

Abstract
Antigen cross-presentation is an adaptation of the cellular process of loading MHC-I molecules with endogenous peptides during their biosynthesis within the endoplasmic reticulum. Cross-presented peptides derive from internalized proteins, microbial pathogens, and transformed or dying cells. The physical separation of internalized cargo from the endoplasmic reticulum, where the machinery for assembling peptide–MHC-I complexes resides, poses a challenge. To solve this problem, deliberate rewiring of organelle communication within cells is necessary to prepare for cross-presentation, and different endocytic receptors and vesicular traffic patterns customize the emergent cross-presentation compartment to the nature of the peptide source. Three distinct pathways of vesicular traffic converge to form the ideal cross-presentation compartment, each regulated differently to supply a unique component that enables cross-presentation of a diverse repertoire of peptides. Delivery of centerpiece MHC-I molecules is the critical step regulated by microbe-sensitive Toll-like receptors. Defining the subcellular sources of MHC-I and identifying sites of peptide loading during cross-presentation remain key challenges.
Keywords: cross-presentation, dendritic cells, endocytosis, phagocytosis, MHC class I, Toll-like receptors
INTRODUCTION
Major histocompatibility complex class I (MHC-I) molecules are polymorphic glycoproteins expressed at the cell surface of all nucleated cells. Their function is to bind short peptides 8–10 amino acids long and present them to CD8 T cells bearing T cell receptors (TCRs) with specificity to the presented peptide (1). An MHC-I molecule consists of a constant light chain called β2-microglobulin that is noncovalently bound to an α chain (the heavy chain) comprised of three domains, α1, α2, and α3 (2). The α1 and α2 domains form four antiparallel β strands and helical regions constituting the peptide-binding groove (3, 4). Within secondary lymphoid organs, peptide presentation by MHC-I on dendritic cells (DCs) either inactivates or primes naive CD8 T cells (5, 6). The decision is made based on the concomitant DC expression of T cell costimulatory molecules. These molecules are expressed on the DC surface as a result of cell-intrinsic signaling from pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) (7–10), that have engaged microbial components (11).
A viral or bacterial infection of cells within tissues leads to MHC-I presentation of foreign microbial peptides through what is termed as the classical pathway of MHC-I presentation. These peptides are recognized by TCRs on experienced antigen-specific cytotoxic CD8 T cells, which target the infected cell for destruction (2, 12) (Figure 1). Cross-presentation refers to the presentation of peptides derived from an extracellular source of proteins, which can include those derived from internalized proteins, microorganisms, or dying cells (Figure 1). During cross-presentation, extracellular proteins delivered into endosomes and phagosomes are physically located within a compartment distinct from the endoplasmic reticulum (ER), where the cornerstone of MHC-I presentation, the peptide-loading complex (PLC), primarily resides (Figure 1). This compartmentalization problem has instigated enormous research into the cell biological and molecular mechanisms that enable cross-presentation.
Figure 1.

The spatial challenge of loading MHC-I with peptide from an extracellular source. Cross-presentation entails loading MHC-I molecules with peptides derived from extracellular cargo such as bacteria, viruses, and dying cells that are internalized through either phagocytosis or endocytosis. Endosomes and phagosomes carrying these cargos are physically distinct from the endoplasmic reticulum (ER), where MHC-I molecules are synthesized, folded, and loaded with peptides. The MHC-I heavy chain polypeptide is cotranslationally translocated into the ER lumen through the Sec61 complex. Its first interaction is with the chaperone calnexin and is followed by assembly with β2-microglobulin (β2m). The MHC-I heavy chain/β2m heterodimer is unstable at this stage and is recruited by calreticulin to the peptide-loading complex (PLC) as part of its folding. Direct association of the empty MHC-I molecules with tapasin along with supporting interactions with calreticulin and ERp57 in the PLC stabilizes the empty MHC-I molecule and favors a conformation of the binding groove that is receptive to binding high-affinity peptides in the ER. Within the PLC, the transporter associated with antigen processing (TAP) translocates into the ER cytosolic peptides that are generated by proteasomal degradation of endogenous proteins, such as those derived from the translation of cellular proteins or viral proteins when cells are virally infected. These peptides are further trimmed by the ER aminopeptidases ERAP1 and ERAP2 to accommodate the peptide length preferred by MHC-I. After peptide loading, MHC-I molecules traffic to the ER-Golgi intermediate compartment (ERGIC) via COPII-coated export vesicles, where they are subjected to quality control (QC) by calreticulin, tapasin, and UDP-glucose:glycoprotein glucosyltransferase (UGT1). MHC-I molecules with low-affinity peptides (depicted as gray ovals bound to MHC-I) accumulate in the ERGIC when peptides with good affinity of loading are absent (for example, viral peptides depicted as green or cellular peptides depicted as orange). MHC-I molecules with suboptimal low-affinity peptides (gray ovals) serve as substrates for UGT1, and some accumulate in the ERGIC and reenter into the PLC for another cycle of peptide loading. Stable optimally loaded MHC-I molecules that pass QC are released and exported to the plasma membrane for recognition by CD8 T cells. The schematic depicts events in human cells. Slight variations, not shown here, apply to mouse cells. The classical pathway of MHC-I presentation takes place in all nucleated cells, whereas cross-presentation is a specialized function conducted predominantly by dendritic cells.
Different facets of cross-presentation have been the subjects of many recent reviews (13–18). This review examines the cell-autonomous endocytic and vesicular trafficking pathways that orchestrate cross-presentation, with a focus, when applicable, on how these pathways are regulated by innate immune receptors. Understanding how cross-presentation is orchestrated is important. Current licensed vaccines have yielded limited success in eliciting CD8 T cell responses and generate mostly neutralizing or opsonizing antibodies effective against pathogens that are extracellular or have a stable antigenic profile (19–22). Defining the mechanisms and regulation of cross-presentation has direct implications for the urgent need to develop T cell vaccines against infectious diseases and cancer (23).
THE CELLS THAT CONDUCT CROSS-PRESENTATION
Unique adaptations of the subcellular pathways of cross-presentation in DCs have highlighted the specialization of this phagocytic subset in cross-presentation (24). Efficient cross-presentation is carried out in vivo by CD24+ conventional DCs requiring the transcription factors IRF8 and BATF3, and by Ly6C+TremL4− monocyte-derived DCs requiring IRF4 but not BATF3 (25). Splenic CD8α+CD24+ DCs and migratory tissue CD103+ DCs excel at cross-presentation under both inflammatory and noninflammatory conditions (26). Langerhans cells, the singular DC type in the epidermis of mice and humans, also cross-present (27). XCR1 and CLEC9A receptors have been proposed to identify cross-presenting populations in humans and mice (28, 29). In humans, the BDCA3+ DCs present in blood and lymphoid and peripheral tissues express both of these receptors and are efficient at cross-presentation (30, 31). Unlike mouse plasmacytoid DCs (pDCs), human pDCs can also cross-present antigen (30, 32). Serpinb9, a serine protease inhibitor that targets the effector molecule granzyme B and protects CD8 T cells from its activity, has been proposed as a unique marker for the cross-presentation-competent CD8α+ splenic DCs (33). However, significant differences in the inhibitory potential of Serpinb9 and the substrate specificity of Serpinb9 between mouse and human cells lend to uncertainty in the functional role of Serpinb9 in cross-presentation (33). In mice, different DC subsets may cooperate to mediate cross-presentation in vivo. During viral infection, activated CD8 T cells at the site of infection produce the chemokines CCL3 and CCL4 and recruit pDCs in a manner dependent on the chemokine receptor CCR5 (34). They also produce the chemokine XCL1 and recruit lymph node–resident XCR1+ DCs (34). Such DC reorganization within the lymph node enables the type I interferon produced by pDCs to optimize cross-presentation by XCR1+ DCs, thereby supporting an optimal CD8 T cell response (34). A similar type of cooperation has been found between pDCs and conventional DCs in cross-priming CD8 T cells specific to the adeno-associated viral capsid (35). TLR9 and its signaling adaptor MyD88 in pDCs license conventional DCs in trans to cross-present capsid antigen to CD8 T cells, and in a manner dependent on signaling through the type I interferon receptor on conventional DCs (35).
Liver sinusoidal endothelial cells, Kupffer cells, and hepatocytes also contribute to cross-presentation and expansion of CD8 T cells within the mouse liver during acute adenoviral infection (36). Hepatocyte expression of collectrin, a membrane protein found to promote vesicle fusion during insulin exocytosis by pancreatic β cells (37), has curiously been linked to the expansion of virus-specific CD8 T cells and viral clearance after adenovirus infection (38). Collectrin augmented hepatocyte and not hematopoietic cell cross-presentation and cross-priming of antigen-specific CD8 T cells in vitro in response to either soluble antigen or remnants of infected necrotic hepatocytes (38). Its role in cross-presentation in these cell types may be related to facilitating vesicular fusion events important for cross-presentation (37, 39).
Many of the cross-presentation pathways have been delineated in murine bone marrow–derived DCs (BMDCs), which also have the ability to process exogenous antigens and cross-present them to CD8 T cells. BMDCs are generated by culturing bone marrow progenitors in the cytokine GM-CSF to yield bona fide DCs that share a transcriptional signature with in vivo migratory DCs (40). GM-CSF-cultured DCs have also been proposed to model inflammatory DCs (41), such as the DC-SIGN/CD209+ monocyte-derived DCs, which actively cross-present peptides derived from bacteria and are recruited to lymph nodes from the blood in a TLR4-dependent manner in response to lipopolysaccharide (LPS) and gram-negative bacteria (42). An important caveat to be aware of when using GM-CSF-cultured BMDCs is the concomitant presence of macrophages expressing the CD11c and MHC-II proteins used to identify DCs within these cultures (40). Thus, delineation of cross-presentation in human DCs and by different tissue-resident DC subtypes is an important goal for future studies.
SECRETORY PATHWAY OF MHC-I TRAFFIC FROM THE ENDOPLASMIC RETICULUM
The heavy chain of MHC-I is cotranslationally inserted into the ER membrane through the ER translocon comprising three polypeptides (Sec61α,β,γ) that make up the Sec61 complex (43) (Figure 1). The molecular chaperones calnexin and immunoglobulin-binding protein (BiP) aid in folding of the nascent heavy chain polypeptide prior to its association with β2-microglobulin (44, 45). Heavy-chain/β2-microglobulin dimers are further stabilized by binding to low-affinity peptides within the ER lumen, but their subsequent interaction with components of the PLC, comprising the peptide transporter associated with antigen processing (TAP), ERp57, calreticulin, and tapasin, enables the binding of high-affinity peptides (2, 46, 47) (Figure 1). MHC-I molecules then associate with transport receptor BAP31, accumulate at ER exit sites, and traffic via COPII-coated export vesicles to the ER-Golgi intermediate compartment (ERGIC) (48–50), a subcompartment of the ER (51) (Figure 1). All forms of heavy-chain/β2-microglobulin dimers, including empty, suboptimally loaded dimers with low-affinity peptide, and optimally loaded dimers with high-affinity peptide, can be exported out of the ER (52). It has been argued that this may even occur with the same efficiency albeit with different exit rates, depending on binding to the PLC (52). A rigorous quality control process follows first in the ERGIC, where certain features are recognized, such as conformational flexibility and folding of the peptide binding groove, and second in the cis-Golgi, where suboptimally loaded dimers accumulate. MHC-I molecules accumulate in the ERGIC when misfolded, for instance, in the absence of peptides with good affinity of loading due to deficiency for TAP or calreticulin (53–55) (Figure 1). Notably, the ERGIC harbors components of the MHC-I PLC such as TAP and calreticulin (56), and the presence of these molecules outside the ER ensures both peptide loading and MHC-I folding (57) during MHC-I recycling between the ER and Golgi (53, 58). Quality control can be mediated by members of the PLC itself, tapasin and calreticulin, which have been reported outside the ER proper, as well as the UDP-glucose:glycoprotein glucosyltransferase (UGT1/UGGT1), which recognizes any conformationally unstable and partly unfolded protein in the ER, ERGIC, and cis-Golgi (52). The end result of quality control is intracellular retention of unstable, empty, and suboptimally loaded dimers that are prevented from reaching the plasma membrane. In resting mouse DCs, fully assembled MHC-I H2-Kb, detected by an antibody called AF6–88.5 (59), do not colocalize with calreticulin, calnexin, TAP2 or the ERGIC-resident lectin ERGIC-53, suggesting that successful export to the plasma membrane takes place after MHC-I molecules have passed the ERGIC quality control (60).
ENDOCYTOSIS, RECYCLING, AND DEGRADATION OF MHC-I MOLECULES
Most of our knowledge of the trafficking of fully assembled MHC-I molecules comes from studies in cells other than professional phagocytes. The cytoplasmic domain of MHC-1 lacks the signals that confer clathrin/AP2 localization of proteins such as transferrin receptor and low-density lipoprotein receptor through clathrin-mediated endocytosis (CME) (61, 62). In HeLa cells, MHC-I molecules are internalized through clathrin (and dynamin)- independent endocytosis (CIE), a process known to mediate the internalization of proteins such as CD59 and β1-integrin (63, 64) and to be regulated by the ADP ribosylation factor 6 (ARF6) (65, 66) (Figure 2). Endocytosis through either CME or CIE delivers proteins intracellularly into early sorting endosomes (67), after which they are routed either to lysosomes for degradation or back to the plasma membrane for recycling.
Figure 2.

Subcellular trafficking routes of MHC-I molecules. Plasma membrane MHC-I molecules undergo clathrin-independent endocytosis (CIE) mediated by the small GTPase ARF6. After endocytosis, MHC-I-carrying endosomes fuse with sorting endosomes marked by RAB5 and EEA1. Once in sorting endosomes, MHC-I molecules can be routed to endolysosomal compartments or the plasma membrane. Other molecules such as transferrin receptors undergo clathrin-mediated endocytosis (CME) once bound to their ligand transferrin. Cargo internalized by CME is also delivered into RAB5+EEA1+ sorting endosomes where it can colocalize with cargo internalized by CIE. A small fraction of MHC-I molecules are routed to RAB7+LAMP1+ late endosome-lysosome compartments in resting bone marrow–derived dendritic cells (DCs). MHC-I molecules have been reported in multivesicular bodies (MVBs), specifically MIIC, because of their colocalization with MHC-II molecules in human Langerhans cells are you pointing out “late endosome-lysosome” as labeled in Fig?. MHC-I molecules recycle back to the plasma membrane through either the fast recycling route, regulated by RAB4 and RAB35, or the slow recycling route, regulated by RAB11a, whose activity is important for trafficking MHC-I molecules to a transitory perinuclear compartment called the endocytic recycling compartment (ERC). The ERC comprises a network of tubular and endosomal structures, some of which appear to be connected by bridges when observed by super-resolution microscopy. MHC-I molecules are returned from the ERC to the plasma membrane with the aid of EHD1 and RAB22a via tubular recycling endosomes (TRE) formed by the EHD1-interacting protein MICAL-L1. During phagocytosis of a bacterium carrying ligands that engage Toll-like receptor (TLR)-MyD88 signals and IKK2 activation, MHC-I molecules from the ERC are diverted to phagosomes through specific IKK2-phosphorylated SNAP23 molecules on phagosomes, which stabilize VAMP8 and syntaxin interactions to initiate fusion of ERC-derived vesicles with the nascent phagosome. Newly delivered phagosomal MHC-I molecules are exported to the plasma membrane after loading with bacterial peptides. Note that different steps in these trafficking routes were studied in different cell types. Please refer to text for details.
The intracellular location of endocytosed MHC-I molecules has been studied in different cell types. In HeLa cells, a fraction of these molecules converges with clathrin-dependent cargo such as transferrin receptor within an early sorting EEA1+ endosome (68) (Figure 2). MHC-I molecules are then diverted to late endosomes and lysosomes, a localization also noted in different types of DCs and presumably reflecting the location of MHC-I molecules that had been internalized from the plasma membrane (Figure 2). A small fraction (10%) of MHC-I molecules colocalize with LAMP-1+ late endosomal/lysosomal compartments in unstimulated BMDCs (60). MHC-I molecules colocalize extensively with LAMP-1 in splenic DCs matured with GM-CSF and TNF-α (69). MHC-I molecules within CD34+ precursor–derived human Langerhans cells colocalize with HLA-DM and HLA-DR in late endosomal and lysosomal compartments termed MIIC (70) (Figure 2). Ultrastructural studies revealed the presence of MHC-I on intraluminal vesicles and limiting membranes of multivesicular structures (70, 71), which also contained MHC-II (71), and late endosomal markers CD63 and mannose-6-phosphate receptor (70). Internalized proteins destined for degradation, exocytosis, or storage are usually incorporated into intraluminal vesicles of multivesicular bodies (72), but the fate of MHC-I molecules that localize to late endosomal/lysosomal compartments in DCs has not been investigated.
Cargo internalized through either CIE or CME to be recycled back to the plasma membrane is routed to sorting endosomes for either fast or slow recycling (Figure 2). Slow recycling entails transport into a transitory endocytic recycling compartment (ERC) (73–77), under control of the small GTPase RAB11a (78, 79) (Figure 2). RAB11a plays a role in transporting proteins to the trans-Golgi network (TGN) in close proximity to the ERC (80). Fast recycling skips the transport step into the ERC and returns endocytosed cargo to the plasma membrane directly from the sorting endosome under control of RAB4 (81–83) or RAB35 (84) (Figure 2). Proteins that enter the slow recycling pathway and transit through the ERC must exit the ERC to return to the plasma membrane. Exit of transferrin receptor from the ERC to the plasma membrane is thought to require GTP hydrolysis of RAB11a, explaining why expression of a constitutively active form of Rab11a (Rab11aQ70L) in CHO cells leads to cargo accumulation in the ERC rather than increased exit back to the plasma membrane (78). In contrast, expression of a dominant negative allele of RAB11a diminished colocalization of transferrin with the ERC (78) and inhibited recycling of β1-integrin (85) and MHC-I molecules in HeLa cells (86).
