

Abstract
Tissue-type plasminogen activator (tPA) activates fibrinolysis and also suppresses innate immune system responses to lipopolysaccharide (LPS) in bone marrow-derived macrophages (BMDMs) and in vivo in mice. The objective of this study was to assess the activity of tPA as a regulator of macrophage physiology in the presence of plasmin. Enzymatically-active and enzymatically-inactive (EI) tPA appeared to comprehensively block the response to LPS in BMDMs, including expression of pro-inflammatory cytokines such as TNFα and interleukin-1β and anti-inflammatory cytokines such as IL-10 and IL-1 receptor-antagonist. The activity of EI-tPA as an LPS response modifier was conserved in the presence of plasminogen. By contrast, in BMDMs treated with tPA and plasminogen or pre-activated plasmin, in the presence or absence of LPS, increased pro-inflammatory cytokine expression was observed and tPA failed to reverse the response. Plasmin independently activated NFκB, ERK1/2, c-Jun N-terminal kinase, and p38 MAP kinase in BMDMs, which is characteristic of pro-inflammatory stimuli. Plasmin-induced cytokine expression was blocked by εACA, aprotinin, and inhibitors of the known plasmin substrate, Protease-activated Receptor-1 (PAR-1), but not by N-methyl-D-aspartate Receptor inhibitor, which blocks the effects of tPA on macrophages. Cytokine expression by BMDMs treated with the PAR-1 agonist, TFLLR, was not inhibited by EI-tPA, possibly explaining why EI-tPA does not inhibit macrophage responses to plasmin and providing evidence for specificity in the ability of tPA to oppose pro-inflammatory stimuli. Regulation of innate immunity by the fibrinolysis system may reflect the nature of the stimulus and a balance between the potentially opposing activities of tPA and plasmin.
Keywords: fibrinolysis, inflammation, tissue-type plasminogen activator, plasmin, NMDA receptor, protease-activated receptor
SUMMARY SENTENCE –
Proteinases in the fibrinolysis system interact with distinct receptors in macrophages to regulate cytokine expression independently and in response to other innate immune system stimuli such as LPS.
Graphical Abstract
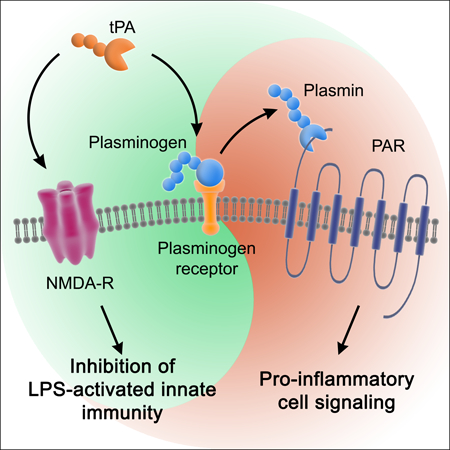
1 │. INTRODUCTION
The response to injury includes four phases: hemostasis, inflammation, proliferation, and remodeling.1–3 By lysing fibrin clots, the fibrinolysis system resolves the hemostasis phase. Tissue-type plasminogen activator (tPA) is one of the two major activators of fibrinolysis in humans and rodents.4,5 We previously demonstrated that in addition to its role in fibrinolysis, tPA suppresses innate immune system responses to lipopolysaccharide (LPS) in bone marrow-derived macrophages (BMDMs) and in vivo in mice.6 The identified pathway did not require tPA proteinase activity but instead, interaction of tPA with the N-methyl-D-aspartate Receptor (NMDA-R) in macrophages. tPA-binding to the NMDA-R activates cell-signaling and regulates cell physiology in multiple cell types.7–12 The ability of tPA to regulate innate immunity in LPS-treated macrophages provides an example in which a gene product that functions mainly in one phase of the response to injury regulates another.
