate KSHV DNA, which was used for real-time PCR. (D) The SLK-iBAC-gfpK52 cells were treated with siRNA for 24 h, and different amounts of doxycycline were added for another 24 h. The total number of cells and the number of reactivated cells (GFP positive) were counted. (E) As for panel D, the whole-cell lysates were used for the Western blot assay to detect the KSHV protein RTA. *, P < 0.001; **, P > 0.05 (the results of three independent experiments were averaged and statistically analyzed using Student's t test).
Then, we wondered if SIRT6 depletion could help with the reactivation of KSHV. The KSHV in SLK-iBAC-gfpK52 cells could not be directly reactivated by siRNA depletion, so SIRT6 is not the only factor that exerts suppressive effects on KSHV. We asked whether the siRNA could help with the reactivation of KSHV from SLK-iBAC-gfpK52 cells. For that purpose, we treated the cells with siRNA for 24 h and added different amounts of doxycycline for another 24 h. The total and reactivated (GFP-positive) cells were counted. As shown in Fig. 7D, the siRNA against SIRT6 significantly enhanced the reactivation of KSHV when a small amount of doxycycline was used but did not significantly enhance the reactivation of KSHV when a larger amount of doxycycline was used. The results were further confirmed by a Western blot assay to detect the KSHV protein RTA, as shown in Fig. 7E. Therefore, depletion of SIRT6 favors the reactivation and infection of KSHV.
SIRT6 interacts with the KSHV genome.
SIRT6 is a member of the HDAC family, which, in general, catalyzes the conversion of the acetylation to the deacetylation of histone or prevents the acetylation of histones to negatively affect gene expression. This procedure requires the interaction of HDAC or SIRT with DNA. Therefore, we hypothesized that SIRT6 interacts with KSHV DNA to exert its regulating functions. To determine that, we performed a DNA chromatin immunoprecipitation (ChIP) assay. SLK-iBAC-gfpK52 cells were stimulated or not stimulated with doxycycline for 24 h and fixed with 4% paraformaldehyde for 20 min. The cross-linked cells were sonicated to break the DNA into fragments 200 to 300 bp in size. Then, the broken DNA-protein complex was pulled down with IgG or anti-SIRT6. The anti-SIRT6 antibody is a ChIP-grade antibody from Abcam, and its quality was confirmed by a Western blot assay, as shown in Fig. 8A. A real-time PCR was used to determine the DNA that was pulled down by IgG or anti-SIRT6 using 2 pairs of primers (one for the ori-Lyt promoter and the other for the pORF50 promoter), as shown in Fig. 8B. We found that SIRT6 interacts with KSHV DNA in both the latent (without Dox) and lytic (with Dox) phases. However, the DNA-SIRT6 interaction was decreased when the KSHV latency was reactivated. This suggests that the interaction of SIRT6 with KSHV DNA is important for the maintenance of viral latency. In summary, cellular gene repressor SIRT6 inhibited KSHV protein production, DNA replication, and reactivation through the DNA-protein interaction.
FIG 8.

Chromatin immunoprecipitation (ChIP) assay to determine the interaction of SIRT6 with KSHV DNA. (A) The anti-SIRT6 antibody was used to detect SIRT6 in SLK-iBAC-gfpK52 cells with or without doxycycline for 48 h. The whole-cell lysates were applied to the Western blot assay to examine the indicated proteins. (B) Cells of the SLK-iBAC-gfpK52 cell line were treated with or without doxycycline for 48 h, fixed with 4% formaldehyde, sonicated to shear the DNA to fragments over a range of 200 to 400 bp in size, and aliquoted into three groups: input, normal mouse IgG incubation, and incubation with anti-SIRT6. The pulled down DNA was detected by PCR using primers designed for the ori-Lyt promoter, the ORF50 promoter, or ORF50 gene-encoding sequences (ORF50 gene). The DNA amount was compared to the input amount, and the result is shown as a percentage.
