

Abstract
Hereditary cancer disorders often provide an important window into novel mechanisms supporting tumor growth. Understanding these mechanisms thus represents a vital goal. Towards this goal, here we report a chemoproteomic map of fumarate, a covalent oncometabolite whose accumulation marks the genetic cancer syndrome hereditary leiomyomatosis and renal cell carcinoma (HLRCC). We applied a fumarate-competitive chemoproteomic probe in concert with LC-MS/MS to discover new cysteines sensitive to fumarate hydratase (FH) mutation in HLRCC cell models. Analysis of this dataset revealed an unexpected influence of local environment and pH on fumarate reactivity, and enabled the characterization of a novel a FH-regulated cysteine residue that lies at a key protein-protein interface in the SWI-SNF tumor suppressor complex. Our studies provide a powerful resource for understanding the covalent imprint of fumarate on the proteome, and lay the foundation for future efforts to exploit this distinct aspect of oncometabolism for cancer diagnosis and therapy.
Introduction
A major finding of modern cancer genomics has been the discovery of driver mutations in primary metabolic enzymes.1–3 Many of these lesions cause the accumulation of “oncometabolites,” endogenous metabolites whose accretion can directly drive malignant transformation. For example, mutation of fumarate hydratase (FH) in the familial cancer syndrome hereditary leiomyomatosis and renal cell carcinoma (HLRCC) leads to high levels of intracellular fumarate.4,5 Fumarate has been hypothesized to promote tumorigenesis both by reversibly inhibiting dioxygenases involved in epigenetic signaling,6,7 as well as by interacting with proteins covalently as an electrophile, forming the non-enzymatic posttranslational modification cysteine S-succination (Fig. 1a).8 This latter mechanism is unique to fumarate, and has been proposed to contribute to the distinct tissue selectivity, gene expression profiles, and clinical outcomes observed in HLRCC relative to other oncometabolite-driven cancers.9,10 Consistent with a functional role, studies have found that S-succination of Keap1 can activate NRF2-mediated transcription in HLRCC.11 Furthermore, global immunohistochemical staining of S-succination has been applied to assess stage and progression of FH-deficient tumors, suggesting the utility of this modification as a biomarker.12
Figure 1.
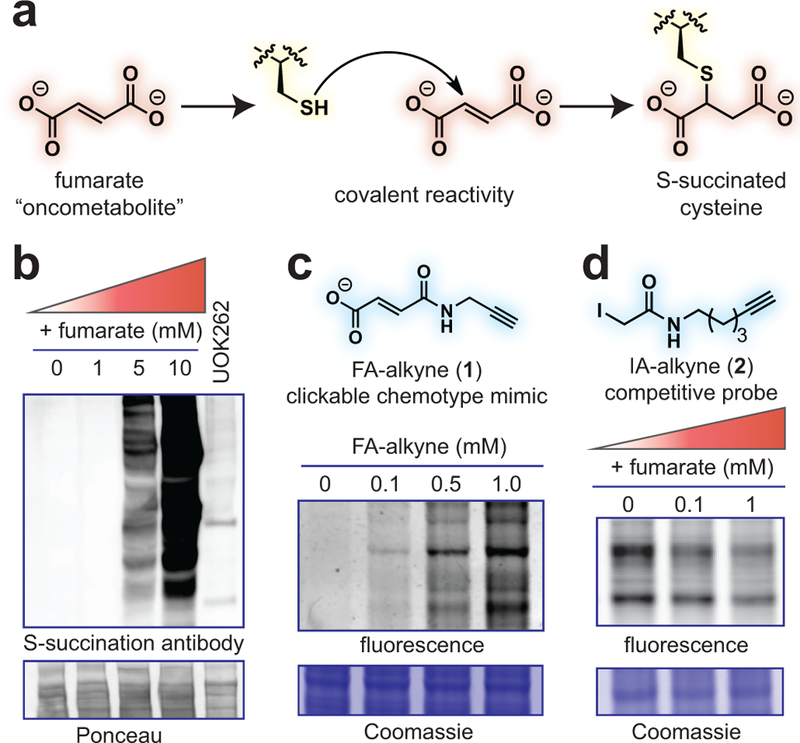
Fumarate is a covalent oncometabolite. (a) Covalent labeling of cysteine residues by fumarate yields the PTM S-succination. (b) Applying S-succinated Cys immunoblotting to establish the concentration of fumarate required for covalent protein labeling. HEK-293 proteomes were treated with fumarate (0, 1, 5, 10 mM) for 15 h prior to western blotting. (c) Applying fumarate alkyne (FA-alkyne, 1) to visualize reactivity of the fumarate chemotype. HEK-293 proteomes were treated with FA-alkyne (0, 0.1, 0.5, 1 mM) for 15 h prior to click chemistry and SDS-PAGE. (d) Applying iodoacetamide alkyne (IA-alkyne, 2) as a competitive probe of covalent fumarate labeling. HEK-293 proteomes were incubated with fumarate for 15 h prior to treatment with 100 μM IA-alkyne for 1 h followed by desalting, click chemistry, and SDS-PAGE. Representative images from two independent experiments are shown in b-d. Uncropped scans of gels and immunoblots are provided in Supplementary Fig. 10.
Despite its potential relevance to HLRCC pathology, our overall understanding of fumarate’s covalent reactivity remains incomplete. Our current knowledge of S-succination is limited to proteins identified by candidate methods, such as Keap1,11 or whole proteome mass spectrometry.13–15 Neither of these approaches report on extent of S-succination, impeding our ability to decipher what structural features drive fumarate’s reactivity and whether these modifications alter protein function. A better understanding of the global scope and stoichiometry of fumarate reactivity has the potential to deliver new insights into HLRCC biology and provide site-specific biomarkers for assessing tumor development and therapeutic response.
Towards this goal, here we report a chemoproteomic map of the covalent targets of the oncometabolite fumarate. First, we establish the utility of chemoproteomic probes to compete for occupancy of fumarate-reactive cysteines. Next, we apply these probes in combination with quantitative mass spectrometry to define the proteome-wide sensitivity of cysteine residues to FH mutation. Functional analysis of this dataset led to the discovery of new molecular determinants of fumarate-sensitivity and the characterization of an FH-sensitive cysteine in SMARCC1, a member of the SWI-SNF tumor suppressor complex. By establishing a new resource for understanding how fumarate reactivity impacts HLRCC biology, our studies provide an essential underpinning for applications seeking to exploit this unique aspect of oncometabolism for clinical benefit.
Results
Comparative affinity profiling of fumarate reactivity
Several recent studies have demonstrated the utility of competitive chemoproteomics to characterize electrophilic drug targets.16,17 To extend these methods to an endogenous oncometabolite, we first characterized fumarate’s reactivity profile. Several pieces of evidence suggest fumarate may exhibit relatively modest reactivity. Most relevantly, studies of the multiple sclerosis drug dimethyl fumarate (Tecfidera; DMF) found its metabolized product monomethyl fumarate (MMF) possesses limited thiol reactivity at micromolar concentrations.18,19 Theoretical calculations indicate this stems from MMF’s higher-lying LUMO, which increases the energetic barrier to covalent bond formation with nucleophilic cysteines.18 Fumarate’s LUMO is even higher in energy than MMF (Supplementary Fig. 1a), suggesting it may possess a distinct reactivity profile. However, the relative reactivity of the drug and oncometabolite have never been directly assessed.
To explore this, we treated HEK-293 cell extracts with increasing amounts of fumarate and analyzed proteomes for covalent labeling using an S-succination antibody (Fig. 1b). An equivalent amount of HLRCC proteome (UOK262, FH−/−) was used to compare levels of S-succination caused by FH mutation. Fumarate proved a relatively mild electrophile, requiring millimolar concentrations to cause S-succination equivalent to HLRCC proteomes (Fig. 1b; Supplementary Fig. 1b).5 We validated this finding using a clickable chemotype mimic fumarate alkyne (FA-alkyne, 1, Fig. 1c). FA-alkyne is more reactive than fumarate due to the analogue’s lower-lying LUMO (Supplementary Fig. 1c). However, consistent with covalent labeling via Michael addition, we observed time- and dose-dependent protein labeling of lysates by FA-alkyne, but not an inert succinate analogue (Supplementary Fig. 1d-e). While FA-alkyne labeling was modestly competed by fumarate, it was completely abrogated by pre-incubation with MMF, DMF, and iodoacetamide, again highlighting the attenuated reactivity of the oncometabolite relative to conventional electrophiles (Supplementary Fig. 1f-g). Low millimolar concentrations of fumarate also impeded cysteine labeling by the established chemoproteomic reagent iodoacetamide alkyne (IA-alkyne, 2; Fig. 1d).16 Pre-treatment of lysates with iodoacetamide reciprocally inhibited fumarate-dependent S-succination, confirming that these chemotypes compete for cysteine occupancy (Supplementary Fig. 1h). These results highlight the distinct reactivity of fumarate relative to DMF and MMF, and suggest this metabolite’s reactivity may be most relevant in pathophysiological contexts such as HLRCC where it accumulates to millimolar levels.
Global chemoproteomic profiling of FH-regulated cysteines
The distinct reactivity of fumarate suggests its accumulation in HLRCC may impart a unique covalent imprint on the proteome. To characterize this effect, we applied IA-alkyne and an LC-MS/MS platform derived from isoTOP-ABPP to map cysteine reactivity changes caused by FH mutation (Fig. 2a).20 Briefly, proteomes were isolated from an immortalized HLRCC cell line (UOK262 FH−/−), and a rescue line in which reintroduction of FH gene reduces S-succination (UOK262WT, FH+/+; Fig. 2b).21 Paired samples from FH−/− and FH+/+ cells were treated with IA-alkyne, conjugated to isotopically distinguishable azide-biotin tags using click chemistry, pooled, and enriched over streptavidin. Following on-bead tryptic digest, IA-alkyne labeled peptides were released by dithionite cleavage of an azobenzene linker. LC-MS/MS was used to identify Cys-containing peptides, with the relative Intensity ratio (R) of light/heavy (L/H) isotopic pairs in the MS1 spectra used as a quantitative readout of relative Cys-labeling stoichiometry (Fig. 2a). R values of ~1 indicates that a cysteine was unaffected by FH mutation, whereas an R value of 2 indicates a cysteine’s reactivity (or abundance) is reduced ~50% by FH mutation (based on the formula “relative modification stoichiometry (%) = [1-(1/R)]*100%”; Fig. 2a). One critical feature of this experiment is that it is mechanism-agnostic, and as such may identify FH-regulated cysteine reactivity changes caused by direct S-succination as well as alternative stimuli such as altered gene expression or oxidative stress (of note, our culture media contained pyruvate, which limits reactive oxygen species in HLRCC cells).9,10,22 We thus term these differentially occupied residues “FH-regulated” cysteines.
Figure 2.