In HeLa cells, approximately 50% of internalized MHC-I molecules divert into ARF6+ ellipsoidal tubules devoid of transferrin receptor (65, 66, 68). These tubules may be components of long tubular recycling endosomes (TREs), which mediate traffic from the ERC to the plasma membrane (66, 87–90). They might also carry MHC-I molecules from sorting endosomes to ERCs, based on the finding that TREs preferentially facilitate trafficking of CIE cargo and that some TREs originate from sorting endosomal membranes (91). A number of C-terminal Eps15 homology domain (EHD) proteins have been implicated in the generation and fission of TREs (92). TRE formation requires generation of phosphatidic acid (93), which recruits molecules interacting with CasL-like 1 (MICAL-L1) and Syndapin 2 to the endosomal membrane to mediate bending and tubulation (88, 89). EHD1 mediates recycling of transmembrane proteins that have been internalized by CME and CIE (87, 94, 95) and has been reported to promote recycling of MHC-I molecules to the plasma membrane (87) (Figure 2). The small GTPase RAB22a has also been associated with MHC-I+ TREs in HeLa cells, and its inactivation is required for final fusion of these tubules with the plasma membrane (86) (Figure 2). In mouse DCs, Rab22a was described to be distributed ubiquitously in endosomes, lysosomes, and Rab11a+ ERCs, to colocalize with intracellular MHC-I molecules as well as internalized soluble antigen, latex beads, and Toxoplasma gondii parasites (96). Rab22a silencing reduced the recycling of MHC-I molecules to the plasma membrane and negatively affected the cross-presentation of soluble and phagocytic antigens (96).
Trafficking of MHC-I molecules from endosomal compartments to the plasma membrane can be controlled by inflammatory signals. CD34+ precursor–derived human Langerhans cells accumulate MHC-I molecules in endolysosomal compartments that mobilize MHC-I to the plasma membrane upon activation of the cells with LPS (70). Over 50% of MHC-I molecules in immature human monocyte-derived DCs are intracellular, and this percentage is reduced to almost 25% following stimulation with LPS (97). In human monocyte-derived DCs, TLR stimulation induces tubulation of late endosomes, but not ERCs unless MHC-I and ICAM-1 molecules on DCs are also ligated by the TCRs and LFA-1 on CD8 T cells (98). ERC tubulation in these human DCs is mediated by MICAL-L1 (99).
INTRACELLULAR STORAGE OF MHC-I MOLECULES IN ENDOSOMAL RECYCLING COMPARTMENTS
An intracellular pool of MHC-I molecules was reported in earlier studies examining mouse BMDCs, a DC-like human cell line, and primary human peripheral blood–derived DCs (97, 100). These MHC-I molecules did not colocalize with tapasin or KDEL motif–containing ER proteins (97, 100). Later work in mouse BMDCs revealed that intracellular MHC-I molecules are concentrated within ERCs marked by Rab11a and the vesicle-associated membrane proteins VAMP3/cellubrevin and VAMP8/endobrevin (60). The ERC pool of MHC-I molecules is also notable specifically within cross-presentation-competent CD8α+ but not CD8α− splenic DCs. A small fraction of intracellular MHC-I molecules also colocalize with Rab11a to the ERC-proximal TGN (60), and with transferrin receptor, which undergoes slow recycling through the ERC (78, 79). In murine BMDCs, MHC-I molecules did not colocalize with EEA1+/Rab5+ early/sorting endosomes or with ERGIC markers, indicating that these MHC-I molecules had passed quality control (60). Expression of the constitutively active Rab11aQ70L in BMDCs led to ERC accumulation of MHC-I molecules, while short-hairpin RNA (shRNA)-mediated silencing of RAB11 abrogated these stores (60). RAB11a is thus important for maintaining the ERC pools of MHC-I molecules in DCs (60).
The ERC is typically perinuclear and localized near the microtubule-organizing center (74). By confocal microscopy with a resolution of ≈300 nm, it appears as a compact perinuclear region, suggesting a structure enclosed by a single limiting membrane. The closely packed nature of the RAB11a-labeled structures making up the ERC was confirmed by three-dimensional structured illumination microscopy (SIM) at ≈110 nm resolution (91). Observation of the ERC by SIM revealed a complex combination of endosomal membranes and independent structures linked by connections up to 500 nm long (91). Direct stochastic optical reconstruction microscopy imaging at ≈10 nm precision of CME cargo transferrin and CIE cargo CD59, chased from the sorting endosomes into the ERC, revealed segregation of these two cargos en route from the sorting endosomes and even within the ERC (91). These data show that while both CME and CIE cargos are internalized into the ERC, they may enter through different routes and remain within distinct subdomains of the ERC (91). It will be important to conduct similar studies on MHC-I molecules in DCs to track their internalization from the plasma membrane into sorting endosomes, their entry into the ERC, and the kinetics of this process. CD1a, a glycoprotein structurally related to MHC-I proteins, is unique among the other CD1 family members in its internalization by CIE (101). Like MHC-I molecules, it has been shown to colocalize with RAB11a in the ERC and not sorting endosomes within freshly isolated human epidermal Langerhans cells (102). Similar to MHC-I molecules, CD1a molecules undergo Rab22a-dependent recycling in HeLa cells (101).
Recycling endosomes and the ERC contribute endomembranes to incoming phagosomes in order to facilitate phagocytosis, especially of large phagocytic cargo, which would otherwise consume a significant portion of the plasma membrane (103). A distinct ERC is lacking in murine macrophages, and the localization of Rab11a to nascent phagosomes in these cells along with the impairment of FcγR-mediated phagocytosis upon expression of a dominant negative allele of Rab11a has led to the proposal that phagosomal Rab11a delivery reflects endomembrane contribution from recycling endosomes to facilitate phagocytosis (104). On the other hand, the absence of Rab11a does not affect phagocytosis in BMDCs (60), which could reflect differences between macrophages and DCs or simply the ability of other endomembranes, perhaps ERGIC derived (56), to compensate in DCs. Indeed, the first observations of ER membrane recruitment to phagosomes in macrophages were attributed to a contribution of endomembranes to forming phagosomes (105).
SURFACE RECEPTOR ENGAGEMENT DURING ANTIGEN INTERNALIZATION AFFECTS CROSS-PRESENTATION
Antigens are delivered into DCs via multiple routes, including macropinocytosis, endocytosis, and phagocytosis. Because cross-presentation involves the presentation of exogenous antigens, it is subject to regulation by extracellular cues perceived through cell surface receptors. The impact on cross-presentation varies depending on the particular receptor or combination of receptors engaged during antigen internalization (106). Receptor-mediated potentiation of cross-presentation might involve intracellular delivery of exogenous antigen and its targeting to subcellular compartments most amenable to cross-presentation (107). Signal transduction during internalization might positively regulate cross-presentation through the induction of costimulation, inflammatory cytokine production, or biogenesis of the peptide–MHC-I complexes on the cell surface (108). All these events affect CD8 T cell activation in response to cross-presented peptide, a process called cross-priming (109).
Understanding the mechanistic basis for the superiority of certain endocytic receptors over others in cross-presentation has important therapeutic applications, particularly in cancer immunotherapy (110–112). Experimental evidence demonstrates that cross-presentation is favored when endocytic receptors deliver antigen into early/late endosomes and not degradative lysosomes. For example, mannosylation of proteins enhances their cross-presentation, a property attributed to engagement of the mannose receptor (113). Use of the antigen ovalbumin (OVA) as a model for mannosylated proteins (114) revealed that unlike pinocytosis, the mannose receptor specifically delivers OVA into an early EEA1+Rab5+ endosomal compartment, which correlates with the ability of mannose receptor–delivered and not pinocytosed OVA to cross-prime antigen-specific CD8 T cells (115, 116). The intracellular trafficking of OVA can be altered through addition of the carbohydrate structure LewisX, which targets the LeX receptor of the murine macrophage galactose-type lectin 1 (MGL-1) (117). MGL-1 has endocytic dileucine-like and YXXØ motifs (X representing any amino acid and Ø representing an amino acid with a bulky hydrophobic side chain) found in transmembrane proteins that undergo internalization from the plasma membrane (118). MGL-1 shuttles OVA from EEA1+Rab11+ to Rab11+ compartments (that are curiously also LAMP1+) where OVA persists, a condition favoring cross-presentation (117).
A number of C-type lectin receptors have been shown to affect cross-presentation. For example, cross-presentation by human DCs is enhanced when antigen is targeted to the C-type lectin receptors langerin, on Langerhans cells (119); CLEC9A, on BDCA3+ DCs (120); DCIR (CLEC4A) (121); and DC-SIGN or DEC-205 on monocyte-derived DCs or dermal DCs (119, 122, 123). In mice, the C-type lectin Clec9a (DNGR-1) detects filamentous actin exposed upon necroptosis (124–126) and is essential for cross-presentation of dying, vaccinia virus–infected cells and protection from viral infection (127–129). In the presence of an adjuvant, Clec9a-targeted antigen is cross-presented in vivo and elicits a cytotoxic CD8 T cell response (130). The change in pH and ionic strength that DNGR-1 encounters upon endocytosis triggers a conformational change mediated by its neck region that is necessary but not sufficient for the function of DNGR-1 in cross-presentation (131). There are also reported differences in the outcome of targeting antigens to C-type lectin receptors in vitro versus in vivo, and in human versus mouse cells. The C-type lectin domain family 12, member A (CLEC12A), is broadly expressed by all human DC subsets and monocytes as well as mouse CD8α+ DCs and pDCs (132). In vitro targeting of antigen to CLEC12A enhances cross-presentation by human DCs because of its ability to retain antigen in early endosomes, and for periods longer than those for DEC-205-targeted antigen (133). Targeting Clec12A in vivo elicits inferior immune responses in mice, which are only moderately improved by the administration of LPS as an adjuvant (132, 134). Similarly, a side-by-side comparison of antigen targeting to murine Clec12A with Clec9A in vitro showed that despite similar levels of surface expression, Clec12A was surprisingly superior to Clec9A in delivering antigen into splenic CD8α+ DCs (135). Antigen targeted to either Clec9A or Clec12A in vitro was poorly cross-presented by murine CD8α+ DCs, and cross-priming was noted only when Clec12A was targeted on mature, previously activated CD8α+ DCs (135). This poor performance in vitro contradicts in vivo performance, which may reflect the ability of Clec9A and Clec12a to work with other factors in vivo in order to cross-prime a CD8 T cell response (135).
Targeting the same antigen with high-affinity antibodies to either mannose receptors, CD40, or DEC-205 on human BDCA1+ and monocyte-derived DCs shows differences in antigen localization and subsequent cross-presentation (136). Mannose receptors and CD40 target antigen to early endosomes, whereas DEC-205 targets antigen to late endosomes (136). These observations are consistent with those reported for mannose receptors and DEC-205 in mouse BMDCs, which had been attributed to the cytosolic domains of these receptors (137). Despite its lowest rate of endocytosis, CD40 is more efficient at inducing cross-presentation by human DCs compared to mannose receptors or DEC-205 (136), perhaps reflecting CD40-dependent activation of NF-κB-inducing kinase (NIK), a central mediator of noncanonical NF-κB signaling (138). DC-specific deletion of NIK in mice impairs CD8 T cell cross-priming, apparently because of intracellular defects in antigen processing and presentation by cross-presenting CD8α+ splenic DCs (139).
Targeting γ chain–containing activating IgG Fc receptors (FcγRs) on DCs is an effective strategy for augmenting the cross-presentation of antigens complexed with IgG (140). In vivo and in vitro studies have shown that this effect is mediated through Fc receptor-associated γ-chain immunoreceptor tyrosine–based activation motif (ITAM) signaling (141). In mice, cross-presentation of peptides derived from immune complexes is impaired in DCs lacking all four FcγRs (i.e., quadruple negative for FcγRI–IV), a defect that can be overcome in vitro by the addition of the complement component C1q—IgG binding to C1q promotes the uptake of immune complexes and activates the classical complement pathway (142). Tracking the internalization and cross-presentation of immune complexes formed in vivo demonstrated an unexpected prominent role for C1q over FcγR (142). These studies suggest a relevant role for C1q targeting in the optimization of CD8 T cell cross-priming.
Complexing antigens with heat shock proteins (HSPs) has also been a strategy to optimize cross-presentation by targeting scavenger receptors on DCs such as SREC1/SCARF1, LOX-1, and SR/CD204 (143, 144). HSPs are transcriptionally induced during cell stress, and dying cells express elevated HSP levels (145). Intracellular HSPs such as HSP70 and HSP90 can participate in cytosolic translocation of endosomal antigens or associate with the proteasome, positioning them to receive peptides as they are generated, and chaperoning them to TAP (144). Extracellular HSPs such as gp96 participate in targeting antigen to relevant innate receptors, such as CD91, on the surface of DCs (144, 146, 147). Recognition of membrane-bound HSPs on the surface of dying cells by the lectin-like oxidized LDL receptor 1 promotes cross-presentation of cellular antigen from these dying cells (148). Chaetocin, a small-molecule thiodioxopiperazine produced by Chaetomium fungi, triggers apoptosis of myeloma cells and their expression of high levels of HSP90 (149). As such, loading of DCs with chaetocin-treated apoptotic myeloma cells elicits potent activation of tumor-specific cytotoxic CD8 T cells (149).
Numerous studies have demonstrated that besides inducing the surface expression of T cell costimulatory molecules, TLR signals can control multiple facets of the assembly and surface delivery of cross-presented peptide–MHC-I complexes. The internalization of microbial antigens by DCs engages TLR signaling and augments antigen cross-presentation (150, 151). Based on studies in BMDCs, the outcome of regulation of cross-presentation by TLRs appears to be dependent on the stage of DC maturation. Immature BMDCs deficient in the expression of TLR signaling adaptors TRIF and MyD88 are significantly impaired in the cross-presentation of antigens from TLR ligand+ cargo (60). Single deficiencies in either MyD88 or TRIF showed that MyD88, and not TRIF, is critical (60). Intermediate-stage BMDCs, at 3–16 h after LPS stimulation, exhibit increased in vitro and in vivo cross-presentation of antigen-antibody complexes that engage FcγRs (152, 153), as well as phagocytic bead-bound, endotoxin-free antigen (153). Mature BMDCs that have been treated with LPS in vitro for a longer period of 24–40 h are unable to cross-present IgG-complexed antigens (152), likely a reflection of their shutdown in antigen uptake (152, 154). Stimulation of human DCs by TLR3 or TLR4 ligands prior to encountering virally infected apoptotic cells, inhibits their subsequent cross-presentation of viral antigens derived from such cells (155). Similarly, systemic pre-treatment of mice with bacterial and viral TLR ligands or infection with a malaria parasite inhibits subsequent cross-priming to cell-associated antigen as a mechanism that might underlie the immunosuppression associated with chronic blood infections (156). It has been proposed that the downregulation of cross-presentation after DC maturation is important for ensuring that only those antigens initially encountered with the signals that activate DCs are favored for cross-presentation (156, 157).
Besides TLRs, other innate immune signaling receptors also affect the outcome of cross-presentation. Recent studies show that protection against adenoviral hepatitis is conferred by vaccination with triphosphate RNA, which mimics viral RNA and serves as the ligand for the cytosolic RNA helicase retinoic acid–inducible gene I (RIG-I) (158). CD8 T cell cross-priming is augmented via signaling through the RIG-I adaptor MAVS and induction of a type I interferon response (158). Deficiency in STAT2, a transcription factor critical in the response to type 1 interferon, impairs the upregulation of MHC-I and costimulatory molecule expression by DCs in response to TLR ligands, and consequent type I interferon production and cross-priming of CD8 T cells (159).
LOGISTICS OF PROCESSING AND CROSS-PRESENTING EXTRACELLULAR PROTEINS
Two major antigen-processing pathways, vacuolar and cytosolic, have emerged to explain how MHC-I molecules are loaded by peptides derived from extracellular sources (Figure 3). The vacuolar pathway is resistant to proteasome inhibitors and proceeds independently of the cytosolic proteasomal degradation of polypeptides (160). Proteins internalized through either endocytosis or phagocytosis are degraded by endosomal or phagosomal proteases, respectively, and resultant peptides are loaded onto MHC-I molecules independently of cytosolic proteasomal degradation and TAP function (161) (Figure 3). The cytosolic pathway of antigen processing is blocked by proteasome inhibitors, suggesting that internalized proteins from endosomes or phagosomes are translocated to the cytoplasm, where they undergo proteasomal degradation (160, 162). Proteasome-generated peptides are imported back into the phagosomes/endosomes to be loaded onto MHC-I molecules. In mouse and human DCs, cross-presented peptides are trimmed by insulin-regulated endopeptidase (IRAP) related to the ER-resident aminopeptidases ERAP1 and ERAP2 (Figure 3) (163, 164). ERAP1 and ERAP2 in humans and an ERAP1 homolog in mice are responsible for trimming proteasome-generated peptides that are translocated by TAP into the ER lumen (164) in preparation for their loading onto newly synthesized MHC-I molecules with the aid of the PLC (2). Notably, the substrate specificity of IRAP is similar to that of ERAP1 and ERAP2, enabling the vacuolar generation of a set of ligands comparable to that generated in the ER by ERAP1 and ERAP2 (164).
Figure 3.