DISCUSSION
KSHV latent infection not only is a strategy of the virus to evade the host's immune surveillance mechanisms but also can have potentially life-threatening cancerous consequences that can occur at any time in the life of the host. KSHV reactivation has been considered a critical step in its pathogenesis (2, 22, 23) because not only does such reactivation expand the viral reservoir but also several lytic KSHV gene products also have antiapoptotic, proliferative, or immunosuppressive properties, increasing the likelihood of malignant transformation (24, 25). The notion that lytic replication has a role in oncogenesis is supported by the fact that KSHV-infected individuals who received long-term antiviral therapy for other (i.e., non-KSHV) infections had lower incidences of KS than their counterparts who did not receive such therapy (26). A dynamic balance between latent and lytic gene expression is required to maintain the persistent infection with KSHV (27). Although lifelong combination antiretroviral therapy (cART) to treat HIV-1 infection has greatly reduced the occurrence of KSHV-related cancer in AIDS patients, KSHV remains in patients for life by connecting its genome to human chromosomes episomally. Therefore, KSHV-infected individuals remain at risk for the development of cancer as a result of the reactivation of latently infected cells.
The gap in the understanding of how KSHV is reactivated has impeded the development of strategies for interfering with KSHV gene expression and viral replication. HDAC family members are generally repressors of cellular and viral genes. Previous studies have demonstrated that HDAC1 interacts with RTA and represses ORF50-mediated viral transcription (28); that the ORF50 promoter is negatively associated with several HDACs (HDACs 1, 5, and 7) (7); that HDAC inhibitors, such as sodium butyrate and trichostatin A, are sufficient to activate the lytic replication of KSHV by activating the ORF50 promoter (29–31); and that SIRT1 regulates KSHV latency by inhibiting the promoter of ORF50 (32, 33). Therefore, a screening of HDACs’ effects on KSHV gene expression and reactivation must provide informative insight into how KSHV reactivation is affected by cellular gene regulators. In this study, we first screened the effects of all 18 HDAC family members (HDACs 1 to 11 and SIRTs 1 to 7) on the KSHV ori-Lyt promoter (Fig. 1) and found that HDACs 2, 7, 8, and 11 and SIRTs 4 and 6 are able to significantly suppress the promoter activity of ori-Lyt. SIRT6 was found to be the strongest inhibitor of ori-Lyt. Further studies revealed that SIRT6 can also repress other KSHV promoters, including pORF50 and pORF59 (Fig. 2 and 3). Using a KSHV BAC DNA-containing cell line (SLK-iBAC-gfpK52), we demonstrated that SIRT6 overexpression is sufficient to repress KSHV gene expression and DNA replication (Fig. 5 and 6). The results were further confirmed by a gene depletion method using siRNA (Fig. 7). In searching for the mechanism of how SIRT6 represses the ori-Lyt and pORF50 promoters, we performed a ChIP assay and found that SIRT6 interacts with KSHV DNA. Therefore, we conclude that SIRT6, a nuclear gene suppressor, has inhibitory effects on KSHV reactivation by interacting with KSHV gene promoters.
siRNA (to SIRT6) could effectively deplete the SIRT6 protein from either SLK-iBAC-gfpK52 or HEK293T cells. The KSHV-harboring cells (SLK-iBAC-gfpK52) were very sensitive to the inducer doxycycline. As can be seen in Fig. 7D, treatment with doxycycline at 0.1 ng per ml for 24 h was enough to reactivate about 4% of the scrambled small RNA-treated cells, but it could reactivate 12% of the siRNA-treated cells. When the concentration of doxycycline was increased to 10 ng/ml or higher, siRNA failed to help the reactivation by doxycycline, indicating that the high-level induction of RTA probably circumvents the suppressive effect of SIRT6. It was also demonstrated that the siRNA enhanced KSHV DNA replication in the context of infection in HEK293T cells (Fig. 7C). However, siRNA alone failed to reactivate KSHV reactivation from SLK-iBAC-gfpK52 cells. This may be explained by the fact that multiple cellular factors are able to repress KSHV reactivation and/or infection.