Global chemoproteomic profiling of FH-regulated cysteine residues. (a) Applying a competitive chemoproteomic platform to study the oncometabolite fumarate. Comparison of proteomes from FH−/− (UOK262) and FH+/+ (UOK262WT) cells are used to define “FH-regulated” Cys residues. (b) S-succination in HLRCC cells is dependent on FH mutation. Representative image from two independent experiments is shown. Uncropped scans of immunoblot is provided in Supplementary Fig. 10. (c) FH-regulated Cys residues identified in UOK262 cells (n ≥2, SD ≤25%). (d) Overlap of FH-regulated Cys residues (R ≥2, n ≥1) with DMF-regulated Cys residues identified in Blewett et al (R ≥2, n≥1). (e) Subcellular localization of FH-regulated and DMF-sensitive Cys residues. (f) Conservation of FH-regulated, FH-neutral, and hyperreactive cysteine residues identified in Weerapana et al.16 Peptides with the highest R values from each dataset (n=50) were used for analysis. Data is presented as box and whiskers plot with box representing 25th-75th percentile, horizontal line representing median, and whiskers representing minimum and maximum values. Statistical significance was assessed using Student’s t-test (two-tailed, unpaired); **P < 0.01. Data for individual proteins is available in supplementary datasets and can be searched via a web interface at ccr2.cancer.gov/resources/Cbl/proteomics/fumarate.
Applying this approach we performed three independent replicate measurements of cysteine reactivity in HLRCC cells leading to the quantification of 1170 cysteine residues (Fig. 2c, Supplementary Dataset 1). This data can be searched via the web at ccr2.cancer.gov/resources/Cbl/proteomics/fumarate. Applying reproducibility metrics (identified in ≥2 datasets, R standard deviation ≤25%) led to the further specification of 684 high confidence FH-regulated cysteines. The reactivity of 105 cysteines was upregulated ≥2-fold by FH rescue, consistent with reduced S-succination or oxidation in FH+/+ cells (Fig. 2c, Supplementary Dataset 1). Identified among these hits were 28 known targets of S-succination including ACO2,15 which was found to be only moderately reactive (Supplementary Dataset 2). Comparing FH-regulated cysteines to those identified in a recent chemoproteomic study of DMF,19 we find only a small fraction (4.2%) with R≥2 overlap, providing additional evidence for the distinct reactivity of these molecules (Supplementary Dataset 1, Figure 2d). A higher percentage of mitochondrial proteins were regulated by FH (41%) versus DMF (8%), suggesting oncometabolite compartmentalization as one driver of this distinct reactivity (Supplementary Dataset 1, Figure 2e). Analysis of the evolutionary conservation of i) FH-regulated cysteines, ii) FH-insensitive cysteines (R~1), and iii) hyperreactive cysteines16 revealed the former to be the least well-conserved (Fig. 2f). This is consistent with the hypothesis that fumarate acts as a covalent metabolite only upon hyperaccumulation, which would limit its reactivity from exerting strong evolutionary pressure.
Interestingly, we found that individual proteins displaying multiple FH-regulated cysteines often exhibited unidirectional changes in reactivity (Supplementary Fig. 2a, Supplementary Dataset 1). This profile may indicate a global change in protein level.23 Therefore, to bolster our analysis, we sought to identify proteins whose fumarate reactivity may be masked by altered protein abundance in FH−/− and FH+/+ rescue HLRCC cells (Fig. 3). We performed whole proteome (MudPIT) LC-MS/MS analyses of FH−/− and FH+/+ cells and used this data to “correct” or normalize reactivity measurements (Supplementary Fig. 2b). Focusing on high confidence FH-regulated cysteines (identified in ≥2 experiments, S.D. ≤25%), we obtained robust protein abundance data (≥10 spectral counts) for 55% of these parent proteins (Supplementary Dataset 1). Correcting for protein abundance led to modestly revised reactivity for the majority of residues analyzed, with 325/376 (86%) showing a less than 2-fold change. Normalization for abundance increased the calculated cysteine reactivity of 7 proteins by ≥2-fold, and decreased the calculated cysteine reactivity of 44 proteins by ≥2-fold (Supplementary Dataset 1, Supplementary Fig. 2c-d, Fig. 3a). To validate our LC-MS/MS identifications, we assessed a subset of targets for fumarate-competitive labeling using the clickable chemotype mimic FA-alkyne (Fig. 3b). Consistent with LC-MS/MS data, capture of OAT, HNRNPL, SMARCC1, and CBX5 was competed by fumarate treatment (Fig. 3c). In contrast, the non-target PKM1 showed no such competition. Overall, these studies demonstrate a strategy for comparing cysteine reactivity profiles between cell lines, and provide an initial glimpse into the sites and stoichiometry of the fumarate-reactive proteome.
Figure 3.

Analyzing the reactivity and abundance of FH-regulated cysteines. (a) Heat map illustrating strategy for correcting Cys reactivity ratios measured in UOK262 (FH-deficient) and UOK262WT (FH rescue) cells using whole proteome MudPIT LC-MS/MS data. Adjusting for protein abundance can lead to an increase in calculated reactivity (left protein subset, red), an insignificant change (middle protein subset, blue), or a decrease in calculated reactivity (right protein subset, green). An extended graphic of protein families harboring high confidence, abundance-corrected FH-regulated cysteine residues is provide in Supplementary Fig. 2d. (b) Validating FH-regulated Cys residues using the clickable chemotype mimic FA-alkyne. (c) FA-alkyne capture of proteins that contain FH-regulated Cys residues is competed by fumarate (3 h pre-incubation with 1 mM fumarate; then 15 h treatment with 100 μM FA-alkyne). Representative images from two independent experiments are shown. Uncropped scans of immunoblots are provided in Supplementary Fig. 10.
Molecular determinants of fumarate-cysteine interactions
Next we sought to utilize our chemoproteomic data to better understand the structural determinants of oncometabolite reactivity. As an initial step, we assessed FH-regulated cysteines for the presence of linear motifs using pLogo (Fig. 4a, Supplementary Dataset 3). Interestingly, FH-regulated cysteines showed an enrichment of acidic residues such as glutamate (E) and aspartate (D) in flanking regions. This was unexpected, as nucleophilic cysteines are typically surrounded by proximal basic residues such as lysine (K) and arginine (R), which can serve as hydrogen bond donors and help stabilize the developing negative charge of the thiolate.24 The atypical nature of the FH-regulated motif was further supported by pLogo analysis of hyperreactive cysteine residues,16 which demonstrated the expected enrichment of basic flanking residues (Fig. 4b, Supplementary Fig. 3a). Fumarate’s cysteine reactivity motif was distinct from that of DMF and HNE (Supplementary Fig. 3a),19,25 with only MMF showing similar enrichment of acidic residues (Supplementary Fig. 3a). Hypothesizing that FH-regulated cysteines may possess a unique local sequence environment, we next asked how fumarate-sensitivity correlated with overall cysteine reactivity. For this, we overlaid hyperreactive cysteines (identified by concentration-dependent IA-alkyne labeling)16 onto our FH-regulated map. This led to the observation that FH-regulated cysteines are strikingly anti-correlated with reactivity (Fig. 4c). To extend this finding, we treated HEK-293 proteomes with exogenous fumarate, and used IA-alkyne analysis to define cysteine residues capable of directly reacting with fumarate (“fumarate-sensitive cysteines”; Supplementary Dataset 4). Of the nine highly FH-regulated cysteine residues quantified in this experiment (R>2, Supplementary Dataset 1) eight were rendered less reactive by fumarate (R>1.5, Supplementary Dataset 4), indicating that exogenous fumarate can influence cysteine occupancy in a manner similar to FH mutation. Overlaying hyperreactive cysteines onto this fumarate dataset again identified an inverse relationship fumarate-sensitivity and cysteine reactivity (Supplementary Fig. 3b, Supplementary Dataset 4). In contrast, stimuli such as DMF19 or GSNO20 were found to target cysteine residues across the fumarate-sensitivity spectrum (Supplementary Fig. 3c-d). Furthermore, in proteins such as NIT2 and GSTO1 that contain nucleophilic active site cysteines, FH mutation and fumarate preferentially reduced the reactivity of distal residues (Fig. 4d, Supplementary Fig. 3e). These analyses define a unique local environment for covalent oncometabolite labeling.
Figure 4.

Establishing the molecular determinants of fumarate-protein interactions. (a) Motif analysis of FH-regulated Cys residues. 50 cysteines found to be most FH-regulated in this study (highest R values, n≥2, SD≤25%) were used for the analysis. (b) Motif analysis of hyperreactive Cys residues identified in Weerapana et al16. 50 cysteines found to be most hyperreactive were used for the analysis. (c) FH-regulated Cys residues are anti-correlated with Cys-reactivity. (d) FH mutation preferentially modulates the reactivity of a distal Cys in NIT2 (n = 3 independent experiments). (e) The reversibility of S-succinated model thiols is slow and dependent on leaving group pKa.. S-succinated thiols (1 mM) were incubated in 100 mM Tris at 37 °C for 24 h, prior to quantification of DMF release by fluorescence assay. Data is presented as mean ± s.e.m.; n = 3/group. (f) Influence of pH on IA-alkyne and FA-alkyne reactivity. Left: IA-alkyne (1h; 100 μM); Right: FA-alkyne (15 h, 1 mM). (g) Influence of pH on proteomic S-succination (16h; 5mM). Representative images from two independent experiments are shown in f-g. Uncropped scans of gels and immunoblots are provided in Supplementary Fig. 10. (h) pH-dependent reaction kinetics of fumarate with thiophenol (n = 3 independent experiments). R2 = goodness of fit. (i) Structures of fumarate and hydrogen fumarate. (j) FH−/− cell lines exhibit decreased intracellular pH relative to FH+/+ lines. pH values were determined using pHrodo Green Statistical significance was assessed using Student’s t-test (two-tailed, unpaired); n = 4/group, *P < 0.05, **P < 0.01.
Fumarate is a conditionally-reactive oncometabolite
To better understand the mechanistic basis for these observations, we first assessed the pKa-dependent reversibility of cysteine S-succination. We reasoned that reversible labeling of low pKa hyperreactive cysteines may enable their irreversible capture by IA-alkyne, explaining the observed anti-correlation. To test this hypothesis, we applied a recently developed fluorescence assay to assess fumarate release from S-succinated thiols of disparate acidities (Fig. 4e, Supplementary Fig. 4a-b).26 While S-succinated thiols did demonstrate pKa-dependent reversibility, the extent of fumarate release was minor, with only 2–4% reversal observed over 24 hours (Fig. 4e, Supplementary Fig. 4c). These studies suggest that reversible S-succination can occur, but makes an overall minor contribution to fumarate’s covalent labeling profile.