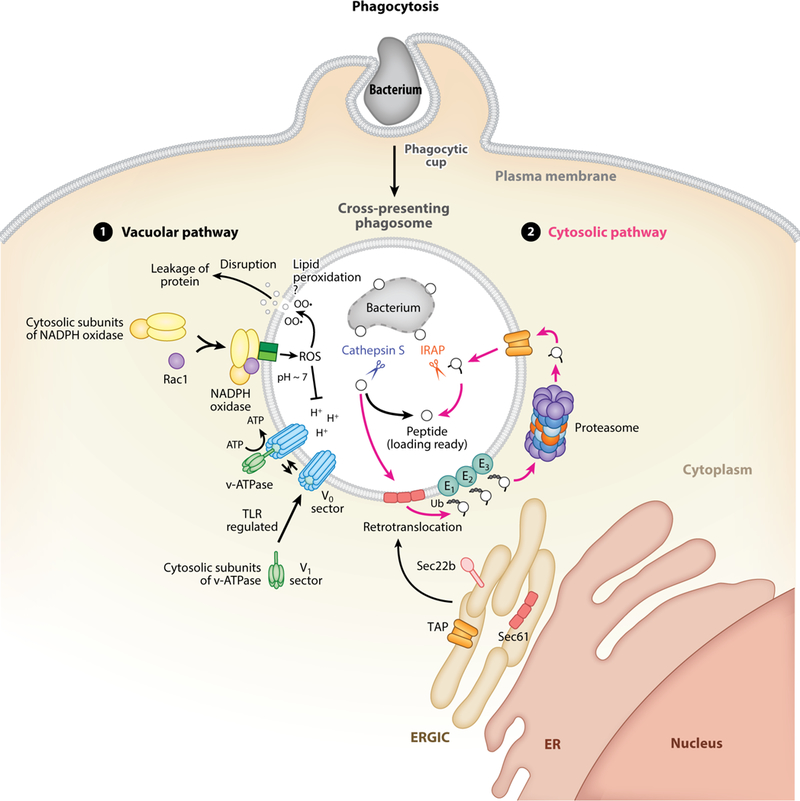
The vacuolar and cytosolic pathways of generating peptides for cross-presentation. Shown are subcellular events within the first 1–4 hours following phagocytosis of bacteria by dendritic cells. Experimental evidence also supports the occurance of similar events around endosomes or parasitophorous vacuoles (see text for details). The nascent phagosome carrying an internalized bacterium matures into a cross-presentation compartment made possible through the activity of several players within both the vacuolar and cytosolic pathways. (1) The vacuolar pathway of cross-presentation contributes to the degradation of proteins, derived from the internalized bacterium in this case, through the activity of vacuolar proteases, most prominent among which is Cathepsin S because of its ability to be functional at a pH that is relatively alkaline compared to the pH optima ~4.5–5 for the majority of vacuolar proteases. In dendritic cells, a pH~7–7.3 most conducive to cross-presentation is maintained for the first few hours through phagosomal reactive oxygen species (ROS) generated by the activity of the NADPH oxidase. A functional NADPH oxidase involves the assembly of its cytosolic subunits and the small GTPase Rac1 with its phagosome membrane integral subunits. Resultant ROS neutralizes the acidic protons (+H) generated through the activity of the v-ATPase, which in turn is assembled by recruitment of its cytosolic V1 sector subunits to its phagosome membrane integral V0 sector subunits, and in a TLR-regulated manner. Counteraction of the v-ATPase by the NADPH oxidase serves to temporarily maintain a neutral phagosomal pH to preserve proteins from excessive degradation by vacuolar proteases and promote cross-presentation. ROS lead to lipid peroxidation (indicated as OO•) and disruption of endosomal membranes, and they may also have the same effects on phagosomal membranes (a possibility indicated with a ‘?’). (2) The cytosolic pathway of cross-presentation relies on recruitment of various players from the endoplasmic reticulum (ER)-Golgi intermediate compartment (ERGIC) to phagosomes through the pairing of the ER soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) Sec22b with syntaxin 4 (not shown) on phagosomes. In this manner, the retrotranslocon Sec61 and the transporter associated with antigen processing (TAP) present in the ERGIC are recruited to the cross-presenting phagosome and function collaboratively to mediate the exit and re-entry, respectively, of polypeptides derived here from the internalized bacterium. Phagosomal Sec61 transports peptides through retrotranslocation to the cytoplasmic side of phagosomes where they have access to the ubiquitin (E1, E2, E3 ligases) and proteasome complex assembled on the cytoplasmic side of phagosomes. This compartmentalization along the phagosomal membrane presumably faciliates translocation of resultant proteasome-degraded peptides back into phagosomes via TAP that had been recruited to phagosomes from the ERGIC. Inside phagosomes, the insulin-regulated aminopeptidase (IRAP) is a trimming aminopeptidase that preferentially acts on those peptides that have been subjected to cytosolic degradation by the proteasome. The combined results of the vacuolar and cytosolic pathways contribute to a diverse repertoire of peptides that are available for binding to MHC-I molecules during cross-presentation. For simplicity, MHC-I molecules are not depicted in this figure. MHC-I molecules become enriched within phagosomes carrying TLR ligands as shown in Figure 2. Their subcellular sources and loading by peptide are shown in Figure 4.
Because phagosomes and endosomes are physically separated from the ER, where many components of the MHC-I PLC are located (Figure 1), vesicular traffic from the ER to phagosomes/endosomes recruits these components to the sites of antigen internalization (Figure 3) (161). The MHC-I PLC, including TAP, is delivered to phagosomes/endosomes by vesicular traffic from the ERGIC through pairing of the ER soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) Sec22b with the plasma membrane SNARE syntaxin 4, which is present on phagosomes and presumably also endosomes (Figure 3) (56, 60, 165, 166). Delivery of the ERGIC to phagosomes originated with observations in macrophages that ER proteins such as calnexin and calreticulin were enriched in phagosomes, and reticular structures staining with the ER enzyme glucose-6-phosphatase were observed connected to phagosomes regardless of their cargo---inert beads, Salmonella typhimurium, Leishmania parasites, or red blood cells (105). These structures were visible when phagocytosis was slowed down with inhibitors of phosphatidylinositol 3-kinase or phagosome acidification (105). A subsequent study contested these findings, showing that ER-phagosome fusion in macrophages is a rare event (167). Nonetheless, multiple lines of evidence in diverse contexts other than cross-presentation, including phagosome biogenesis, calcium signaling within phagosomes, phagolysosomal fusion, and infection with intracellular pathogens, now strongly support communication between the ER and phagosomes (168).
Cross-presentation is designed to ensure priming of a CD8 T cell response to viral or tumor antigens by DCs that are not themselves infected or transformed. However, it also underlies the priming of CD8 T cells to microbial antigens derived from intracellular pathogens residing within intracellular compartments distinct from phagosomes. As such, a protective CD8 T cell response to cross-presented antigens has been demonstrated for pathogens such as Brucella abortus, Leishmania major, T. gondii, Trypanosoma cruzi, and Mycobacterium tuberculosis, all of which establish a specialized intracellular niche within cells (169–174). CD8 T cell responses to these pathogens are clinically relevant. For example, healing lesions in cutaneous leishmaniasis are characterized by the presence of CD8 T cells and their production of IFN-γ (175). In humans, M. tuberculosis infection generates specific CD8 T cell responses, and cytokine-producing CD8 T cells are present in tuberculosis granulomas in the Rhesus macaque model where CD8 T cell depletion reverses protection after bacillus Calmette-Guérin vaccination (176).
ERGIC components have been noted around B. abortus–containing phagosomes (177). The ER has been reported to be important for the cross-presentation of T. gondii–derived antigens to CD8 T cells (178, 179), where actively infected host cells are required for mobilizing a CD8 T cell response (179, 180). Recruitment of host ER membranes to live parasite-containing parasitophorous vacuoles (PV) that actively avoid fusion with the endolysosomal pathway of infected cells (181), directly correlated with CD8 T cell cross-priming (179). This process was later shown to be dependent on Sec22b-mediated TAP delivery to T. gondii PVs (56). Since these studies, experimental evidence has shown that Sec22b-mediated vesicular traffic to phagosomes emanates from the ERGIC and not the ER proper. TAP, calnexin, and calreticulin are also present in the ERGIC and can be detected along with Sec22b and ERGIC-53 in protein extracts from phagosomes containing latex beads (56, 60). In contrast, the ER-resident protein ERp72 or the cis-Golgi-resident ER-Golgi SNARE YKT6 could not be detected in phagosomal proteins, indicating that the presence of ER-associated proteins on phagosomes is selective (60). Only ER proteins that are also in the ERGIC, and not proteins from the ER proper, have been detected on phagosomes (60).
The MHC-I PLC is delivered to phagosomes from the ERGIC within the first few hours following phagocytosis (Figure 3), coinciding with the time when cross-presentation in vitro reaches a plateau, within 3 h (182, 183). TAP is functional on those phagosomes, as shown by the ability to import peptides into purified phagosomes in vitro. N-glycosylation of the peptides in purified phagosomes demonstrated that besides TAP, the ER N-glycosylation machinery was recruited to those phagosomes (182). Silencing Sec22b in BMDCs abrogates cross-presentation (56, 60). Notably, generation of mice where Sec22b is specifically deleted in CD11c+ DCs have shown conflicting results, with either impairment (184)—consistent with the silencing data—or no impairment (185) in cross-presentation of soluble or phagocytic antigen to antigen-specific CD8 T cells in vitro and in vivo (184, 185). The basis for this discrepancy potentially relates to technical differences (186). While the reduction in DC cross-presentation upon Sec22b knockdown has been consistent in independent studies (56, 60, 184, 185), discrepancy in cross-presentation by Sec22 knockout DCs versus Sec22b knockdown DCs has been narrowed down to varying effects of Sec22b shRNA targeting on 23 genes (185). Some of these genes are involved in phagosome biology and could potentially harbor new modulators of cross-presentation (185). A similar strategy of Cre-lox-mediated deletion of Sec22b specifically in DCs validated the critical role of Sec22b in cross-presentation and extended that role to impaired cross-presentation of cell-associated antigen derived from virus-infected or necroptotic cells, as well as impaired priming of tumor-specific CD8 T cells leading to exacerbation of tumor growth (184). A notable observation in this study was the importance of Sec22b expression by DCs for successful anti-PD1 treatment against a tumor model that is well controlled by such checkpoint blockade immunotherapy (184). Thus, while contradictory, each study offers new insight: potential new modulators of cross-presentation (185) and an unexpected Sec22b-dependent function for anti-PD1 in cross-priming (184).
ADVERSE EFFECTS OF VACUOLAR ACIDIFICATION ON CROSS-PRESENTATION
Vacuolar acidification is the result of regulated assembly of a large multiprotein proton pump, the vacuolar (v)-ATPase comprising the transmembrane V0 sector that forms a proton channel and the cytosolic V1 sector responsible for ATPase activity (Figure 3) (187). Efficient cross-presentation necessitates protection of antigens from excessive degradation by lysosomal enzymes, many of which have low-pH optima (13). Compared to macrophages, DCs have limited lysosomal proteolysis, preventing internalized antigens from being degraded prior to encountering antigen-specific T cells in lymph nodes (188). Increased phagosomal acidification and proteolysis induced by overexpressing the master regulator of lysosome biogenesis transcription factor EB (TFEB) (189, 190) in immature BMDCs reduce cross-presentation in vitro and in vivo (191). Notably, TFEB expression is lower in splenic CD8α+ DCs compared to other splenic DCs and macrophages (191). Besides decreased antigen uptake, the blockade in cross-presentation by mature DCs can also be due to the LPS-dependent upregulation in the levels of TFEB (191). The glucocorticoid-induced leucine zipper (GILZ) protein is expressed by many DC subsets, but its highest expression is in CD8α+ DCs, where it limits soluble antigen degradation, perhaps accounting for its ability to augment cross-presentation (192).
Activity of the v-ATPase is countered by the NADPH oxidase 2 complex (NOX2) comprising four cytosolic proteins, p47phox, p40phox, p67phox, and the small GTPase Rac, and membrane-integral cytochrome b558, which is a heterodimer of p22phox and gp91phox proteins (Figure 3) (193). Human monocyte-derived DCs and cross-presentation-competent CD8α+ murine DCs assemble a functional NOX2 complex on phagosomal membranes through recruitment of p47phox, gp91phox, and Rac2 (194–196). NOX2 leads to the production of reactive oxygen species (ROS) and alkalinization of the phagosomal pH (Figure 3) (194, 195, 197, 198). ROS have several other effects on the phagosomal compartment that aid in antigen preservation; ROS inhibit lysosomal proteases with low-pH optima (194, 197, 199) and reversibly oxidize lysosomal proteases of the cysteine cathepsin family (200–202).
Several reports have demonstrated that TLR engagement increases CD8 T cell activation by cross-presented peptide (150, 151, 203, 204). Early work showed that TLRs control cross-presentation by increasing the efficiency of MHC-I peptide loading in human monocyte-derived DCs through promotion of NOX2 activity (205), which counteracts the v-ATPase and contributes to the preservation of peptides as explained above (194, 198, 199). The cross-presentation of phagocytic antigen by BMDCs that had been stimulated with LPS 3–16 h earlier (so-called intermediate DCs, in reference to their state of maturation) is dependent on TLR4-mediated preservation of phagosomal antigen due to a slower rate of phagolysosomal fusion (and thus slower acidification) (153). This contrasts with the TLR-inducible rate of phagolysosomal fusion during phagocytosis of bacteria by macrophages (206), or the LPS-induced increase in v-ATPase assembly and lysosomal acidification in BMDCs driven by increased V1 sector recruitment to lysosomal membranes (Figure 3) (207). The GTPase RAB34 (208, 209) mediated reorganization and clustering of lysosomes in intermediate DCs, preventing their fusion with phagosomes (153). These in vitro observations led to the proposal that activated DCs enter a TLR-induced surveillance state that allows continuous sampling of the tissue microenvironment and efficient cross-presentation in vivo even after maturation (210). This presumably happens during the initial phases of infection before tissue destruction and release of self-antigens, thereby posing no risk of autoreactivity.
DCs from mice deficient in gp91phox, Rac2, and VAMP8, and human DCs knocked down for VAMP8, all show defects in cross-presentation (194, 195, 211, 212). Leishmania promastigotes remodel their phagosomes by selectively cleaving VAMP8 to block NOX2 assembly and evade subsequent cross-presentation (212). During intracellular infection with Listeria monocytogenes, Siglec G, a member of the sialic acid–binding immunoglobulin-like lectin family, recruits the phosphatase SHP-1 to phagosomes through interaction with its cytosolic immunoreceptor tyrosine-based inhibitory motif (ITIM) (213). Phagosomal SHP-1 dephosphorylates p47phox, inhibits phagosomal NOX2 activation, and impairs the formation of specific peptide-MHC-I complexes on CD8α+ DC (213). These effects were independent of the reported ability of Siglec-G to suppress type I interferon production (214), as both Siglec G–sufficient and Siglec G–deficient splenic DCs produced similar levels of type I interferon upon infection with L. monocytogenes (213). Monocyte-derived DCs from patients with chronic granulomatous disease (CGD), who have a genetic deficiency in gp91phox, also show defects in cross-presentation (198). The rare X-linked immunodeficiency Wiskott-Aldrich syndrome is caused by loss-of-function mutations in the hematopoietically expressed Wiskott-Aldrich syndrome protein (WASP), which promotes nucleation of branched actin filaments (215). Murine DC-specific deletion of WASP leads to increased Rac2 activation and cross-presentation (216). This effect was specific to soluble antigen and not immune complexes, and to CD8α− and not CD8α+ DCs, likely reflecting the already high cross-presentation capacity of CD8α+ DCs (216). Increased cross-presentation was attributed to increased localization and activation of Rac2 to phagosomal membranes in WASP-deficient CD8α− DCs, and increased ROS production preventing endosomal/phagosomal acidification (216, 217).
ROLE OF CYTOSOLIC TRANSLOCATION IN CROSS-PRESENTATION
The preservation from proteolytic degradation necessary for cross-presentation does not exclude proteolysis by vacuolar proteases that function best under alkaline conditions, most notable among which is cathepsin S, with a pH optimum between 6.0 and 7.5 (Figure 3) (218, 219). The cytosolic proteasome is also involved in generating peptides for cross-presentation, and this adds to the repertoire of peptides that can be cross-presented by MHC-I molecules. For internalized antigens to be accessible to the cytosolic proteasome, translocation from phagosomes or endosomes is necessary (Figure 3), and this has been experimentally demonstrated mainly through assays using luciferase, β-lactamase, or cytochrome c designed to trigger a specific readout once they access the cytosol in cross-presenting DCs—and notably in the absence of bacterial pore-forming toxins or other virulence factors (56, 220–222).
Cytosolic translocation of antigen can be regulated by surface receptor ligation. Upon binding to OVA, the mannose receptor undergoes polyubiquitination of the single lysine residue in its cytoplasmic domain, an event that mediates cytosolic transport of endocytosed OVA into the cytoplasm and enables cross-presentation (223, 224). ROS production by NOX2 can also induce leakage of antigens from endosomes into the cytosol as a result of endosomal lipid peroxidation and disruption of endosomal membranes (Figure 3) (225). Translocation to the cytosol during cross-presentation might involve the Sec61 translocon, which imports newly synthesized proteins into the ER (Figure 3), but also mediates reverse transport (retrotranslocation) of misfolded proteins from the ER to the cytosol for degradation by the proteasome (226, 227), a process called ER-associated degradation (ERAD) (228, 229). siRNA-mediated knockdown of Sec61 or its entrapment within the ER reduces cross-presentation (230, 231), and closure of the Sec61 translocon by exotoxin A (232) inhibits cytosolic transport of antigen (230, 233). Notably, endosomal recruitment of Sec61 and subsequent cytosolic translocation of antigen depend on signaling from the TLR adaptor TRIF (Figure 3) (231). The AAA ATPase p97, which serves as the final force in pulling ERAD substrates into the cytosol through the translocon (234), has been shown to play an important role in mouse BMDC cross-presentation of mannose receptor–endocytosed soluble antigen and monocyte-derived human DC cross-presentation of a melanoma CD8 T cell epitope derived from a long synthetic peptide (223, 235). Notably, cross-presentation of the same melanoma epitope was not affected by exotoxin A treatment, suggesting a dominant role for p97 over Sec61 during processing of long peptides (235).