KSHV ORF50 is the key protein for reactivation of KSHV not only because it is required for the KSHV life cycle switch from latency to the lytic phase but also because the exogenous expression of the ORF50 protein is sufficient to reactivate latent KSHV from cells (18, 29, 34). The ORF50 protein activates KSHV gene expression and binds to ori-Lyt to initiate the DNA replication program (20, 21). Hence, our present studies focused on the interaction of SIRT6 with ori-Lyt and pORF50. Future studies will aim to reveal whether SIRT6 interacts with other KSHV DNA elements at a global level. No specific SIRT6 inhibitor (small compound) is available; the SIRT6-KSHV DNA might be an effective target for developing an interfering molecule for infection prevention or therapeutic purposes. Interestingly, a recent study found that the KSHV-encoded protein viral interferon regulatory factor 3, which interacts with HDAC5, contributes to KSHV-induced lymphangiogenesis, which is related to KSHV-caused malignancy (35). Therefore, studies on the relationship between HDAC/SIRT and KSHV might also result in an understanding of how KSHV causes cancer.
MATERIALS AND METHODS
Tissue culture and virus.
The SLK-iBAC-gfpK52 (36) and HEK293T (ATCC CRL-11268) cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS), 100 U/ml penicillin, 100 μg/ml streptomycin, and amphotericin B (2.5 μg/ml) (37). For immunohistochemical staining, cells were grown on round coverslips (Corning Incorporated, Corning, NY) in 24-well plates (Becton, Dickinson and Company, Franklin Lakes, NJ). KSHV for infection was obtained from induced BCBL-1 cells as previously described (38).
Antibodies.
Monoclonal antibodies against FLAG (M2) were purchased from Sigma-Aldrich (St. Louis, MO). The monoclonal antibodies against tubulin (sc-58666) and GFP (sc-9996) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-RTA monoclonal antibody was a gift from Yan Yuan (University of Pennsylvania) (21). Rabbit anti-SIRT6 antibody (catalog number ab62739) was bought from Abcam (Cambridge, MA).
Plasmids, siRNA, and transfection.
Plasmids expressing FLAG-tagged HDAC1 to HDAC11 and SIRT1 to SIRT7 were gifts from Edward Seto. Plasmids expressing luciferase under the control of different KSHV promoters, pori-Lyt-Luc, pORF50-luc, and pORF59-luc, were gifts from Yan Yuan (University of Pennsylvania) (20, 39). siRNA to SIRT6 and a scrambled small RNA, used as a negative control, were purchased from Fisher Life Technologies (catalog numbers AM16708 and ID 116148, respectively). Plasmids expressing luciferase under the control of other promoters included pRSV-luc, pTK-luc, pMCMV-luc, and pHCMV-luc. These plasmids were generated by changing the promoter from pORF50-luc and were stored in Q. Tang's laboratory. The transfection of plasmids and siRNA was performed using the Lipofectamine 3000 reagent (Life Technologies, Grand Island, NY), according to the manufacturer's instructions.
Luciferase assay.
HEK293T cells were seeded in a 24-well plate and were cotransfected with HDAC- or SIRT-expressing plasmids with a luciferase reporter gene directed by the promoter of ORF50 (genomic location, nucleotides 70561 to 71598; GenBank accession number U75698) [25]; pORF50-luc), ORF59 (genomic location, nucleotides 96737 to 98034; GenBank accession number U75698; pORF59-luc), or ori-Lyt (genomic location, nucleotides 24093 to 24342; GenBank accession number U75698; pori-Lyt-luc). Each assay was performed in triplicate, and the luciferase activity was normalized by the amounts of total protein. At 24 h or 48 h after transfection, we added the d-luciferin solution (catalog number P/N 122796; PerkinElmer Inc., Waltham, MA) to the cells at a final concentration of 150 μg/ml and incubated the cells for 10 min. The cells were imaged with an IVIS Spectrum imaging system with LivingImage software (PerkinElmer Inc.) to quantitatively measure the number of photons emitted by the luciferase activity. This measurement is represented as a color map and overlaid on a monochrome photograph of the 24-well plate for identification. Regions of interest (ROI) were drawn to quantify the luciferase activity.
Viral DNA isolation from cells and viral DNA assay by regular or real-time PCR.
