

Summary
PARN loss-of-function mutations cause a severe form of the hereditary disease dyskeratosis congenita (DC). PARN deficiency affects the stability of non-coding RNAs such as human telomerase RNA (hTR), but these effects do not explain the severe disease in patients. We demonstrate that PARN deficiency affects the levels of numerous miRNAs in human cells. PARN regulates miRNA levels by stabilizing either mature or precursor miRNAs by removing oligo(A) tails added by the poly(A) polymerase PAPD5, which if remaining recruit the exonuclease DIS3L or DIS3L2 to degrade the miRNA. PARN knockdown destabilizes multiple miRNAs that repress p53 translation, which leads to an increase in p53 accumulation in a Dicer-dependent manner, thus explaining why PARN defective patients show p53 accumulation. This work also reveals that DIS3L and DIS3L2 are critical 3’ to 5’ exonucleases that regulate miRNA stability, with the addition and removal of 3’ end extensions controlling miRNA levels in the cell.
Graphical Abstract
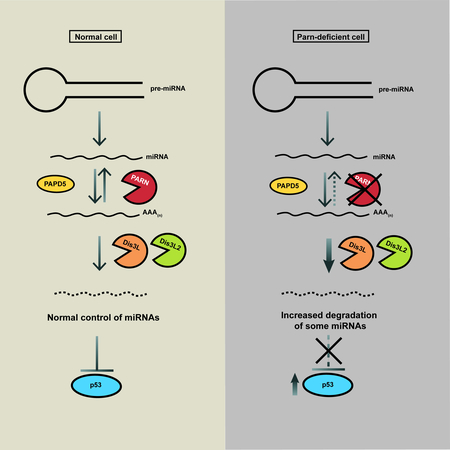
eTOC blurb:
LOF mutations in the 3’ to 5’ exoribonuclease PARN lead to a severe form of Dyskeratosis Congenita (DC). Shukla et al found that PARN regulates the levels of specific miRNAs in the cell which modulate p53 protein levels. PARN inhibition increases p53 levels in cancer cells providing a therapeutic opportunity.
Introduction
The adenylation of 3’ ends of cellular RNAs by poly(A) polymerases modulates the function and stability of both mRNAs and non-coding RNAs. PARN is a processive mammalian poly(A)-specific ribonuclease proposed to remove poly(A) tails from the 3’ ends of mRNAs (Dehlin et al., 2000; Körner and Wahle, 1997). Recent work has shown that PARN regulates the stability of several ncRNAs in mammalian cells, including scaRNAs, human telomerase RNA (hTR), piRNAs and Y RNAs (Berndt et al., 2012; Izumi et al., 2016; Moon et al., 2015; Nguyen et al., 2015; Shukla and Parker, 2017; Shukla et al., 2016; Son et al., 2018; Tang et al., 2016; Tseng et al., 2015). This suggests that the deadenylation activity of PARN is important for regulating the stability of a variety of RNAs in mammalian cells.
miRNAs are small 21-23 nt non-coding RNAs that regulate gene expression in eukaryotic cells through base pairing with their target mRNAs (Ha and Kim, 2014). miRNAs are transcribed as long primary transcripts (pri-miRNA), which are trimmed by to generate the precursor miRNA (pre-miRNA) containing the miRNA stem-loop (Finnegan and Pasquinelli, 2013). The pre-miRNA is subsequently cleaved by Dicer to generate the mature miRNA, which assembles with Argonaute and GW182 along with other proteins to form the RNA-induced silencing complex (RISC) (Finnegan and Pasquinelli, 2013). While the role of miRNAs in regulating gene expression is well studied, the mechanism(s) that regulate the stability of miRNAs in mammalian cells are not well understood. Previous work has suggested that XRN2-mediated 5’ to 3’ degradation can regulate the stability of some miRNAs in model organisms (Chatterjee and Großhans, 2009) and Tudor S/N mediated endonucleolytic degradation of some miRNAs occurs in mammalian cells (Elbarbary et al., 2017).
miRNAs can be modified by non-templated U or A additions at the 3’ end in diverse cell types and organisms (Burroughs et al., 2010; Landgraf et al., 2007). In plants, Hen1-mediated 3’ end methylation of the 2’-OH moiety has been shown to protect endogenous plant siRNAs and miRNAs from uridylation and degradation by SND1 (Li et al., 2005; Ramachandran and Chen, 2008; Yu et al., 2005). In black cottonwood plant, adenylation of the 3’ end is a feature of miRNA degradation products, and adenylation can also reduce the degradation of plant miRNAs (Lu et al., 2009). In the alga Chlamydomonas, Mut68 uridylates the 3’ ends of endogenous siRNAs and miRNAs, suggesting a conserved function of 3’ end modification of small RNAs in different organisms (Ibrahim et al., 2010).
The best studied example of 3’ end non-templated addition of miRNAs in mammalian cell is the uridylation of the let-7 pre-miRNA by non-canonical uridylases TUT4/TUT7 (Hagan et al., 2009; Heo et al., 2009). TUT4/TUT7 are recruited by the RNA binding protein LIN28 to pre-let-7, which leads to polyuridylation of the pre-let-7 3’ end and affects its processing into mature let-7, thereby playing a role in regulating let-7 miRNA levels and function in animal development (Heo et al., 2008). It has also been proposed that monouridylation of some let-7 pre-miRNAs as opposed to polyuridylation is important for their processing to mature let-7 miRNA in HeLa cells, suggesting that the activity of Tut4/Tut7 may be regulated in mammalian cells in order to maintain a balance between let-7 processing and degradation (Heo et al., 2012). Uridylation of pre-let-7 leads to the recruitment of the 3’ to 5’ exonuclease DIS3L2, and DIS3L2 degrades polyuridylated pre-let-7 in undifferentiated stem cells (Chang et al., 2013; Ustianenko et al., 2013). Uridylation of pre-miRNAs and miRNAs has also been shown to occur on other families of miRNAs in diverse cell types, suggesting that uridylation of pre-miRNAs and mature miRNAs is a general feature of miRNA regulation (Berezikov et al., 2011; Jones et al., 2012; 2009; Thornton et al., 2014; Wyman et al., 2011).
Adenylation at the 3’ end has also been shown to occur for some miRNAs, although it is suggested to be less frequent compared to uridylation (Burroughs et al., 2010; Wyman et al., 2011). The most well understood example of miRNA adenylation is GLD2-mediated monoadenylation of miR-122, which has been shown to enhance the stability and function of miR-122 in mammalian cells (Burns et al., 2011; Katoh et al., 2009). Similarly, monoadenylation of the 3’ end by GLD2 also enhanced the stability of some other miRNAs in human fibroblasts (D’Ambrogio et al., 2012). In contrast, PAPD5-mediated adenylation has been proposed to destabilize miR-21 in human cancer cell lines (Boele et al., 2014), and in Drosophila, wisp-mediated 3’ adenylation of maternal miRNAs promotes their clearance (Lee et al., 2014). However, the mechanisms that regulate miRNA 3’ end adenylation and deadenylation, and the role of PARN in this process, are not well understood. Because PARN-mediated deadenylation has been shown to protect many non-coding RNAs from degradation by 3’ to 5’ exonucleases (Moon et al., 2015; Nguyen et al., 2015; Shukla and Parker, 2017; Shukla et al., 2016; Tseng et al., 2015), we hypothesized that PARN might stabilize some miRNAs by removing oligo(A) tails that would otherwise recruit processive 3’ to 5’ exonucleases to degrade the miRNA. Further, the loss of miRNA stability might explain some of the phenotypes observed in PARN deficient patients with dyskeratosis congenita.
Dyskeratosis Congenita (DC) is caused by genetic defects in components of the telomerase holoenzyme in human cells and leads to bone marrow failure and cancer (Kirwan and Dokal, 2009; Mason and Bessler, 2011). While most mutations associated with DC pathogenesis are in genes important for human telomerase RNA (hTR) assembly (DKC1) or telomerase RNA stability (TERC), mutations in PARN were shown to cause a severe form of DC known as Hoyeraal-Hreidarsson syndrome, which causes abnormally short telomeres and congenital defects (Burris et al., 2016; Dhanraj et al., 2015; Stuart et al., 2015; Tummala et al., 2015). Subsequently, it was shown that loss of PARN leads to defective 3’ end maturation of hTR, leading to oligoadenylation by PAPD5 and 3’ to 5’ degradation by EXOSC10 in the nucleus (Boyraz et al., 2016; Nguyen et al., 2015; Shukla et al., 2016; Tseng et al., 2015), or cytoplasmic export and decapping and 5’ to 3’ degradation by DCP2/XRN1 (Shukla et al., 2016). While loss of telomerase RNA function explains telomere shortening in DC patients, it doesn’t explain the pleiotropic and severe phenotype of the disease caused by PARN mutations.
We hypothesized that PARN deficiency affects the stability of miRNAs in human cells, which could explain the severe phenotype of PARN deficiency in DC patients. We show that in HeLa cells, PARN affects the levels of multiple miRNAs. PARN protects miRNAs from degradation by removing adenosines from their 3’ ends that are added by the poly(A) polymerase PAPD5. In absence of PARN, 3’ end adenylation leads to recruitment of the cytoplasmic exonucleases DIS3L or DIS3L2 which degrade miRNAs. Moreover, several miRNAs decreased in PARN depleted cells target the p53 mRNA, and PARN knockdown leads to upregulation of p53 protein levels in multiple cell lines with or without DNA damage. PARN knockdown also sensitizes HeLa cells to chemotherapeutic agents, which leads to cell cycle arrest and apoptosis. This may explain why PARN mutations lead to a severe phenotype of DC in patients, since chronic upregulation of p53 signaling would negatively affect cell growth and development in these patients. Further, the use of PARN inhibitors combined with chemotherapy could be a therapeutic strategy to treat a subset of cancers that are caused by repressed wild-type p53 protein.
Results
PARN regulates the stability of diverse miRNAs in human cells
To investigate whether PARN affects miRNAs in human cells, we sequenced miRNA populations from control and PARN knockdown HeLa cells. From four biological replicates, we identified 86 miRNAs that were upregulated more than 1.5-fold in PARN knockdown cells, and 157 miRNAs that were downregulated more than 0.7-fold out of 807 miRNAs (Fig. 1A and Data S1), demonstrating that PARN affects the levels of multiple miRNAs.
Figure 1.