We next considered an alternative hypothesis: that protonated hydrogen fumarate may function as the active electrophile in S-succination reactions. The plausibility of hydrogen fumarate as a reactive species has precedence in previous studies of MMF,18 and would be expected to increase fumarate’s reactivity by lowering its LUMO energy (Supplementary Fig. 4d) and reducing repulsive electrostatic interactions with the cysteine thiolate. Consistent with this premise, we found that while IA-alkyne exhibits increased protein labeling at higher pH (presumably due to higher thiolate concentrations; Fig. 4f), labeling of proteins by FA-alkyne and fumarate proceeds to a greater extent at lower pHs which favor hydrogen fumarate formation (Fig. 4f-g, Supplementary Fig. 4e). Analysis of S-succination kinetics using model thiols further confirmed the increased reaction rate of fumarate at acidic pH (Fig. 4h-i, Supplementary Fig. 4f-h). The second-order rate constants for S-succination of thiophenol (10−2 to 10−3 M−1 s−1) were slow relative to rates of thiol adduction measured for iodoacetamide (0.15 M−1 s−1),27 hydroxynonenal (1.2 M−1 s−1),28 and hydrogen peroxide (2.9 M−1 s−1),29 consistent with the negligible concentrations of hydrogen fumarate available for reaction at neutral pH (Supplementary Fig. 4h). Accelerated product formation at acidic pH was specifically observed with fumarate and not DMF, presumably because the latter is a competent electrophile under neutral conditions (Supplementary Fig. 4i). FH-deficient HLRCC cells exhibited decreased intracellular pH relative to FH rescue and HEK-293 cells, consistent with an environment conducive to S-succination (Fig. 4j). These studies reveal a paradoxical influence of pH on fumarate reactivity, and suggest hydrogen fumarate as a novel molecular entity responsible for covalent protein modification in HLRCC.
FH-regulated cysteines intersect kidney cancer pathways
To identify novel biology affected by covalent oncometabolism, we performed pathway analysis of FH-regulated cysteines. To focus on S-succination events likely to functionally impact protein activity we employed the informatics tool Mutation Assessor,30 which uses a conservation-based analysis to estimate the probability that a mutation will alter protein function. FH-regulated cysteines were analyzed as C to E mutations to mimic of the negative charge and steric bulk introduced by S-succination. Analyzing raw and abundance-corrected FH-regulated cysteines (R≥2, 50% relative stoichiometry) we identified 74 proteins whose modification was expected to have a high or moderate impact on protein function (Fig. 5a, Supplementary Dataset 3). Extending this workflow to lower stoichiometry FH-regulated cysteines (R≥1.5) highlighted an additional 63 proteins. Gene ontology analysis found these candidate functionally S-succinated proteins clustered in pathways related to mitochondria, metabolism, RNA processing, and gene expression, many of which play known roles in kidney cancer pathogenesis (Fig. 5b, Supplementary Dataset 3). For mechanistic analysis we prioritized targets 1) for whom loss-of-function genetic lesions had been identified in kidney cancer patient genomes, and 2) that mapped to predicted cofactor or protein binding sites (Fig. 5c-d). These analyses led us to investigate SMARCC1 as a case study to understand the functional consequences of fumarate reactivity.
Figure 5.

Functional analyses of FH-regulated Cys residues. (a) Percentage of FH-regulated Cys residues predicted to be functional using the informatics tool Mutation Assessor (R ≥1.5, n ≥2). (b) Gene ontology analysis of FH-regulated Cys residues. (c) Correlation between genes containing FH-regulated Cys residues and genetic alterations in kidney cancer (RCC). Statistical significance was assessed using Student’s t-test (two-tailed, unpaired); **P < 0.01. (d) SMARCC1 C520 lies in the SWIRM domain and is highly conserved. (e) SMARCC1 C520 lies at the SNF5 subunit interface. (f) SMARCC1 C520 undergoes FH-regulated changes in occupancy in HLRCC cells (n = 3 independent experiments). (g) SMARCC1 C520E mutation limits co-immunoprecipitation with SNF5 in HEK-293 cells co-overexpressing FLAG-tagged SNF5 with Myc-tagged SMARCC1. (h) SMARCC1 S-succination can be detected in FH-deficient and FH WT HLRCC cell lines after IP of endogenous SMARCC1. (i) SNF5 demonstrates decreased co-immunoprecipitation and decreased levels in FH−/− HLRCC cells. Left: Results from SWI/SNF complex co-immunoprecipitation with BRG1 antibody. Right: Endogenous levels of SWI/SNF complex members in HLRCC cells. Representative images from two independent experiments are shown in g-i. Uncropped scans of immunoblots are provided in Supplementary Fig. 10. (j) EZH2 inhibitors exhibit modest selectivity for FH-deficient HLRCC cells. UOK262 FH −/− or FH WT spheroids were treated with GSK126 (21 days; 1, 2, 4, 8, 16 μM) and % viability plotted relative to the vehicle treated spheroids. Data is presented as mean ± s.d.; n = 3/group. (k) EZH2 inhibitors are toxic to HLRCC spheroids. UOK262 FH −/− spheroids were treated with vehicle or EPZ6438 (14 days; 5 μM). Figure is representative of 6 replicates.
Analysis of an FH-regulated cysteine in the SWI/SNF complex
SMARCC1 is a core member of the SWI-SNF chromatin remodeling complex, a known tumor suppressor in many cancers.31 In clear cell renal cell carcinoma (ccRCC) SMARCC1 is commonly deleted due to its position on chromosome 3, which lies adjacent to the VHL tumor suppressor. Of note, SMARCC1 does not exhibit coordinate mutation and deletion in VHL-deficient ccRCC, suggesting an intact genomic copy of SMARCC1 is required for survival.32 The FH-regulated residue Cys520 (C520; Fig. 5e) lies in SMARCC1’s SWIRM domain, the most common location of SMARCC1 somatic mutation in cancer. Studies of SMARCC1’s mouse ortholog (Srg3) have found the SWIRM domain regulates the stability of SNF5, a tumor suppressive subunit of SWI/SNF.33 A recent crystal structure revealed C520 lies within a solvent-exposed helix residing directly at the SMARCC1-SNF5 interface, suggesting its modification may obstruct this protein-protein interaction (Fig. 5d).34 To test this hypothesis, we performed co-immunoprecipitation experiments in HEK-293 cells transfected with plasmids encoding FLAG-tagged SNF5 and either wild-type SMARCC1, a C520E, or a C520S mutant. Mutation of C520 to a S-succination mimic (glutamate) abrogated the ability of SNF5 to capture SMARCC1 (Fig. 5f, Supplementary Fig. 5a). In contrast, a less obtrusive serine mutation was completely tolerated (Supplementary Fig. 5b). Similarly, while ectopic expression of wild type SMARCC1 stabilized SNF5, the C520E mutation had less of an effect (Supplementary Fig. 5c).35 Additionally, treatment of cells co-overexpressing SMARCC1/SNF5 with cell-permeable ethyl fumarate also reduced SNF5 stability (Supplementary Fig. 5d).
Analysis of HLRCC cells revealed greater labeling of SMARCC1 C520 by IA-alkyne in FH+/+ rescue as compared to FH−/− cells, consistent with covalent modification of this residue by endogenous fumarate (Fig. 5e). FH mutation also reduced the reactivity of the homologous cysteine in SMARCC2, whose SWIRM domain is nearly identical to SMARCC1’s (Supplementary Fig. 5e-f). However, direct detection of C520 S-succination proved more challenging. MudPIT analysis of FH−/− cell lines validated S-succination of several fumarate-sensitive proteins (GCLM, PCBP1, TCP1, Supplementary Dataset 5, Supplementary Fig. 6), but not SMARCC1. S-succination blots of SMARCC1 immunoprecipitated from FH−/− and FH+/+ HLRCC cells were characterized by high background, with slightly increased signal in FH-deficient cells (Fig. 5g). To further explore this phenomenon, we next examined HLRCC cells for evidence of a disrupted SMARCC1-SNF5 interaction. Co-immunoprecipitation of SWI/SNF complex indicated a modest decrease in SMARCC1’s interaction with SNF5 in FH−/− relative to FH+/+ rescue cells (Fig. 5h). In line with decreased interaction, SNF5 protein, but not transcript levels, are also lower in these cells (Fig. 5h, Supplementary Fig. 7a-b). While the interaction between SMARCC1 and SNF5 is weakened, it is not fully disrupted, as glycerol gradient fractionation indicated that the core SWI/SNF complex remains intact (Supplementary Fig. 8a). HLRCC cells have a transcriptional profile consistent with SNF5 inhibition, with FH- and SNF5-dependent transcripts from published RNA-Seq datasets exhibiting significant overlap (Supplementary Dataset 6, Supplementary Fig S8b-c). Knockdown studies indicate SNF5-dependent regulation of one of these genes is lost upon FH mutation, consistent with the hypothesis that FH activity may regulate SNF5 (Supplementary Fig. 8d). Finally, we examined the sensitivity of HLRCC cells to EZH2 inhibitors, whose lethality is known to be potentiated by SNF5 disruption.36 Multiple EZH2 inhibitors exhibited FH-dependent disruption of HLRCC spheroid growth (Fig. 5i-j, Supplementary Fig. 8e), consistent with a measurable, but minor, impact of fumarate on SNF5 function. These studies demonstrate a novel cysteine-dependent protein-protein interaction in the SWI-SNF complex that may be modulated by oncometabolite accumulation.
Proteomic ID of ligandable cysteines upregulated in HLRCC
Although reactive metabolites are largely expected to exert negative effects on protein function,37 as a final experiment we wondered whether our dataset may also be capable of identifying pathways activated by FH mutation. To explore this concept we reassessed our chemoproteomic data from FH−/− and FH+/+ rescue HLRCC cell lines, focusing on FH-regulated cysteines with R values <1 (blue region, Fig. 2c). These residues possess greater reactivity in FH−/− cells, and are expected to originate from proteins whose abundance or activity is increased by FH loss. Applying gene set enrichment analysis (GSEA)38 to this cysteine subset identified an enrichment in proteins activated by the transcription factors HIF-1α and NRF2 (Fig. 6a), pathways which have previously been shown to be overactive in HLRCC.4,9,11 This suggests chemoproteomic analyses may provide a useful complement to traditional methods such as gene expression for the discovery of novel cancer pathways. However, an additional feature chemoproteomics is that it also has the potential to identify leads for covalent ligands targeting these pathways (Fig. 6b). To explore this concept, we cross referenced cysteines activated by FH mutation with a recently reported covalent fragment library whose proteome-wide targets were characterized.17 This analysis identified ligandable cysteines in several pathways that exhibit increased IA-alkyne reactivity upon FH loss, including glycolysis, hypoxia, and reactive-oxygen stress (Supplementary Fig. 9). These studies highlight a strategy for mining chemoproteomic data to identify novel targets and lead fragments for pathway disruption in HLRCC.
Figure 6.

(a) Gene set enrichment analysis (GSEA) of FH-regulated Cys residues highlights pathways whose activity is functionally upregulated in HLRCC cells. (b) Concept of applying chemoproteomic pathway discovery data to identify new candidate ligands for pathway targeting.