In order to examine the effects of tPA on the response to LPS independently of plasmin, in our previous study,6 we performed most of our experiments using enzymatically-inactive tPA (EI-tPA). Our results were replicated with enzymatically-active, non-mutated tPA; however, we were careful to exclude sources of plasminogen. Others have shown that plasmin activates NFκB and induces expression of pro-inflammatory cytokines in monocytes and macrophages13–16 Plasmin also may contribute to the resolution of inflammation.17 The protease activity of plasmin appears essential for its effects on cytokine expression. Plasminogen receptors, such as annexin A2/S100A10 complex, annexin A1, α-enolase, and Plg-RTK, play a critical role in mediating the effects of plasmin in inflammation.15,17–20 One function of plasminogen receptors may be to facilitate plasminogen activation and then deliver plasmin to cell-signaling receptors in the Protease-activated Receptor (PAR) family.18,21–24 Alternative pathways by which plasmin may induce inflammation also have been described, such as by proteolytic activation of the chemokine, monocyte chemoattractant protein-1 (MCP-1/CCL2).24,25
In this study, we demonstrate that the activity of tPA as an inhibitor of the LPS response in BMDMs is apparently comprehensive; not only does tPA block expression of pro-inflammatory cytokines but also interleukin-10 (IL-10) and IL-1 receptor-antagonist (IL-1RA), which demonstrate anti-inflammatory activity.26,27 In the presence of plasminogen, the previously reported indistinguishable effects of EI-tPA and enzymatically-active tPA on cytokine expression6 are no longer observed because plasmin independently promotes expression of cytokines, including pro-inflammatory cytokines, by a pathway that is independent of the NMDA-R, and instead, dependent on PAR activation. tPA failed to inhibit the effects of plasmin on gene regulation in BMDMs; this result was probably explained by the inability of tPA to neutralize pro-inflammatory events mediated by PAR activation. This study provides the first evidence of specificity in the activity of tPA as an inhibitor of pro-inflammatory macrophage stimuli. The ability of plasmin to promote inflammatory cytokine expression, even in the presence of tPA, justifies testing EI-tPA, as the preferred form of tPA, as a candidate inhibitor of innate immunity.
2 │. MATERIALS AND METHODS
2.1 │. Proteins and reagents
Enzymatically-active human tPA, which is produced in CHO cells and 95% in the two-chain form, and human EI-tPA, which carries the S478→A mutation and is 90% in the single-chain form, were from Molecular Innovations. Glu-Plasminogen was purified from human plasma as previously described.28 The purified plasminogen preparations studied here were unresolved mixtures of the two major glycoforms. Plasmin (>10 International units/ mL), which was pre-activated with immobilized low molecular weight urokinase, was from Molecular Innovations. LPS serotype 055:B5 from E. coli was from Sigma-Aldrich. Dizocilpine (MK-801) was from Cayman Chemical. ε-aminocaproic acid (ƐACA) was from MP Biomedicals. Aprotinin was from PanReac AppliChem. SCH 79797 was from Cayman Chemicals and RWJ 56110 from R&D Systems. The PAR1 agonist peptide, TFLLR, and the control peptide, RLLFT, were from R&D Systems. The plasmin-specific substrate, H-D-Val-Leu-Lys p-nitroanilide (S-2251), was from Molecular Innovations.
2.2 │. BMDM cultures
Bone marrow cells were isolated from the femurs of 16-week-old wild-type C57BL/6J male mice, as previously described.29 Cells were plated in non-tissue culture-treated dishes and cultured in DMEM/F-12 medium (Gibco) containing 10% FBS (Gibco) and 20% L929 cell-conditioned medium for 10 days. Non-adherent cells were eliminated on day 10. Adherent cells included >95% BMDMs as determined by F4/80 and CD11b immunoreactivity.
BMDMs were cultured in serum-free medium (SFM) for 30 min and then treated simultaneously for 1 or 3 h with various proteins and reagents, including LPS (0.1 μg/mL), vehicle (20 mM sodium phosphate, 150 mM NaCl, pH 7.4), enzymatically-active tPA (12 nM), EI-tPA (12 nM), plasminogen (20–500 nM), plasmin (10–50 nM), ƐACA (10 mM), aprotinin (33 units/ml), SCH 79797 (2 μM), TFLLR (10 μM), and RLLFT (10 μM). MK-801 (1 μM) was added to cultures 30 min before other reagents. RWJ 56110 (30 μM) was added 2 h before other reagents. Maintaining the BMDMs in SFM while performing these experiments assured that bovine plasminogen was not inadvertently present during the treatments. All reagents used to assess pathways involved in the response to tPA and plasmin had no effect on cell viability at the concentrations studied, as determined by MTT assay (Invitrogen).