Other evidence argues against a role for Sec61 as a retrotranslocon in cross-presentation. Treatment of immortalized mouse DCs with mycolactone, a polyketide-derived macrolide produced by Mycobacterium ulcerans that was recently found to potently inhibit Sec61 (236–239), suppressed both cross-presentation and classic presentation (240). Mycolactone blocked protein import into the ER, but surprisingly it had no effect on protein export to the cytosol from either endosomes or the ER lumen (240). Accessory factors that associate with Sec61, the ERAD ubiquitin ligase HMG-coA reductase degradation 1 homolog (Hrd1) and the pseudorhomboid protease Der1, do not play a role in cross-presentation (228, 235). Other ER-derived translocons may exist, given that Sec22b-mediated ERGIC recruitment to the internalization pathway is critical for export of antigens into the cytosol (56). One candidate is IRGM3, a member of the 47-kDa immunity-related GTPases (p47 GTPases) that resides in both the ER and lipid bodies thought to derive from the ER and that is important for cross-presentation likely by facilitating access of internalized antigens to the cytosol (241). IRGM3 has been proposed to disrupt T. gondii PVs (242, 243), but direct evidence for IRGM3-dependent cytosolic antigen translocation is lacking (241).
Several principles related to the cytosolic pathway of antigen processing have emerged from studying cross-presentation of T. gondii–derived antigens. Secreted proteins such as SAG1, released from T. gondii tachyzoites into PVs, are potent inducers of a CD8 T cell response (244). Infection of mice with T. gondii tachyzoites engineered to express CD8 T cell epitopes within either intracellular or secreted antigens, favored a CD8 T cell response specific to secreted antigens (245, 246) and suggested escape of the secreted antigen from the PVs to the cytosol. Interestingly, the requirement for Sec22b-dependent host ERGIC recruitment differs for cross-presentation of different antigens as demonstrated by comparing the soluble SAG1 antigen to another immunodominant T. gondii GRA6 antigen. GRA6 is secreted into the PV but inserts into the PV membrane through its hydrophobic domain, and only the membrane-bound GRA6 is presented by MHC-I molecules (247). GRA6 cross-presentation is independent of Sec22b, which might indicate that Sec22b-dependent recruitment of ERGIC components does not deliver the AAA ATPase p97 necessary for the proteasomal degradation of integral membrane proteins such as GRA6 (248). p97 may thus be recruited to PVs independently of Sec22b. TAP-independent cross-presentation of Leishmania major–derived antigen and protection of mice had suggested vacuolar confinement of L. major antigens (249), although Sec22b recruitment to L. major PVs was later shown (250). These observations are an example where vacuolar cross-presentation independently of TAP can coexist with ERGIC recruitment to phagosomes, and both pathways contribute to cross-presentation to broaden the repertoire of peptides presented to CD8 T cells.
THE SITES OF PEPTIDE LOADING ONTO MHC-I MOLECULES DURING CROSS-PRESENTATION
The ER is the primary residence of the MHC-I PLC, but ERGIC recruitment to phagosomes and endosomes delivers all the components necessary for potential loading of MHC-I molecules at those non-ER locations (161). Several lines of evidence support phagosomal/endosomal loading: Detection of the MHC-I PLC in phagosomes or endosomes (56, 182, 183, 251); the presence of specific peptide–MHC-I complexes within the phagosomal lumen containing peptide from a parent protein opsonized onto beads (182, 183); demonstration of the phagosomal origin of cross-presented peptide–MHC-I complexes based on their resistance to the secretory pathway inhibitor brefeldin-A (70, 173, 183); and successful reconstitution of peptide loading in vitro using purified phagosomes (182). These studies argue against loading of MHC-I molecules in the ER and subsequent export to nascent phagosomes. Collectively, the evidence implicates phagosomes as both the source of peptide (derived from phagocytosed cargo) and the site of peptide loading during cross-presentation.
Evidence supporting the entry of exogenously derived proteasome-generated peptides into the ER for loading onto MHC-I molecules through the conventional secretory pathway of MHC-I presentation is limited. It would entail experimental demonstration of direct access of internalized antigen to the ER proper, which has been difficult given the reported Sec22b-dependent recruitment of proteins from the ERGIC (some of which are also in the ER) to endosomes/phagosomes (56). An ultrasensitive flow cytometry–based assay, which relies on loading of semipermeabilized cells with fluorescein-labeled reporter peptides and monitoring of their ER compartmentalization in real time in the presence of ATP (functional TAP) or ADP (inactive TAP) (252), might be adapted to track whether proteasome-generated peptides from exogenous sources can enter the ER via TAP (252). This assay has revealed that among human immune cells, subsets within the peripheral blood, monocytes, and dendritic cells have the highest levels of TAP expression and capacity for TAP-dependent peptide translocation (252).
Nevertheless, there are reports describing intracellular trafficking of exogenous antigens to the ER in DCs, a scenario that might be dependent on the nature of the antigenic cargo and the surface receptors it engages during internalization. The molecular chaperone function of HSPs (discussed above) and glucose-regulated proteins (GRPs), which maintain proteostasis during cellular stress, has been exploited in enhancing cross-presentation (143, 253). GRPs are related to HSPs in function but differ in their unique possession of ER localization KDEL signals (253). Inclusion of GRP170 as a chaperone in a gp100 melanoma antigen-based cancer vaccine controls its cross-presentation by directing the antigen into EEA1+ or transferrin receptor+ endosomes, which transiently acquire Sec61 and KDEL and are dependent on TAP and the ERAD machinery for optimal induction of a CD8 T cell response (254). These data were interpreted to support a model where GRP170-gp100 complexes access the ER proper after endocytosis and before the steps of retrotranslocation, cytosolic proteasomal degradation, and TAP-mediated import into the ER for loading MHC-I molecules (254). A similar pathway of retrograde transport where exogenous antigen can access the ER has been described specifically during DC cross-presentation of soluble and not particulate antigen (255). These pathways might use the same machinery that proteins, lipids, certain viruses, and bacterial toxins use in retrograde transport from endosomes to secretory compartments such as the Golgi and ER (256).
Finally, there may be compartmentalization constraints that favor entry of peptides into the endosomes or phagosomes where their parent polypeptide was originally present. The cytosolic subunits of the proteasome have been detected on phagosomes and found to transiently associate with phagosomal membranes during a distinct time window after phagosome biogenesis (Figure 3) (182, 183). Ubiquitinated proteins, including those that had been phagocytosed by macrophages, were also detected on the cytoplasmic side of phagosomes, and their amount increased with time upon the inhibition of proteasome activity linking peptide ubiquitination (presumably by E1, E2 and E3 ligases) to proteasomal degradation (Figure 3) (183). TAP has been localized to endosomes and phagosomes; thus, peptides generated by proteasomal degradation could potentially gain access to the phagosomal/endosomal lumen (Figure 3) (183, 257). The ability to block cross-presentation by targeted inhibition of TAP specifically on endosomal but not ER membranes has implicated endosomes as the sites of MHC-I loading, with cross-presented antigen being transported to the plasma membrane from those endosomes (257). Import of peptides into phagosomes has also been demonstrated by measuring not only intraphagosomal accumulation of peptides in isolated phagosomes (albeit crude preparations) but also their binding directly to beads inside isolated phagosomes, which makes the contribution of TAP from contaminating ER membranes less likely (258). In these assays, TAP mediates peptide import, although some peptides, as exemplified by the SIINFEKL peptide derived from OVA, can also enter phagosomes through a second TAP-independent step that is presently undefined (258).
SUBCELLULAR SOURCES OF MHC-I MOLECULES FOR CROSS-PRESENTATION
While the generation of peptides by vacuolar or cytosolic processing would not be expected to have bearing on the subcellular source of MHC-I molecules used for peptide loading, definitive evidence for this is lacking, in part because of the difficulty in pinpointing where peptide loading occurs. The latter is presently associated primarily, but not exclusively, with the sites where TAP is present, and because ER-resident TAP can also be recruited to endosomes and phagosomes, pinpointing the sites of loading of MHC-I molecules becomes tricky. Loading in the ER would involve newly synthesized MHC-I molecules, while loading within vacuolar compartments would be predicted to involve MHC-I molecules trafficking between the plasma membrane and endocytic recycling compartments. However, this is not necessarily always the case, as discussed below.
In BMDCs, the ERC serves as a source of MHC-I molecules for cross-presentation of peptides derived from phagocytic cargo (60). Human pDCs have been reported to contain a major intracellular pool of MHC-I molecules that colocalizes exclusively with the transferrin receptor. Because transferrin receptors can also recycle through the ERC, the colocalization of MHC-I molecules with transferrin receptors suggests their presence within the ERC (259). This pool may underlie the ability of pDCs to serve as primary mediators of antiviral responses by allowing prompt cross-presentation of peptides derived from internalized viruses (259). Based on the recycling paths of CIE cargo reviewed above, the most likely source of MHC-I molecules within the ERC of DCs is the plasma membrane. ERC-derived MHC-I molecules were found to be enriched specifically on phagosomes carrying TLR ligand+ cargo, peaking at 4.5 h after phagocytosis and dependent on MyD88 but not TRIF expression in the DC (60). The differential reliance of phagosomal Sec22b and MHC-I accumulation on TLR signaling along with the inability to colocalize MHC-I molecules within the ERGIC of DCs (60, 97, 100) supported the notion that MHC-I molecules were recruited from a source other than the ERGIC. Concomittant accumulation of Rab11a, and the ERC SNAREs VAMP3 and VAMP8 (261), but not the non-ERC SNAREs VAMP2 and VAMP7, provided experimental support for the ERC origin of the MHC-I molecules associated with TLR ligand–bearing phagosomes (60). An intact ERC is critical for the positive edge that TLR signals impart on cross-presentation (60). Dispersion of the ERC from its perinuclear region with nocodazole as well as lentiviral-mediated silencing of Rab11a severely impair cross-presentation of peptides derived from phagocytic cargo (60). Disintegration of the tubular ERC in human DCs with nocodazole also reduces their ability to activate antigen-specific CD8 T cells (98). On the other hand, increasing MHC-I traffic to the ERC via expression of a constitutively active Rab11a in BMDCs shows enhanced kinetics of MHC-I recruitment to TLR ligand+ phagosomes, but it fails to rescue MHC-I recruitment and cross-presentation in the absence of TLR signals (60). Therefore, while RAB11a expression is critical for TLR-regulated cross-presentation, its role is primarily to traffic MHC-I molecules into the ERC and not to phagosomes (79). A similar scenario may apply to the reported Rab11a-dependent trafficking of TLR4 to Escherichia coli–carrying phagosomes (262), which likely relates to the dissolution of the ERC source of TLR4 rather than a direct impairment of TLR4 trafficking to phagosomes upon Rab11a suppression. Collectively, the evidence from these studies suggests that coregulated trafficking of TLR4 and MHC-I molecules from the ERC to bacteria-containing phagosomes might serve to further remodel cross-presenting phagosomes for additional functions such as TRIF-dependent type I interferon production (262), which further stimulates CD8 T cell cross-priming (263).
Directed trafficking of MHC-I molecules from the ERC to TLR ligand+ phagosomes is orchestrated in two steps (Figure 2). First, MyD88 signals from phagosomes carrying TLR ligands phosphorylate the inhibitor of NF-κB kinase subunit 2 (IKK2, also known as IKKβ), a subunit of the IκB kinase (IKK) (60). Second, activated IKK2 phosphorylates phagosomal SNAP23, which serves as a docking site for recruitment of ERC SNAREs to phagosomal membranes (60) and when phosphorylated stabilizes SNARE complexes to mediate membrane fusion (264, 265). Vesicular trafficking from the ERC delivers to phagosomes the critical numbers of MHC-I molecules once TLRs signal infection and the increased need for cross-presentation (60) (Figure 3).
The cytoplasmic tail of the MHC-I protein contains a conserved tyrosine residue at position 320 that is part of a YXXA motif reminiscent of the tyrosine-based YXXØ endocytic motifs (118). Y320 is responsible for trafficking MHC-I molecules into the endolysosomal pathway in both murine bone marrow– and spleen–derived cultured DCs (69). Its replacement by phenylalanine disrupts cross-presentation in vitro and in vivo during vesicular stomatitis virus or Sendai virus infection, and without affecting classical MHC-I antigen presentation (69). Based on the shared roles of Y320 in the MHC-I protein and YXXØ in other transmembrane proteins in the endocytosis of these proteins, the MHC-I molecules that undergo peptide exchange within endolysosomal compartments are most likely recycled from the plasma membrane (Figure 3). Curiously, however, the Y320 mutation reduces surface MHC-I levels and leads to the accumulation of MHC-I molecules in a giantin+ Golgi compartment (69). This suggests delivery of MHC-I molecules to endolysosomes from a Golgi-like compartment (69), with the possibility of either the stalling of MHC-I molecules in this compartment en route to the plasma membrane or their endocytosis into this compartment prior to routing to cross-presenting endolysosomes (Figure 3). Lending support to these possibilities, immature primary monocyte-derived human DCs and a DC-like cell line KG-1 have been reported to contain intracellular stores of MHC-I molecules that colocalize with the Golgi marker GM-130 (97), and some MHC-I localization with the TGN marker TGN38 has been noted in unstimulated BMDCs (60). Absence of TGN38 from phagosomes in BMDCS has excluded the TGN as a likely source of MHC-I molecules that are recruited to phagosomes (60).
Rab43a is a small GTPase selectively expressed by BATF3-dependent CD8α+ DCs and CD103+ DCs, where it colocalizes with TGN38 and the Golgi marker giantin (266). CD8α+ DCs from Rab43-deficient mice showed defects in the cross-presentation of cell-associated and soluble antigens in vivo and in vitro (266). The cross-presentation of cell-associated antigen by GM-CSF– and IL-4–cultured, monocyte-derived, Rab43-deficient DCs, on the other hand, was unaffected, highlighting distinct mechanisms of cross-presentation in these two DC subsets (266). In HeLa cells, overexpression of the E3 ubiquitin ligase membrane-associated RING-CH 9 (MARCH9), which mediates ubiquitination of lysine residues within the MHC-I cytoplasmic tail, leads to the appearance of MHC-I within endosomes marked by syntaxin-6 (267), a protein involved in vesicular trafficking from the TGN to endosomes (268) (Figure 3). MARCH9 transcript levels are upregulated over time after treatment by LPS (267). The consequences of this remain to be formally tested, but it may serve to increase TGN export of newly synthesized MHC-I molecules to cross-presentation-competent endosomes. This possibility is consistent with the observations that murine MARCH9-deficient DCs are impaired in the cross-presentation of soluble antigen (267). On the other hand, MARCH9 overexpression also led to the appearance of MHC-I molecules in a smaller fraction of endosomes marked by secretory carrier membrane protein 3 (SCAMP3) (267), which in HeLa and baby hamster kidney (BHK) cells has been shown to sort epidermal growth factor receptor (EGFR) to multivesicular endosomes for eventual lysosomal degradation (269) (Figure 3). The delayed kinetics for peak MARCH9 induction with LPS (267) may also serve to divert TGN traffic of MHC-I molecules to multivesicular bodies through SCAMP3+ endosomes, perhaps for MHC-I degradation and downmodulation of CD8 T cell activation.
Another trafficking route of MHC-I to endolysosomes that does not involve the plasma membrane or recycling compartments is mediated by CD74 (also known as the invariant chain). CD74 associates with MHC-II molecules in the ER to prevent peptide binding and chaperones MHC-II from the ER to endosomal compartments (270, 271) (Figure 3). Older studies had reported colocalization and association of CD74 with MHC-I molecules (71, 272, 273), and MHC-I and CD74 interact intracellularly within BMDCs, forming a protein complex within a pre-Golgi compartment (274). MHC-I internalization from the plasma membrane was unaffected upon CD74 deficiency, whereas endolysosomal localization was reduced leading to profound defects in cross-presentation in vitro and diminished virus-specific CD8 T cell responses to vesicular stomatitis virus infection (274). Deletion of the cytosolic CD74 endosomal trafficking motif abrogated soluble antigen cross-presentation (274), suggesting that trafficking of the MHC-I/CD74 complex is dictated by CD74. Fewer peptide-loaded MHC-I molecules colocalized with LAMP-1+ endolysosomal compartments in CD74-deficient splenic DCs (274), but whether MHC-I molecules are loaded in these LAMP-1+ endolysosomal compartments and exit from these compartments to the plasma membrane is unknown.
EMERGING PORTRAIT OF THE CROSS-PRESENTATION COMPARTMENT
The studies reviewed here collectively paint the picture of a cross-presentation compartment created de novo, and most possibly at the delivery sites of extracellular antigen. Depending on the nature of the cargo, be it a bacterium, parasite, immune complex, apoptotic cell, necroptotic cell, or other, specific interactions between cargo ligands and cellular receptors further control remodeling of the maturing phagosome. This is most rapidly achieved by transcription-independent phosphorylation of key components that govern communication among cellular organelles—for example, the phosphorylation of SNARE components, like SNAP23, that stabilize SNARE pins and orchestrate vesicular fusion. Other than cellular receptors, microbial virulence factors also shape phagosome maturation as a means of establishing an intracellular microbial niche while evading lysosomal degradation and antigen presentation. Thus, the cross-presentation compartment that emerges is one that is dynamic and multifaceted, where no one pathway prevails or applies. Experimental evidence from multiple labs point to the convergence of three vesicular trafficking pathways coming together to remodel the cross-presentation compartment (Figure 4). These pathways are defined here as the ERGIC pathway, the ERC pathway, and the lysosome-related organelle (LRO) pathway (Figure 4). They differ by origin, molecular mediators, and signal requirements that dictate their recruitment, as well as the type of component they bring to the cross-presentation compartment.
Figure 4.