Discussion
The discovery of hereditary cancers driven by TCA cycle mutations provided some of the first evidence that metabolites themselves could fuel tumorigenic signaling.1,2 Recent data indicates that “oncometabolic” signaling is not unique to these contexts, but may instead represent a broader aspect of malignancy.3 While this provides another powerful example of how studying genetic disorders can illuminate general cancer mechanisms, 39 understanding precisely how oncometabolites drive malignant transformation and developing diagnostics to track this process remain critical goals. Here we have applied chemoproteomics to define a novel complement of protein cysteines sensitive to the oncometabolite fumarate in the genetic cancer syndrome HLRCC. Competitive labeling using IA-alkyne facilitates the discovery of FH-regulated cysteines while circumventing the necessity for antibody-generation or direct detection of poorly ionizable, negatively charged S-succinated peptides. To facilitate future applications of this resource, we have created an accessible web interface where this data may be easily browsed and queried (ccr2.cancer.gov/resources/Cbl/proteomics/fumarate). Our study builds on previous proteome-wide studies of reactive lipids and oxidants,25,40 and represents the first application of isoTOP-ABPP to study a hereditary cancer disorder driven by a reactive metabolite. In the future, we anticipate the analyses presented here should provide a useful model for studying biological contexts marked by the production of other endogenous electrophiles including oxidized lipids,25 itaconate,41 and acyl-CoAs.42,43
An unanticipated finding of our study was that FH-regulated cysteines demonstrate an anti-correlation with cysteine reactivity. Furthermore, fumarate reactivity is increased at acidic pH. This suggests fumarate requires both hyperaccumulation as well as protonation to function as a covalent metabolite. In this regard HLRCC tumors constitute a “perfect storm” for S-succination, due to their potent production of fumarate (driven by FH loss) as well as lactic acid (due to increased glycolysis).4,21 Fumarate, lactate, and protein S-succination are also coordinately upregulated in diabetes and mitochondrial respiratory chain disorders,13,44 suggesting additional disease contexts where this conditional reactivity may play a role. Pathway analyses led to identification of a FH-regulated cysteine in SMARCC1 that is critical for protein-protein interactions with the tumor suppressor SNF5. Interestingly, a recent histopathology study identified an FH-deficient HLRCC patient sample (1 of 31 analyzed) in which SNF5 was depressed in the absence of a mutation, leading the authors to hypothesize SNF5 may be regulated by non-genetic mechanisms in HLRCC.45 Further study will be required to determine the prevalence of this phenomenon and its correlation with covalent S-succination of SMARCC1. Besides defining a minor contribution of fumarate to SWI/SNF dysfunction in HLRCC, our findings also illustrate the potential of covalent metabolites to regulate protein-protein interactions (PPIs) in the nucleus. FH-regulated cysteines with high R values are characterized by a large degree of surface exposure (Supplementary Dataset 7), and it is tempting to speculate this may predispose them towards disruption of biomolecular interactions. While relatively few examples of reactive metabolite-dependent PPIs exist,37 the role of cysteine oxidation in regulating such interactions is well-precedented.40,46 Our studies suggest further investigation of metabolite-regulated protein-protein and protein-nucleic acid interactions may be warranted.
Finally, it is important to point out some limitations of the current study. One drawback of our competitive profiling method is that it does not directly identify S-succinated cysteines, but rather cysteines whose reactivity is altered by FH mutation. This leads to the caveat that the observed reactivity changes could be due to direct modification by fumarate, or reactive oxygen species that are known to be produced as a consequence of FH mutation. Therefore, an important future goal will be the development of methods for direct S-succination analysis, including immunoprecipitation-grade antibodies and/or techniques analogous to the biotin-switch protocols used to investigate other cysteine modifications.47 In addition, our work illustrates the difficulty of differentiating between changes in reactivity and expression in FH mutant and wild-type cell lines. This issue was also encountered in a recent study of NRF2 mutant cancers,23 and illustrates the importance of integrating chemoproteomic data with whole proteome and transcriptome analyses to provide a more complete picture of cysteine reactivity. A final critical challenge exemplified by the current study is high throughput validation. We identified >100 cysteines predicted to be functionally impacted by FH mutation (Supplementary Dataset 3), which exceeds the bandwidth of most laboratories for mechanistic follow-up. In the future we anticipate chemoproteomic studies of protein-metabolite interactions will benefit from marriage to orthogonal high-throughput methods, such as pooled CRISPR screening approaches,48 to enable the rapid validation of functional oncometabolite targets. The data presented here provide an information-rich resource for such studies, and as such constitute a critical step towards defining fumarate as a signaling molecule and biomarker in HLRCC and other pathophysiological settings.
Online Methods
General materials and methods
Streptavidin agarose resin was purchased from ThermoFisher Scientific (20353). Protein A/G plus agarose resin was purchased from Sigma (20423). Fumaric acid (A10976) and ethyl fumarate (A12545) were purchased from Alfa Aesar. Maleic acid (M0375), dimethyl fumarate (242926), mono-methyl fumarate (651419), and thiophenol (240249) were purchased from Sigma. Cycloheximide (14126) and EPZ6438 (16174) were purchased from Cayman Chemical. Fmoc-Cys-OH (FAA1362) was purchased from Iris Biotech. JQEZ-05 (S8607) and GSK126 (S7061) were purchased from Selleck Chemical. Anti-FLAG pulldown was performed using immunoprecipitation kit (KBA-319–383) from Rockland Immunochemicals, Inc. SDS-PAGE was performed using Bis-Tris NuPAGE gels (4–12%, Invitrogen #NP0322), and MES running buffer (Life technologies #NP0002) in Xcell SureLock MiniCells (Invitrogen) according to the manufacturer’s instructions. SDS-PAGE fluorescence was visualized using an ImageQuant Las4010 Digitial Imaging System (GE Healthcare). Total protein content on SDS-PAGE gels was visualized by Blue-silver Coomassie stain, made according to the published procedure.49 For western blotting, SDS-PAGE gels were transferred to nitrocellulose membranes (Novex, Life Technologies # LC2001) by electroblotting at 30 volts for 1 hour using a XCell II Blot Module (Novex). Membranes were blocked using StartingBlock (PBS) Blocking Buffer (Thermo Scientific) for 20 minutes, then incubated overnight at 4 °C in a solution containing the primary antibody of interest (1:3000 dilution for S-succinated-Cys antibody and 1:1000 dilution for all other antibodies) in the above blocking buffer with 0.05% Tween 20. The membranes were next washed with TBST buffer, and incubated with a secondary HRP-conjugated antibody (anti-rabbit IgG, HRP-linked [7074], Cell Signaling, 1:1000 dilution) for 1 hour at room temperature. The membranes were again washed with TBST, treated with chemiluminescence reagents (Western Blot Detection System, Cell Signaling) for 1 minute, and imaged for chemiluminescent signal using an ImageQuant Las4010 Digitial Imaging System (GE Healthcare). Fluorimetric analysis of fumarate using hydrazonyl chloride 4 was performed on Photon Technology International QuantMaster fluorimeter using 1-cm path length, 0.13 mL quartz microcuvettes (Helma #101-015-40) at ambient temperature (22 ± 2 °C), using an excitation wavelength of 390 nm, slit width of 3.5 nm, and monitoring emission from 410 nm to 615 nm.
Cell lines, plasmids, and antibodies
HEK-293 cells were obtained from the NCI tumor cell repository. UOK262 (FH−/−), UOK262WT (FH +/+ rescue), UOK268 (FH−/−) and UOK268WT (FH +/+ rescue) cells were obtained from Linehan lab.21 Plasmids encoding FLAG-tagged SNF5, Myc-tagged SMARCC1 and Myc-tagged GFP were obtained as a gift from Trevor Archer (Epigenetics & Stem Cell Biology Laboratory, NIEHS). C520E and C520S mutations were introduced to Myc-SMARCC1 entry clone using custom oligos along with the Quick Change Site-Directed Mutagenesis Kit (Agilent #200515) and transformed into DH10B cells. The insert was fully sequenced to confirm the mutation. Transfection-quality plasmid DNA was generated using the GenElute HP Maxiprep Kit. Qubit Protein Assay kit was purchased from Life Technologies (Q33212). S-succinated-Cys antibody was kindly provided by Prof. Norma Frizzell (University of South Carolina). SMARCC1 (11956), SNF5 (8745), BRG1 (3508), PKM1 (7067), Myc-Tag (2278), FLAG-Tag (14793) and HA-Tag (3724) antibodies were purchased from Cell Signaling Technologies. OAT (A305–355A), HNRNP-L (A303–895A), CBX5 (A300–877A), EEF2 (A301–688A) and MAP2K4 (A302–658A) antibodies were purchased from Bethyl Laboratories, Inc. IP-grade antibodies for SMARCC1 (sc-32763) and BRG1 (sc-17796) were obtained from Santa Cruz Biotechnology.
Chemicals
Synthesis of compounds are described in Supplementary Note 1.
Cell culture and isolation of whole-cell lysates
HEK-293 cells were cultured at 37 °C under 5% CO2 atmosphere in a growth medium of DMEM supplemented with 10% FBS and 2 mM glutamine. UOK262 and UOK268 cell lines were cultured in DMEM supplemented with 10% FBS, 2 mM glutamine, 1 mM pyruvate. UOK262WT and UOK268WT cell lines were cultured in DMEM supplemented with 10% FBS, 2 mM glutamine, 1 mM pyruvate and 0.3 mg/mL of G418. Unfractionated proteomes were harvested from cell lines (80–90% confluency) by washing adherent cells 3x with ice cold PBS, scraping cells into a Falcon tube, and centrifuging (1400 rcf x 3 min, 4 °C) to form a cell pellet. After removal of PBS supernatant, cell pellets were either stored at −80 °C or immediately lysed by sonication. For lysis, cells were first resuspended in 1–2 mL ice cold PBS (10–20 × 106 cells/mL) containing protease inhibitor cocktail (1x, EDTA-free, Cell Signaling Technology # 5871S) and PMSF (1 mM, Sigma # 78830). These samples were then lysed by sonication using a 100 W QSonica XL2000 sonicator (3 × 1s pulse, amplitude 1, 60s resting on ice between pulses). Lysates were pelleted by centrifugation (14,000 rcf x 30 min, 4 °C) and quantified on a Qubit 2.0 Fluorometer using a Qubit Protein Assay Kit. Quantified proteomes were diluted to 2 mg/mL and stored in 1 mg aliquots at −80 °C for chemoproteomic or enzyme activity analyses. For the studies involving pH-dependence, cells were lysed in a lysis buffer containing 50 mM potassium phosphate buffer at specified pH, 1 mM PMSF and 1x protease inhibitor cocktail.
Calculation of metabolite LUMO energies
LUMO energies were calculated using the program Spartan ‘10 as previously described26. Briefly, models of each metabolite or drug were drawn using the Build command. Carboxylic acid hydrogens were deleted for fumarate, mono-methyl fumarate, and hydrogen fumarate to render them as di- and monoanions as appropriate. Calculations of the ground state equilibrium geometry for each molecule were performed using the the Hartree-Fock 3–21G model in vacuum. All calculations were performed starting from the AM1 geometry, with Global Calculations and Orbitals and Energies boxes checked.. LUMO energies were obtained by selecting Display and Orbital Energies, and are specified in units of eV.