2.3 │. Plasminogen activation in association with BMDMs
BMDMs were seeded in 48-well plates at a density of 105 cells/well. The cells were washed 3 times with Hanks buffered-salt solution with calcium and magnesium, 10 mM HEPES, pH 7.4, and 1.0 mg/ml bovine serum albumin, and incubated in the same buffer for 30 minutes at 37° C. Plasminogen (20–500 nM), S-2251 (0.2 mM), enzymatically-active tPA (12 nM) and EI-tPA (12 nM) were added as indicated. S-2251 hydrolysis was determined by monitoring the absorbance at 405 nm as a function of time at 37° C using a Spectramax M2 ELISA plate reader.
2.4 │. Analysis of cytokines in conditioned medium
BMDMs were transferred to SFM for 30 min and then treated with plasminogen and/or tPA as indicated for 3 h. Serum-free conditioned medium was harvested and assayed for mouse tumor necrosis factor-α (TNFα) protein and interleukin-6 (IL-6) protein using Quantikine ELISA systems (R&D System).
2.5 │. Analysis of cytokine mRNA expression
RNA was isolated using the NucleoSpin RNA kit (Macherey-Nagel) and reverse-transcribed using the iScript cDNA synthesis kit (Bio-Rad). RT-qPCR was performed to determine the relative abundance of mRNAs encoding TNFα, interleukin-1β (IL-1β), IL-6, CCL2, IL-10, and IL-1RA using TaqMan gene expression products (Thermo Scientific), as previously described.29 Changes in mRNA expression were calculated using the 2-ΔΔCt method and GAPDH mRNA as an internal normalizer.
2.6 │. Analysis of cell-signaling
Cell extracts were prepared in RIPA buffer containing Protease Inhibitors and Phosphatase Inhibitor Cocktail (Pierce). Protein concentrations were determined by DC Protein Assay (Bio-Rad). Equivalent amounts of cellular protein were subjected to SDS PAGE and electrotransferred to polyvinylidene difluoride membranes. Membranes were blocked and probed with primary antibodies from Cell Signaling Technology, including antibodies that target phospho-p38 MAPK (Thr180/Tyr182) (Catalog number 9215), total p38 MAPK (9212) phospho-ERK1/ERK2 (Thr202/Tyr204) (4370), total ERK1/ERK2 (9102), phospho-c-Jun N-terminal kinase (Thr183/Tyr185) (9251), total c-Jun N-terminal kinase (9252), phospho-IκBα (Ser32) (2859), total IκBα (9242) and β-actin (3700), followed by horseradish peroxidase conjugated secondary antibodies. Immunoblots were developed using ProSignal Pico ECL Reagent substrate (Prometheus) and imaged using the Azure Biosystems C300 imaging system.
2.7 │. Phosphoprotein proteome profiling
BMDMs were transferred to SFM for 30 min and treated with active tPA (12 nM) and plasminogen (200 nM) or with vehicle for 1 h. Cell extracts were prepared using lysis buffer provided in the Proteome Profiler Human Phospho-Kinase Array kit (R&D Systems). An equivalent amount of cellular protein (400 μg) was incubated with the 2 nitrocellulose membranes. Phosphorylated proteins were detected using biotinylated detection antibodies. Although this array was initially developed to identify phosphoproteins in human proteins, we demonstrated excellent cross-reactivity with rodent proteins by comparing human and rat Schwann cells.30
2.8 │. Statistics
Statistical analysis was performed using GraphPad Prism 5.0. Results are presented as the mean ± SEM. Each replicate was performed using BMDM cultures from a separate mouse. RT-qPCR data were analyzed by one-way ANOVA followed by Bonferroni’s multiple comparison test (*p<0.05, **p<0.01, ***p<0.001).
3 │. RESULTS
3.1 │. EI-tPA blocks LPS-induced cytokine expression in the presence of plasminogen
The first objective of this study was to determine whether plasminogen affects the ability of tPA to regulate pro-inflammatory cytokine expression by BMDMs in response to LPS. When enzymatically-active tPA (12 nM) and increasing concentrations of plasminogen were incubated with BMDMs in SFM supplemented with plasmin-specific chromogenic substrate, plasminogen activation was observed and the amount of plasmin formed was a function of the plasminogen concentration (Fig. 1A). In the absence of tPA, plasminogen activation was extremely weak or entirely absent over the 70 min monitoring period, indicating that our plasminogen preparations were not contaminated with plasmin and the BMDMs do not independently generate significant levels of plasminogen activators (PAs) in the absence of offsetting PA inhibitors, such as PAI-1 (Fig. 1B). Plasminogen activation also was not detected when cells were incubated with 12 nM EI-tPA and increasing concentrations of plasminogen (Fig. 1C).