KSHV DNA from SLK-iBAC-gfpK52 cells was isolated using Hirt's method (40). In brief, the SLK-iBAC-gfpK52 cells were treated or not treated with doxycycline and were lysed in Hirt buffer (0.6% SDS, 10 mM EDTA, pH 7.5). The cell debris and cellular chromosome were pelleted by adding 1/4 volume of 5 M NaCl. The DNA-containing supernatant was treated with RNase, extracted with phenol-chloroform, and mixed with 70% isopropanol, and the viral DNA was precipitated by centrifugation. To examine the KSHV DNA amount, a real-time PCR was performed in a 25-μl reaction mixture using the primers Ori-Lyt_Forward (CCT ACA TGG GCA GCT TGT CC) and Ori-Lyt_Reverse (TGC TGC CGG GGC TCC TCG TT). All samples were analyzed in triplicate using SYBR Green 2× master mix (Applied Biosystems, Foster City, CA) on an Applied Biosystems 7900HT Fast real-time PCR system. After an initial denaturation incubation of 5 min at 95°C, 40 cycles of three-step cycling were performed with an annealing temperature of 55°C. Melt curve analysis was then performed to verify product specificity.
Immunocytochemistry and fluorescence in situ hybridization.
Cells were grown on coverslips, and immunostaining was performed after fixation with 1% paraformaldehyde (10 min at room temperature) and permeabilization in 0.2% Triton X-100 (20 min on ice) by sequential incubation with primary and Texas Red (TR)-labeled secondary antibodies (Vector Laboratories, Burlingame, CA) for 30 min each (all solutions were in phosphate-buffered saline [PBS]). For the simultaneous detection of KSHV DNA, cells were first immunostained for cellular or viral proteins and then treated for 1 h at 37°C with DNase-free RNase (200 U/ml in PBS containing 25 mM MgCl2; Roche, Indianapolis, IN). After refixation in 4% paraformaldehyde (10 min at room temperature), samples were equilibrated in 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), dehydrated in ethanol (70, 80, and 100% ethanol for 3 min each at −20°C), air dried, and incubated overnight at 37°C with the hybridization mixture. To detect DNA, the probe DNA and the cellular DNA were denatured at 94°C for 5 min. After hybridization, samples were washed at 37°C with 55% formamide in 2× SSC (twice for 15 min each time), 2× SSC (10 min), and 0.25× SSC (twice for 5 min each time). The hybridized probes were labeled with fluorescein isothiocyanate (FITC)-avidin (1:500 in 4× SSC plus 0.5% bovine serum albumin; Vector Laboratories), and the signals were amplified by using biotinylated antiavidin (1:250; Vector Laboratories), followed by another round of FITC-avidin staining. Finally, the cells were equilibrated in PBS, stained for DNA with Hoechst 33258 (0.5 μg/ml), and mounted in Fluoromount G mounting medium (Fisher Scientific, Newark, DE).
Probe preparation-nick translation.
The BACmid of KSHV (39) containing whole KSHV DNA was labeled with biotin-11-dUTP by nick translation. The DNase concentration for nick translation was adjusted to yield probe DNA fragments 200 to 500 bp in length. Probe DNA was dissolved at 10 ng/μl in Hybrisol VII hybridization solution (Oncor, Gaithersburg, MD) containing 100 ng of salmon sperm DNA (Gibco-BRL), 1 μg of yeast tRNA (Sigma), and 0.5 mg of Cot1 DNA (Gibco-BRL)/μl.
Immunoblot analysis.
Proteins were separated by sodium dodecyl sulfate-7.5% polyacrylamide gel electrophoresis (10 to 20 μg loaded in each lane), transferred to nitrocellulose membranes (GE Healthcare), and blocked with 5% nonfat milk for 60 min at room temperature. The membranes were incubated overnight at 4°C with primary antibody, followed by incubation with a horseradish peroxidase-coupled secondary antibody (Amersham Inc.) and detection with enhanced chemiluminescence (Pierce, Rockford, IL) using standard methods. Membranes were stripped with stripping buffer (100 mM beta-mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl, pH 6.8) for 30 min at 50°C, washed with PBS–0.1% Tween 20, and used to detect additional proteins.
ChIP assay and real-time PCR.