The different subcellular sources of MHC-I molecules for cross-presentation. Published evidence supports the use of four distinct sources of MHC-I for cross-presentation by dendritic cells (DCs): (1) the endoplasmic reticulum (ER), (2) the trans-Golgi network (TGN), (3) the plasma membrane, and (4) the endosomal recycling compartment (ERC). (1) Direct trafficking of MHC-I molecules from the ER to endolysosomal compartments has been reported in bone marrow–derived DCs mediated by the chaperone CD74, which associates with MHC-I molecules and chaperones them to late endosomal compartments marked by LAMP-1. This compartment may contain endocytosed antigen delivered from the plasma membrane. Deficiency in CD74 impairs cross-presentation, suggesting that in this case ER-delivered MHC-I and not plasma membrane MHC-I is loaded within the late endosomal/lysosomal compartment, from which they may traffic directly to the plasma membrane for recognition by CD8 T cells. (2) The secretory pathway of newly synthesized MHC-I molecules (see Figure 1) is diverted by MARCH9-mediated ubiquitination of MHC-I in the TGN and their diversion via SCAMP3+ vesicles to multivesicular bodies (MVBs) (perhaps for degradation) or syntaxin-6+ vesicles to late endosomes (perhaps for loading with antigen delivered from the sorting endosome). (3) A tyrosine residue at position 320 (Y320) within the MHC-I cytoplasmic domain is critical for delivery of MHC-I to endosomal cross-presentation compartments, where peptide loading presumably takes place. Y320 might traffic MHC-I from the Golgi compartment to late endosomes, similar to its role in trafficking MHC-I from the plasma membrane to late endosomes, perhaps during clathrin-independent endocytosis. The ER, TGN, and plasma membrane sources of MHC-I are presumably used preferably for the cross-presentation of endocytic antigen, but this remains to be formally tested. (4) During phagocytosis, plasma membrane MHC-I might become internalized as phagosomes form, but when cargo such as a bacterium carries TLR ligands, additional numbers of MHC-I are recruited from the MHC-I-rich ERC under control of TLR-MyD88-IKK2 signaling (designated in blue letters). This mobilization serves to augment cross-presentation by increasing the number of ERC-resident MHC-I molecules available for loading with bacterial peptides. It is assumed that loading takes place in the phagosome followed by export of peptide-MHC-I complexes to the plasma membrane.
The ERGIC pathway of vesicular traffic originates from the ERGIC, is mediated by the SNARE Sec22b, and delivers components of the MHC-I PLC, most notably TAP, in preparation for cross-presentation (56) (Figure 4). This pathway likely also delivers Sec61, present not only in the ER but also in the ERGIC (275). MyD88 signaling from TLR4 has been reported to increase localization of TAP to Rab5+ endosomes and to compartments staining for endocytosed soluble antigen, thus stimulating endocytic antigen cross-presentation (257). As mentioned above, TRIF signaling has been reported to deliver Sec61 to endosomes (231). In the case of phagosomes, ERGIC recruitment was found to be independent of TLR signals, as demonstrated by the abundance of Sec22b, ERGIC-53, and calnexin on DC phagosomes regardless of their content of TLR ligands and irrespective of the ability of DCs to conduct TLR signaling (60). The role of Sec22b in cross-presentation (56, 60) is linked to its ability to mediate delivery of the MHC-I PLC to phagosomes/endosomes and equip these compartments for cytosolic translocation (56).
The ERC pathway of vesicular traffic originates from the ERC, and its recruitment to phagosomes is controlled by compartmentalized TLR signals that uniquely phosphorylate SNAP23 on phagosomes carrying TLR ligands, to mediate stable fusion between that specific phagosome and the ERC (Figure 4). The ERC pathway serves a purpose different from the ERGIC pathway. The ERC is the source of MHC-I molecules that are recruited to microbial antigen-carrying phagosomes (60). While there were no notable differences in the patterns of Sec22b staining around phagosomes with or without TLR ligands, MHC-I molecules colocalized with Sec22b on phagosomes only when phagosomes concurrently carried TLR ligands (60). Unlike the role of Sec22b in cross-presentation, Rab11a silencing impairs cross-presentation because it destroys the ERC stores of MHC-I molecules (60).
The LRO pathway of vesicular traffic originates from LROs and serves a third purpose in cross-presentation (Figure 4). It delivers to phagosomes the membrane-integral component of the NOX2 complex, cytochrome b558, comprising gp91phox and p22phox, to ensure the protection of antigens from lysosomal proteases and their preservation for cross-presentation. The existence of this pathway can be deduced from studies that have examined the subcellular localization of components of the NOX2 complex. gp91phox has been colocalized with the ERC marker Rab11a in CHO and RAW 264.7 murine macrophage cell lines, and a tagged version of p22phox was localized in primary bone marrow–derived macrophages to recycling endosomes labeled with transferrin (276). However, in resting DCs, gp91phox is present in LAMP-1/2+ LROs (60, 199) and not Rab11a+ ERCs (60). gp91phox is initially recruited to nascent phagosomes from the plasma membrane (196), followed by replenishment from LROs (196, 199) with the aid of the small GTPase Rab27a (199) and the Ca2+-sensing protein synaptotagmin 11 (277). gp91phox recruitment to phagosomes in murine BMDCs is dependent on the SNARE VAMP8/endobrevin (212), and formation of a stable SNARE complex comprising VAMP8, SNAP23, and syntaxin-7 mediates fusion of gp91phox-containing LROs with phagosomes/endosomes (196, 211). The delivery of TAP1 and MHC-I proteins to endosomes appears to be independent of VAMP8 (211), highlighting the LRO pathway as distinct from the ERGIC and ERC pathways. Furthermore, while Rab11a and MHC-I proteins colocalize in BMDCs, NOX2 colocalizes with neither and instead colocalizes almost completely with LAMP-1 in resting BMDCs and LAMP-1+ phagosomes irrespective of the TLR ligand content of those phagosomes (60). These findings highlight the differences between macrophages and DCs (24), but they also reveal that trafficking of MHC-I and trafficking of NOX2 to phagosomes are subject to different rules.
The ERGIC, ERC, and LRO pathways of vesicular traffic coexist with the vacuolar and cytosolic pathways of cross-presentation such that ERC-derived MHC-I molecules can be loaded by peptides generated through the activity of vacuolar proteases such as cathepsin S (the vacuolar pathway) and/or be loaded by proteasome-TAP-dependent peptides (the cytosolic pathway), as described above (Figure 4). Longer peptides, such as those derived from melanoma antigens gp100 and MelanA/MART1, appear to have a preference for vacuolar processing based on the lack of involvement of Sec22b and TAP (278), although the ERAD component p97 indicating retrotranslocation and cytosolic processing—has been reported to be important for their cross-presentation (235). Nevertheless, the use of peptides generated through both the vacuolar and the cytosolic pathways allows MHC-I molecules to be loaded by a broader repertoire of peptides that contribute to a diverse CD8 T cell response. The ERC pathway adds one more significant dimension to cross-presentation. It is critical for the positive edge that TLR signals impart on cross-presentation by delivering rate-limiting molecules of MHC-I. Instead of assembling new correctly folded MHC-I molecules in the ER, the ERC maintains preassembled stores of MHC-I molecules that can readily be recruited to phagosomes containing microbial cargo under the guidance of TLR signals.
CONCLUDING REMARKS
As the review comes to its conclusion, it is worthwhile noting the knowledge gaps that remain in our understanding of cross-presentation: (a) defining the subcellular sources of MHC-I molecules used for cross-presentation of soluble versus phagocytic antigens, (b) pinpointing the subcellular sites where MHC-I molecules are loaded by peptides from exogenous sources and defining the biochemistry of loading at these sites, (c) dissecting the regulation of cross-presentation according to the extracellular and tissue-specific cues at steady state and during infection. Continued research into the mechanisms of cross-presentation in BMDCs as well as DCs isolated from both mouse and human tissues should close these gaps and pave the way for the therapeutic exploitation of these pathways to cure different types of cancer and infectious diseases, such as tuberculosis, HIV, and malaria.
Figure 5.
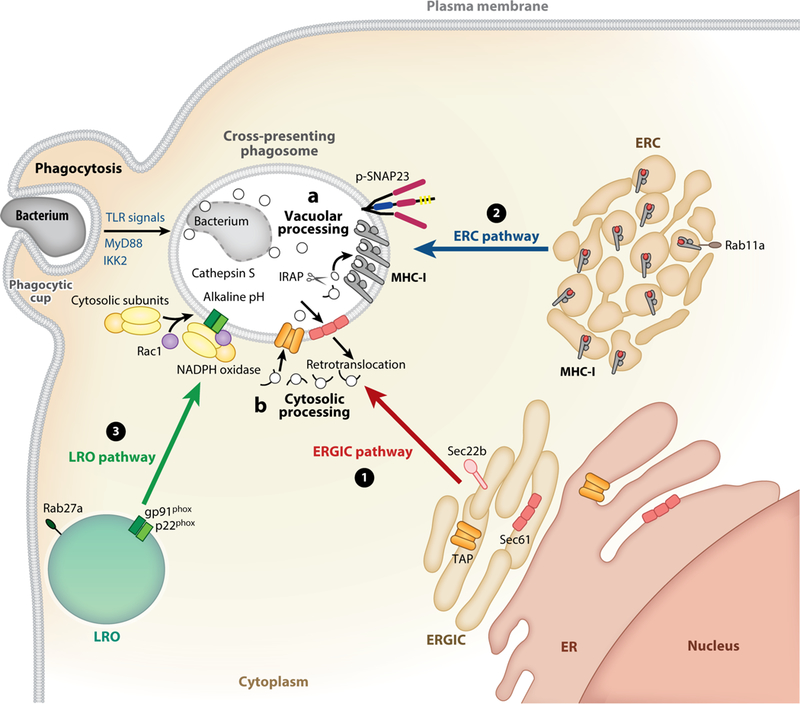
Convergence of three vesicular pathways of endocytic traffic on the cross-presentation compartment in dendritic cells. A series of events is depicted leading to the formation of a cross-presentation-competent compartment after phagocytosis of bacteria. In dendritic cells, three pathways of vesicular traffic converge on nascent phagosomes carrying bacteria: the (1) ERGIC, (2) ERC, and (3) LRO pathways. (1) The ERGIC pathway traffics vesicles from the ER-Golgi intermediate compartment (ERGIC) and is dependent on pairing of the ER SNARE Sec22b with syntaxin-4 on the phagosomal membrane. This pathway delivers TAP, other components of the peptide-loading complex (not shown), and possibly also the ER retrotranslocon Sec61. The ERGIC pathway is mobilized independently of Toll-like receptor (TLR) signals during phagocytosis, regardless of the nature of the phagocytosed cargo, and in response to undefined signals likely associated with the process of phagocytosis. In the case of endocytosis, TLR signaling can increase TAP and Sec61 localization to endosomes in a MyD88 and TRIF dependent manner, respectively (not shown). (2) The ERC pathway traffics vesicles from the ERC to phagosomes marked by phosphorylated synaptosomal-associated protein SNAP23. This pathway is regulated by TLR signals (designated in blue letters). When internalized cargo carries microbial structures that engage TLRs, resultant MyD88-IKK2 signals phosphorylate SNAP23 on phagosomes. SNAP23 phosphorylation stabilizes interactions between ERC VAMP3/8 and a putative phagosomal syntaxin (not shown) to mediate fusion of ERC-derived vesicles with the bacterium-containing phagosome. ERC-phagosome fusion delivers MHC-I molecules from the ERC. Activity of the small GTPase Rab11a (shown) is important for formation of the MHC-I-rich ERC. (3) The LRO pathway traffics vesicles from lysosome-related organelles (LRO), is dependent on activity of the small GTPase Rab27a and formation of a stable SNARE complex comprising VAMP8, SNAP23, and syntaxin-7 (not shown). The LROs deliver to phagosomes the membrane-integral subunits of the NADPH oxidase, gp91phox and p22phox. Recruitment of the small GTPase Rac1 and cytosolic subunits of the NADPH oxidase to gp91phox and p22phox on the phagosomal membrane leads to the assembly of an active NADPH oxidase complex that raises phagosomal pH and protects antigens from complete degradation. The regulation of the LRO pathway is poorly understood and might occur at the point of trafficking from LROs or assembly of the cytosolic and membrane components of the NADPH oxidase. Concurrently with these events, proteins derived from bacterial degradation are subjected to one of two routes of proteolysis: (A) vacuolar processing relying on the activity of phagosomal cathepsin S, which remains active at the relatively alkaline pH of the cross-presentation compartment, and (B) cytosolic processing relying on retrotranslocation of antigen to the cytosol (perhaps through ERGIC delivered Sec61), degradation by the proteasome, and transport back into phagosomes through TAP. Peptides are trimmed by insulin-regulated endopeptidase (IRAP) in preparation for MHC-I loading. The combined activity of these two pathways (A and B) would serve to diversify the peptide repertoire that can be generated from exogenous cargo, loaded onto MHC-I and exported to the plasma membrane for cross-presentation to CD8 T cells (not shown). Together with the vacuolar and cytosolic pathways of antigen processing (A and B), the ERGIC (1), ERC (2), and LRO (3) pathways of vesicular traffic equip phagosomes for cross-presentation of microbial antigen.
ACKNOWLEDGMENTS
The author is grateful to Priyanka Nair-Gupta and Gaetan Barbet for their valuable contributions. J.M.B. thanks her funding sources: National Institutes of Health grants AI123284, AI073899, AI127658, DK072201, DK111862, the Burroughs Wellcome Fund, and the Leukemia & Lymphoma Society.