Figure 1. Plasminogen activation in association with cultured BMDMs.
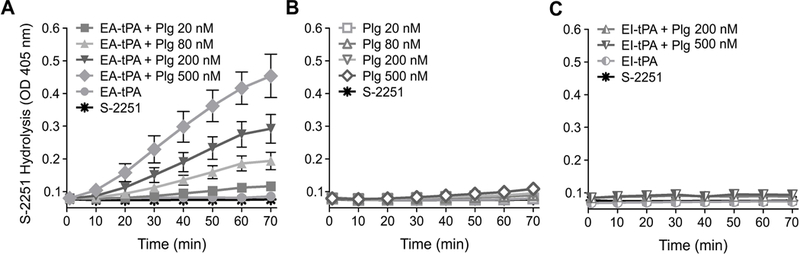
(A) BMDMs were serum-starved for 30 min and then treated with 12 nM enzymatically-active tPA (EA-tPA), plasminogen (Plg) at the indicated concentrations, and S-2251 (0.2 mM). The plots marked “EA-tPA” and “S-2251” show substrate hydrolysis in cultures treated with EA-tPA and S-2251 in the absence of Plg and with S-2251 alone, respectively. (B) Serum-starved BMDMs were incubated with Plg at the indicated concentrations and S-2251. As a control, S-2251 was added to cultures alone. (C) BMDMs were incubated with EI-tPA, Plg (200 nM or 500 nM), and S-2251. The graphs marked “EI-tPA” and “S-2251” show substrate hydrolysis in cultures treated with EI-tPA and S-2251 in the absence of Plg or S-2251 alone, respectively. The absorbance at 405 nm was determined every 10 min for 70 min. Overlapping data points preclude distinguishing individual points when substrate hydrolysis did not occur (mean ± SEM, n = 6).
Because EI-tPA did not activate plasminogen, to begin, we studied the effects of plasminogen on the activity of EI-tPA in cytokine mRNA expression experiments. BMDMs were treated with EI-tPA alone or in the presence of 20–500 nM plasminogen for 3 h. As a control, BMDMs were exposed to LPS (0.1 μg/mL). The 3 h exposure time was selected, as opposed to 6–8 h, as in our previous studies,6,29 to avoid changes in cell adhesion or survival that may result from prolonged exposure to plasminogen or plasmin. LPS significantly increased expression of TNFα, IL-1β, IL-6, and CCL2, as anticipated (Fig. 2A). In the absence of LPS, EI-tPA did not significantly increase cytokine expression and this result was conserved in cells treated with EI-tPA and plasminogen.
Figure 2. EI-tPA neutralizes the effects of LPS on cytokine expression in BMDMs in the presence of plasminogen.