The SLK-iBAC-gfpK52 cells were seeded in a 100-mm dish, and 24 h later, the cells were either induced or not induced with doxycycline for 48 h. Then, the cells were fixed with 1% formaldehyde and then sonicated to shear the DNA into 100- to 300-bp fragments. The anti-SIRT6 antibody or normal IgG was incubated with the DNA-protein mixture, and chromatin immunoprecipitation (ChIP) assays were performed using an EZ-ChIP kit (Millipore, Billerica, MA), according to the manufacturer’s instructions. The amount of DNA immunoprecipitated by antibodies (normal IgG or anti-SIRT6) was examined by regular or real-time PCR in a 25-μl reaction mixture using the primers Ori-Lyt_Forward (CCT ACA TGG GCA GCT TGT CC) and Ori-Lyt_Reverse (TGC TGC CGG GGC TCC TCG TT), ORF50 promoter_forward (5′-CAT GAC TCC AAC CGT CT-3′) and ORF50 promoter reverse (5′-CCG TGA GGA TCC GAA T-3′), and ORF50 encoding gene forward (5′-CGCAATGCGTTACGTTGTTG-3′) and ORF50 encoding gene reverse (5′-GCCCGGACTGTTGAATCG-3′). All samples were analyzed in triplicate using SYBR Green 2× master mix (Applied Biosystems, Foster City, CA) on an Applied Biosystems 7900HT Fast real-time PCR system. After an initial denaturation incubation of 5 min at 95°C, 40 cycles of three-step cycling were performed with an annealing temperature of 55°C. Melt curve analysis was then performed to verify product specificity. The ChIP efficiency for each antibody was calculated as the percentage of the input using the threshold cycle method, and the standard deviation was generated from the triplicate PCRs.
Confocal microscopy.
Cells were examined at ×10, ×40, and ×60 magnification with a Leica TCS SPII confocal laser scanning system equipped with a water-cooled argon-krypton laser. Two wavelength channels (495 and 590 nm) were recorded simultaneously or sequentially. Power and integration were adjusted to minimize bleed-through between the green and far red channels prior to data acquisition. The digital images obtained were cropped and adjusted for contrast with Photoshop software (Adobe Systems, San Jose, CA).
ACKNOWLEDGMENTS
We acknowledge the instrumental support of the PSM Molecular Biology Core Laboratory. We thank Yan Yuan for providing us with the anti-RTA antibody and the luciferase-expressing plasmids. We thank Fayuan Wen for technical assistance and Stephen Lin for English editing.
This study was supported by NIH/NIAID SC1AI112785 (to Q.T.); the National Institute on Minority Health and Health Disparities of the National Institutes of Health under award numbers G12MD007597 (to Q.T.), R01DE016680 (to F.Z.), and R01DE026101 (to F.Z.); a China postdoctoral science fund project (2016M592037 to M.H.); and a 2016 Anhui postdoctoral fund project (2016B096 to M.H.).
REFERENCES
- 1.Ganem D. 2010. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Invest 120:939–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cannon MJ, Laney AS, Pellett PE. 2003. Human herpesvirus 8: current issues. Clin Infect Dis 37:82–87. doi: 10.1086/375230. [DOI] [PubMed] [Google Scholar]
- 3.Chapple IL, Hamburger J. 2000. The significance of oral health in HIV disease. Sex Transm Infect 76:236–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crum-Cianflone N, Hullsiek KH, Satter E, Marconi V, Weintrob A, Ganesan A, Barthel RV, Fraser S, Agan BK. 2009. Cutaneous malignancies among HIV-infected persons. Arch Intern Med 169:1130–1138. doi: 10.1001/archinternmed.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Speck SH, Ganem D. 2010. Viral latency and its regulation: lessons from the gamma-herpesviruses. Cell Host Microbe 8:100–115. doi: 10.1016/j.chom.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaw RN, Arbiser JL, Offermann MK. 2000. Valproic acid induces human herpesvirus 8 lytic gene expression in BCBL-1 cells. AIDS 14:899–902. [DOI] [PubMed] [Google Scholar]