Footnotes
DISCLOSURE STATEMENT
The author is not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Bjorkman PJ. 1997. MHC restriction in three dimensions: a view of T cell receptor/ligand interactions. Cell 89:167–70 [DOI] [PubMed] [Google Scholar]
- 2.Blum JS, Wearsch PA, Cresswell P. 2013. Pathways of antigen processing. Annu. Rev. Immunol 31:443–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. 1987. Structure of the human class I histocompatibility antigen, HLA-A2. Nature 329:506–12 [DOI] [PubMed] [Google Scholar]
- 4.Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. 1987. The foreign antigen binding site and T cell recognition regions of class I histocompatibility antigens. Nature 329:512–18 [DOI] [PubMed] [Google Scholar]
- 5.Schwartz RH. 1992. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell 71:1065–68 [DOI] [PubMed] [Google Scholar]
- 6.Mueller DL, Jenkins MK, Schwartz RH. 1989. Clonal expansion versus functional clonal inactivation: a costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu. Rev. Immunol 7:445–80 [DOI] [PubMed] [Google Scholar]
- 7.Liu Y, Janeway CA Jr. 1991. Microbial induction of co-stimulatory activity for CD4 T-cell growth. Int. Immunol 3:323–32 [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Janeway CA Jr. 1992. Cells that present both specific ligand and costimulatory activity are the most efficient inducers of clonal expansion of normal CD4 T cells. PNAS 89:3845–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. 1997. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388:394–97 [DOI] [PubMed] [Google Scholar]
- 10.Janeway CA Jr. 1989. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol 54(Part 1):1–13 [DOI] [PubMed] [Google Scholar]
- 11.Takeuchi O, Akira S. 2010. Pattern recognition receptors and inflammation. Cell 140:805–20 [DOI] [PubMed] [Google Scholar]
- 12.Perreault C 2010. The origin and role of MHC class I-associated self-peptides. Prog. Mol. Biol. Transl. Sci 92:41–60 [DOI] [PubMed] [Google Scholar]
- 13.Alloatti A, Kotsias F, Magalhaes JG, Amigorena S. 2016. Dendritic cell maturation and cross-presentation: Timing matters! Immunol. Rev 272:97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blander JM. 2016. The comings and goings of MHC class I molecules herald a new dawn in cross-presentation. Immunol. Rev 272:65–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cruz FM, Colbert JD, Merino E, Kriegsman BA, Rock KL. 2017. The biology and underlying mechanisms of cross-presentation of exogenous antigens on MHC-I molecules. Annu. Rev. Immunol 35:149–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grotzke JE, Sengupta D, Lu Q, Cresswell P. 2017. The ongoing saga of the mechanism(s) of MHC class I-restricted cross-presentation. Curr. Opin. Immunol 46:89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gutierrez-Martinez E, Planes R, Anselmi G, Reynolds M, Menezes S, et al. 2015. Cross-presentation of cell-associated antigens by MHC class I in dendritic cell subsets. Front. Immunol 6:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Endert P 2016. Intracellular recycling and cross-presentation by MHC class I molecules. Immunol. Rev 272:80–96 [DOI] [PubMed] [Google Scholar]
- 19.Delany I, Rappuoli R, De Gregorio E. 2014. Vaccines for the 21st century. EMBO Mol. Med 6:708–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McMichael A, Picker LJ, Moore JP, Burton DR. 2013. Another HIV vaccine failure: where to next? Nat. Med 19:1576–77 [DOI] [PubMed] [Google Scholar]
- 21.Gilbert SC. 2012. T-cell-inducing vaccines—what’s the future. Immunology 135:19–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stoll-Keller F, Barth H, Fafi-Kremer S, Zeisel MB, Baumert TF. 2009. Development of hepatitis C virus vaccines: challenges and progress. Expert Rev. Vaccines 8:333–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fehres CM, Unger WW, Garcia-Vallejo JJ, van Kooyk Y. 2014. Understanding the biology of antigen cross-presentation for the design of vaccines against cancer. Front. Immunol 5:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Savina A, Amigorena S. 2007. Phagocytosis and antigen presentation in dendritic cells. Immunol. Rev 219:143–56 [DOI] [PubMed] [Google Scholar]
- 25.Briseno CG, Haldar M, Kretzer NM, Wu X, Theisen DJ, et al. 2016. Distinct transcriptional programs control cross-priming in classical and monocyte-derived dendritic cells. Cell Rep 15:2462–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Segura E, Villadangos JA. 2009. Antigen presentation by dendritic cells in vivo. Curr. Opin. Immunol 21:105–10 [DOI] [PubMed] [Google Scholar]
- 27.Igyarto BZ, Kaplan DH. 2013. Antigen presentation by Langerhans cells. Curr. Opin. Immunol 25:115–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poulin LF, Reyal Y, Uronen-Hansson H, Schraml BU, Sancho D, et al. 2012. DNGR-1 is a specific and universal marker of mouse and human Batf3-dependent dendritic cells in lymphoid and nonlymphoid tissues. Blood 119:6052–62 [DOI] [PubMed] [Google Scholar]
- 29.Bachem A, Hartung E, Guttler S, Mora A, Zhou X, et al. 2012. Expression of XCR1 characterizes the Batf3-dependent lineage of dendritic cells capable of antigen cross-presentation. Front. Immunol 3:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Segura E, Amigorena S. 2014. Cross-presentation by human dendritic cell subsets. Immunol. Lett 158:73–78 [DOI] [PubMed] [Google Scholar]
- 31.van der Aa E, van Montfoort N, Woltman AM. 2015. BDCA3+CLEC9A+ human dendritic cell function and development. Semin. Cell Dev. Biol 41:39–48 [DOI] [PubMed] [Google Scholar]
- 32.Tel J, Schreibelt G, Sittig SP, Mathan TS, Buschow SI, et al. 2013. Human plasmacytoid dendritic cells efficiently cross-present exogenous Ags to CD8+ T cells despite lower Ag uptake than myeloid dendritic cell subsets. Blood 121:459–67 [DOI] [PubMed] [Google Scholar]
- 33.Mangan MS, Vega-Ramos J, Joeckel LT, Mitchell AJ, Rizzitelli A, et al. 2017. Serpinb9 is a marker of antigen cross-presenting dendritic cells. Mol. Immunol 82:50–56 [DOI] [PubMed] [Google Scholar]
- 34.Brewitz A, Eickhoff S, Dahling S, Quast T, Bedoui S, et al. 2017. CD8+ T cells orchestrate pDC-XCR1+ dendritic cell spatial and functional cooperativity to optimize priming. Immunity 46:205–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rogers GL, Shirley JL, Zolotukhin I, Kumar SRP, Sherman A, et al. 2017. Plasmacytoid and conventional dendritic cells cooperate in crosspriming AAV capsid-specific CD8+ T cells. Blood 129:3184–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ebrahimkhani MR, Mohar I, Crispe IN. 2011. Cross-presentation of antigen by diverse subsets of murine liver cells. Hepatology 54:1379–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fukui K, Yang Q, Cao Y, Takahashi N, Hatakeyama H, et al. 2005. The HNF-1 target collectrin controls insulin exocytosis by SNARE complex formation. Cell Metab 2:373–84 [DOI] [PubMed] [Google Scholar]
- 38.Dolina JS, Cechova S, Rudy CK, Sung SJ, Tang WW, et al. 2017. Cross-presentation of soluble and cell-associated antigen by murine hepatocytes is enhanced by collectrin expression. J. Immunol 198:2341–51 [DOI] [PubMed] [Google Scholar]
- 39.Akpinar P, Kuwajima S, Krutzfeldt J, Stoffel M. 2005. Tmem27: a cleaved and shed plasma membrane protein that stimulates pancreatic beta cell proliferation. Cell Metab 2:385–97 [DOI] [PubMed] [Google Scholar]
- 40.Helft J, Bottcher J, Chakravarty P, Zelenay S, Huotari J, et al. 2015. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c+MHCII+ macrophages and dendritic cells. Immunity 42:1197–211 [DOI] [PubMed] [Google Scholar]
- 41.Shortman K, Naik SH. 2007. Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol 7:19–30 [DOI] [PubMed] [Google Scholar]
- 42.Cheong C, Matos I, Choi JH, Dandamudi DB, Shrestha E, et al. 2010. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209+ dendritic cells for immune T cell areas. Cell 143:416–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rapoport TA, Li L, Park E. 2017. Structural and mechanistic insights into protein translocation. Annu. Rev. Cell Dev. Biol 33:369–90 [DOI] [PubMed] [Google Scholar]
- 44.Cresswell P, Ackerman AL, Giodini A, Peaper DR, Wearsch PA. 2005. Mechanisms of MHC class I-restricted antigen processing and cross-presentation. Immunol. Rev 207:145–57 [DOI] [PubMed] [Google Scholar]
- 45.Paulsson K, Wang P. 2003. Chaperones and folding of MHC class I molecules in the endoplasmic reticulum. Biochim. Biophys. Acta 1641:1–12 [DOI] [PubMed] [Google Scholar]
- 46.Hulpke S, Tampe R. 2013. The MHC I loading complex: a multitasking machinery in adaptive immunity. Trends Biochem. Sci 38:412–20 [DOI] [PubMed] [Google Scholar]
- 47.Neefjes J, Jongsma ML, Paul P, Bakke O. 2011. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol 11:823–36 [DOI] [PubMed] [Google Scholar]
- 48.Spiliotis ET, Manley H, Osorio M, Zuniga MC, Edidin M. 2000. Selective export of MHC class I molecules from the ER after their dissociation from TAP. Immunity 13:841–51 [DOI] [PubMed] [Google Scholar]
- 49.Paquet ME, Cohen-Doyle M, Shore GC, Williams DB. 2004. Bap29/31 influences the intracellular traffic of MHC class I molecules. J. Immunol 172:7548–55 [DOI] [PubMed] [Google Scholar]
- 50.Abe F, Van Prooyen N, Ladasky JJ, Edidin M. 2009. Interaction of Bap31 and MHC class I molecules and their traffic out of the endoplasmic reticulum. J. Immunol 182:4776–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Appenzeller-Herzog C, Hauri HP. 2006. The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J. Cell Sci 119:2173–83 [DOI] [PubMed] [Google Scholar]
- 52.Springer S 2015. Transport and quality control of MHC class I molecules in the early secretory pathway. Curr. Opin. Immunol 34:83–90 [DOI] [PubMed] [Google Scholar]
- 53.Donaldson JG, Williams DB. 2009. Intracellular assembly and trafficking of MHC class I molecules. Traffic 10:1745–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raposo G, van Santen HM, Leijendekker R, Geuze HJ, Ploegh HL. 1995. Misfolded major histocompatibility complex class I molecules accumulate in an expanded ER-Golgi intermediate compartment. J. Cell Biol 131:1403–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Hateren A, James E, Bailey A, Phillips A, Dalchau N, Elliott T. 2010. The cell biology of major histocompatibility complex class I assembly: towards a molecular understanding. Tissue Antigens 76:259–75 [DOI] [PubMed] [Google Scholar]
- 56.Cebrian I, Visentin G, Blanchard N, Jouve M, Bobard A, et al. 2011. Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell 147:1355–68 [DOI] [PubMed] [Google Scholar]
- 57.Wearsch PA, Cresswell P. 2008. The quality control of MHC class I peptide loading. Curr. Opin. Cell Biol 20:624–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ghanem E, Fritzsche S, Al-Balushi M, Hashem J, Ghuneim L, et al. 2010. The transporter associated with antigen processing (TAP) is active in a post-ER compartment. J. Cell Sci 123:4271–79 [DOI] [PubMed] [Google Scholar]
- 59.Kuhns ST, Pease LR. 1998. A region of conformational variability outside the peptide-binding site of a class I MHC molecule. J. Immunol 161:6745–50 [PubMed] [Google Scholar]
- 60.Nair-Gupta P, Baccarini A, Tung N, Seyffer F, Florey O, et al. 2014. TLR signals induce phagosomal MHC-I delivery from the endosomal recycling compartment to allow cross-presentation. Cell 158:506–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Neefjes JJ, Stollorz V, Peters PJ, Geuze HJ, Ploegh HL. 1990. The biosynthetic pathway of MHC class II but not class I molecules intersects the endocytic route. Cell 61:171–83 [DOI] [PubMed] [Google Scholar]
- 62.Kirchhausen T, Owen D, Harrison SC. 2014. Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harb. Perspect. Biol 6: a016725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sandvig K, Torgersen ML, Raa HA, van Deurs B. 2008. Clathrin-independent endocytosis: from nonexisting to an extreme degree of complexity. Histochem. Cell Biol 129:267–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mayor S, Parton RG, Donaldson JG. 2014. Clathrin-independent pathways of endocytosis. Cold Spring Harb. Perspect. Biol 6:a016758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Naslavsky N, Weigert R, Donaldson JG. 2004. Characterization of a nonclathrin endocytic pathway: membrane cargo and lipid requirements. Mol. Biol. Cell 15:3542–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Radhakrishna H, Donaldson JG. 1997. ADP-ribosylation factor 6 regulates a novel plasma membrane recycling pathway. J. Cell Biol 139:49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jovic M, Sharma M, Rahajeng J, Caplan S. 2010. The early endosome: a busy sorting station for proteins at the crossroads. Histol. Histopathol 25:99–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Naslavsky N, Weigert R, Donaldson JG. 2003. Convergence of non-clathrin- and clathrin-derived endosomes involves Arf6 inactivation and changes in phosphoinositides. Mol. Biol. Cell 14:417–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lizee G, Basha G, Tiong J, Julien JP, Tian M, et al. 2003. Control of dendritic cell cross-presentation by the major histocompatibility complex class I cytoplasmic domain. Nat. Immunol 4:1065–73 [DOI] [PubMed] [Google Scholar]
- 70.MacAry PA, Lindsay M, Scott MA, Craig JI, Luzio JP, Lehner PJ. 2001. Mobilization of MHC class I molecules from late endosomes to the cell surface following activation of CD34-derived human Langerhans cells. PNAS 98:3982–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kleijmeer MJ, Escola JM, UytdeHaag FG, Jakobson E, Griffith JM, et al. 2001. Antigen loading of MHC class I molecules in the endocytic tract. Traffic 2:124–37 [DOI] [PubMed] [Google Scholar]
- 72.Hanson PI, Cashikar A. 2012. Multivesicular body morphogenesis. Annu. Rev. Cell Dev. Biol 28:337–62 [DOI] [PubMed] [Google Scholar]
- 73.Maxfield FR, McGraw TE. 2004. Endocytic recycling. Nat. Rev. Mol. Cell Biol 5:121–32 [DOI] [PubMed] [Google Scholar]
- 74.Grant BD, Donaldson JG. 2009. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol 10:597–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gruenberg J 2001. The endocytic pathway: a mosaic of domains. Nat. Rev. Mol. Cell Biol 2:721–30 [DOI] [PubMed] [Google Scholar]
- 76.Hopkins CR. 1983. Intracellular routing of transferrin and transferrin receptors in epidermoid carcinoma A431 cells. Cell 35:321–30 [DOI] [PubMed] [Google Scholar]
- 77.Yamashiro DJ, Tycko B, Fluss SR, Maxfield FR. 1984. Segregation of transferrin to a mildly acidic (pH 6.5) para-Golgi compartment in the recycling pathway. Cell 37:789–800 [DOI] [PubMed] [Google Scholar]
- 78.Ren M, Xu G, Zeng J, De Lemos-Chiarandini C, Adesnik M, Sabatini DD. 1998. Hydrolysis of GTP on rab11 is required for the direct delivery of transferrin from the pericentriolar recycling compartment to the cell surface but not from sorting endosomes. PNAS 95:6187–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ullrich O, Reinsch S, Urbe S, Zerial M, Parton RG. 1996. Rab11 regulates recycling through the pericentriolar recycling endosome. J. Cell Biol 135:913–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wilcke M, Johannes L, Galli T, Mayau V, Goud B, Salamero J. 2000. Rab11 regulates the compartmentalization of early endosomes required for efficient transport from early endosomes to the trans-Golgi network. J. Cell Biol 151:1207–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Choudhury A, Sharma DK, Marks DL, Pagano RE. 2004. Elevated endosomal cholesterol levels in Niemann-Pick cells inhibit rab4 and perturb membrane recycling. Mol. Biol. Cell 15:4500–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.van der Sluijs P, Hull M, Webster P, Male P, Goud B, Mellman I. 1992. The small GTP-binding protein rab4 controls an early sorting event on the endocytic pathway. Cell 70:729–40 [DOI] [PubMed] [Google Scholar]
- 83.Daro E, van der Sluijs P, Galli T, Mellman I. 1996. Rab4 and cellubrevin define different early endosome populations on the pathway of transferrin receptor recycling. PNAS 93:9559–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Klinkert K, Echard A. 2016. Rab35 GTPase: a central regulator of phosphoinositides and F-actin in endocytic recycling and beyond. Traffic 17:1063–77 [DOI] [PubMed] [Google Scholar]
- 85.Powelka AM, Sun J, Li J, Gao M, Shaw LM, et al. 2004. Stimulation-dependent recycling of integrin β1 regulated by ARF6 and Rab11. Traffic 5:20–36 [DOI] [PubMed] [Google Scholar]
- 86.Weigert R, Yeung AC, Li J, Donaldson JG. 2004. Rab22a regulates the recycling of membrane proteins internalized independently of clathrin. Mol. Biol. Cell 15:3758–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Caplan S, Naslavsky N, Hartnell LM, Lodge R, Polishchuk RS, et al. 2002. A tubular EHD1-containing compartment involved in the recycling of major histocompatibility complex class I molecules to the plasma membrane. EMBO J 21:2557–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Giridharan SS, Cai B, Vitale N, Naslavsky N, Caplan S. 2013. Cooperation of MICAL-L1, syndapin2, and phosphatidic acid in tubular recycling endosome biogenesis. Mol. Biol. Cell 24:1776–90, S1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sharma M, Giridharan SS, Rahajeng J, Naslavsky N, Caplan S. 2009. MICAL-L1 links EHD1 to tubular recycling endosomes and regulates receptor recycling. Mol. Biol. Cell 20:5181–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maldonado-Baez L, Williamson C, Donaldson JG. 2013. Clathrin-independent endocytosis: a cargo-centric view. Exp. Cell Res 319:2759–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xie S, Bahl K, Reinecke JB, Hammond GR, Naslavsky N, Caplan S. 2016. The endocytic recycling compartment maintains cargo segregation acquired upon exit from the sorting endosome. Mol. Biol. Cell 27:108–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Naslavsky N, Caplan S. 2011. EHD proteins: key conductors of endocytic transport. Trends Cell Biol 21:122–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xie S, Naslavsky N, Caplan S. 2014. Diacylglycerol kinase alpha regulates tubular recycling endosome biogenesis and major histocompatibility complex class I recycling. J. Biol. Chem 289:31914–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Grant BD, Caplan S. 2008. Mechanisms of EHD/RME-1 protein function in endocytic transport. Traffic 9:2043–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lin SX, Grant B, Hirsh D, Maxfield FR. 2001. Rme-1 regulates the distribution and function of the endocytic recycling compartment in mammalian cells. Nat. Cell Biol 3:567–72 [DOI] [PubMed] [Google Scholar]
- 96.Cebrian I, Croce C, Guerrero NA, Blanchard N, Mayorga LS. 2016. Rab22a controls MHC-I intracellular trafficking and antigen cross-presentation by dendritic cells. EMBO Rep 17:1753–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ackerman AL, Cresswell P. 2003. Regulation of MHC class I transport in human dendritic cells and the dendritic-like cell line KG-1. J. Immunol 170:4178–88 [DOI] [PubMed] [Google Scholar]