Gel-based detection of FA-alkyne labeled proteomes
20 μg proteome was incubated with specified concentration of FA-alkyne at room temperature for the specified time. For competition experiments, 1 mg proteome (0.5 mL, 2 mg/mL) was pre-incubated with the competitor (1 mM) for 3 h, followed by 15 h treatment with FA-alkyne (100 μM). Proteomes were then desalted using Illustra™ NAP-5 columns (GE Healthcare # 17085301) to remove unreacted reagents and 20 μg proteomes were used for analysis. Proteins labeled by FA-alkyne were visualized by SDS-PAGE via Cu(I)-catalyzed [3 + 2] cycloaddition with a fluorescent azide as previously reported42 Briefly, TAMRA-azide (100 μM; 5 mM stock solution in DMSO), TCEP (1 mM; 100 mM stock in H2O), tris-(benzyltriazolylmethyl)amine ligand (TBTA; 100 μM ; 1.7 mM stock in DMSO:tert-butanol 1:4), and CuSO4 (1 mM; 50 mM stocks in H2O) were sequentially added to the labeled proteome. Reactions were vortexed, incubated at room temperature for 1 hour, quenched by addition of 4x SDS-loading buffer (strongly reducing) and analyzed by SDS-PAGE. Gels were fixed and destained in a solution of 50% MeOH/40% H2O/10% AcOH overnight to remove excess probe fluorescence, rehydrated with water, and visualized using an ImageQuant Las4010 (GE Healthcare) with green LED excitation (λmax 520–550 nm) and a 575DF20 filter.
Chemoproteomic labeling and enrichment of FH-regulated cysteines
For identification of FH-regulated cysteines (Dataset S1), 2 mg of UOK262 or UOK262WT proteomes (1 mL, 2 mg/mL) were labeled with 100 µM IA-alkyne (10 μL, 10 mM stock in DMSO) for 1 h at room temperature. For enrichment of FH-regulated cysteines, probe labeled proteins were then conjugated to light (low fumarate proteomes: vehicle-treated HEK-293 or UOK262WT) or heavy (high fumarate proteomes: fumarate-treated HEK-293 or UOK262) diazobenzene biotin-azide (azo) tag by Cu(I)-catalyzed [3 + 2] cycloaddition as previously reported.20 Briefly, azo-tag (100 μM), TCEP (1 mM), TBTA (100 μM), and CuSO4 (1 mM) were sequentially added to the labeled proteome. Reactions were vortexed and incubated at room temperature for 1 h. Proteomes labeled with heavy and light azo-tags were then combined pairwise and centrifuged (6500 rcf x 10 min, 4 °C) to collect precipitated protein. Supernatant was discarded, and protein pellets were resuspended in 500 μL of methanol (dry-ice chilled) with sonication, and centrifuged (6500 rcf x 10 min, 4 °C). This step was repeated, and the resulting washed pellet was redissolved (1.2% w/v SDS in PBS; 1 mL); sonication followed by heating at 80–95 °C for 5 min was used to ensure complete solubilization. Samples were cooled to room temperature, diluted with PBS (5.0 mL), and incubated with Streptavidin beads (100 μL of 50% aqueous slurry per enrichment) overnight at 4 °C. Samples were allowed to warm to room temperature, pelleted by centrifugation (1400 rcf x 3 min), and supernatant discarded. Beads were then sequentially washed with 0.2% SDS in PBS (5 mL x 1), PBS (5 mL x 3) and H2O (5 mL x 3) for a total of 7 washes. For identification of fumarate-sensitive cysteines (Dataset S4), 2 mg of HEK-293 proteomes (1 mL, 2 mg/mL) were incubated with 1 mM fumaric acid (10 μL, 100 mM stock in DMSO) or vehicle (DMSO, 10 μL) overnight at room temperature, followed by labeling with 100 μM IA-alkyne (10 μL, 10 mM stock in DMSO) for 1 h, desalted using NAP-5 columns to remove unreacted reagents, and further processed via click chemistry and streptavidin enrichment as noted above. An identical experiment performed using 1 mM succinic acid (10 μL, 100 mM stock in DMSO) was used to identify and control for potential non-covalent effects of fumarate, such as metabolism or non-specific cysteine reactivity losses occurring during the 15 h incubation.
On bead reductive alkylation, tryptic digest and diazobenzene cleavage of proteomic samples
Following the final wash, protein-bound streptavidin beads were resuspended 6 M urea in PBS (500 μL) and reductively alkylated by sequential addition of 10 mM DTT (25 μL of 200 mM in H2O, 65 °C for 20 min) and 20 mM iodoacetamide (25 μL of 400 mM in H2O, 37 °C for 30 min) to each sample. Reactions were then diluted by addition of PBS (950 μL), pelleted by centrifugation (1400 rcf x 3 min), and the supernatant discarded. Samples were then subjected to tryptic digest by addition of 200 μL of a pre-mixed solution of 2M urea in PBS, 1 mM CaCl2 (2 μL of 100 mM in H2O), and 2 μg of Trypsin Gold (Promega, 4 μL of 0.5 μg/μL in 1% acetic acid). Samples were shaken overnight at 37 °C and pelleted by centrifugation (1400 rcf x 3 min). Beads were then washed sequentially with PBS (500 µL x 3) and H2O (500 µL x 3). Labeled peptides were eluted from the beads by sodium dithionite mediated cleavage of the diazobenze of the azo-tag. For this, beads were incubated with freshly prepared 50 mM sodium dithionite in PBS (50 µL) for 1 h at room temperature. Beads were pelleted by centrifugation (1400 rcf x 3 min) and supernatant was transferred to a new Eppendorf tube. The cleavage process was repeated twice more with 50 mM sodium dithionite (75 µL) and supernatants were combined with the previous. The beads were additionally washed two times with water (75 µL) and supernatants were collected and combined with previous. Formic acid (17.5 µL) was added to the combined supernatants and samples were stored at −20 °C until ready for LC-MS/MS analysis.
LC-MS/MS and data analysis for quantitative cysteine reactivity profiling
Mass spectrometry was performed using a Thermo LTQ Orbitrap Discovery mass spectrometer coupled to an Agilent 1200 series HPLC. Labeled peptide samples were pressure loaded onto 250 mm fused silica desalting column packed with 4 cm of Aqua C18 reverse phase resin (Phenomenex). Peptides were eluted onto a 100 mm fused silica biphasic column packed with 10 cm C18 resin and 4 cm Partisphere strong cation exchange resin (SCX, Whatman), using a five step multidimensional LC-MS protocol (MudPIT). Each of the five steps used a salt push (0%, 50%, 80%, 100%, and 100%), followed by a gradient of buffer B in Buffer A (Buffer A: 95% water, 5% acetonitrile, 0.1% formic acid; Buffer B: 20% water, 80% acetonitrile, 0.1% formic acid) as outlined previously.16 The flow rate through the column was ~0.25 µL/min, with a spray voltage of 2.75 kV. One full MS1 scan (400–1800 MW) was followed by 8 data dependent scans of the nth most intense ion. Dynamic exclusion was enabled. The tandem MS data, generated from the 5 MudPIT runs, was analyzed by the SEQUEST algorithm.50 Static modification of cysteine residues (+57.0215 m/z, iodoacetamide alkylation) was assumed with no enzyme specificity. The precursor-ion mass tolerance was set at 50 ppm while the fragment-ion mass tolerance was set to 0 (default setting). Data was searched against a human reverse-concatenated non-redundant FASTA database containing Uniprot identifiers. MS datasets were independently searched with light and heavy azo-tag parameter files; for these searches differential modifications on cysteine of +456.2849 (light) or +462.2987 (heavy) were used. MS2 spectra matches were assembled into protein identifications and filtered using DTASelect2.051, to generate a list of protein hits with a peptide false-discovery rate of <5%. With the –trypstat and –modstat options applied, peptides were restricted to fully tryptic (-y 2) with a found modification (-m 0) and a delta-CN score greater than 0.06 (-d 0.06). Single peptides per locus were also allowed (-p 1) as were redundant peptides identifications from multiple proteins, but the database contained only a single consensus splice variant for each protein. Quantification of L/H ratios were calculated using the cimage quantification package described previously16.
Whole proteome protein abundance analysis
100 µg of UOK262 or UOK262WT proteomes (100 µL, 1 mg/mL) were precipitated by the addition of 5 µL 100% trichloroacetic acid in PBS, vortexed and frozen at −80 °C overnight. Samples were thawed, and proteins were pelleted by centrifugation (17000 rcf x 10 min). Each protein pellet was washed by resuspension in acetone (500 µL) using sonication, followed by centrifugation (2200 rcf x 10 min). Supernatant was discarded, pellet was allowed to dry and then resuspended thoroughly by sonication in 30 µL 8M urea in PBS. Reductive alkylation was then performed by sequential addition of 70 µL of 100 mM ammonium bicarbonate and 1.5 µL of 1M DTT (65 °C for 15 min) and iodoacetamide (2.5 μL of 400 mM in H2O, room temperature for 30 min). Reactions were then diluted by addition of PBS (120 μL) and tryptic digest was performed by addition of 2 μg of Trypsin Gold and and 2.5 µL of 100 mM CaCl2, followed by overnight incubation at 37 °C. Trypsin was quenched by addition of 10 µL formic acid (~5% final volume) and undigested protein was pelleted by centrifugation (17000 rcf x 20 min). Supernatant was collected and stored at −20 °C until ready for LC-MS/MS analysis which was performed using ~50 µL of each sample. LC-MS/MS was performed as described above with slight modification to MudPIT protocol. Here salt pushes of 0%, 25%, 50%, 80%, and 100% were employed. Tandem MS data analysis was performed as described above, without the –modstat option applied. Spectral counting was used for calculating the UOK262WT:UOK262 protein abundance ratios for those proteins which had >10 spectral counts in at least one of the two cell lines and these ratios were used to correct the FH-regulated cysteine ratios wherever possible.
Bioinformatic analysis of FH-regulated cysteines
Annotation of protein subcellular localization as well as cysteine function and conservation was generated from the Uniprot Protein Knowledgebase (UniProtKB) as described previously.52 Analysis of linear sequences flanking FH-regulated cysteines was performed using the informatics tool pLogo, accessible at: https://plogo.uconn.edu. Input sequences are listed in Dataset S3, and were derived from the 50 cysteines found to be most FH-regulated in this study, (highest R values, n≥2, SD≤25%), the 50 cysteines found to be most hyperreactive,16 DMF-sensitive,19 MMF-sensitive,19 and HNE-sensitive25 in literature datasets, and the 50 cysteines found to be most sensitive to addition of exogenous fumarate (Dataset S4). Protein sequences for motif analysis were derived from their tryptic peptide sequences using Peptide Extender (schwartzlab.uconn.edu/pepextend). Conservation and functional impact of FH-regulated cysteines identified in chemoproteomic experiments was analyzed using the informatics tool Mutation Assessor, accessible at: http://mutationassessor.org/r3. Conservation analysis depicted in Fig. 2d represents the output of the variant conservation (VC) score for the 50 cysteines found to be most FH-regulated in this study (highest L/H ratio [R] values, n≥2, SD≤25%).), the 50 cysteines found to be most FH-neutral in this study (R values closest to 1), and the 50 cysteines found to be most hyperreactive in a previous chemoproteomic study performed by Weerapana et al.16 Solvent accessible surface area (SASA) calculations were performed using Naccess (Hubbard, S.J. & Thornton, J.M. (1993), ‘NACCESS’, Computer Program, Department of Biochemsitry and Molecular Biology, University College London.). Naccess calculates atomic accessible surface areas using a previously reported method53 in which a spherical probe is rolled over the entire Van der Waal’s surface of the macromolecule. The accessible surface area is then calculated from the path traced out from the center of the spherical probe, using a radius of 1.4 A, which is equivalent to water. Available pdb structures for the top 50 FH-regulated (n=22), fumarate-sensitive (n = 20), fumarate-insensitive (n = 23), and hyperreactive16 (n = 22) cysteine residues were used to generate data for analysis. Potential functional impact of fumarate modifications (Fig. 5a, Dataset S3) reflects the effect of C to E mutations on the functional impact (FI) output of Mutation Assessor. Gene ontology analysis was performed using the bioinformatics tool DAVID, accessible at: http://david.ncifcrf.gov/. Output tables in Dataset S3 reflect DAVID analysis of FH-regulated cysteines predicted to have a medium or high impact on protein function by Mutation Assessor. Candidate functional fumarate targets were assessed for cases of genomic alteration in renal cell carcinoma (clear cell and non-clear cell) using cBioPortal (http://cbioportal.org). Structural analysis of candidate functional fumarate targets known to undergo genomic alteration in renal cell carcinoma was performed using Chimera. For gene set enrichment analysis (GSEA, Fig. 6), R values for FH-regulated peptides were log2-transformed and analyzed for 1000 permutations using the Broad Institute’s javaGSEA desktop application (http://software.broadinstitute.org/gsea/ downloads.jsp). For proteins in which R values were measured for more than one cysteine-containing peptide, the peptide with the greatest absolute R value was used for GSEA analysis. GSEA outputs were re-plotted for graphics using a variant of ReplotGSEA package, accessible at: https://github.com/PeeperLab/Rtoolbox/blob/ master/R/ReplotGSEA.R.