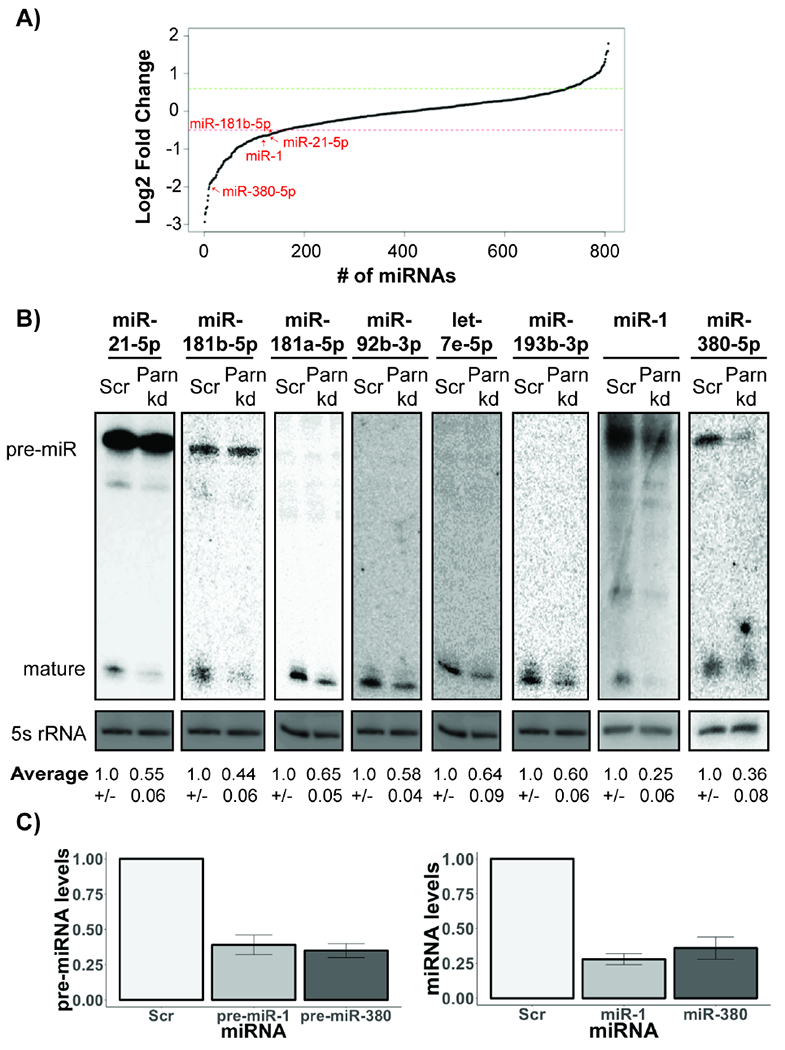
PARN regulates the levels and stability of diverse miRNAs in HeLa cells. A) Line plot depicting changes in miRNA levels in PARN knockdown cells compared to control for four biological replicates for 807 total miRNAs. Each dot represents an individual miRNA. Red and green intercepts depict lower and upper cutoffs for differentially expressed miRNAs (<0.7× or >1.5X in PARN KD compared to control). B) Representative northern blots of indicated miRNAs in control and PARN knockdown cells (Average +/− S.D. for five biological replicates). C) Relative pre- and mature miRNA levels in control and PARN knockdown cells quantified using qRT-PCR to 5s rRNA (Average +/− S.D. for three biological replicates).
To verify our sequencing results, and to determine if PARN knockdown was reducing mature miRNAs or pre-miRNAs, we examined the levels of mature and pre-miRNAs by other methods. We found that miR-181b-5p and miR-21-5p were not affected at precursor level, and only the mature forms were reduced upon PARN knockdown (Fig. 1B). In contrast, we found that miR-1 and miR-380-5p are reduced at the pre-miRNA form, with the levels of the mature miRNA similar to the pre-miRNA upon PARN knockdown (Fig. 1B and C). This argues that the reduction in these miRNAs occurs at the precursor stage. The reduction in miR-181b-5p and miR-21-5p miRNA levels upon PARN knockdown was stronger when detected by northern blotting as compared to sequencing (Fig. 1A and B). To investigate whether miRNA sequencing underrepresented the number of miRNAs decreased upon PARN knockdown, we measured the levels of six additional miRNAs which were above the 0.7-fold threshold we used for estimating the number of miRNAs regulated positively by PARN (Fig. 1A). Four out of these six miRNAs, miR-181a-5p, miR-193b-3p, miR-92b-3p and let-7e-5p, were also reduced between 0.5-fold and 0.35-fold upon PARN knockdown (Fig. 1B). These results suggest that PARN knockdown reduces the levels of several miRNAs in HeLa cells, and that miRNA sequencing underestimates the effect of PARN knockdown on miRNA levels.
To determine if miRNA levels were reduced in the PARN knockdown due to increased degradation, we measured the stability of miRNAs in control and PARN knockdown following transcriptional repression with actinomycin D. We observed that PARN knockdown led to a reduction in miRNA stability of all six miRNAs compared to control cells, with miR-181b-5p and miR-181a-5p most affected by PARN knockdown and let-7e-5p the least affected (Fig. 2A and B). Consistent with miRNA having half-lives ranging from >8 hours to several days (Bail et al., 2010), we observed miRNAs are barely degraded in control cells. The reduction in miRNA stability upon PARN knockdown argues that PARN functions to stabilize some miRNAs.
Figure 2.
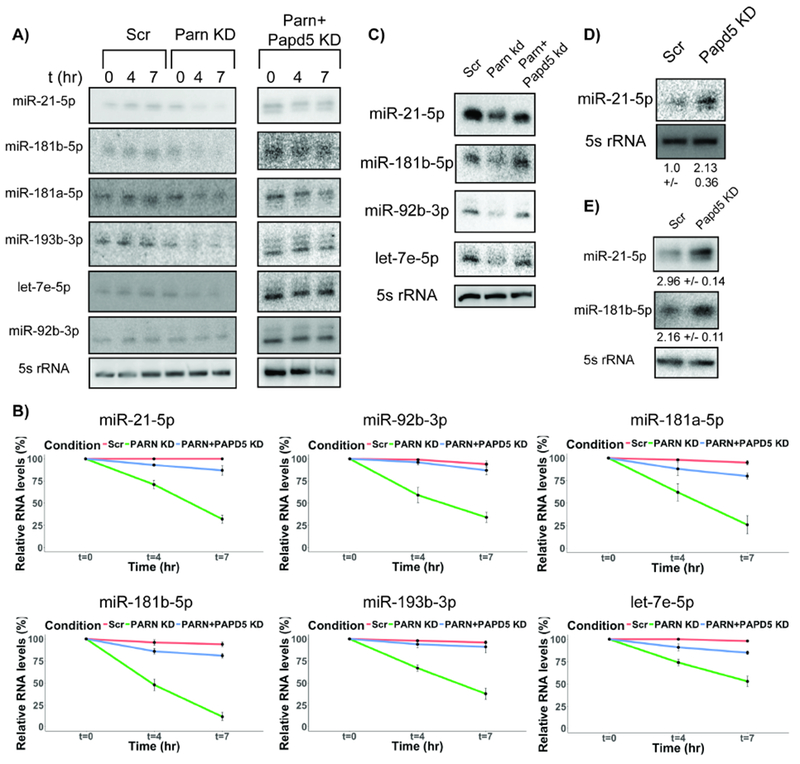
PARN inhibition leads to decrease in miRNA stability and can be recused by a co-knockdown of PAPD5. A) Representative northern blots for miRNA decay rates in control, PARN KD and PARN & PAPD5 co-KD cells at 0, 4 & 7 hours after transcription shutoff. B) Quantification of miRNA decay rates in PARN KD and PARN and PAPD5 co- KD cells compared to control cells (Average +/− S.D. for four biological replicates). C) Representative northern blots for miRNA levels in PARN and PAPD5 co-knockdown cells compared to control and PARN knockdown cells. D) and E) Representative northern blots for miRNA levels in patient fibroblast and HeLa cells respectively upon PAPD5 KD (Average +/− S.D. for three biological replicates).
A previous study showed that PARN knockdown led to an increase in an adenylated form of miR-21-5p in human cells (Boele et al., 2014), which led to the interpretation that PARN might degrade miR-21-5p. In contrast, we observed a reduction in miR-21-5p upon PARN knockdown in our sequencing data, which we verified by northern blots (Figure 1B). This suggests that PARN actually stabilizes miR-21-5p by deadenylating an adenylated intermediate which would otherwise be degraded by 3’ to 5’ exonucleases (see below).
PAPD5 destabilizes miRNAs in the absence of PARN
Previous studies on PARN-mediated stabilization of other ncRNAs suggests that PARN removes oligo(A) tails added by PAPD5 from the 3’ end of its substrates, which would otherwise lead to the degradation of the ncRNA by a competing 3’ to 5’ exonuclease recruited by the oligo(A) tail (Berndt et al., 2012; Nguyen et al., 2015; Shukla and Parker, 2017; Shukla et al., 2016; Tseng et al., 2015). We therefore asked whether PAPD5 co-knockdown rescues the reduction in miRNA levels upon PARN knockdown.
We found that PAPD5 co-knockdown was sufficient to both rescue the reduced levels of miR-21-5p, miR-181b-5p, miR-92b-3p and let-7e-5p, and the increased decay rates in PARN knockdown cells (Fig. 2A, B and C, Fig. S1). Similarly, in a patient fibroblast cell line with loss-of-function PARN mutation, we observed that PAPD5 knockdown leads to a 2-fold increase in the levels of miR-21-5p (Fig. 2D). Moreover, we also found that PAPD5 knockdown by itself led to an average 2.5-fold increase in the levels of miR-181b-5p and miR-21-5p (Fig. 2E).
These observations suggest that PAPD5-mediated 3’ end adenylation of miRNAs modulates miRNA stability and in the absence of PARN recruits 3’ to 5’ exonucleases to degrade miRNAs.
DIS3L and DIS3L2 are 3’-to’5’ exonucleases that modulate miRNA stability
To identify the enzymes that degrade adenylated miRNAs, we focused on the two predominant cytoplasmic 3’ to 5’ exonucleases, DIS3L and DIS3L2, which degrade a variety of ncRNA substrates in human cells (Zinder and Lima, 2017). DIS3L2 has also been shown to degrade uridylated pre-miRNA in human cells, although whether it can also target adenylated miRNAs has not been examined (Chang et al., 2013; Ustianenko et al., 2013).
We found that DIS3L knockdown led to an eleven-fold increase in the levels of miR-1, a four-fold increase in the levels of miR-181b-5p, but only minor effects on miR-380-5p (1.1×) or miR-21-5p (1.4×) (Fig. 3A). In contrast, DIS3L2 knockdown increased miR-21-5p (4×), miR-181b-5p (3×), miR-380-5p (4.5×), but had no effect on miR-1 (Fig. 3A). This demonstrates that both DIS3L and DIS3L2 modulate miRNA levels in a miRNA-specific manner.