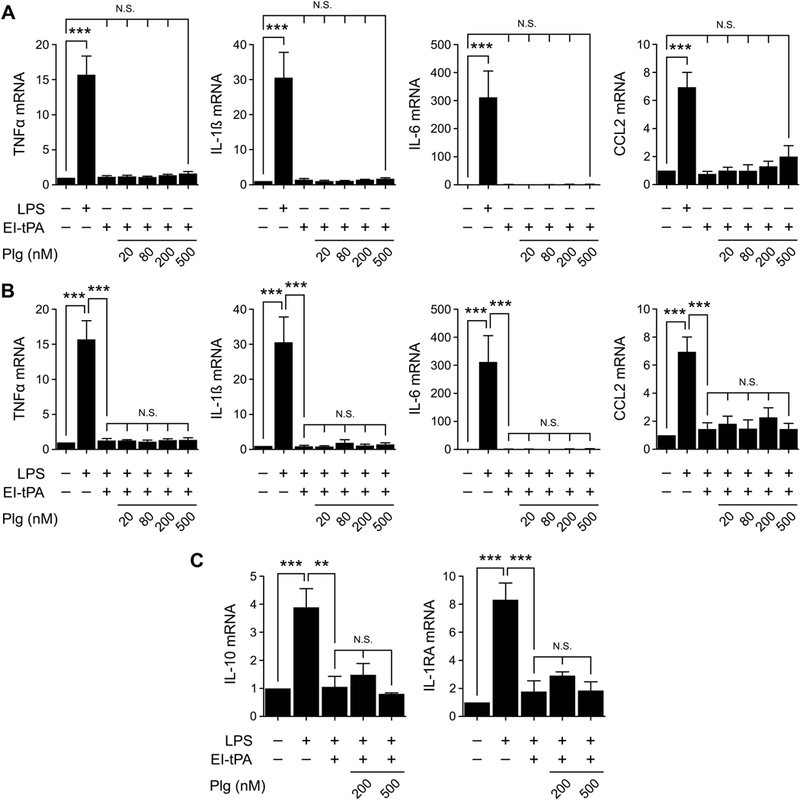
(A) BMDMs were treated with LPS (0.1 μg/mL) or with EI-tPA (12 nM) alone and in the presence of increasing concentrations (20–500 nM) of plasminogen (Plg) for 3 h. (B) BMDMs were treated with LPS (0.1 μg/ml) alone or with LPS plus EI-tPA (12 nM) in the presence of increasing concentrations of Plg for 3 h. (C) BMDMs were treated with LPS, LPS plus EI-tPA (12 nM), or with LPS plus EI-tPA and Plg (200 or 500 nM). RT-qPCR was performed to compare mRNA levels for TNFα, IL-1β, IL-6, CCL2, IL-10, and IL-1RA (mean ± SEM; n = 5 in panels A and B and 3 in panel C; ***p<0.001, N.S., not statistically significant; one-way ANOVA with Bonferroni’s post hoc test). The presented results show “fold increase” in mRNA expression compared with cultures treated with vehicle (no LPS, EI-tPA, or Plg).
Next, we studied the effects of EI-tPA on cytokine expression in cells treated simultaneously with LPS. In the absence of plasminogen, EI-tPA (12 nM) reversed the effects of LPS (0.1 μg/mL) on expression of TNFα, IL-1β, IL-6, and CCL2 (Fig. 2B), confirming our earlier results.6 EI-tPA also reversed the effects of LPS on expression of IL-10 and IL-1RA (Fig. 2C), gene products with known anti-inflammatory activities.26,27 It thus appears that EI-tPA neutralizes the response to LPS comprehensively, and not just the effects of LPS on expression of pro-inflammatory mediators. Plasminogen at concentrations up to 500 nM failed to affect the efficacy of EI-tPA in reversing LPS-induced gene regulatory events. These results demonstrate that the activity of EI-tPA as an antagonist of the macrophage response to LPS is conserved in the presence of plasminogen.
3.2 │. Plasminogen activation reverses the effects of enzymatically-active tPA on LPS-induced cytokine expression
To determine whether plasminogen regulates cytokine expression by BMDMs in the absence of tPA, cells were treated with increasing concentrations of plasminogen or with LPS (0.1 μg/mL) as a positive control. Fig. 3A shows that, in the absence of tPA, plasminogen did not significantly regulate cytokine expression. When BMDMs were treated simultaneously with LPS and 20–500 nM plasminogen, plasminogen did not regulate the LPS response (Fig. 3B).
Figure 3. Plasminogen activation reverses the effects of tPA on LPS-induced cytokine expression.