Validation of FH-regulated targets using FA-alkyne
5 mg of HEK-293 proteome (2.5 mL, 2 mg/mL) was pre-treated with 1 mM fumaric acid (25 μL, 100 mM stock in DMSO) or DMSO for 3 hours prior to incubation with 100 μM FA-alkyne (25 μL, 10 mM stock in DMSO) for 15 hours. Proteomes were then desalted using Illustra™ NAP-25 columns (GE Healthcare # 17085201) to remove unreacted reagents. Labeled proteomes were enriched via Cu(I)-catalyzed [3 + 2] cycloaddition with biotin-azide as described above for chemoproteomic analysis. Following the final wash, enriched resin was collected on top of centrifugal filters (VWR, 82031–256). Proteins were eluted from resin via addition of 40 μL 1x SDS sample buffer, followed by boiling for 10 min at 95 °C. Following repetition of the elution step, both eluents were combined and 20 μL of the combined eluent was loaded onto a 4–12% SDS-PAGE gel and analyzed by western blotting.
Fluorescent quantification of fumarate release from S-succinated thiols
S-succinated thiols (1 mM final concentration, 5 μL of 20 mM stock in DMSO) were incubated in TRIS buffer (100 mM; pH 7, 7.5, and 8) at 37 °C for 24 h. After incubation, reactions were developed by treatment with equal volume of hydrazonyl chloride 4 from Zengeya et al.26 (150 μM final concentration, 300 μM stock in CH3CN) for 1 h at room temperature. Fluorescence produced was then measured on Photon Technology International QuantMaster fluorimeter using 1-cm path length, 0.13 mL quartz microcuvettes (Helma #101–015-40) at ambient temperature (22 ± 2 °C), using an excitation wavelength of 390 nm, slit width of 3.5 nm, and monitoring emission from 410 nm to 615 nm. Fluorescence emission values at 530 nm were used to calculate percent DMF released by interpolating into a standard curve of DMF reacting hydrazonyl chloride 4 under identical conditions.
Determination of pH-dependent S-succination kinetics
The influence of pH on S-succination reaction kinetics were determined using the model nucleophile thiophenol. Reactions consisted of 3 mM thiophenol (10 μL, 60 mM stock in CH3CN), 15–45 mM fumaric acid (10 μL, 1200–300 mM stock in DMSO), 0.2 mM 7-diethylamino-4-methylcoumarin (2 μL, 10 mM stock in DMSO), and 100 mM tris(hydroxymethyl)phosphine (THP; 5 μL, 400 mM stock in water) in a final volume of 200 μL PBS pH 7.2. pH was adjusted to 5.5, 6.5, 7, and 7.5 using 2 M NaOH or 1 M HCl as necessary and checked with pH strips (Millipore MColorpHast pH 2.0–9.0). Reactions were initiated by addition of thiophenol (t=0) and 10 μL reaction aliquots were analyzed using an Agilent Technologies 1260 Infinity HPLC equipped with a Phenomenex Kinetex C18 column (2.6 μm, 100 Angstrom, 100 × 2.1 mm inner diameter) and UV detector with monitoring at 254 nm. Solvents used were 0.1% TFA in H2O (Solvent A) and CH3CN (solvent B). Compounds were eluted at a flow rate of 0.5 mL/minute. The method utilized an isocratic step from 0 to 2 minutes with 100% A, followed by a linear gradient to 50% B over 15 minutes, followed by an increasing gradient with solution B until at 20 minutes the solvent composition was 100% solution B. A representative HPLC trace is provided in the Supplementary Information. DMF alkylation reaction kinetics were analyzed similarly, substituting 1 mM DMF as an electrophile. Cysteine S-succination kinetics of were determined similarly, substituting 3 mM Fmoc-Cys-OH (Iris Biotech) as a nucleophile. The retention times of S-succinated thiophenol, DMF-modified thiophenol, and Fmoc-Cys-OH S-succinated were determined using synthetic standards. Reactions were monitored for 15–24 hour. The rate of S-succinated product formation was assessed by calculating the integrated absorbance of the product peak at 254 nm relative to the integrated absorbance of the 7-diethylamino-4-methylcoumarin internal standard at 254 nm. Kinetic traces are available in the Supplementary Information. Data were fit to a one phase exponential association equation Y = (Ymax*e(-k*t) where Y is the normalized absorbance at a given time, Ymax is the maximum absorbance, t is time in minutes, and k is the apparent rate constant (kapp) for the S-succination reaction (in units of h−1).. Data values were constrained to a plateau of 100%. kapp for each reaction condition was plotted against fumarate concentration and a linear regression was performed, the slope of which was converted to M−1 s−1 and considered the estimated second order rate constant for the reaction at a given pH.
Intracellular pH measurement
For cytosolic pH measurement, cells were plated in quadruplicates in Corning BioCoat Poly-D-Lysine 96-well plates (Corning, 354640) at plating densities of 3 ×104 cells perwell for HEK-293 cells and 1 ×104 cells per well for UOK262 FH −/− and UOK262 FH rescue cells. Cells were allowed to adhere for 24 h and incubated with 10 µM of pHrodo™ green AM (Life Technologies, P35373) intracellular pH indicator at 37 °C for 30 mins as per manufacturer’s protocol. Standard curves of cytosolic pH were generated using pHrodo™ green AM intracellular pH calibration buffer kit (Life Technologies, P35379) that contains buffers at pH 4.5, 5.5, 6.5 and 7.5, as well as valinomycin and nigericin to help equilibriate the pH inside and outside the cells. The fluorescence was then measured using a Biotek Synergy MX plate reader, with pHrodo™ green AM detected using λex of 509 nm and λem of 533 nm as per the manufacturer’s instructions.
Ectopic expression and co-immunoprecipitation of SMARCC1 and SNF5
HEK-293 cells were plated in 10 cm dishes (3 ×106 cells/dish in 10 ml DMEM media/well), and allowed to adhere and grow for 24 h. FLAG-tagged SNF5 was co-overexpressed with Myc-tagged GFP, SMARCC1, SMARCC1-C520E, or SMARCC1-C520S using Lipofectamine 2000 (Invitrogen # 11668019) according to the manufacturer’s instructions. Co-overexpressions were carried out by incubating the cells for 48 h at 37 °C under 5% CO2 atmosphere, after which the cells were harvested, soluble proteome isolated and quantified as described above. Anti-FLAG pulldown was performed using immunoprecipitation kit (KBA-319–383) according to the manufacturer’s instruction. 1 mg of the lysate was incubated with the anti-FLAG resin overnight at 4 °C. Purified protein was ran on SDS-PAGE and immunoblotted against anti-Myc-tag and anti-FLAG-tag.
Analysis of effects of SMARCC1 overexpression on endogenous SNF5
HEK-293 cells were plated in 6-well dishes (6 ×105 cells/well in 3 ml DMEM media/well), and allowed to adhere and grow for 24 h. At this point, transient transfection of plasmids encoding for Myc-tagged GFP, SMARCC1 or SMARCC1(C520E) was performed using lipofectamine 2000 (Invitrogen # 11668019) according to the manufacturer’s instructions. Overexpression was carried out by incubating the cells for 48 h at 37 °C under 5% CO2 atmosphere. For the cycloheximide treatment experiment, overexpression was carried out for 96 h. After 96 h, media was changed and cells were incubated with 200 μg/mL cycloheximide or vehicle for additional 24 h. After the treatment, cells were harvested, and soluble proteome was isolated and quantified as described above. 10 μg of lysates were loaded per lane of the gel for the western blot analysis of endogenous SNF5 and expression levels of Myc-tagged GFP, SMARCC1 or SMARCC1(C520E).
Co-immunoprecipitation of endogenous SMARCC1 and BRG1 in HLRCC cells
For co-immunoprecipitation of endogenous SMARCC1 and BRG1, whole cell lysates from HLRCC cells were first prepared by resuspending cell pellets in IP-buffer containing 50 mM Tris pH 8, 400 mM NaCl, 2 mM EDTA, 10% glycerol, 1% NP-40 (Ipegal® CA-630, Sigma # I8896), 1 mM PMSF and 1X protease inhibitor cocktail. The lysates were pelleted by centrifugation (14,000 rcf x 30 min, 4 °C) and pre-cleared by incubating with protein A/G plus agarose resin (30 μL) for 1 h at 4 °C. Pre-cleared supernatant was collected by centrifugation (10,000 rcf x 5 min, 4 °C) and diluted to 1 mg/mL concentration. For each co-immunoprecipitation, 2 mg of whole cell proteome was incubated with 2.5 μg/mL of SMARCC1 (sc-32763) or BRG1 (sc-17796) antibody at 4 °C for 1 h. Protein A/G plus agarose resin (100 μL) was added to each sample and incubated overnight at 4 °C. Samples were pelleted by centrifugation, supernatant was discarded, and beads were then washed with IP-buffer (1 mL x 3). Enriched proteins were eluted from resin via addition of 40 μL 1× SDS sample buffer, followed by boiling for 10 min at 95 °C. Following repetition of the elution step, both eluents were combined and 20 μL of the combined eluent was loaded onto a 4–12% SDS-PAGE gel and analyzed by western blotting.