- 7.Lu F, Zhou J, Wiedmer A, Madden K, Yuan Y, Lieberman PM. 2003. Chromatin remodeling of the Kaposi's sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J Virol 77:11425–11435. doi: 10.1128/JVI.77.21.11425-11435.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller G, Heston L, Grogan E, Gradoville L, Rigsby M, Sun R, Shedd D, Kushnaryov VM, Grossberg S, Chang Y. 1997. Selective switch between latency and lytic replication of Kaposi's sarcoma herpesvirus and Epstein-Barr virus in dually infected body cavity lymphoma cells. J Virol 71:314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fouladi M. 2006. Histone deacetylase inhibitors in cancer therapy. Cancer Invest 24:521–527. doi: 10.1080/07357900600814979. [DOI] [PubMed] [Google Scholar]
- 10.Marks PA, Xu WS. 2009. Histone deacetylase inhibitors: potential in cancer therapy. J Cell Biochem 107:600–608. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. 2003. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seto E, Yoshida M. 2014. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol 6:a018713. doi: 10.1101/cshperspect.a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kouzarides T. 2007. SnapShot: histone-modifying enzymes. Cell 128:802. doi: 10.1016/j.cell.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 14.Kouzarides T. 2007. Chromatin modifications and their function. Cell 128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 15.Lukac DM, Kirshner JR, Ganem D. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J Virol 73:9348–9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lukac DM, Renne R, Kirshner JR, Ganem D. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304–312. doi: 10.1006/viro.1998.9486. [DOI] [PubMed] [Google Scholar]
- 17.Zhu FX, Cusano T, Yuan Y. 1999. Identification of the immediate-early transcripts of Kaposi's sarcoma-associated herpesvirus. J Virol 73:5556–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun R, Lin SF, Staskus K, Gradoville L, Grogan E, Haase A, Miller G. 1999. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J Virol 73:2232–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.AuCoin DP, Colletti KS, Cei SA, Papouskova I, Tarrant M, Pari GS. 2004. Amplification of the Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 lytic origin of DNA replication is dependent upon a cis-acting AT-rich region and an ORF50 response element and the trans-acting factors ORF50 (K-Rta) and K8 (K-bZIP). Virology 318:542–555. doi: 10.1016/j.virol.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 20.Lin CL, Li H, Wang Y, Zhu FX, Kudchodkar S, Yuan Y. 2003. Kaposi's sarcoma-associated herpesvirus lytic origin (ori-Lyt)-dependent DNA replication: identification of the ori-Lyt and association of K8 bZip protein with the origin. J Virol 77:5578–5588. doi: 10.1128/JVI.77.10.5578-5588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Tang Q, Maul GG, Yuan Y. 2006. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: dual role of replication and transcription activator. J Virol 80:12171–12186. doi: 10.1128/JVI.00990-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bubman D, Cesarman E. 2003. Pathogenesis of Kaposi's sarcoma. Hematol Oncol Clin North Am 17:717–745. [DOI] [PubMed] [Google Scholar]
- 23.Janier M, Agbalika F, de La Salmoniere P, Lassau F, Lagrange P, Morel P. 2002. Human herpesvirus 8 seroprevalence in an STD clinic in Paris: a study of 512 patients. Sex Transm Dis 29:698–702. doi: 10.1097/00007435-200211000-00013. [DOI] [PubMed] [Google Scholar]
- 24.Staskus KA, Sun R, Miller G, Racz P, Jaslowski A, Metroka C, Brett-Smith H, Haase AT. 1999. Cellular tropism and viral interleukin-6 expression distinguish human herpesvirus 8 involvement in Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease. J Virol 73:4181–4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li DJ, Verma D, Mosbruger T, Swaminathan S. 2014. CTCF and Rad21 act as host cell restriction factors for Kaposi's sarcoma-associated herpesvirus (KSHV) lytic replication by modulating viral gene transcription. PLoS Pathog 10:e1003880. doi: 10.1371/journal.ppat.1003880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin DF, Kuppermann BD, Wolitz RA, Palestine AG, Li H, Robinson CA. 1999. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. Roche Ganciclovir Study Group. N Engl J Med 340:1063–1070. doi: 10.1056/NEJM199904083401402. [DOI] [PubMed] [Google Scholar]