Figure 3.
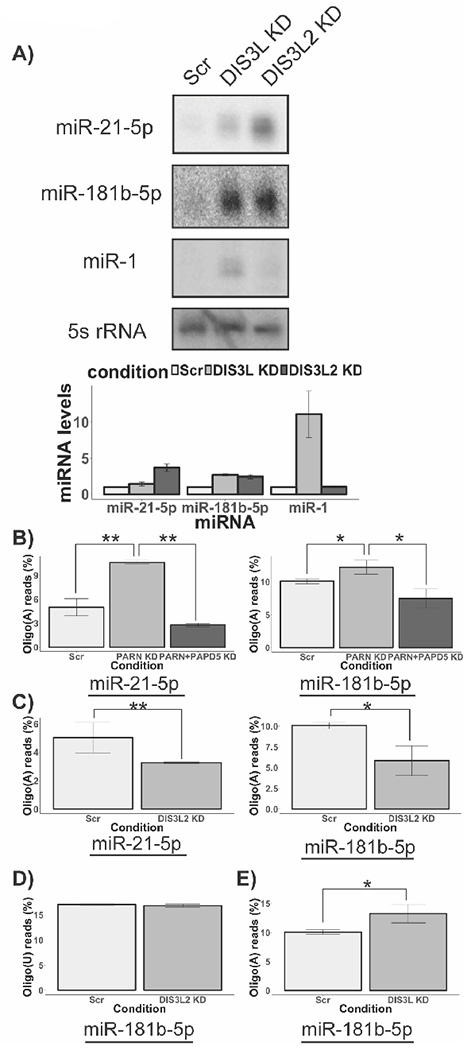
DIS3L and DIS3L2 are 3’ to 5’ exonucleases involved in miRNA processing and degradation. A) Representative northern blots for miRNA levels in DIS3L and DIS3L2 knockdown cells. Histogram shows miRNA levels in the indicated knockdown normalized to 5s rRNA (Average +/− S.D. for four biological replicates). B) Histogram depicting proportion of oligoadenylated species for miR-21–5p and miR-181b-5p respectively in PARN knockdown and PARN and PAPD5 co-knockdown cells for two biological replicates (Average +/− S.D.). Z scores were calculated using two proportion Z-test and converted to P values. (*** P<0.001). C) Histogram depicting proportion of oligoadenylated species for miR-21–5p and miR-181b-5p respectively in control knockdown and DIS3L2 knockdown cells for two biological replicates (Average +/− S.D.). Z scores were calculated using two proportion Z-test and converted to P values. (*** P<0.001). D) Histogram depicting proportion of oligouridylated species for miR-21– 5p and miR-181b-5p respectively in control knockdown and DIS3L2 knockdown cells for two biological replicates (Average +/− S.D.). E) Histogram depicting proportion of oligoadenylated species for miR-181b-5p in control and DIS3L knockdown cells for two biological replicates (Average +/− S.D.). Z scores were calculated using two proportion Z-test and converted to P values. (*** P<0.001).
To investigate whether DIS3L or DIS3L2 regulate the stability of miRNAs globally in human cells, we sequenced miRNA libraries from DIS3L and DIS3L2 knockdown cells. We found that DIS3L knockdown led to global changes in miRNA levels; out of 746 miRNAs, 129 miRNAs were upregulated more than 1.5-fold in DIS3L knockdown cells, and 185 miRNAs were downregulated more than 0.7-fold in DIS3L knockdown cells compared to control cells (Fig. S2 and Data S1). Similarly, DIS3L2 knockdown also led to global changes in miRNA population in HeLa cells; out of 598 miRNAs, 106 miRNAs were upregulated more than 1.5-fold, and 194 miRNAs were downregulated more than 0.7-fold in DIS3L2 knockdown cells (Fig. S3 and Data S1). These observations suggest that DIS3L and DIS3L2 affect the stability of many human miRNAs.
PARN deadenylates miRNAs to protect them from PAPD5-mediated adenylation and degradation by DIS3L or DIS3L2
Our analysis of miRNA steady state levels in PARN knockdown and PARN and PAPD5 co-knockdown cells suggests that for a subset of miRNAs, 3’ end adenylation is controlled by the activities of PARN and PAPD5. To assess the effect of PARN and PAPD5 activity on miRNA 3’ ends directly, we sequenced the 3’ end of miR-21-5p and miR-181b-5p in control, PARN knockdown and PARN and PAPD5 co-knockdown cells. In control cells, 5% of the reads represented adenylated miR-21-5p species at the canonical 3’ end (Fig. 3B and Data S2). In PARN knockdown cells, we observed a two-fold increase in the levels of adenylated ends, demonstrating PARN deadenylates miR-21-5p (Fig. 3B). Finally, PARN and PAPD5 co-knockdown led to a strong decrease in the levels of adenylated reads at the 3’ end of miR-21-5p compared to control cells (Fig. 3B). We observed a similar trend for the 3’ end of miR-181b-5p. In PARN knockdown cells, we observed an increase in the proportion of adenylated reads compared to control cells (Fig. 3B and Data S3). Conversely, in PARN and PAPD5 co-knockdown cells, the percentage of adenylated reads decreased two-fold compared to control cells (Fig. 3B). This provides evidence for PAPD5-mediated oligoadenylation of miRNA 3’ ends and argues that PARN removes oligo(A) tails from the 3’ ends of miRNAs, thereby protecting them from degradation.
Since DIS3L2 knockdown leads to an increase in the steady state levels of several miRNAs (Fig. 3A), we sequenced the 3’ ends of miR-21-5p and miR-181b-5p in DIS3L2 knockdown cells to determine how DIS3L2 affects their 3’ ends. In contrast to previous data that showed that DIS3L2 degrades uridylated let-7 miRNA precursors (Chang et al., 2013; Ustianenko et al., 2013), we found that DIS3L2 knockdown did not lead to an increase in oligo(U) tails at the 3’ end of miR-21-5p; in fact, we were unable to detect any uridylated tails at the 3’ end of miR-21-5p in either control or DIS3L2 knockdown cells. Similarly, the fraction of uridylated miR-181b-5p species remains unchanged between control and DIS3L2 knockdown cells, suggesting that DIS3L2’s activity on miRNAs is not restricted to uridylated ends (see below).
We observed that DIS3L2 knockdown led to a 2× decrease in the fraction of oligo(A) tails at the 3’ end of miR-21-5p and miR-181b-5p (Fig. 3C). In contrast, the fraction of oligo(U) tails at the 3’ end of miR-181b-5p remains unchanged (Fig. 3D). The reduction in oligoadenylation of miRNA 3’ ends upon DIS3L2 knockdown suggests that DIS3L2 actually impedes PARN from removing oligo(A) tails from the 3’ ends of substrate miRNAs. One possibility is that DIS3L2 outcompetes PARN and ‘commits’ the adenylated miRNA species for degradation (Fig. S4). In the absence of DIS3L2, PARN can deadenylate the 3’ ends of miRNAs, which leads to a reduction in the fraction of oligoadenylated species for miRNAs in DIS3L2 depleted cells.
miR-181b-5p levels, but not miR-21-5p levels, were affected by the activity of DIS3L. We found that DIS3L knockdown led to a 1.3-fold increase in the fraction of oligo(A) reads at the 3’ end of miR-181b-5p (Fig. 3E). Since DIS3L prefers oligoadenylated substrates for its activity (see below), this observation suggests that DIS3L recognizes oligo(A) tails at the 3’ end of miR-181b-5p and degrades the miRNA through this 3’ to 5’ degradation pathway. These results demonstrate that PARN, DIS3L and DIS3L2 affect the stability of subsets of miRNAs in human cells, although the basis for miRNA-specific effects remains to be established.
PARN, DIS3L and DIS3L2 act on miRNA substrates in vitro
The above results suggest that PARN, DIS3L and DIS3L2 can remove oligo(A) tails from miRNAs, which would be consistent with the known poly(A) specificity of PARN. To directly determine if these enzymes can act on adenylated miRNAs, we immunopurified each nuclease from HeLa cells and examined their activity on 5’-radiolabeled miR-21-5p without any tails (N), a 3’ tail of six adenosines (oA) or 6 uridines (oU). Consistent with PARN preferring poly(A) or oligo(A) tails, we observed that immunopurified PARN protein from wild type cells, but not PARN knockdown cells, preferentially deadenylated oA-miR-21, with reduced activity on oU-miR-21 (Fig. 4A and B). This demonstrates that PARN can deadenylate oligo(A) miRNAs, consistent with our in vivo results.
Figure 4.
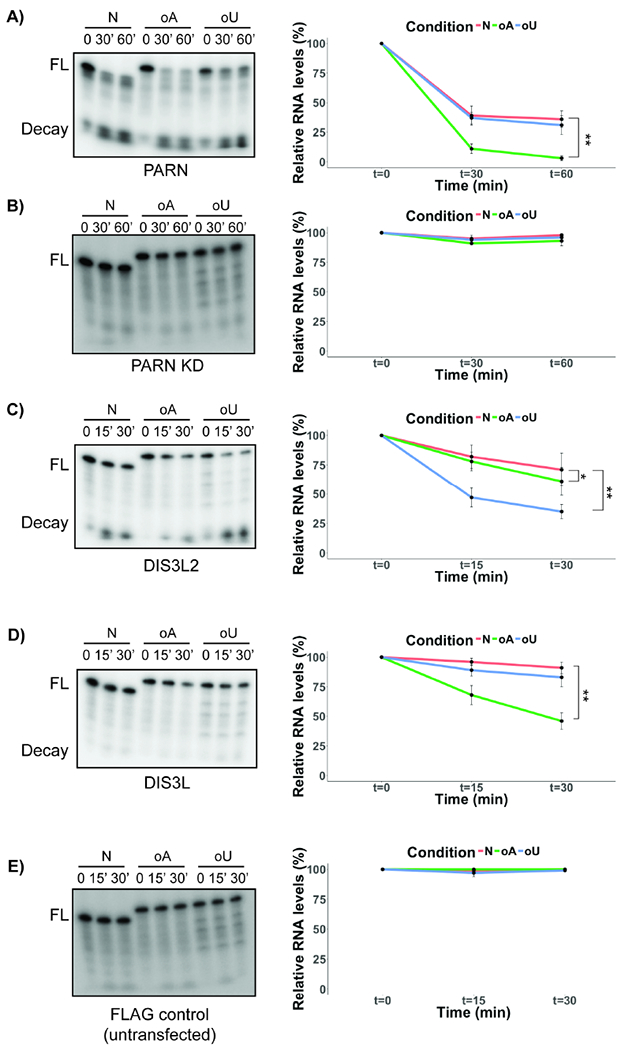
PARN, DIS3L2 and DIS3L degrade various miR-21-5p species in vitro with different affinities. In vitro degradation assay for miR-21-5p incubated with immunopurified PARN from A) wild type or B) PARN knockdown cells for indicated time points. FL: Full-length miRNA; Decay: Decay product of the reaction. C), D) and E) In vitro degradation assay for miR-21–5p incubated with immunopurified FLAG-DIS3L2, FLAG-DIS3L and FLAG control respectively for indicated time points. FL: Full-length miRNA; Decay: Decay product of the reaction (Line plot in all cases indicates quantification of average +/− S.D. for three biological replicates). *P<0.05, **P<0.01, two-tailed unpaired Student’s T-test.