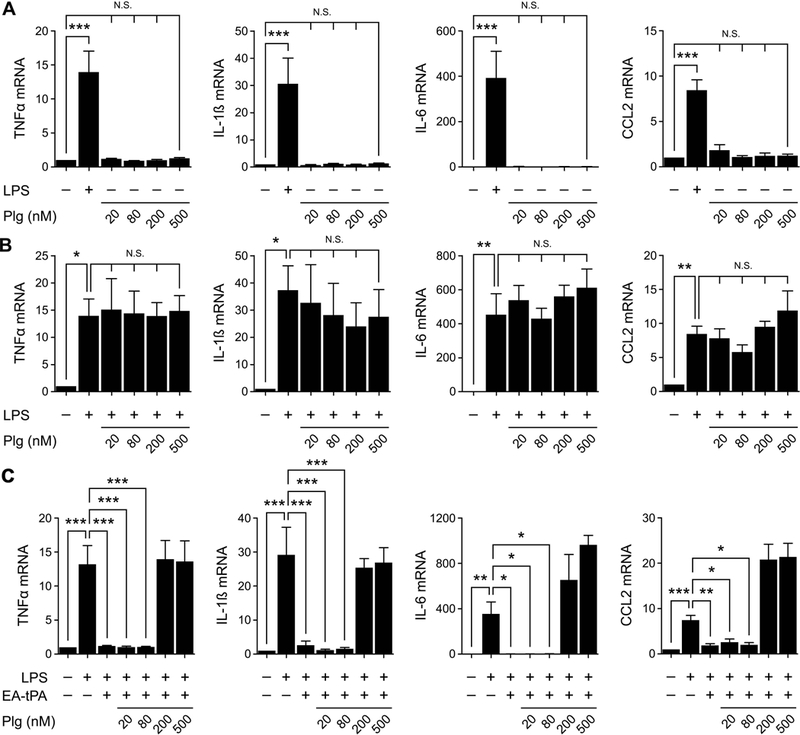
(A) BMDMs were treated with LPS (0.1 μg/ml) or with increasing concentrations (20–500 nM) of plasminogen (Plg) in the absence of LPS for 3 h. (B) BMDMs were treated with LPS (0.1 μg/ml) alone or together with increasing concentrations (20–500 nM) of Plg for 3 h. (C) BMDMs were treated with LPS (0.1 μg/ml), LPS plus 12 nM enzymatically-active tPA (EA-tPA), or LPS plus 12 nM EA-tPA and increasing concentrations of plasminogen (Plg) for 3 h. RT-qPCR was performed to compare mRNA expression of TNFα, IL-1β, IL-6 and CCL2 (mean ± SEM; n = 4–6; *p<0.05, **p<0.01, ***p<0.001, N.S., not statistically significant; one-way ANOVA with Bonferroni’s post hoc analysis). The presented results show “fold increase” in mRNA expression compared with cultures treated with vehicle.
Next, BMDMs were treated with 0.1 μg/mL LPS, in the presence and absence of enzymatically-active tPA and increasing concentrations of plasminogen. LPS induced expression of the mRNAs encoding TNFα, IL-1β, IL-6, and CCL2 and, in the absence of plasminogen, tPA reversed the effects of LPS on cytokine expression (Fig. 3C). tPA retained its activity when 20–80 nM plasminogen was present. By contrast, when 200–500 nM plasminogen was present, the ability of active tPA to block cytokine expression in response to LPS was reversed. In these cells, expression of TNFα, IL-1β, IL-6, and CCL2 was unchanged, compared with cells treated with LPS alone, or slightly increased.
Next, we treated BMDMs with active tPA and 20–500 nM plasminogen, in the absence of LPS. When the plasminogen concentration was 200–500 nM, expression of pro-inflammatory cytokines increased to about the level observed with 0.1 μg/mL LPS alone (Fig. 4A-D). Because, plasminogen failed to induce cytokine expression in the absence of tPA, we hypothesized that plasminogen activation was necessary. Figs. 4E and 4F show that 12 nM tPA and 200 nM plasminogen increased expression of IL-10 and IL-1RA, similarly to LPS.
Figure 4. Plasminogen activation increases cytokine expression.