Validation of S-succination of fumarate-sensitive cysteine residues by MUDPIT LC-MS/MS
TCA-precipitated protein samples from whole cell extracts from UOK262 and UOK268 FH −/− cells were analyzed independently in triplicate by Multidimensional Protein Identification Technology (MudPIT), as described previously.54 After recombinant endoproteinase LysC and trypsin digestions, peptide mixtures were pressure-loaded onto 100 µm fused silica microcapillary columns packed first with 9 cm of reverse phase material (Aqua; Phenomenex), followed by 3 cm of 5-μm Strong Cation Exchange material (Luna; Phenomenex), followed by 1 cm of 5-μm C18 RP. The loaded microcapillary columns were placed in-line with a 1260 Quartenary HPLC (Agilent). The application of a 2.5 kV distal voltage electrosprayed the eluting peptides directly into Orbitrap-Velos Pro or Elite hybrid mass spectrometers (Thermo Scientific) equipped with a custom-made nano-LC electrospray ionization source. Full MS spectra were recorded on the eluting peptides over a 400 to 1600 m/z range in the Orbitrap at 60K resolution, followed by fragmentation in the ion trap (at 35% collision energy) on the first to fifteenth most intense ions selected from the full MS spectrum. Dynamic exclusion was enabled for 90 sec.55 Mass spectrometer scan functions and HPLC solvent gradients were controlled by the XCalibur data system (Thermo Scientific). RAW files were extracted into .ms2 file format using RawDistiller v. 1.0.56 RawDistiller D(g, 6) settings were used to abstract MS1 scan profiles by Gaussian fitting and to implement dynamic offline lock mass using six background polydimethylcyclosiloxane ions as internal calibrants56. MS/MS spectra were first searched using ProLuCID57 with a peptide mass tolerance of 10 ppm and 500 ppm for fragment ions. Trypsin specificity was imposed on both ends of candidate peptides during the search against a protein database combining 36,628 human proteins (NCBI 2016–06-10 release), as well as 193 usual contaminants such as human keratins, IgGs and proteolytic enzymes. To estimate false discovery rates (FDR), each protein sequence was randomized (keeping the same amino acid composition and length) and the resulting “shuffled” sequences were added to the database, for a total search space of 73,642 amino acid sequences. Masses of 57.0215 Da and 116.0112 Da were differentially added to cysteine residues to account for alkylation by CAM and succination, respectively, while 15.9949 Da were differentially added to methionine residues. DTASelect v.1.951 was used to select and sort peptide/spectrum matches (PSMs) passing the following criteria set: PSMs were only retained if they had a DeltCn of at least 0.08; minimum XCorr values of 1.8 for singly-, 2.0 for doubly-, and 3.0 for triply-charged spectra; peptides had to be at least 7 amino acids long. Results from each sample were merged and compared using CONTRAST51. Combining all six runs, proteins had to be detected by at least 2 peptides and/or 4 spectral counts. Proteins that were subsets of others were removed using the parsimony option in DTASelect on the proteins detected after merging all runs. Proteins that were identified by the same set of peptides (including at least one peptide unique to such protein group to distinguish between isoforms) were grouped together, and one accession number was arbitrarily considered as representative of each protein group. NSAF7 58 was used to create the final reports on all detected peptides and non-redundant proteins identified across the different runs. Spectral and protein level FDRs were, on average, 0.29 ± 0.04% and 2.7 ± 0.4%, respectively. NSAF7 was also used to generate a list of all peptide to spectrum matches (PSMs) leading to the identification of succinylated proteins. NSAF7 was used to create PDF files displaying fully annotated MS/MS spectra (shown below) matching the modified peptides listed in Dataset S5.
Analysis of SWI/SNF complex composition by glycerol gradient fractionation in HLRCC cells
UOK262 FH −/− and FH+/+ rescue cells were grown to 90% confluency in 2 × 15 cm dishes per cell line. Cells were harvested by trypsinization and washed once in ice-cold PBS. Nuclei were isolated by incubating the cell pellets in Buffer A (20 mM HEPES pH 7.9, 25 mM KCl, 10% glycerol, 0.1% NP-40, 1 mM DTT with PMSF, aprotinin, leupeptin and pepstatin) for 7 min. Nuclei were pelleted and washed in buffer A without NP-40. Washed and pelleted nuclei were resuspended in Buffer C (10 mM HEPES, pH 7.6, 3 mM MgCl2, 100 mM KCl, 0.1 mM EDTA, 10% glycerol, 1 mM DTT with PMSF, aprotinin, leupeptin and pepstatin). Ammonium sulfate was added to 0.3 M final concentration. Samples were incubated in a rotating wheel at 4 °C for 30 min and cleared by ultracentrifugation (150000 rcf x 30 min). 300 mg of ammonium sulfate powder was introduced per mL of cleared lysate. After ice incubation for 20 min, proteins were precipitated by ultracentrifugation (150000 rcf x 30 min). Pelleted proteins were resuspended in 100 μL HEMG1000 buffer (25 mM HEPES pH 7.6, 0.1 mM EDTA, 12.5 mM MgCl2, 100 mM KCl, 1 mM DTT with PMSF, aprotinin, leupeptin and pepstatin). 400 μg of resuspended proteins were layered over 10 mL, 10–30% glycerol gradient, prepared with HMG1000 buffer without glycerol or with 30% glycerol, and separated by centrifugation at 40000 rpm (Beckman Coulter XL-100K, Brea, CA) for 16 h using SW32Ti rotor (Beckman Coulter, Brea, CA). 500 μL-fractions were collected and analyzed by western blotting using antibodies against BRG1 (Abcam, ab110641) and SNF5 (Santa Cruz Biotechnology, sc-166165).
Identification and validation of genes co-regulated by FH and SNF5
To identify SNF5-sensitive genes from renal carcinoma cell lines, RNA-Seq data was analyzed from published datasets (GEO: GSE71505) which examined the Wilms’ kidney tumor cell line G401 with and without SNF5 re-expression (n = 2 for each condition), and the renal leiomyoblastoma cell line G402 with and without SNF5 re-expression (n = 3 for each condition). To identify FH-sensitive genes from renal carcinoma cell lines, RNA-Seq data was analyzed from published datasets (GEO: GSE77542) which examined the HLRCC cell line UOK262 with and without FH re-expression (n = 17 for control cells, n = 10 for FH re-expression). The resulting reads were trimmed using Trimmomatic utility (Bolger et al. 2014) and mapped to hg19 using STAR (Dobin et al. 2013) using default parameters. Read counts were obtained using HTSeq-count (Anders et al. 2015) in conjunction with a standard gene annotation files from UCSC (University of California Santa Cruz; http://genome.ucsc.edu) and differential expression was determined using DESeq2 pipeline (Love et al. 2014). Differentially expressed genes were filtered using a false discovery rate threshold of < 0.05 and a fold change threshold of > 1.5-fold relative to the reference sample. To determine whether similar sets of genes may be regulated by FH in UOK262 as is regulated by SNF5 in G401 and G402, two pediatric renal cancers, overlapping gene sets were compared. Significant overlap between datasets was obtained by comparing the observed number of overlapping genes to the number of overlapping genes predicted by chance (based on the total number of expressed genes in each cell line). The largest overlap was observed for genes upregulated by SNF5 re-expression in G401 and G402 cell lines, with the next most significant overlaps between genes upregulated by FH re-expression in UOK262 and SNF5 re-expression in G402 and G401 cell lines, respectively (Fig. S5j and Dataset S7).
Comparing genes upregulated by tumor suppressor re-expression in all three datasets resulted in identification of 46 genes upregulated by SNF5 in both G401 and G402 cell lines, and FH in UOK262 cells (see figure for Venn diagram and excel spreadsheet for a list of genes). Out of these, 9 genes were selected for validation in the UOK262 and UOK268 cell lines with FH re-expression (Fig. S5k). LRRC15 was upregulated by FH re-expression in both cells lines, and was one of the most robustly increased by SNF5 re-expression in both G401 and G402 cells. Therefore, we next analyzed whether SNF5 knockdown in the UOK262 cell line would similarly regulate the expression of this gene. Using qRT-PCR in three independent experiments (3 replicates per experiment), two independent SNF5 knockdown constructs decreased both SNF5 expression and LRRC15 expression (Figure S5l). This supports a mechanism by which FH regulates at least a subset of overlapping genes as SNF5.
SNF5 knockdown in UOK262 cells
Two SNF5 targeting short hairpin vectors (TRCN0000295966 as sh47#2; TRCN0000298820 as sh47#4) were purchased from Sigma. Control vector (pLKO.1 puro) was a gift from Bob Weinberg (Addgene plasmid # 8453). The vectors were packaged into lentivirus using HEK293T cells, and the viral particles were concentrated by ultracentrifugation. A hundred thousand UOK262 or UOK262WT cells were plated on 12-well plate and transduced. Following 24 hour-post transduction, medium was refreshed. Forty-eight hours post transduction cells were selected using puromycin (2 μg/ml) for three days.
qRT-PCR analysis of SWI/SNF expression and SNF5-regulated genes in HLRCC cells
RNA was extracted from transduced and selected cells using Trizol. Five hundred nanograms of total RNA was reverse-transcribed using Verso cDNA synthesis kit (Thermo Scientific, Waltham, MA). qPCR was performed with Maxima SYBR Green Master Mix (Thermo Scientific). qPCR primers are SNF5 forward: 5’- ATCGTCACATGLCATCACGGATAC, reverse: 5’- GGACACAGCCTTGTACTTCTC; SMARCC1 forward: 5’- CACCCCAGCCAGGTCAGAT, reverse: TGCAACAGTGGGAATCATGC; ACTB forward: CATGTACGTTGCTATCCAGGC, reverse: CTCCTTAATGTCACGCACGAT. UBC forward: 5’-ATTTGGGTCGCGGTT CTTG, reverse: 5’-TGCCTTGACATTCTCGATGGT; LRRC15 forward: 5’- GGGCTTCTGAAGATGGACTTAC, reverse: 5’-CCTGTCCACACCCACATATTC; CX3CL1 forward: 5’- GAAAGGAAAGAGGGAGGTAAGG, reverse: 5’-CTAAGGTGCTCTGCTGGTAAG; DOCK11 forward: 5’- CTCAGAAGGGTGGTGTGATAAA, reverse: 5’- GTCAGGAAGTTGGGTCAAGTAA; TGM2 forward: 5’-TGTTGGTCAGAGGAGTGATTG, reverse: 5’-GGAGTGGACCTTGTGGTTATT; CEACAM1 forward: 5’- ATCTCCATCCGTTGGTTCTTC, reverse: 5’-CTCCCTCTTGACAGGGTTTATG; IRAK2 forward: 5’- CCGGTTTACCTGAAGGACTTAC, reverse: 5’-TCCTTTGCCATCACGTTCTC; PLAT forward: 5’- GAGGCCTTGTCTCCTTTCTATTC, reverse: GTCGGTGACTGTTCTGTTAAGT; LRRC32 forward: 5’- CTTCATACTGGTCTCTGCCATC, reverse: CCCGGCTTCTTTAGGCTTTA; IL6R forward: 5’
Inhibition of HLRCC spheroid growth by EZH2 inhibitors
For tumor spheroid formation, a total of 5000 single cell suspensions were plated in 100 μL of complete media (DMEM supplemented with 20% FBS, 1x MEM non-essential amino acids, and 1x Anti-Anti) into each well of a 96-well ultra-low attachment plates (Corning 3603). After 3 days in culture, tumor spheroid formation was confirmed visually using the EVOS XL Core Cell Imaging System (Thermo Fisher Scientific). On day 0, 100 μL of media containing 2x concentration of drug was added to the wells diluting the compound to the indicated concentration. Every 3 or 4 days, 100 μL of media was removed and replaced with 100 μL of media with 2x concentration of drug. The spheroids were treated for 21 days. The spheroids where then dissociated with Cell Titer Glo 3D (Promega # G9681) following manufacturer’s instructions. The plates were then read on an Enspire Mulitmode Plate Reader (Perkin-Elmer).