In contrast, immunoprecipitated FLAG-DIS3L2 preferentially degrades o(U)-miR-21, but also has some activity on oA-miR-21 (Fig. 4C). This suggests that while oligo(U) modified miRNA is the preferred substrate for DIS3L2, it can also act on oligoadenylated miRNAs, albeit with reduced activity. Finally, immunoprecipitated FLAG-DIS3L showed activity on oA-miR-21, but little activity on unmodified or oU-miR-21 (Fig. 4D). As a control, immunoprecipitation with a FLAG antibody from untransfected cells showed no activity on any substrate (Fig. 4E). The ability of immunopurified DIS3L to deadenylate o(A)-miR-21-5p in vitro is consistent with the increase in oligoadenylated miRNA species seen in vivo with DIS3L knockdown (Fig. 3E), and provides evidence that DIS3L can deadenylate and possibly degrade adenylated miRNAs. Together, these results indicate that DIS3L and DIS3L2 have the ability to deadenylate miRNAs in vitro.
PARN-regulated miRNAs modulate the p53 signaling pathway in human cells
To determine a biological role of PARN mediated regulation of miRNAs, we examined whether the miRNAs reduced in PARN knockdown cells targeted distinct biological pathways in the cell. KEGG analysis identified several pathways affected by miRNAs altered negatively in PARN knockdown cells, most notably the p53 signaling pathway (Fig. S5). Several miRNAs downregulated upon PARN knockdown, such as miR-380-5p, miR-1285, miR-92, miR-214, miR-485, miR-331, miR-665, miR-3126 and miR-25, have either been shown, or are predicted, to target the TP53 mRNA (Agarwal et al., 2015; Liu et al., 2016; Swarbrick et al., 2010; Vlachos et al., 2015), which codes for the tumor suppressor protein p53 (Fig. 5A). Similarly, other miRNAs such as miR-660 and miR-32, which repress p53 inhibitors such as MDM2, were upregulated in PARN knockdown cells (Fig. 5A). These observations suggest that PARN-mediated regulation of miRNA levels might regulate p53 protein levels in human cells.
Figure 5.
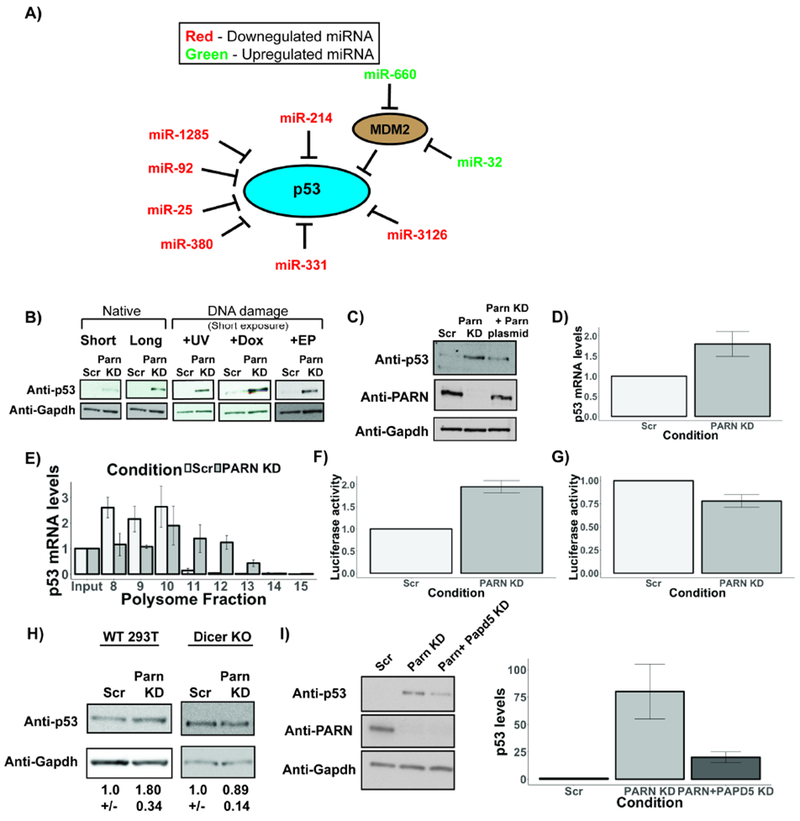
PARN regulates a miRNA circuit that controls p53 levels in human cells. A) miRNAs that affect p53 signaling pathway are affected in PARN knockdown cells. miRNAs in red are downregulated in PARN knockdown cells, while miRNAs in green are upregulated in PARN knockdown cells. B) Representative western blots depicting p53 protein levels in PARN knockdown cells under various conditions. C) Representative western blots depicting p53 and PARN protein levels in PARN knockdown and rescued cells. D) p53 mRNAs levels as measured using RT-qPCR in PARN knockdown cells (Average +/− S.D. for three biological replicates). E) p53 mRNA levels in various polysome fractions measured using qRT-PCR and normalized to input (Average +/− S.D. for two biological replicates). F) and G) Luciferase activity of fluc-p53 fusion construct or fluc alone respectively in control and PARN knockdown cells. Firefly luciferase activity was normalized to renilla luciferase internal control. Luciferase activity in control cells was set to 1 (Average +/− S.D. for four biological replicates). H) Representative western blots depicting p53 protein levels in 293T wild type and Dicer knockout cells. (Average +/− S.D. for three biological replicates). I) Representative western blots depicting p53 levels in control, PARN knockdown and PARN & PAPD5 co-knockdown HeLa cells. Bar graph depicts average +/− S.D. for five biological replicates.
Strikingly, we found that p53 levels were upregulated >20-fold in PARN knockdown cells by western blotting (Fig. 5B). Consistent with this observation, PARN knockdown led to an increase in the proportion of cells in the G0/G1 cell cycle phase compared to control cells, which is consistent with p53’s role in causing cell cycle arrest in G1 phase (Agarwal et al., 1995; Di Leonardo et al., 1994) (Fig. S6A). Moreover, p53 upregulation upon exposure to DNA damage using UV (25 J/m2), Doxorubicin (Dox) or Etoposide (EP) treatment was increased in PARN knockdown cells (~12-fold upon UV, ~16-fold upon Dox, and ~15-fold upon EP) compared to control (Fig. 5B). Re-introduction of PARN through a plasmid in PARN knockdown cells rescued p53 levels to ~70% of PARN knockdown cells, demonstrating that p53 increase is directly dependent on loss of PARN in cells (Fig. 5C).
Five observations argue that PARN knockdown increases p53 levels by relieving miRNA mediated translation repression of the p53 mRNA. First, while p53 mRNA levels increased ~1.9 fold in PARN knockdown HeLa cells (Zhang and Yan, 2015) (Fig. 5D), p53 protein levels increased >20-fold, which is consistent with enhanced p53 mRNA translation (Fig. 5B and S6B). Moreover, upregulation of p53 was not caused by a decrease in the HPV E6 oncoprotein, known to target p53 for degradation in HeLa cells (Fig. S6G) (Scheffner et al., 1990). Second, in HeLa cells we observed that PARN knockdown caused the endogenous p53 mRNA to move deeper into polysomes, consistent with increased translation (Figure 5E and S6D). Third, PARN knockdown leads to increased luciferase expression from a reporter firefly luciferase mRNA with the p53 3’ UTR (Chen and Kastan, 2010), while an unrelated 3’ UTR shows decreased luciferase expression (Figure 5 F and G). Fourth, while PARN knockdown increased p53 levels in Hek293T cells (Figure 5H), Hek293T cells lacking Dicer (Bogerd et al., 2014) no longer showed an increase in p53 levels (Figure 5H). It should be noted that Hek293T cells already accumulate high amounts of the p53 protein due to the formation of the LTag-p53 complex (Deppert et al., 1989; Lilyestrom et al., 2006), and this explains why we see a smaller effect of PARN knockdown on p53 levels. Fifth, PAPD5 co-knockdown, which rescues miRNA levels upon PARN depletion, also rescued p53 levels compared to PARN knockdown with or without DNA damaging agents (Fig. 5I and S6C).
These observations argue that PARN knockdown increases p53 levels by relieving miRNA mediated translation repression. This provides a molecular explanation for why DC patient cells containing LOF mutations in PARN exhibit p53 upregulation and cell cycle abnormalities (Dhanraj et al., 2015; Tummala et al., 2015). The increase in p53 levels may contribute to the severe phenotype of the disease in these patients.
PARN depletion sensitizes cancer cells to chemotherapeutic agents
A large number of human cancers downregulate the p53 pathway for increased proliferation and resistance to DNA damaging agents (Bieging et al., 2014; Joerger and Fersht, 2016). Therefore, upregulation of p53 levels by PARN depletion might make cells more sensitive to chemotherapeutic agents such as Doxorubicin or Etoposide, which both upregulated p53 levels in PARN knockdown HeLa cells (Fig. 5B). We found that PARN knockdown led to a strong reduction in cell viability upon Doxorubicin treatment compared to control cells (Fig. 6A and B). Similarly, Etoposide treatment also led to a reduction in HeLa cell viability upon PARN knockdown compared to control cells (Fig. 6C).
Figure 6.