- 27.Kang H, Wiedmer A, Yuan Y, Robertson E, Lieberman PM. 2011. Coordination of KSHV latent and lytic gene control by CTCF-cohesin mediated chromosome conformation. PLoS Pathog 7:e1002140. doi: 10.1371/journal.ppat.1002140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gwack Y, Byun H, Hwang S, Lim C, Choe J. 2001. CREB-binding protein and histone deacetylase regulate the transcriptional activity of Kaposi's sarcoma-associated herpesvirus open reading frame 50. J Virol 75:1909–1917. doi: 10.1128/JVI.75.4.1909-1917.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye J, Gradoville L, Daigle D, Miller G. 2007. De novo protein synthesis is required for lytic cycle reactivation of Epstein-Barr virus, but not Kaposi's sarcoma-associated herpesvirus, in response to histone deacetylase inhibitors and protein kinase C agonists. J Virol 81:9279–9291. doi: 10.1128/JVI.00982-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shin HJ, DeCotiis J, Giron M, Palmeri D, Lukac DM. 2014. Histone deacetylase classes I and II regulate Kaposi's sarcoma-associated herpesvirus reactivation. J Virol 88:1281–1292. doi: 10.1128/JVI.02665-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhatt S, Ashlock BM, Toomey NL, Diaz LA, Mesri EA, Lossos IS, Ramos JC. 2013. Efficacious proteasome/HDAC inhibitor combination therapy for primary effusion lymphoma. J Clin Invest 123:2616–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He M, Gao SJ. 2014. A novel role of SIRT1 in gammaherpesvirus latency and replication. Cell Cycle 13:3328–3330. doi: 10.4161/15384101.2014.968431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He M, Yuan H, Tan B, Bai R, Kim HS, Bae S, Che L, Kim JS, Gao SJ. 2016. SIRT1-mediated downregulation of p27Kip1 is essential for overcoming contact inhibition of Kaposi's sarcoma-associated herpesvirus transformed cells. Oncotarget 7:75698–75711. doi: 10.18632/oncotarget.12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gradoville L, Gerlach J, Grogan E, Shedd D, Nikiforow S, Metroka C, Miller G. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J Virol 74:6207–6212. doi: 10.1128/JVI.74.13.6207-6212.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee HR, Li F, Choi UY, Yu HR, Aldrovandi GM, Feng P, Gao SJ, Hong YK, Jung JU. 2018. Deregulation of HDAC5 by viral interferon regulatory factor 3 plays an essential role in Kaposi's sarcoma-associated herpesvirus-induced lymphangiogenesis. mBio 9:e02217-17. doi: 10.1128/mBio.02217-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Avey D, Tepper S, Li W, Turpin Z, Zhu F. 2015. Phosphoproteomic analysis of KSHV-infected cells reveals roles of ORF45-activated RSK during lytic replication. PLoS Pathog 11:e1004993. doi: 10.1371/journal.ppat.1004993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Bruyn Kops A, Knipe DM. 1988. Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell 55:857–868. [DOI] [PubMed] [Google Scholar]
- 38.Cheng B, Martinez FP, Katano H, Tang Q. 2009. Evidence of inability of human cytomegalovirus to reactivate Kaposi's sarcoma-associated herpesvirus from latency in body cavity-based lymphocytes. J Clin Virol 46:244–248. doi: 10.1016/j.jcv.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martinez FP, Tang Q. 2012. Leucine zipper domain is required for Kaposi sarcoma-associated herpesvirus (KSHV) K-bZIP protein to interact with histone deacetylase and is important for KSHV replication. J Biol Chem 287:15622–15634. doi: 10.1074/jbc.M111.315861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hou W, Torres L, Cruz-Cosme R, Arroyo F, Irizarry L, Luciano D, Marquez A, Rivera LL, Sala AL, Luo MH, Tang Q. 2016. Two polypyrimidine tracts in intron 4 of the major immediate early gene are critical for gene expression switching from IE1 to IE2 and for replication of human cytomegalovirus. J Virol 90:7339–7349. doi: 10.1128/JVI.00837-16. [DOI] [PMC free article] [PubMed] [Google Scholar]