- 98.Compeer EB, Flinsenberg TW, Boon L, Hoekstra ME, Boes M. 2014. Tubulation of endosomal structures in human dendritic cells by Toll-like receptor ligation and lymphocyte contact accompanies antigen cross-presentation. J. Biol. Chem 289:520–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Compeer EB, Boes M. 2014. MICAL-L1-related and unrelated mechanisms underlying elongated tubular endosomal network (ETEN) in human dendritic cells. Commun. Integr. Biol 7: e994969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Delamarre L, Holcombe H, Mellman I. 2003. Presentation of exogenous antigens on major histocompatibility complex (MHC) class I and MHC class II molecules is differentially regulated during dendritic cell maturation. J. Exp. Med 198:111–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Barral DC, Cavallari M, McCormick PJ, Garg S, Magee AI, et al. 2008. CD1a and MHC class I follow a similar endocytic recycling pathway. Traffic 9:1446–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Salamero J, Bausinger H, Mommaas AM, Lipsker D, Proamer F, et al. 2001. CD1a molecules traffic through the early recycling endosomal pathway in human Langerhans cells. J. Investig. Dermatol 116:401–8 [DOI] [PubMed] [Google Scholar]
- 103.Aderem A 2002. How to eat something bigger than your head. Cell 110:5–8 [DOI] [PubMed] [Google Scholar]
- 104.Cox D, Lee DJ, Dale BM, Calafat J, Greenberg S. 2000. A Rab11-containing rapidly recycling compartment in macrophages that promotes phagocytosis. PNAS 97:680–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gagnon E, Duclos S, Rondeau C, Chevet E, Cameron PH, et al. 2002. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell 110:119–31 [DOI] [PubMed] [Google Scholar]
- 106.Burgdorf S, Kurts C. 2008. Endocytosis mechanisms and the cell biology of antigen presentation. Curr. Opin. Immunol 20:89–95 [DOI] [PubMed] [Google Scholar]
- 107.Moretti J, Blander JM. 2014. Insights into phagocytosis-coupled activation of pattern recognition receptors and inflammasomes. Curr. Opin. Immunol 26:100–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Iwasaki A, Medzhitov R. 2010. Regulation of adaptive immunity by the innate immune system. Science 327:291–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bevan MJ. 2006. Cross-priming. Nat. Immunol 7:363–65 [DOI] [PubMed] [Google Scholar]
- 110.Caminschi I, Maraskovsky E, Heath WR. 2012. targeting dendritic cells in vivo for cancer therapy. Front. Immunol 3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kreutz M, Tacken PJ, Figdor CG. 2013. Targeting dendritic cells---why bother? Blood 121:2836–44 [DOI] [PubMed] [Google Scholar]
- 112.van Dinther D, Stolk DA, van de Ven R, van Kooyk Y, de Gruijl TD, den Haan JMM. 2017. Targeting C-type lectin receptors: a high-carbohydrate diet for dendritic cells to improve cancer vaccines. J. Leukoc. Biol 102:1017–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rauen J, Kreer C, Paillard A, van Duikeren S, Benckhuijsen WE, et al. 2014. Enhanced cross-presentation and improved CD8+ T cell responses after mannosylation of synthetic long peptides in mice. PLOS ONE 9:e103755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Harvey DJ, Wing DR, Kuster B, Wilson IB. 2000. Composition of N-linked carbohydrates from ovalbumin and co-purified glycoproteins. J. Am. Soc. Mass Spectrom 11:564–71 [DOI] [PubMed] [Google Scholar]
- 115.Burgdorf S, Kautz A, Bohnert V, Knolle PA, Kurts C. 2007. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 316:612–16 [DOI] [PubMed] [Google Scholar]
- 116.Burgdorf S, Lukacs-Kornek V, Kurts C. 2006. The mannose receptor mediates uptake of soluble but not of cell-associated antigen for cross-presentation. J. Immunol 176:6770–76 [DOI] [PubMed] [Google Scholar]
- 117.Streng-Ouwehand I, Ho NI, Litjens M, Kalay H, Boks MA, et al. 2016. Glycan modification of antigen alters its intracellular routing in dendritic cells, promoting priming of T cells. eLife 5:e11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lizee G, Basha G, Jefferies WA. 2005. Tails of wonder: endocytic-sorting motifs key for exogenous antigen presentation. Trends Immunol 26:141–49 [DOI] [PubMed] [Google Scholar]
- 119.Fehres CM, Kalay H, Bruijns SC, Musaafir SA, Ambrosini M, et al. 2015. Cross-presentation through langerin and DC-SIGN targeting requires different formulations of glycan-modified antigens. J. Control Release 203:67–76 [DOI] [PubMed] [Google Scholar]
- 120.Schreibelt G, Klinkenberg LJ, Cruz LJ, Tacken PJ, Tel J, et al. 2012. The C-type lectin receptor CLEC9A mediates antigen uptake and (cross-)presentation by human blood BDCA3+ myeloid dendritic cells. Blood 119:2284–92 [DOI] [PubMed] [Google Scholar]
- 121.Klechevsky E, Flamar AL, Cao Y, Blanck JP, Liu M, et al. 2010. Cross-priming CD8+ T cells by targeting antigens to human dendritic cells through DCIR. Blood 116:1685–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Saluja SS, Hanlon DJ, Sharp FA, Hong E, Khalil D, et al. 2014. Targeting human dendritic cells via DEC-205 using PLGA nanoparticles leads to enhanced cross-presentation of a melanoma-associated antigen. Int. J. Nanomed 9:5231–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fehres CM, van Beelen AJ, Bruijns SC, Ambrosini M, Kalay H, et al. 2015. In situ delivery of antigen to DC-SIGN+CD14+ dermal dendritic cells results in enhanced CD8+ T-cell responses. J. Investig. Dermatol 135:2228–36 [DOI] [PubMed] [Google Scholar]
- 124.Ahrens S, Zelenay S, Sancho D, Hanc P, Kjaer S, et al. 2012. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 36:635–45 [DOI] [PubMed] [Google Scholar]
- 125.Zhang JG, Czabotar PE, Policheni AN, Caminschi I, Wan SS, et al. 2012. The dendritic cell receptor Clec9A binds damaged cells via exposed actin filaments. Immunity 36:646–57 [DOI] [PubMed] [Google Scholar]
- 126.Hanc P, Fujii T, Iborra S, Yamada Y, Huotari J, et al. 2015. Structure of the complex of F-actin and DNGR-1, a C-type lectin receptor involved in dendritic cell cross-presentation of dead cell-associated antigens. Immunity 42:839–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Iborra S, Izquierdo HM, Martinez-Lopez M, Blanco-Menendez N, Reis e Sousa C, Sancho D. 2012. The DC receptor DNGR-1 mediates cross-priming of CTLs during vaccinia virus infection in mice. J. Clin. Investig 122:1628–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sancho D, Joffre OP, Keller AM, Rogers NC, Martinez D, et al. 2009. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 458:899–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zelenay S, Keller AM, Whitney PG, Schraml BU, Deddouche S, et al. 2012. The dendritic cell receptor DNGR-1 controls endocytic handling of necrotic cell antigens to favor cross-priming of CTLs in virus-infected mice. J. Clin. Investig 122:1615–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sancho D, Mourao-Sa D, Joffre OP, Schulz O, Rogers NC, et al. 2008. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J. Clin. Investig 118:2098–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hanc P, Schulz O, Fischbach H, Martin SR, Kjaer S, Reis ESC. 2016. A pH- and ionic strength-dependent conformational change in the neck region regulates DNGR-1 function in dendritic cells. EMBO J 35:2484–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lahoud MH, Proietto AI, Ahmet F, Kitsoulis S, Eidsmo L, et al. 2009. The C-type lectin Clec12A present on mouse and human dendritic cells can serve as a target for antigen delivery and enhancement of antibody responses. J. Immunol 182:7587–94 [DOI] [PubMed] [Google Scholar]
- 133.Hutten TJ, Thordardottir S, Fredrix H, Janssen L, Woestenenk R, et al. 2016. CLEC12A-mediated antigen uptake and cross-presentation by human dendritic cell subsets efficiently boost tumor-reactive T cell responses. J. Immunol 197:2715–25 [DOI] [PubMed] [Google Scholar]
- 134.Lahoud MH, Ahmet F, Kitsoulis S, Wan SS, Vremec D, et al. 2011. Targeting antigen to mouse dendritic cells via Clec9A induces potent CD4 T cell responses biased toward a follicular helper phenotype. J. Immunol 187:842–50 [DOI] [PubMed] [Google Scholar]
- 135.Macri C, Dumont C, Panozza S, Lahoud MH, Caminschi I, et al. 2017. Antibody-mediated targeting of antigen to C-type lectin-like receptors Clec9A and Clec12A elicits different vaccination outcomes. Mol. Immunol 81:143–50 [DOI] [PubMed] [Google Scholar]
- 136.Chatterjee B, Smed-Sorensen A, Cohn L, Chalouni C, Vandlen R, et al. 2012. Internalization and endosomal degradation of receptor-bound antigens regulate the efficiency of cross presentation by human dendritic cells. Blood 120:2011–20 [DOI] [PubMed] [Google Scholar]
- 137.Mahnke K, Guo M, Lee S, Sepulveda H, Swain SL, et al. 2000. The dendritic cell receptor for endocytosis, DEC-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J. Cell Biol 151:673–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sun SC. 2011. Non-canonical NF-κB signaling pathway. Cell Res 21:71–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Katakam AK, Brightbill H, Franci C, Kung C, Nunez V, et al. 2015. Dendritic cells require NIK for CD40-dependent cross-priming of CD8+ T cells. PNAS 112:14664–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Platzer B, Stout M, Fiebiger E. 2014. Antigen cross-presentation of immune complexes. Front. Immunol 5:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Boross P, van Montfoort N, Stapels DA, van der Poel CE, Bertens C, et al. 2014. FcRγ-chain ITAM signaling is critically required for cross-presentation of soluble antibody-antigen complexes by dendritic cells. J. Immunol 193:5506–14 [DOI] [PubMed] [Google Scholar]
- 142.Ho NI, Camps MGM, de Haas EFE, Trouw LA, Verbeek JS, Ossendorp F. 2017. C1q-dependent dendritic cell cross-presentation of in vivo-formed antigen-antibody complexes. J. Immunol 198:4235–43 [DOI] [PubMed] [Google Scholar]
- 143.Murshid A, Gong J, Calderwood SK. 2012. The role of heat shock proteins in antigen cross presentation. Front. Immunol 3:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Binder RJ. 2014. Functions of heat shock proteins in pathways of the innate and adaptive immune system. J. Immunol 193:5765–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Calderwood SK, Mambula SS, Gray PJ Jr., Theriault JR. 2007. Extracellular heat shock proteins in cell signaling. FEBS Lett 581:3689–94 [DOI] [PubMed] [Google Scholar]
- 146.Basu S, Binder RJ, Ramalingam T, Srivastava PK. 2001. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 14:303–13 [DOI] [PubMed] [Google Scholar]
- 147.Binder RJ, Srivastava PK. 2004. Essential role of CD91 in re-presentation of gp96-chaperoned peptides. PNAS 101:6128–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhu H, Fang X, Zhang D, Wu W, Shao M, et al. 2015. Membrane-bound heat shock proteins facilitate the uptake of dying cells and cross-presentation of cellular antigen. Apoptosis 21:96–109 [DOI] [PubMed] [Google Scholar]
- 149.Vo MC, Nguyen-Pham TN, Lee HJ, Jung SH, Choi NR, et al. 2017. Chaetocin enhances dendritic cell function via the induction of heat shock protein and cancer testis antigens in myeloma cells. Oncotarget 8:46047–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mantegazza AR, Magalhaes JG, Amigorena S, Marks MS. 2013. Presentation of phagocytosed antigens by MHC class I and II. Traffic 14:135–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Nair-Gupta P, Blander JM. 2013. An updated view of the intracellular mechanisms regulating cross-presentation. Front. Immunol 4:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gil-Torregrosa BC, Lennon-Dumenil AM, Kessler B, Guermonprez P, Ploegh HL, et al. 2004. Control of cross-presentation during dendritic cell maturation. Eur. J. Immunol 34:398–407 [DOI] [PubMed] [Google Scholar]
- 153.Alloatti A, Kotsias F, Pauwels AM, Carpier JM, Jouve M, et al. 2015. Toll-like receptor 4 engagement on dendritic cells restrains phago-lysosome fusion and promotes cross-presentation of antigens. Immunity 43:1087–100 [DOI] [PubMed] [Google Scholar]
- 154.Garrett WS, Chen LM, Kroschewski R, Ebersold M, Turley S, et al. 2000. Developmental control of endocytosis in dendritic cells by Cdc42. Cell 102:325–34 [DOI] [PubMed] [Google Scholar]
- 155.Weck MM, Grunebach F, Werth D, Sinzger C, Bringmann A, Brossart P. 2007. TLR ligands differentially affect uptake and presentation of cellular antigens. Blood 109:3890–94 [DOI] [PubMed] [Google Scholar]
- 156.Wilson NS, Behrens GM, Lundie RJ, Smith CM, Waithman J, et al. 2006. Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat. Immunol 7:165–72 [DOI] [PubMed] [Google Scholar]
- 157.Villadangos JA, Schnorrer P, Wilson NS. 2005. Control of MHC class II antigen presentation in dendritic cells: a balance between creative and destructive forces. Immunol. Rev 207:191–205 [DOI] [PubMed] [Google Scholar]
- 158.Hochheiser K, Klein M, Gottschalk C, Hoss F, Scheu S, et al. 2016. Cutting edge: The RIG-I ligand 3pRNA potently improves CTL cross-priming and facilitates antiviral vaccination. J. Immunol 196:2439–43 [DOI] [PubMed] [Google Scholar]
- 159.Xu J, Lee MH, Chakhtoura M, Green BL, Kotredes KP, et al. 2016. STAT2 is required for TLR-induced murine dendritic cell activation and cross-presentation. J. Immunol 197:326–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rock KL, Shen L. 2005. Cross-presentation: underlying mechanisms and role in immune surveillance. Immunol. Rev 207:166–83 [DOI] [PubMed] [Google Scholar]
- 161.Joffre OP, Segura E, Savina A, Amigorena S. 2012. Cross-presentation by dendritic cells. Nat. Rev. Immunol 12:557–69 [DOI] [PubMed] [Google Scholar]
- 162.Wagner CS, Grotzke JE, Cresswell P. 2012. Intracellular events regulating cross-presentation. Front. Immunol 3:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Saveanu L, Carroll O, Weimershaus M, Guermonprez P, Firat E, et al. 2009. IRAP identifies an endosomal compartment required for MHC class I cross-presentation. Science 325:213–17 [DOI] [PubMed] [Google Scholar]
- 164.Weimershaus M, Evnouchidou I, Saveanu L, van Endert P. 2013. Peptidases trimming MHC class I ligands. Curr. Opin. Immunol 25:90–96 [DOI] [PubMed] [Google Scholar]
- 165.Becker T, Volchuk A, Rothman JE. 2005. Differential use of endoplasmic reticulum membrane for phagocytosis in J774 macrophages. PNAS 102:4022–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.McNew JA, Parlati F, Fukuda R, Johnston RJ, Paz K, et al. 2000. Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature 407:153–59 [DOI] [PubMed] [Google Scholar]
- 167.Touret N, Paroutis P, Terebiznik M, Harrison RE, Trombetta S, et al. 2005. Quantitative and dynamic assessment of the contribution of the ER to phagosome formation. Cell 123:157–70 [DOI] [PubMed] [Google Scholar]
- 168.Nunes-Hasler P, Demaurex N. 2017. The ER phagosome connection in the era of membrane contact sites. Biochim. Biophys. Acta 1864:1513–24 [DOI] [PubMed] [Google Scholar]
- 169.Oliveira SC, Splitter GA. 1995. CD8+ type 1 CD44hi CD45 RBlo T lymphocytes control intracellular Brucella abortus infection as demonstrated in major histocompatibility complex class I- and class II-deficient mice. Eur. J. Immunol 25:2551–57 [DOI] [PubMed] [Google Scholar]
- 170.Turner J, Dockrell HM. 1996. Stimulation of human peripheral blood mononuclear cells with live Mycobacterium bovis BCG activates cytolytic CD8+ T cells in vitro. Immunology 87:339–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Canaday DH, Ziebold C, Noss EH, Chervenak KA, Harding CV, Boom WH. 1999. Activation of human CD8+ alpha beta TCR+ cells by Mycobacterium tuberculosis via an alternate class I MHC antigen-processing pathway. J. Immunol 162:372–79 [PubMed] [Google Scholar]
- 172.Belkaid Y, Von Stebut E, Mendez S, Lira R, Caler E, et al. 2002. CD8+ T cells are required for primary immunity in C57BL/6 mice following low-dose, intradermal challenge with Leishmania major. J. Immunol 168:3992–4000 [DOI] [PubMed] [Google Scholar]
- 173.Pfeifer JD, Wick MJ, Roberts RL, Findlay K, Normark SJ, Harding CV. 1993. Phagocytic processing of bacterial antigens for class I MHC presentation to T cells. Nature 361:359–62 [DOI] [PubMed] [Google Scholar]
- 174.da Conceicao-Silva F, Perlaza BL, Louis JA, Romero P. 1994. Leishmania major infection in mice primes for specific major histocompatibility complex class I-restricted CD8+ cytotoxic T cell responses. Eur. J. Immunol 24:2813–17 [DOI] [PubMed] [Google Scholar]
- 175.da Silva Santos C, Brodskyn CI. 2014. The role of CD4 and CD8 T cells in human cutaneous leishmaniasis. Front. Public Health 2:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lin PL, Flynn JL. 2015. CD8 T cells and Mycobacterium tuberculosis infection. Semin. Immunopathol 37:239–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Celli J, de Chastellier C, Franchini DM, Pizarro-Cerda J, Moreno E, Gorvel JP. 2003. Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J. Exp. Med 198:545–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Blanchard N, Gonzalez F, Schaeffer M, Joncker NT, Cheng T, et al. 2008. Immunodominant, protective response to the parasite Toxoplasma gondii requires antigen processing in the endoplasmic reticulum. Nat. Immunol 9:937–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Goldszmid RS, Coppens I, Lev A, Caspar P, Mellman I, Sher A. 2009. Host ER–parasitophorous vacuole interaction provides a route of entry for antigen cross-presentation in Toxoplasma gondii–infected dendritic cells. J. Exp. Med 206:399–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Dupont CD, Christian DA, Selleck EM, Pepper M, Leney-Greene M, et al. 2014. Parasite fate and involvement of infected cells in the induction of CD4+ and CD8+ T cell responses to Toxoplasma gondii. PLOS Pathog 10:e1004047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mordue DG, Hakansson S, Niesman I, Sibley LD. 1999. Toxoplasma gondii resides in a vacuole that avoids fusion with host cell endocytic and exocytic vesicular trafficking pathways. Exp. Parasitol 92:87–99 [DOI] [PubMed] [Google Scholar]
- 182.Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, van Endert P, Amigorena S. 2003. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature 425:397–402 [DOI] [PubMed] [Google Scholar]
- 183.Houde M, Bertholet S, Gagnon E, Brunet S, Goyette G, et al. 2003. Phagosomes are competent organelles for antigen cross-presentation. Nature 425:402–6 [DOI] [PubMed] [Google Scholar]
- 184.Alloatti A, Rookhuizen DC, Joannas L, Carpier JM, Iborra S, et al. 2017. Critical role for Sec22b-dependent antigen cross-presentation in antitumor immunity. J. Exp. Med 214:2231–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wu SJ, Niknafs YS, Kim SH, Oravecz-Wilson K, Zajac C, et al. 2017. A critical analysis of the role of SNARE protein SEC22B in antigen cross-presentation. Cell Rep 19:2645–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Montealegre S, van Endert P. 2017. MHC class I cross-presentation: stage lights on Sec22b. Trends Immunol 38:618–21 [DOI] [PubMed] [Google Scholar]