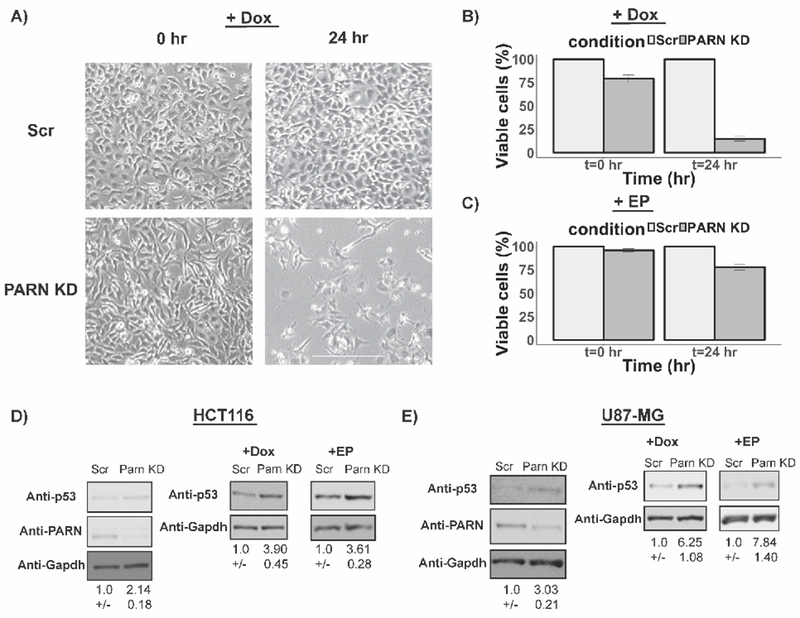
PARN knockdown increases sensitivity of HeLa cells to chemotherapeutic agents. A) Representative bright field images of control and PARN knockdown HeLa cells at indicated time points after Doxorubicin treatment (Scale bar: 300 μm). B) and C) Viable cells under various knockdown and treatment conditions as measured using trypan blue staining assay (Average +/− S.D for three biological replicates). D) Representative western blots for p53 and PARN protein levels in HCT116 cells with or without Dox or EP treatment (Average +/− S.D. for four biological replicates). E) Representative western blots for p53 and PARN protein levels in U87 cells with or without Dox or EP treatment (Average +/− S.D. for five biological replicates for Dox treatment and three biological replicates for EP treatment).
To determine the generality of these effects, we examined PARN depletion in the colon cancer cell line HCT116, and a glioblastoma cell line U87, both of which contain mutations in other oncogenes that leads to repression of wild type p53 levels. In both HCT116 and U87 cell lines PARN depletion led to a two to three-fold increase in the levels of p53 protein (Fig. 6D and E), which was further increased by treatment with Dox or EP (Fig. 6D and E). These results indicate that selective PARN depletion increases sensitivity of cancer cells to chemotherapeutic drugs such as Doxorubicin and Etoposide, and PARN could be a therapeutic target to reduce the proliferation of tumors that express a reduced level of functional p53 protein.
Discussion
PARN modulates miRNA stability in human cells
Herein we describe a pathway of miRNA regulation where PAPD5-mediated adenylation competes with PARN-mediated deadenylation to limit the levels of a subset of miRNAs in human cells. Several observations suggest that PARN regulates the stability of miRNAs in human cells. First, PARN knockdown leads to a change in levels of many human miRNAs both positively and negatively in human cells (Fig. 1A), including decreased levels of miR-21-5p, miR-181b-5p and miR-92b-3p (Fig. 1B and C, S6E). Second, PARN knockdown leads to a decrease in miRNA stability following transcription shutoff (Fig. 2A and B). Third, PARN knockdown leads to an increase in the oligoadenylation of miR-21-5p and miR-181b-5p, which suggests that PARN plays a role in deadenylating miRNAs (Fig. 3B). These observations suggest that PARN can act on some miRNA substrates in human cells.
We provide evidence for the role of PARN in protecting miR-21-5p from degradation. A previous study suggested that PARN plays a role in trimming miR-21-5p leading to its degradation, since PARN depletion led to an increase in the adenylated form of miR-21-5p (Boele et al., 2014), which we also observed (Fig. 3B). However, we suggest that this adenylated miR-21-5p is a degradation intermediate, which is consistent with the observation that PARN knockdown leads to a ~45% decrease in miR-21-5p levels without affecting the pre-miR-21 (Fig. 1B). Since miR-21 overexpression is a hallmark of many cancers, the regulation of miR-21 stability by PARN may represent a pathway for modulation of miR-21 levels in human disease (Pfeffer et al., 2015).
PAPD5-mediated adenylation and PARN-mediated deadenylation modulate miRNA levels in mammalian cells
Our data suggest that PAPD5-mediated 3’ end adenylation is a key event in controlling the stability of several miRNAs in human cells. PARN-mediated deadenylation competes with PAPD5 to limit the availability of miRNAs for degradation. The levels of miRNAs that decrease upon PARN knockdown can be rescued by a co-knockdown of PAPD5 (Fig. 2C and S6F). Similarly, PAPD5 co-knockdown rescues the loss in stability of miRNAs affected by PARN knockdown (Fig. 2A and B). Moreover, PAPD5 co-knockdown also reduces the adenylation of miR-21-5p and miR-181b-5p in PARN knockdown cells (Fig. 3B). These observations suggest that PARN and PAPD5 compete to limit the effect of 3’ end adenylation on miRNA stability. This competition can be regulated by the availability of PARN or PAPD5, thereby serving as a tunable switch for miRNA levels and function in the cell. Furthermore, the role of PARN in limiting PAPD5-mediated miRNA degradation expands the repertoire of cellular non-coding RNAs that are stabilized by PARN in human cells.
DIS3L and DIS3L2 are exonucleases responsible for the degradation of adenylated miRNAs in human cells
We identified a role for the 3’ to 5’ exonucleases DIS3L and DIS3L2 in degrading adenylated miRNAs in human cells. A knockdown of DIS3L led to an increase in the levels of miR-1 and miR-181b-5p (Fig. 3A). Similarly, DIS3L2 knockdown led to an increase in the levels of miR-21-5p and miR-181b-5p (Fig. 3A). 3’ end deep sequencing of DIS3L2 substrates revealed a mechanism where DIS3L2 competes with PARN and restricts its ability to deadenylate miRNAs and protect them from degradation by DIS3L2 (Fig. 3C and S4). Further, the activity of DIS3L2 is not restricted to uridylated miRNAs as revealed by the absence of U resides on the 3’ end of miR-21-5p and unchanged oligo(U) tails on miR-181b-5p (Fig. 3D). In vitro analysis of DIS3L2’s degradation activity on various miR-21-5p substrates revealed that DIS3L2 has a higher affinity for uridylated miR-21-5p, but can also degrade adenylated miR-21-5p substrates (Fig. 4C). Together, this data suggests that DIS3L2 can process and degrade adenylated miRNAs in a miRNA-specific manner.
DIS3L’s preference for adenylated substrates explains its activity on adenylated miRNA 3’ ends, and DIS3L knockdown led to an increase in the levels of adenylated miR-181b-5p due to a block in deadenylation and degradation (Fig. 3E). Further, DIS3L only shows activity on adenylated miR-21-5p in vitro, suggesting that DIS3L is selective in its affinity for adenylated miRNA substrates for degradation (Fig. 4D). These observations provide a framework for the regulation of subsets of miRNAs by conserved exonucleases in mammalian cells.
Specific miRNAs are regulated by a network comprising of the enzymes PARN, PAPD5, DIS3L and DIS3L2
Our data suggests that not all miRNAs are sensitive to the activity of PARN, DIS3L or DIS3L2; rather, specific subsets of miRNAs are substrates for these enzymes. Several factors could play a role in modulating the sensitivity of specific miRNAs to adenylation and degradation (Fig. 7). First, specific co-factors could recruit different enzymes to modify the 3’ end of miRNAs, as is the case for let-7 and LIN28 (Hagan et al., 2009; Heo et al., 2009). Second, the 3’ end of miRNAs could be exposed through its binding to target mRNAs. It has previously been shown that perfect complementarity between miRNAs and mRNAs leads to 3’ end non-templated addition and degradation of these miRNAs (Ameres et al., 2010; Cazalla et al., 2010). Therefore, expression of target mRNAs with appropriate miRNA base-pairing sites will target miRNAs to this pathway of adenylation and degradation. Third, miRNA release from the RISC complex might make specific miRNAs sensitive to 3’ end modification, although to our knowledge there is currently no evidence for this mechanism. It also remains to be determined why individual miRNAs are differentially sensitive to DIS3L and DIS3L2.
Figure 7.
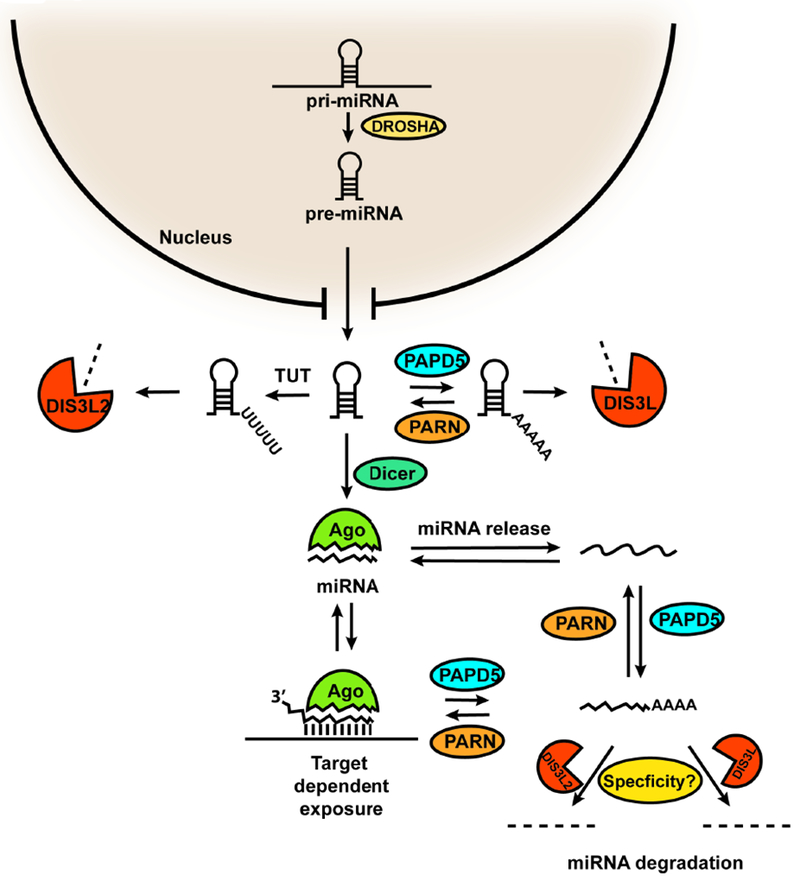
Model for the regulation of miRNA stability by 3’ end adenylation and deadenylation by PARN and PAPD5, and degradation by DIS3L or DIS3L2. pri-miRNAs are processed to pre-miRNAs by Drosha, which are exported to the cytoplasm and matured by Dicer. The pre-miRNA could be subjected to adenylation and deadenylation at the 3’ end by the competing activities of PARN and PAPD5 in the cytoplasm leading to degradation by DIS3L. Pre-miRNA is also uridylated and degraded by the activities of TUT/DIS3L2. Mature miRNA could be targeted to adenylation by PAPD5 either through release from RISC or due to binding to complementary target mRNAs. PARN limits the adenylation of mature miRNA strands, and in PARN deficient cells, DIS3L or DIS3L2 can degrade specific miRNAs.