Supplementary Material
Acknowledgements
The authors thank Dr. Carissa Grose (Protein Expression Laboratory) for cloning and preparation of plasmid DNA, Dr. Trevor Archer (NIEHS) for the gift of the SMARCC1 and SNF5 plasmids, Allison Roberts, Julie Garlick, and Dr. Thomas. Zengeya (NCI) for assisting with preliminary studies. This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (ZIA BC011488–05, ZIA BC011038–10) bnd the CCR FLEX Program. Support for E.W. was provided by the NIH (1R01GM117004 and 1R01GM118431–01A1).
Footnotes
Materials and Methods
Detailed materials and methods, including cell lines, plasmids, antibodies, chemicals, and proteomic protocols are available in the Online Methods.
Additional Resources
All of the data are accessible in the supplemental datasets (Tables S1-S6) and can further be explored using our web-based resource (ccr2.cancer.gov/resources/Cbl/proteomics/fumarate).
Competing interests
The authors declare no competing financial interests.
Data availability
The authors declare that all data supporting the findings of this study are available within the paper and its supplementary information files. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the identifiers PXD009378 (Datasets S1–S4) and PXD009202 (Dataset S5).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
References
- 1.Launonen V et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A 98, 3387–3392, 10.1073/pnas.051633798 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomlinson IP et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30, 406–410, 10.1038/ng849 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Meier JL Metabolic mechanisms of epigenetic regulation. ACS Chem Biol 8, 2607–2621, 10.1021/cb400689r (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Isaacs JS et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8, 143–153, 10.1016/j.ccr.2005.06.017 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Pollard PJ et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 14, 2231–2239, 10.1093/hmg/ddi227 (2005). [DOI] [PubMed] [Google Scholar]
- 6.MacKenzie ED et al. Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol Cell Biol 27, 3282–3289, 10.1128/MCB.01927-06 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sciacovelli M et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537, 544–547, 10.1038/nature19353 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alderson NL et al. S-(2-Succinyl)cysteine: a novel chemical modification of tissue proteins by a Krebs cycle intermediate. Arch Biochem Biophys 450, 1–8, 10.1016/j.abb.2006.03.005 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Sullivan LB et al. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell 51, 236–248, 10.1016/j.molcel.2013.05.003 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sudarshan S et al. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol 29, 4080–4090, 10.1128/MCB.00483-09 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kinch L, Grishin NV & Brugarolas J Succination of Keap1 and activation of Nrf2-dependent antioxidant pathways in FH-deficient papillary renal cell carcinoma type 2. Cancer Cell 20, 418–420, 10.1016/j.ccr.2011.10.005 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bardella C et al. Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J Pathol 225, 4–11, 10.1002/path.2932 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Piroli GG et al. Succination is Increased on Select Proteins in the Brainstem of the NADH dehydrogenase (ubiquinone) Fe-S protein 4 (Ndufs4) Knockout Mouse, a Model of Leigh Syndrome. Mol Cell Proteomics 15, 445–461, 10.1074/mcp.M115.051516 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adam J et al. Fumarate Hydratase Deletion in Pancreatic beta Cells Leads to Progressive Diabetes. Cell Rep 20, 3135–3148, 10.1016/j.celrep.2017.08.093 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ternette N et al. Inhibition of Mitochondrial Aconitase by Succination in Fumarate Hydratase Deficiency. Cell Rep 3, 689–700, 10.1016/j.celrep.2013.02.013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weerapana E et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795, 10.1038/nature09472 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Backus KM et al. Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574, 10.1038/nature18002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt TJ, Ak M & Mrowietz U Reactivity of dimethyl fumarate and methylhydrogen fumarate towards glutathione and N-acetyl-L-cysteine--preparation of S-substituted thiosuccinic acid esters. Bioorg Med Chem 15, 333–342, 10.1016/j.bmc.2006.09.053 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Blewett MM et al. Chemical proteomic map of dimethyl fumarate-sensitive cysteines in primary human T cells. Sci Signal 9, rs10, 10.1126/scisignal.aaf7694 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Y et al. Chemoproteomic Strategy to Quantitatively Monitor Transnitrosation Uncovers Functionally Relevant S-Nitrosation Sites on Cathepsin D and HADH2. Cell Chem Biol 23, 727–737, 10.1016/j.chembiol.2016.05.008 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Y et al. UOK 262 cell line, fumarate hydratase deficient (FH-/FH-) hereditary leiomyomatosis renal cell carcinoma: in vitro and in vivo model of an aberrant energy metabolic pathway in human cancer. Cancer Genet Cytogenet 196, 45–55, 10.1016/j.cancergencyto.2009.08.018 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng L et al. Fumarate induces redox-dependent senescence by modifying glutathione metabolism. Nat Commun 6, 6001, 10.1038/ncomms7001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bar-Peled L et al. Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell 171, 696–709 e623, 10.1016/j.cell.2017.08.051 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poole LB The basics of thiols and cysteines in redox biology and chemistry. Free Radic Biol Med 80, 148–157, 10.1016/j.freeradbiomed.2014.11.013 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang C, Weerapana E, Blewett MM & Cravatt BF A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat Methods 11, 79–85, 10.1038/nmeth.2759 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zengeya TT et al. Co-opting a Bioorthogonal Reaction for Oncometabolite Detection. J Am Chem Soc 138, 15813–15816, 10.1021/jacs.6b09706 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reisz JA, Bechtold E, King SB, Poole LB & Furdui CM Thiol-blocking electrophiles interfere with labeling and detection of protein sulfenic acids. FEBS J 280, 6150–6161, 10.1111/febs.12535 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doorn JA & Petersen DR Covalent modification of amino acid nucleophiles by the lipid peroxidation products 4-hydroxy-2-nonenal and 4-oxo-2-nonenal. Chem Res Toxicol 15, 1445–1450 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Winterbourn CC & Hampton MB Thiol chemistry and specificity in redox signaling. Free Radic Biol Med 45, 549–561, 10.1016/j.freeradbiomed.2008.05.004 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Reva B, Antipin Y & Sander C Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 39, e118, 10.1093/nar/gkr407 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DelBove J et al. Identification of a core member of the SWI/SNF complex, BAF155/SMARCC1, as a human tumor suppressor gene. Epigenetics 6, 1444–1453, 10.4161/epi.6.12.18492 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brugarolas J PBRM1 and BAP1 as novel targets for renal cell carcinoma. Cancer J 19, 324–332, 10.1097/PPO.0b013e3182a102d1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sohn DH et al. SRG3 interacts directly with the major components of the SWI/SNF chromatin remodeling complex and protects them from proteasomal degradation. J Biol Chem 282, 10614–10624, 10.1074/jbc.M610563200 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Yan L, Xie S, Du Y & Qian C Structural Insights into BAF47 and BAF155 Complex Formation. J Mol Biol 429, 1650–1660, 10.1016/j.jmb.2017.04.008 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Keppler BR & Archer TK Ubiquitin-dependent and ubiquitin-independent control of subunit stoichiometry in the SWI/SNF complex. J Biol Chem 285, 35665–35674, 10.1074/jbc.M110.173997 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knutson SK et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 110, 7922–7927, 10.1073/pnas.1303800110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harmel R & Fiedler D Features and regulation of non-enzymatic post-translational modifications. Nat Chem Biol 14, 244–252, 10.1038/nchembio.2575 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550, 10.1073/pnas.0506580102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Erez A & DeBerardinis RJ Metabolic dysregulation in monogenic disorders and cancer - finding method in madness. Nat Rev Cancer 15, 440–448, 10.1038/nrc3949 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Yang J, Gupta V, Carroll KS & Liebler DC Site-specific mapping and quantification of protein S-sulphenylation in cells. Nat Commun 5, 4776, 10.1038/ncomms5776 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strelko CL et al. Itaconic acid is a mammalian metabolite induced during macrophage activation. J Am Chem Soc 133, 16386–16389, 10.1021/ja2070889 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kulkarni RA et al. Discovering Targets of Non-enzymatic Acylation by Thioester Reactivity Profiling. Cell Chem Biol 24, 231–242, 10.1016/j.chembiol.2017.01.002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wagner GR et al. A Class of Reactive Acyl-CoA Species Reveals the Non-enzymatic Origins of Protein Acylation. Cell Metab 25, 823–837 e828, 10.1016/j.cmet.2017.03.006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frizzell N, Thomas SA, Carson JA & Baynes JW Mitochondrial stress causes increased succination of proteins in adipocytes in response to glucotoxicity. Biochem J 445, 247–254, 10.1042/BJ20112142 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Agaimy A et al. SWI/SNF protein expression status in fumarate hydratase-deficient renal cell carcinoma: immunohistochemical analysis of 32 tumors from 28 patients. Hum Pathol 77, 139–146, 10.1016/j.humpath.2018.04.004 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Reddie KG & Carroll KS Expanding the functional diversity of proteins through cysteine oxidation. Curr Opin Chem Biol 12, 746–754, 10.1016/j.cbpa.2008.07.028 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Li R & Kast J Biotin Switch Assays for Quantitation of Reversible Cysteine Oxidation. Methods Enzymol 585, 269–284, 10.1016/bs.mie.2016.10.006 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Jost M & Weissman JS CRISPR Approaches to Small Molecule Target Identification. ACS Chem Biol 13, 366–375, 10.1021/acschembio.7b00965 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Candiano G et al. Blue silver: a very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis 25, 1327–1333, 10.1002/elps.200305844 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Eng JK, McCormack AL & Yates JR An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spec 5, 976–989 (1994). [DOI] [PubMed] [Google Scholar]
- 51.Tabb DL, McDonald WH & Yates JR DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteom Res 1, 21–26 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bak DW, Pizzagalli MD & Weerapana E Identifying Functional Cysteine Residues in the Mitochondria. ACS chemical biology 12, 947–957, 10.1021/acschembio.6b01074 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee B & Richards FM The interpretation of protein structures: estimation of static accessibility. J Mol Biol 55, 379–400 (1971). [DOI] [PubMed] [Google Scholar]
- 54.Florens L & Washburn MP Proteomic analysis by multidimensional protein identification technology. Methods Mol Biol 328, 159–175 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Zhang Y, Wen Z, Washburn MP & Florens L Effect of dynamic exclusion duration on spectral count based quantitative proteomics. Anal Chem 81, 6317–6326 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Zhang Y, Wen Z, Washburn MP & Florens L Improving proteomics mass accuracy by dynamic offline lock mass. Anal Chem 83, 9344–9351 (2011). [DOI] [PubMed] [Google Scholar]
- 57.Xu T et al. ProLUCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J Proteomics 129, 16–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, Wen Z, Washburn MP & Florens L Refinements to label free proteome quantitation: how to deal with peptides shared by multiple proteins. Anal Chem 2010, 2272–2281 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.