- 187.Cotter K, Stransky L, McGuire C, Forgac M. 2015. Recent insights into the structure, regulation, and function of the V-ATPases. Trends Biochem. Sci 40:611–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES. 2005. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science 307:1630–34 [DOI] [PubMed] [Google Scholar]
- 189.Settembre C, Fraldi A, Medina DL, Ballabio A. 2013. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol 14:283–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, et al. 2009. A gene network regulating lysosomal biogenesis and function. Science 325:473–77 [DOI] [PubMed] [Google Scholar]
- 191.Samie M, Cresswell P. 2015. The transcription factor TFEB acts as a molecular switch that regulates exogenous antigen-presentation pathways. Nat. Immunol 16:729–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Calmette J, Bertrand M, Vetillard M, Ellouze M, Flint S, et al. 2016. Glucocorticoid-induced leucine zipper protein controls macropinocytosis in dendritic cells. J. Immunol 197:4247–56 [DOI] [PubMed] [Google Scholar]
- 193.Lam GY, Huang J, Brumell JH. 2010. The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Semin. Immunopathol 32:415–30 [DOI] [PubMed] [Google Scholar]
- 194.Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, et al. 2006. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 126:205–18 [DOI] [PubMed] [Google Scholar]
- 195.Savina A, Peres A, Cebrian I, Carmo N, Moita C, et al. 2009. The small GTPase Rac2 controls phagosomal alkalinization and antigen crosspresentation selectively in CD8+ dendritic cells. Immunity 30:544–55 [DOI] [PubMed] [Google Scholar]
- 196.Dingjan I, Linders PT, van den Bekerom L, Baranov MV, Halder P, et al. 2017. Oxidized phagosomal NOX2 complex is replenished from lysosomes. J. Cell Sci 130:1285–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kotsias F, Hoffmann E, Amigorena S, Savina A. 2013. Reactive oxygen species production in the phagosome: impact on antigen presentation in dendritic cells. Antioxid. Redox Signal 18:714–29 [DOI] [PubMed] [Google Scholar]
- 198.Mantegazza AR, Savina A, Vermeulen M, Perez L, Geffner J, et al. 2008. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood 112:4712–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Jancic C, Savina A, Wasmeier C, Tolmachova T, El-Benna J, et al. 2007. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat. Cell Biol 9:367–78 [DOI] [PubMed] [Google Scholar]
- 200.Rybicka JM, Balce DR, Chaudhuri S, Allan ER, Yates RM. 2012. Phagosomal proteolysis in dendritic cells is modulated by NADPH oxidase in a pH-independent manner. EMBO J 31:932–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hari A, Ganguly A, Mu L, Davis SP, Stenner MD, et al. 2015. Redirecting soluble antigen for MHC class I cross-presentation during phagocytosis. Eur. J. Immunol 45:383–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Allan ER, Tailor P, Balce DR, Pirzadeh P, McKenna NT, et al. 2014. NADPH oxidase modifies patterns of MHC class II-restricted epitopic repertoires through redox control of antigen processing. J. Immunol 192:4989–5001 [DOI] [PubMed] [Google Scholar]
- 203.Datta SK, Raz E. 2005. Induction of antigen cross-presentation by Toll-like receptors. Springer Semin. Immunopathol 26:247–55 [DOI] [PubMed] [Google Scholar]
- 204.Nair P, Amsen D, Blander JM. 2011. Co-ordination of incoming and outgoing traffic in antigen-presenting cells by pattern recognition receptors and T cells. Traffic 12:1669–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Vulcano M, Dusi S, Lissandrini D, Badolato R, Mazzi P, et al. 2004. Toll receptor-mediated regulation of NADPH oxidase in human dendritic cells. J. Immunol 173:5749–56 [DOI] [PubMed] [Google Scholar]
- 206.Blander JM, Medzhitov R. 2004. Regulation of phagosome maturation by signals from Toll-like receptors. Science 304:1014–18 [DOI] [PubMed] [Google Scholar]
- 207.Trombetta ES, Ebersold M, Garrett W, Pypaert M, Mellman I. 2003. Activation of lysosomal function during dendritic cell maturation. Science 299:1400–3 [DOI] [PubMed] [Google Scholar]
- 208.Wang T, Hong W. 2002. Interorganellar regulation of lysosome positioning by the Golgi apparatus through Rab34 interaction with Rab-interacting lysosomal protein. Mol. Biol. Cell 13:4317–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kasmapour B, Gronow A, Bleck CK, Hong W, Gutierrez MG. 2012. Size-dependent mechanism of cargo sorting during lysosome-phagosome fusion is controlled by Rab34. PNAS 109:20485–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Drutman SB, Trombetta ES. 2010. Dendritic cells continue to capture and present antigens after maturation in vivo. J. Immunol 185:2140–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Dingjan I, Paardekooper LM, Verboogen DRJ, von Mollard GF, Ter Beest M, van den Bogaart G. 2017. VAMP8-mediated NOX2 recruitment to endosomes is necessary for antigen release. Eur. J. Cell Biol 96:705–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Matheoud D, Moradin N, Bellemare-Pelletier A, Shio MT, Hong WJ, et al. 2013. Leishmania evades host immunity by inhibiting antigen cross-presentation through direct cleavage of the SNARE VAMP8. Cell Host Microbe 14:15–25 [DOI] [PubMed] [Google Scholar]
- 213.Ding Y, Guo Z, Liu Y, Li X, Zhang Q, et al. 2016. The lectin Siglec-G inhibits dendritic cell cross-presentation by impairing MHC class I-peptide complex formation. Nat. Immunol 17:1167–75 [DOI] [PubMed] [Google Scholar]
- 214.Chen W, Han C, Xie B, Hu X, Yu Q, et al. 2013. Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell 152:467–78 [DOI] [PubMed] [Google Scholar]
- 215.Thrasher AJ, Burns SO. 2010. WASP: a key immunological multitasker. Nat. Rev. Immunol 10:182–92 [DOI] [PubMed] [Google Scholar]
- 216.Baptista MA, Keszei M, Oliveira M, Sunahara KK, Andersson J, et al. 2016. Deletion of Wiskott-Aldrich syndrome protein triggers Rac2 activity and increased cross-presentation by dendritic cells. Nat. Commun 7:12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Baptista MAP, Westerberg LS. 2017. Activation of compensatory pathways via Rac2 in the absence of the Cdc42 effector Wiskott-Aldrich syndrome protein in dendritic cells. Small GTPases Jan 27:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Shen L, Sigal LJ, Boes M, Rock KL. 2004. Important role of cathepsin S in generating peptides for TAP-independent MHC class I crosspresentation in vivo. Immunity 21:155–65 [DOI] [PubMed] [Google Scholar]
- 219.Shi GP, Munger JS, Meara JP, Rich DH, Chapman HA. 1992. Molecular cloning and expression of human alveolar macrophage cathepsin S, an elastinolytic cysteine protease. J. Biol. Chem 267:7258–62 [PubMed] [Google Scholar]
- 220.Lin ML, Zhan Y, Proietto AI, Prato S, Wu L, et al. 2008. Selective suicide of cross-presenting CD8+ dendritic cells by cytochrome c injection shows functional heterogeneity within this subset. PNAS 105:3029–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Baleeiro RB, Walden P. 2017. Immature human DCs efficiently translocate endocytosed antigens into the cytosol for proteasomal processing. Mol. Immunol 88:148–54 [DOI] [PubMed] [Google Scholar]
- 222.Giodini A, Cresswell P. 2008. Hsp90-mediated cytosolic refolding of exogenous proteins internalized by dendritic cells. EMBO J 27:201–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Zehner M, Chasan AI, Schuette V, Embgenbroich M, Quast T, et al. 2011. Mannose receptor polyubiquitination regulates endosomal recruitment of p97 and cytosolic antigen translocation for cross-presentation. PNAS 108:9933–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zehner M, Burgdorf S. 2013. Regulation of antigen transport into the cytosol for cross-presentation by ubiquitination of the mannose receptor. Mol. Immunol 55:146–48 [DOI] [PubMed] [Google Scholar]
- 225.Dingjan I, Verboogen DR, Paardekooper LM, Revelo NH, Sittig SP, et al. 2016. Lipid peroxidation causes endosomal antigen release for cross-presentation. Sci. Rep 6:22064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Tretter T, Pereira FP, Ulucan O, Helms V, Allan S, et al. 2013. ERAD and protein import defects in a sec61 mutant lacking ER-lumenal loop 7. BMC Cell Biol 14:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Kaiser ML, Romisch K. 2015. Proteasome 19S RP binding to the Sec61 channel plays a key role in ERAD. PLOS ONE 10:e0117260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Grotzke JE, Cresswell P. 2015. Are ERAD components involved in cross-presentation? Mol. Immunol 68:112–15 [DOI] [PubMed] [Google Scholar]
- 229.Romisch K 2017. A case for Sec61 channel involvement in ERAD. Trends Biochem. Sci 42:171–79 [DOI] [PubMed] [Google Scholar]
- 230.Imai J, Hasegawa H, Maruya M, Koyasu S, Yahara I. 2005. Exogenous antigens are processed through the endoplasmic reticulum-associated degradation (ERAD) in cross-presentation by dendritic cells. Int. Immunol 17:45–53 [DOI] [PubMed] [Google Scholar]
- 231.Zehner M, Marschall AL, Bos E, Schloetel JG, Kreer C, et al. 2015. The translocon protein Sec61 mediates antigen transport from endosomes in the cytosol for cross-presentation to CD8+ T cells. Immunity 42:850–63 [DOI] [PubMed] [Google Scholar]
- 232.Schauble N, Cavalie A, Zimmermann R, Jung M. 2014. Interaction of Pseudomonas aeruginosa Exotoxin A with the human Sec61 complex suppresses passive calcium efflux from the endoplasmic reticulum. Channels 8:76–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Ackerman AL, Giodini A, Cresswell P. 2006. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity 25:607–17 [DOI] [PubMed] [Google Scholar]
- 234.Ye Y, Meyer HH, Rapoport TA. 2001. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414:652–56 [DOI] [PubMed] [Google Scholar]
- 235.Menager J, Ebstein F, Oger R, Hulin P, Nedellec S, et al. 2014. Cross-presentation of synthetic long peptides by human dendritic cells: a process dependent on ERAD component p97/VCP but not sec61 and/or Derlin-1. PLOS ONE 9:e89897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Baron L, Paatero AO, Morel JD, Impens F, Guenin-Mace L, et al. 2016. Mycolactone subverts immunity by selectively blocking the Sec61 translocon. J. Exp. Med 213:2885–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.George KM, Chatterjee D, Gunawardana G, Welty D, Hayman J, et al. 1999. Mycolactone: a polyketide toxin from Mycobacterium ulcerans required for virulence. Science 283:854–57 [DOI] [PubMed] [Google Scholar]
- 238.McKenna M, Simmonds RE, High S. 2016. Mechanistic insights into the inhibition of Sec61-dependent co- and post-translational translocation by mycolactone. J. Cell Sci 129:1404–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.McKenna M, Simmonds RE, High S. 2017. Mycolactone reveals the substrate-driven complexity of Sec61-dependent transmembrane protein biogenesis. J. Cell Sci 130:1307–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Grotzke JE, Kozik P, Morel JD, Impens F, Pietrosemoli N, et al. 2017. Sec61 blockade by mycolactone inhibits antigen cross-presentation independently of endosome-to-cytosol export. PNAS 114(29):E5910–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Bougneres L, Helft J, Tiwari S, Vargas P, Chang BH, et al. 2009. A role for lipid bodies in the cross-presentation of phagocytosed antigens by MHC class I in dendritic cells. Immunity 31:232–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Martens S, Parvanova I, Zerrahn J, Griffiths G, Schell G, et al. 2005. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLOS Pathog 1:e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Ling YM, Shaw MH, Ayala C, Coppens I, Taylor GA, et al. 2006. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J. Exp. Med 203:2063–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Khan IA, Ely KH, Kasper LH. 1991. A purified parasite antigen (p30) mediates CD8+ T cell immunity against fatal Toxoplasma gondii infection in mice. J. Immunol 147:3501–6 [PubMed] [Google Scholar]
- 245.Kwok LY, Lutjen S, Soltek S, Soldati D, Busch D, et al. 2003. The induction and kinetics of antigen-specific CD8 T cells are defined by the stage specificity and compartmentalization of the antigen in murine toxoplasmosis. J. Immunol 170:1949–57 [DOI] [PubMed] [Google Scholar]
- 246.Gregg B, Dzierszinski F, Tait E, Jordan KA, Hunter CA, Roos DS. 2011. Subcellular antigen location influences T-cell activation during acute infection with Toxoplasma gondii. PLOS ONE 6:e22936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Lopez J, Bittame A, Massera C, Vasseur V, Effantin G, et al. 2015. Intravacuolar membranes regulate CD8 T cell recognition of membrane-bound Toxoplasma gondii protective antigen. Cell Rep 13:2273–86 [DOI] [PubMed] [Google Scholar]
- 248.Carlson EJ, Pitonzo D, Skach WR. 2006. p97 functions as an auxiliary factor to facilitate TM domain extraction during CFTR ER-associated degradation. EMBO J 25:4557–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Bertholet S, Goldszmid R, Morrot A, Debrabant A, Afrin F, et al. 2006. Leishmania antigens are presented to CD8+ T cells by a transporter associated with antigen processing-independent pathway in vitro and in vivo. J. Immunol 177:3525–33 [DOI] [PubMed] [Google Scholar]
- 250.Ndjamen B, Kang BH, Hatsuzawa K, Kima PE. 2010. Leishmania parasitophorous vacuoles interact continuously with the host cell’s endoplasmic reticulum; parasitophorous vacuoles are hybrid compartments. Cell Microbiol 12:1480–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Ackerman AL, Kyritsis C, Tampe R, Cresswell P. 2003. Early phagosomes in dendritic cells form a cellular compartment sufficient for cross presentation of exogenous antigens. PNAS 100:12889–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Fischbach H, Doring M, Nikles D, Lehnert E, Baldauf C, et al. 2015. Ultrasensitive quantification of TAP-dependent antigen compartmentalization in scarce primary immune cell subsets. Nat. Commun 6:6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Zuo D, Subjeck J, Wang XY. 2016. Unfolding the role of large heat shock proteins: new insights and therapeutic implications. Front. Immunol 7:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Wang H, Yu X, Guo C, Zuo D, Fisher PB, et al. 2013. Enhanced endoplasmic reticulum entry of tumor antigen is crucial for cross-presentation induced by dendritic cell-targeted vaccination. J. Immunol 191:6010–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Ackerman AL, Kyritsis C, Tampe R, Cresswell P. 2005. Access of soluble antigens to the endoplasmic reticulum can explain cross-presentation by dendritic cells. Nat. Immunol 6:107–13 [DOI] [PubMed] [Google Scholar]
- 256.Johannes L, Popoff V. 2008. Tracing the retrograde route in protein trafficking. Cell 135:1175–87 [DOI] [PubMed] [Google Scholar]
- 257.Burgdorf S, Scholz C, Kautz A, Tampe R, Kurts C. 2008. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat. Immunol 9:558–66 [DOI] [PubMed] [Google Scholar]
- 258.Lawand M, Abramova A, Manceau V, Springer S, van Endert P. 2016. TAP-dependent and -independent peptide import into dendritic cell phagosomes. J. Immunol 197:3454–63 [DOI] [PubMed] [Google Scholar]
- 259.Di Pucchio T, Chatterjee B, Smed-Sorensen A, Clayton S, Palazzo A, et al. 2008. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nat. Immunol 9:551–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Stow JL, Manderson AP, Murray RZ. 2006. SNAREing immunity: the role of SNAREs in the immune system. Nat. Rev. Immunol 6:919–29 [DOI] [PubMed] [Google Scholar]
- 261.Husebye H, Aune MH, Stenvik J, Samstad E, Skjeldal F, et al. 2010. The Rab11a GTPase controls Toll-like receptor 4-induced activation of interferon regulatory factor-3 on phagosomes. Immunity 33:583–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, et al. 2003. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol 4:1009–15 [DOI] [PubMed] [Google Scholar]
- 263.Snyder DA, Kelly ML, Woodbury DJ. 2006. SNARE complex regulation by phosphorylation. Cell Biochem. Biophys 45:111–23 [DOI] [PubMed] [Google Scholar]
- 264.Wickner W, Schekman R. 2008. Membrane fusion. Nat. Struct. Mol. Biol 15:658–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Kretzer NM, Theisen DJ, Tussiwand R, Briseno CG, Grajales-Reyes GE, et al. 2016. RAB43 facilitates cross-presentation of cell-associated antigens by CD8α+ dendritic cells. J. Exp. Med 213:2871–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.De Angelis Rigotti F, De Gassart A, Pforr C, Cano F, N’Guessan P, et al. 2017. MARCH9-mediated ubiquitination regulates MHC I export from the TGN. Immunol. Cell Biol 95:753–64 [DOI] [PubMed] [Google Scholar]
- 267.Bock JB, Klumperman J, Davanger S, Scheller RH. 1997. Syntaxin 6 functions in trans-Golgi network vesicle trafficking. Mol. Biol. Cell 8:1261–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Falguieres T, Castle D, Gruenberg J. 2012. Regulation of the MVB pathway by SCAMP3. Traffic 13:131–42 [DOI] [PubMed] [Google Scholar]
- 269.Roche PA, Marks MS, Cresswell P. 1991. Formation of a nine-subunit complex by HLA class II glycoproteins and the invariant chain. Nature 354:392–94 [DOI] [PubMed] [Google Scholar]
- 270.Lamb CA, Cresswell P. 1992. Assembly and transport properties of invariant chain trimers and HLA-DR-invariant chain complexes. J. Immunol 148:3478–82 [PubMed] [Google Scholar]
- 271.Sugita M, Brenner MB. 1995. Association of the invariant chain with major histocompatibility complex class I molecules directs trafficking to endocytic compartments. J. Biol. Chem 270:1443–48 [DOI] [PubMed] [Google Scholar]
- 272.Vigna JL, Smith KD, Lutz CT. 1996. Invariant chain association with MHC class I: preference for HLA class I/beta 2-microglobulin heterodimers, specificity, and influence of the MHC peptide-binding groove. J. Immunol 157:4503–10 [PubMed] [Google Scholar]
- 273.Basha G, Omilusik K, Chavez-Steenbock A, Reinicke AT, Lack N, et al. 2012. A CD74-dependent MHC class I endolysosomal cross-presentation pathway. Nat. Immunol 13:237–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Greenfield JJ, High S. 1999. The Sec61 complex is located in both the ER and the ER-Golgi intermediate compartment. J. Cell Sci 112(Part 10):1477–86 [DOI] [PubMed] [Google Scholar]
- 275.Casbon AJ, Allen LA, Dunn KW, Dinauer MC. 2009. Macrophage NADPH oxidase flavocytochrome B localizes to the plasma membrane and Rab11-positive recycling endosomes. J. Immunol 182:2325–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Arango Duque G, Fukuda M, Descoteaux A. 2013. Synaptotagmin XI regulates phagocytosis and cytokine secretion in macrophages. J. Immunol 190:1737–45 [DOI] [PubMed] [Google Scholar]
- 277.Ma W, Zhang Y, Vigneron N, Stroobant V, Thielemans K, et al. 2016. Long-peptide cross-presentation by human dendritic cells occurs in vacuoles by peptide exchange on nascent MHC class I molecules. J. Immunol 196:1711–20 [DOI] [PubMed] [Google Scholar]