PARN deficiency causes misregulation of p53 signaling via a miRNA pathway
The effect of PARN on miRNAs explains why PARN mutations in DC patients leads to the upregulation of p53 levels in patient cells (Tummala et al., 2015). We show that several miRNAs that are downregulated in PARN knockdown cells target the TP53 mRNA which codes for the p53 protein (Fig. 5A). Further, we show that PARN knockdown leads to significant upregulation of the p53 protein in HeLa cells, which normally contain very low levels of the p53 protein due to the HPV E6 oncoprotein-mediated degradation of p53 (Fig. 5B). Moreover, the effect on p53 protein levels is without corresponding changes in p53 mRNAs levels, which argues that PARN deficiency affects the translational regulation of the p53 mRNA by miRNAs that are downregulated in PARN knockdown cells (Fig. 5D). PARN knockdown shifts p53 mRNA deeper in polysome fractions compared to control cells (Fig. 5E). Further, PARN knockdown leads to an increase in luciferase activity when fused to the 3’ UTR of p53, but not luciferase alone (Fig. 5F and G). Previous studies have suggested that PARN might regulate p53 mRNA levels via CUGBP1 and miR-125b/miR-504, as well as a negative feedback loop during DNA damage (Devany et al., 2013; Zhang et al., 2015). However, miR-125b levels were not affected by PARN knockdown, and miR-504 was absent in our libraries. The fact the p53 protein was upregulated in wild-type Hek293T cells, but not Dicer knockout HEK293T cells, further suggests that loss of miRNA-mediated regulation of p53 translation leads to p53 upregulation in PARN knockdown cells (Fig. 5H). Further, PARN knockdown also leads to an increase in p53 levels in HCT116 and U87 cell lines, suggesting that PARN-mediated regulation of p53 levels is a general mechanism for controlling p53 signaling (Fig. 6D and E).
Since miRNA levels in PARN knockdown cells are rescued by co-knockdown of PAPD5, we hypothesized that PAPD5 co-knockdown should also rescue p53 induction in PARN knockdown cells. We show that PAPD5 co-knockdown rescues p53 protein levels in PARN knockdown cells with or without DNA damage (Fig. 5I and S6C). This suggests that not only can PARN modulation be used to induce the expression of p53 protein, but also that PAPD5 modulation can be used to reduce the accumulation of p53 in PARN deficient cells. We and others have previously shown that PAPD5 knockdown rescues telomerase RNA levels and function in PARN deficient cells (Boyraz et al., 2016; Shukla et al., 2016; Tseng et al., 2015). We propose that PAPD5 depletion should also rescue p53 accumulation observed in DC patients, thus making PAPD5 an attractive target for DC therapy, of which there are no treatment options currently available apart from bone marrow transplants.
PARN-mediated p53 regulation plays a role in growth and proliferation of some cancers
The effect of PARN and PAPD5 on p53 regulation suggests that genetic changes in the expression of these genes should alter tumor progression in humans by either increasing or decreasing the expression of p53. PARN overexpression, which would increase miRNA-mediated repression of p53, occurs in acute leukemia and squamous cell lung cancer (Maragozidis et al., 2015; 2012). As expected, PARN overexpression is associated with poor prognosis of human cancers according to the Human Protein Atlas Project analysis of available human tumors in the repository (Uhlén et al., 2015; 2017). Conversely, PAPD5 overexpression, which increases the degradation of p53 repressing miRNAs leading to the induction of p53, is associated with improved prognosis of human cancers (Uhlén et al., 2017). The effect of PARN and PAPD5 expression on human cancer prognosis suggests that the miRNA-mediated p53 pathway can be targeted to reduce the growth of human cancers.
STAR Methods
Cell culture
HeLa, U87 and HCT116 cells were purchased from ATCC and verified for mycoplasma contamination. Hek293T wild-type and Dicer knockout cells were a kind gift from Prof. Christopher Sullivan’s lab at University of Texas at Austin. TERT-immortalized PARN patient fibroblast were a kind gift from Prof. Suneet Agarwal’s lab at Boston Children’s hospital. HeLa and Hek293T cells were cultured in DMEM containing 10% FBS, 1% Pen/Strep, 1× Glutamax and Normocin at 37°C under ambient conditions. HCT116 cells were cultured in McCoy’s 5A medium containing 10% FBS, 1% Pen/Strep, 1× Glutamax and Normocin at 37°C under ambient conditions. U87 cells were cultured in Eagle’s Modified Essential Medium Cells containing 10% FBS, 1% Pen/Strep, 1× Glutamax and Normocin 37°C under ambient conditions. Patient fib roblasts were cultured in Eagle’s Modified Essential Medium Cells containing 15% FBS, 1% Pen/Strep, 1× Glutamax and Normocin 37°C under ambient conditions. Cells were sub-cultured upon reaching 80% confluency.
RNA interference in cells
HeLa, Hek293T, U87 and HCT116 cells were seeded ~100,000 cells/well in a six-well plate 24 hours before transfection. Patient fibroblast were seeded at ~50,000 cells/well in a six well plate. siRNA transfection was performed using Interferin (Polyplus) to a final concentration of 5 nM per well as per manufacturer’s protocol. 72 hours after transfection, cells were collected for either RNA or protein analysis.
Transcription shutoff in HeLa cells
72 hours after siRNA transfection as described above, cells were treated with Actinomycin D (Sigma Aldrich) at 5 μg/ml. Cells were harvested at 0, 4 and 7 hours after Act D treatment for RNA analysis.
PARN plasmid co-transfection in HeLa cells
siRNA transfection of PARN siRNA was repeated as above. 24 hours after siRNA transfection, 1 μg of a GFP or PARN plasmid was transfected using JetPrime (Polyplus) as per manufacturer’s protocol. Cells were harvested for protein analysis 48 hours after plasmid transfection.
siRNAs and plasmid
siRNAs targeting PARN (siGenome), PAPD5 (On-Target plus), DIS3L (On-Target plus) and DIS3L2 (On-Target plus) were purchased from Dharmacon in the Smartpool formulation. Secondary siRNA for PARN and PAPD5 were purchased from Dharmacon in individual and Smartpool formulation respectively. All-stars negative control siRNA from Qiagen was used as negative control. PARN plasmid was a kind gift from Prof. Yukihide Tomari’s lab at University of Tokyo, Japan (Katoh et al., 2015). FLAG-DIS3L and FLAG-DIS3L2 plasmids were a kind gift from Prof. Stepanka Vanacova at Masaryk University, Czech Republic. p53-luciferase plasmid was a kind gift from Prof. Michael Kastan at Duke University.
RNA extraction and northern blotting
Total RNA was extracted from cell lysates using Quick RNA mini-prep kit from Zymo Research as per manufacturer’s protocol. After quantification on Nanodrop, 10 μg of total RNA was separated on a 10% acrylamide 7 M Urea gel. RNA was transferred to a nylon membrane (Nytran SPC, GE Healthcare) using wet transfer at 4°3. After UV crosslinking, the blot was pre-hybridized and hybridized in PerfectHyb Plus Hybridization Buffer (Sigma Aldrich) at 42°C. miRNA LNA probes for each target miRNA were purchased from Exiqon. Probe against 5s rRNA has been described previously (Shukla et al., 2016). After hybridization and washing in 2x SSC 0.1% SDS wash buffer, blots were exposed to a cassette and imaged on a Typhoon FLA 9500 Phosphoimager. Band intensities were quantified using ImageQuant TL.
RT-qPCR for mRNA and miRNA
For quantification of PARN or DIS3L2 mRNA levels, 5 μg of total RNA was used for first strand cDNA synthesis using RNA-to-cDNA Ecodry premix (Double primed) from Takara Biotech. 1:5 dilution of cDNA was used for RT-qPCR using iQ SYBR Green Supermix (Bio-Rad) and primer pairs using the manufacturer’s protocol on the Bio-Rad CFX96 Real-Time PCR system. For quantification of indicated precursor or mature miRNAs, 1 μg of total RNA was used for cDNA preparation using miScript II RT Kit from Qiagen. 1:5 dilution of cDNA was used for RT-qPCR using the miScript SYBR Green PCR Kit from Qiagen as per manufacturer’s protocol on a Bio-Rad CFX96 Real-Time PCR system. 5s rRNA was used as a loading control in all quantifications for normalization and fold-change calculation using the 2^(−ΔΔCt) method.
Western blotting
20 μg of total protein was separated on a 4%-12% Bis-Tris NuPage gel (ThermoFisher) and transferred to protran membrane (Amersham). After blocking in 5% non-fat milk in 1×TBST, blots were probed with appropriate primary and secondary antibodies for one hour each. Antibodies used in this study are described in Key Resources table.
DNA damage and imaging
Doxorubicin and Etoposide were purchased from Sigma Aldrich. For DNA damage treatment, Dox or EP was added to a final concentration of 1 μM or 10 μM respectively, 48 hours after siRNA transfection. Chemical treatment was allowed to take place for 24 hours, after which cells were either harvested for protein analysis, or imaged on an EVOS FL cell imaging system. For cell viability measurement, equal number of cells were stained with trypan blue, and viable cells were counted on a hemocytometer.
Luciferase activity assay
Cells transfected with control or PARN siRNA were transfected with 1 μg of plasmid mix containing either 0.8 μg of p53-Fluc and 0.2 μg of Rluc plasmids, or 0.8 μg of Fluc and 0.2 μg of Rluc plasmids 48 hours after siRNA transfection. After 24 hours, cells were lysed in 1× passive lysis buffer as per manufacturer’s protocol (Dual-Glo Luciferase activity system by Promega). Equal amount of cell lysate was used for the measurement of luciferase activities on a Glomax Multi+ Detection System (Promega).
Cell cycle analysis of HeLa cells
HeLa cells were seeded in a six-well plate and siRNA transfections were performed as described above. Cells were gently washed once in 1×PBS, making sure that no cells were discarded during media removal or washing. After trypsinization, cells were fixed in 66% cold ethanol at 4°C for at least two hours. Propidium Iodide staining of the cell suspension was performed as per manufacturer’s protocol for the Propidium Iodide Flow Cytometry Kit from Abcam (ab139418). After staining, stained particles were counted on a BD Accuri C6 Plus flow cytometer. Percentage of cells in each cell cycle phase was calculated using volume of respective peaks (2N: G0/G1; 2N-4N: S; >4N: G2/M).
In vitro degradation assays of miR-21-5p substrates
PARN was immunopurified from ~2 million HeLa cells (control or PARN knockdown) using the PARN antibody (ab27778) pre-conjugated to Protein G plus agarose beads (Pierce). FLAG-DIS3L and FLAG-DIS3L2 were immunopurified from HeLa cells transfected with 5 μg of respective plasmids using Anti-FLAG M2 agarose affinity gel (Sigma Aldrich). miR-21-5p substrates were radiolabeled with 32P at 5’ end using T4 PNK (NEB). Degradation assays were performed as previously described (Ustianenko et al., 2013). Reactions were stopped at indicated time points by mixing with equal volume of stop buffer (95% formamide, 25 mM EDTA (pH 8.0), 0.03% bromophenol blue and xylene cyanol). Reactions were separated on 15% acrylamide 8M Urea gel in 1× TBE buffer and imaged on a Typhoon FLA 9500 phosphoimager. Loss of the full-length band was quantified for each time point using ImageQuant TL.
Polysome fractionation and RNA analysis
Cells were lysed at 4°C in lysis buffer containing 20 mM Tris-HCl pH 7.4, 5 mm MgCl2, 100 mM KCl, 0.5% NP-40, 1 mM DTT, 5 mM Heparin and protease inhibitor tablet without EDTA (Roche). Lysis was done by 10× passages through a 23g needle, spun 3 min at 16xg. The supernatent was collected and Triton-X and deoxycholate were added to 0.5% final concentration. 20 OD 260 units were loaded onto a 15-50% sucrose gradient in 20 mM HEPES 7.6, 100 mM KCl, 5 mM MgCl2, 1 mM DTT and spun in aSW40TI swinging bucket rotor for 2 hours at 36K RPM at 4°C. Gradients were collected on an ISCO gradient fractionator.
RNA was extracted from indicated fractions and one fraction equivalent input using TriZol (Ambion) as per manufacturer’s protocol. cDNA was prepared from 1 μg of RNA using the RNA-to-cDNA EcoDry premix (oligo dT) from Takara Biotech. p53 mRNA levels in each sample was quantified using RT-qPCR as described above. RNA levels were normalized to the input for control and PARN knockdown cells.
microRNA sequencing
1 μg of total RNA was used as input for library preparation using the NEXTflex Small RNA Library prep kit V3 for Illumina from Bioo Scientific. Libraries were sequenced on an Illumina Next Seq sequencer using the 1×150 cycle kit. Approximately 8 million unpaired reads were obtained for each library. After quality filtering and adapter trimming, including trimming of 4N bases from the 5’ and 3’ ends of reads, reads were mapped to the mature miRNA database (release 21) from miRbase using the blastn tool from NCBI Blast software. Best matches were selected according to the lowest E-value and counted using a custom python script. Abundance of miRNAs was calculated and normalized internally to reads per million. miRNA fold differences were plotted on R using the ggplot2 library.
3’ end sequencing of miRNAs
3’ end sequencing was performed using a previously described protocol (Goldfarb and Cech, 2013). Reverse primer for 3’ RACE was selected as the first 20 bases of the mature miR-21-5p or miR-181b-5p. Libraries were sequenced on an Illumina Next Seq sequencer using the 1×150 cycle kit. Approximately 3 million raw reads were obtained for each library. Reads of interest were selected using the search sequence corresponding to the miRNA and the 3’ appendix. Canonical 3’ end for miR-21-5p was defined as described previously (Boele et al., 2014). Canonical 3’ end for miR-181b-5p was defined as listed on miRbase. Statistics were performed using two-proportion Z-score test, and Z scores were converted to P values to identify statistically significant differences.
Supplementary Material
Data S1 (Related to Figures 1 and 3).
Fold change values for miRNA libraries in PARN KD, DIS3L KD and DIS3L2 KD HeLa cells compared to control.
Data S2 (Related to Figure 3).
Raw counts corresponding to each 3’ end for miR-21-5p in Scr, PARN KD, PARN and PAPD5 KD and DIS3L2 KD HeLa cells.
Data S3 (Related to Figure 3).
Raw counts corresponding to each 3’ end for miR-181b-5p in Scr, PARN KD, PARN and PAPD5 KD, DIS3L KD and DIS3L2 KD HeLa cells.
Highlights.
PARN regulates the levels of selected miRNAs in cells by deadenylating 3’ ends.
DIS3L and DIS3L2 are critical 3’ to 5’ exonucleases that degrade adenylated miRNAs.
Some PARN-regulated miRNAs control p53 levels in human cells.
PARN inhibition induces p53 accumulation and decreases cancer cell viability.
Acknowledgements:
The authors thank members of the Parker lab for their suggestions and feedback, Anne Webb for help with figures, Jonathan Rubin for computational assistance, and BioFrontiers Sequencing and Cell Culture Cores for technical assistance. This work was supported by funds from NIH R01 GM45443 to R. P and HHMI. R.P. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: S.S. and R.P. are co-inventors on a provisional patent to develop PARN inhibitors for cancer therapy. Other authors declare no competing interests.
References
- Agarwal ML, Agarwal A, Taylor WR, and Stark GR (1995). p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proceedings of the National Academy of Sciences 92, 8493–8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal V, Bell GW, Nam J-W, and Bartel DP (2015). Predicting effective microRNA target sites in mammalian mRNAs. eLife 4, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameres SL, Horwich MD, Hung J-H, Xu J, Ghildiyal M, Weng Z, and Zamore PD (2010). Target RNA-directed trimming and tailing of small silencing RNAs. Science 328, 1534–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bail S, Swerdel M, Liu H, Jiao X, Goff LA, Hart RP, and Kiledjian M (2010). Differential regulation of microRNA stability. Rna 16, 1032–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezikov E, Robine N, Samsonova A, Westholm JO, Naqvi A, Hung J-H, Okamura K, Dai Q, Bortolamiol-Becet D, Martin R, et al. (2011). Deep annotation of Drosophila melanogaster microRNAs yields insights into their processing, modification, and emergence. Genome Research 21, 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt H, Harnisch C, Rammelt C, Stohr N, Zirkel A, Dohm JC, Himmelbauer H, Tavanez JP, Huttelmaier S, and Wahle E (2012). Maturation of mammalian H/ACA box snoRNAs: PAPD5-dependent adenylation and PARN-dependent trimming. Rna 18,> 958–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieging KT, Mello SS, and Attardi LD (2014). Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 14, 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boele J, Persson H, Shin JW, Ishizu Y, Newie IS, Sokilde R, Hawkins SM, Coarfa C, Ikeda K, Takayama KI, et al. (2014). PAPD5-mediated 3’ adenylation and subsequent degradation of miR-21 is disrupted in proliferative disease. Proceedings of the National Academy of Sciences 111, 11467–11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Whisnant AW, Kennedy EM, Flores O, and Cullen BR (2014). Derivation and characterization of Dicer- and microRNA-deficient human cells. Rna 20, 923–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyraz B, Moon DH, Segal M, Muosieyiri MZ, Aykanat A, Tai AK, Cahan P, and Agarwal S (2016). Posttranscriptional manipulation of TERC reverses molecular hallmarks of telomere disease. J. Clin. Invest. 126, 3377–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns DM, D’Ambrogio A, Nottrott S, and Richter JD (2011). CPEB and two poly(A) polymerases control miR-122 stability and p53 mRNA translation. Nature 473, 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris AM, Ballew BJ, Kentosh JB, Turner CE, Norton SA, NCI DCEG Cancer Genomics Research Laboratory, NCI DCEG Cancer Sequencing Working Group, Giri N, Alter BP, Nellan A, et al. (2016). Hoyeraal-Hreidarsson Syndrome due to PARN Mutations: Fourteen Years of Follow-Up. Pediatr. Neurol. 56, 62–68.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs AM, Ando Y, de Hoon MJL, Tomaru Y, Nishibu T, Ukekawa R, Funakoshi T, Kurokawa T, Suzuki H, Hayashizaki Y, et al. (2010). A comprehensive survey of 3” animal miRNA modification events and a possible role for 3” adenylation in modulating miRNA targeting effectiveness. Genome Research 20, 1398–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazalla D, Yario T, Steitz JA, and Steitz J (2010). Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science 328, 1563–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H-M, Triboulet R, Thornton JE, and Gregory RI (2013). A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature 497, 244–248.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, and Groβhans H (2009). Active turnover modulates mature microRNA activity in Caenorhabditis elegans. Nature 461, 546–549. [DOI] [PubMed] [Google Scholar]
- Chen J, and Kastan MB (2010). 5”−3-”UTR interactions regulate p53 mRNA translation and provide a target for modulating p53 induction after DNA damage. Genes & Development 24, 2146–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehlin E, Wormington M, Korner CG, and Wahle E (2000). Cap-dependent deadenylation of mRNA. Embo J 19, 1079–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deppert W, Steinmayer T, and Richter W (1989). Cooperation of SV40 large T antigen and the cellular protein p53 in maintenance of cell transformation. Oncogene 4, 1103–1110. [PubMed] [Google Scholar]
- Devany E, Zhang X, Park JY, Tian B, and Kleiman FE (2013). Positive and negative feedback loops in the p53 and mRNA 3’ processing pathways. Proc. Natl. Acad. Sci. U.S.a. 110, 3351–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanraj S, Gunja SMR, Deveau AP, Nissbeck M, Boonyawat B, Coombs AJ, Renieri A, Mucciolo M, Marozza A, Buoni S, et al. (2015). Bone marrow failure and developmental delay caused by mutations in poly(A)-specific ribonuclease (PARN). J Med Genet 52, 738–748. [DOI] [PubMed] [Google Scholar]
- Di Leonardo A, Linke SP, Clarkin K, and Wahl GM (1994). DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes & Development 8, 2540–2551. [DOI] [PubMed] [Google Scholar]
- D’Ambrogio A, Gu W, Udagawa T, Mello CC, and Richter JD (2012). Specific miRNA stabilization by Gld2-catalyzed monoadenylation. CellReports 2, 1537–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbarbary RA, Miyoshi K, Myers JR, Du P, Ashton JM, Tian B, and Maquat LE (2017). Tudor-SN-mediated endonucleolytic decay of human cell microRNAs promotes G 1/S phase transition. Science 356, 859–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan EF, and Pasquinelli AE (2013). MicroRNA biogenesis: regulating the regulators. Crit. Rev. Biochem. Mol. Biol 48, 51–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb KC, and Cech TR (2013). 3’ terminal diversity of MRP RNA and other human noncoding RNAs revealed by deep sequencing. BMC Mol. Biol. 14, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha M, and Kim VN (2014). Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15, 509–524. [DOI] [PubMed] [Google Scholar]
- Hagan JP, Piskounova E, and Gregory RI (2009). Lin28 recruits the TUTase Zcchc11 to inhibit let-7 maturation in mouse embryonic stem cells. Nature Structural & Molecular Biology 16, 1021–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo I, Ha M, Lim J, Yoon M-J, Park J-E, Kwon SC, Chang H, and Kim VN (2012). Mono-uridylation of pre-microRNA as a key step in the biogenesis of group II let-7 microRNAs. Cell 151, 521–532. [DOI] [PubMed] [Google Scholar]
- Heo I, Joo C, Cho J, Ha M, Han J, and Kim VN (2008). Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Molecular Cell 32, 276–284. [DOI] [PubMed] [Google Scholar]
- Heo I, Joo C, Kim Y-K, Ha M, Yoon M-J, Cho J, Yeom K-H, Han J, and Kim VN (2009). TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell 138, 696–708. [DOI] [PubMed] [Google Scholar]
- Ibrahim F, Rymarquis LA, Kim E-J, Becker J, Balassa E, Green PJ, and Cerutti H (2010). Uridylation of mature miRNAs and siRNAs by the MUT68 nucleotidyltransferase promotes their degradation in Chlamydomonas. Proc. Natl. Acad. Sci. U.S.a. 107, 3906–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi N, Shoji K, Sakaguchi Y, Honda S, Kirino Y, Suzuki T, Katsuma S, and Tomari Y (2016). Identification and Functional Analysis of the Pre-piRNA 3′ Trimmer in Silkworms. Cell 164, 962–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joerger AC, and Fersht AR (2016). The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu. Rev. Biochem. 85, 375–404. [DOI] [PubMed] [Google Scholar]
- Jones MR, Blahna MT, Kozlowski E, Matsuura KY, Ferrari JD, Morris SA, Powers JT, Daley GQ, Quinton LJ, and Mizgerd JP (2012). Zcchc11 uridylates mature miRNAs to enhance neonatal IGF-1 expression, growth, and survival. PLoS Genet 8, e1003105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MR, Quinton LJ, Blahna MT, Neilson JR, Fu S, Ivanov AR, Wolf DA, and Mizgerd JP (2009). Zcchc11-dependent uridylation of microRNA directs cytokine expression. Nat. Cell Biol. 11, 1157–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh T, Sakaguchi Y, Miyauchi K, Suzuki T, Kashiwabara SI, Baba T, and Suzuki T (2009). Selective stabilization of mammalian microRNAs by 3’ adenylation mediated by the cytoplasmic poly(A) polymerase GLD-2. Genes & Development 23, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh T, Hojo H, and Suzuki T (2015). Destabilization of microRNAs in human cells by 3’ deadenylation mediated by PARN and CUGBP1. Nucleic Acids Research 43, 7521–7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirwan M, and Dokal I (2009). Dyskeratosis congenita, stem cells and telomeres. BBA - Molecular Basis of Disease 1792, 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korner CG, and Wahle E (1997). Poly(A) tail shortening by a mammalian poly(A)-specific 3’-exoribonuclease. Journal of Biological Chemistry 272, 10448–10456. [DOI] [PubMed] [Google Scholar]
- Landgraf P, Rusu M, Sheridan R, Sewer A, lovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. (2007). A Mammalian microRNA Expression Atlas Based on Small RNA Library Sequencing. Cell 129, 1401–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Choi Y, Kim K, Jin H, Lim J, Nguyen TA, Yang J, Jeong M, Giraldez AJ, Yang H, et al. (2014). Adenylation of maternally inherited microRNAs by Wispy. Molecular Cell 56, 696–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yang Z, Yu B, Liu J, and Chen X (2005). Methylation Protects miRNAs and siRNAs from a 3’-End Uridylation Activity in Arabidopsis. Current Biology 15, 1501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilyestrom W, Klein MG, Zhang R, Joachimiak A, and Chen XS (2006). Crystal structure of SV40 large T-antigen bound to p53: interplay between a viral oncoprotein and a cellular tumor suppressor. Genes & Development 20, 2373–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zhang C, Zhao Y, and Feng Z (2016). MicroRNA Control of p53. J. Cell. Biochem. 118, 7–14. [DOI] [PubMed] [Google Scholar]
- Lu S, Sun Y-H, and Chiang VL (2009). Adenylation of plant miRNAs. Nucleic Acids Research 37, 1878–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragozidis P, Papanastasi E, Scutelnic D, Totomi A, Kokkori I, Zarogiannis SG, Kerenidi T, Gourgoulianis KI, and Balatsos NAA (2015). Poly(A)-specific ribonuclease and Nocturnin in squamous cell lung cancer: prognostic value and impact on gene expression. Mol. Cancer 14, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragozidis P, Karangeli M, Labrou M, Dimoulou G, Papaspyrou K, Salataj E, Pournaras S, Matsouka P, Gourgoulianis KI, and Balatsos NAA (2012). Alterations of deadenylase expression in acute leukemias: evidence for poly(a)-specific ribonuclease as a potential biomarker. Acta Haematol. 128, 39–46. [DOI] [PubMed] [Google Scholar]
- Mason PJ, and Bessler M (2011). The genetics of dyskeratosis congenita. Cancer Genetics 204, 635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon DH, Segal M, Boyraz B, Guinan E, Hofmann I, Cahan P, Tai AK, and Agarwal S (2015). Poly(A)-specific ribonuclease (PARN) mediates 3’-end maturation of the telomerase RNA component. Nature Genetics 47, 1482–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D, St-Sauveur VG, Bergeron D, Dupuis-Sandoval F, Scott MS, and Bachand F (2015). A Polyadenylation-Dependent 3′ End Maturation Pathway Is Required for the Synthesis of the Human Telomerase RNA. CellReports 13, 2244–2257. [DOI] [PubMed] [Google Scholar]
- Pfeffer SR, Yang CH, and Pfeffer LM (2015). The Role of miR-21 in Cancer. Drug Dev. Res. 76, 270–277. [DOI] [PubMed] [Google Scholar]
- Ramachandran V, and Chen X (2008). Degradation of microRNAs by a family of exoribonucleases in Arabidopsis. Science 321, 1490–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, and Howley PM (1990). The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63, 1129–1136. [DOI] [PubMed] [Google Scholar]
- Shukla S, and Parker R (2017). PARN Modulates Y RNA Stability and Its 3’-End Formation. Molecular and Cellular Biology 37, e00264–17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, Schmidt JC, Goldfarb KC, Cech TR, and Parker R (2016). Inhibition of telomerase RNA decay rescues telomerase deficiency caused by dyskerin or PARN defects. Nature Structural & Molecular Biology 23, 286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son A, Park J-E, and Kim VN (2018). PARN and TOE1 Constitute a 3′ End Maturation Module for Nuclear Non-coding RNAs. CellReports 23, 888–898. [DOI] [PubMed] [Google Scholar]
- Stuart BD, Choi J, Zaidi S, Xing C, Holohan B, Chen R, Choi M, Dharwadkar P, Torres F, Girod CE, et al. (2015). Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nature Genetics 47, 512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarbrick A, Woods SL, Shaw A, Balakrishnan A, Phua Y, Nguyen A, Chanthery Y, Lim L, Ashton LJ, Judson RL, et al. (2010). miR-380-5p represses p53 to control cellular survival and is associated with poor outcome in MYCN-amplified neuroblastoma. Nat. Med. 16, 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Tu S, Lee H-C, Weng Z, and Mello CC (2016). The RNase PARN-1 Trims piRNA 3′ Ends to Promote Transcriptome Surveillance in C. elegans. Cell 164, 974–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton JE, Du P, Jing L, Sjekloca L, Lin S, Grossi E, Sliz P, Zon LI, and Gregory RI (2014). Selective microRNA uridylation by Zcchc6 (TUT7) and Zcchcll (TUT4). Nucleic Acids Research 42, 11777–11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng C-K, Wang H-F, Burns AM, Schroeder MR, Gaspari M, and Baumann P (2015). Human Telomerase RNA Processing and Quality Control. CellReports 13, 2232–2243. [DOI] [PubMed] [Google Scholar]
- Tummala H, Walne A, Collopy L, Cardoso S, la Fuente, de J, Lawson S, Powell J, Cooper N, Foster A, Mohammed S, et al. (2015). Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J. Clin. Invest. 125, 2151–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419–1260419. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, et al. (2017). A pathology atlas of the human cancer transcriptome. Science 357, eaan2507. [DOI] [PubMed] [Google Scholar]
- Ustianenko D, Hrossova D, Potesil D, Chalupnikova K, Hrazdilova K, Pachernik J, Cetkovska K, Uldrijan S, Zdrahal Z, and Vaňáčová Š (2013). Mammalian DIS3L2 exoribonuclease targets the uridylated precursors of let-7 miRNAs. Rna 19, 1632–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachos IS, Zagganas K, Paraskevopoulou MD, Georgakilas G, Karagkouni D, Vergoulis T, Dalamagas T, and Hatzigeorgiou AG (2015). DIANA-miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Research 43, W460–W466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman SK, Knouf EC, Parkin RK, Fritz BR, Lin DW, Dennis LM, Krouse MA, Webster PJ, and Tewari M (2011). Post-transcriptional generation of miRNA variants by multiple nucleotidyl transferases contributes to miRNA transcriptome complexity. Genome Research 21, 1450–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Yang Z, Li J, Minakhina S, Yang M, Padgett RW, Steward R, and Chen X (2005). Methylation as a crucial step in plant microRNA biogenesis. Science 307, 932–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L-N, and Yan Y-B (2015). Depletion of poly(A)-specific ribonuclease (PARN) inhibits proliferation of human gastric cancer cells by blocking cell cycle progression. BBA - Molecular Cell Research 1853, 522–534. [DOI] [PubMed] [Google Scholar]
- Zhang X, Devany E, Murphy MR, Glazman G, Persaud M, and Kleiman FE (2015). PARN deadenylase is involved in miRNA-dependent degradation of TP53 mRNA in mammalian cells. Nucleic Acids Research 43, 10925–10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinder JC, and Lima CD (2017). Targeting RNA for processing or destruction by the eukaryotic RNA exosome and its cofactors. Genes & Development 31, 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 (Related to Figures 1 and 3).
Fold change values for miRNA libraries in PARN KD, DIS3L KD and DIS3L2 KD HeLa cells compared to control.
Data S2 (Related to Figure 3).
Raw counts corresponding to each 3’ end for miR-21-5p in Scr, PARN KD, PARN and PAPD5 KD and DIS3L2 KD HeLa cells.
Data S3 (Related to Figure 3).
Raw counts corresponding to each 3’ end for miR-181b-5p in Scr, PARN KD, PARN and PAPD5 KD, DIS3L KD and DIS3L2 KD HeLa cells.