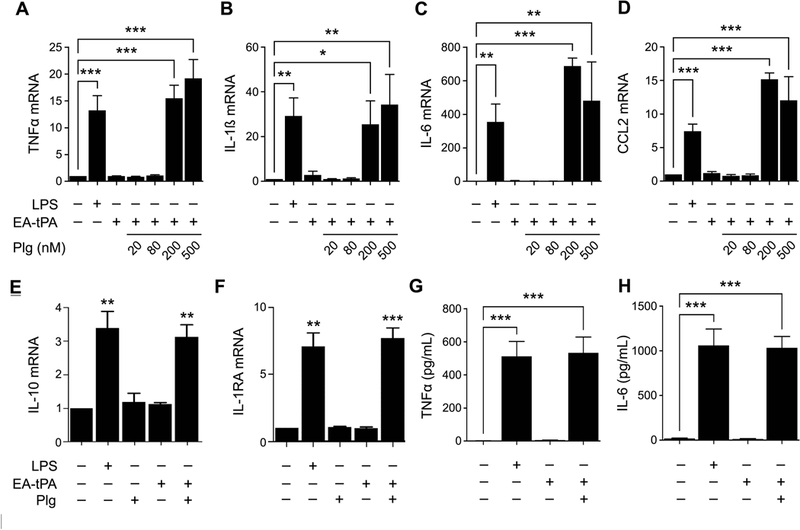
(A-D) BMDMs were treated with LPS (0.1 μg/ml), 12 nM enzymatically-active tPA (EA-tPA) alone, or with EA-tPA and increasing concentrations of plasminogen (Plg) in the absence of LPS for 3 h. Expression of the mRNAs encoding TNFα, IL-1β, IL-6 and CCL2 was determined (mean ± SEM; n = 3–5; *p<0.05, **p<0.01, ***p<0.001; one-way ANOVA with Bonferroni’s post hoc test). (E, F) BMDMs were treated with LPS (0.1 μg/ml), Plg (200 nM), EA-tPA (12 nM), EA-tPA plus 200 nM Plg, or vehicle. RT-qPCR was performed to determine mRNA expression of IL-10 and IL-1RA (n=3). The presented results show “fold increase” in mRNA expression compared with cultures treated with vehicle. (G, H) BMDMs were treated with LPS (0.1 μg/ml), EA-tPA (12 nM), EA-tPA plus plasminogen (Plg) (200 nM), or vehicle for 3 h. TNFα protein (pg/ml) and IL-6 protein (pg/ml) in conditioned medium was determined by ELISA (mean ± SEM; n = 3; ***p<0.001; one-way ANOVA with Bonferroni’s post hoc analysis).
To confirm that active tPA and plasminogen induce cytokine expression in BMDMs at the protein level, we analyzed conditioned medium samples by ELISA. Significantly increased levels of TNFα (Fig. 4G) and IL-6 (Fig. 4H) were detected in conditioned medium from cells treated with tPA and 200 nM plasminogen. We did not examine IL-1β protein given the complex system of inflammasome proteins that regulate IL-1β protein secretion.31
εACA binds to the lysine-binding sites of plasminogen and inhibits binding of plasminogen to cell-surface receptors.32 In doing so, ƐACA may disrupt interactions that promote plasminogen activation. We treated BMDMs with active tPA and plasminogen (200 nM) in the presence and absence of 10 mM εACA. As shown in Fig. 5A-D, the increase in expression of TNFα, CCL2, IL-1β, and IL-6 induced by tPA plus 200 nM plasminogen was blocked by εACA. In control experiments, εACA alone failed to significantly affect cytokine expression by BMDMs.
Figure 5. Plasmin enzyme activity is necessary for increasing pro-inflammatory cytokine expression.
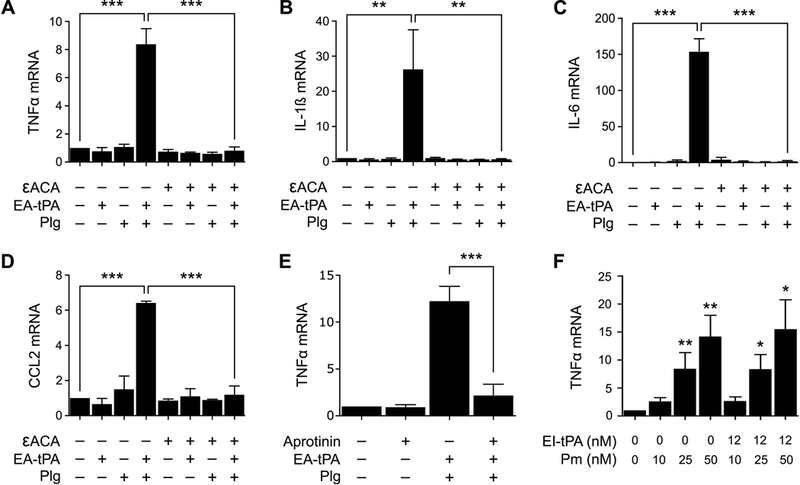
(A-D) BMDMs were treated for 3 h with 12 nM enzymatically-active tPA (EA-tPA), Plg (200 nM), EA-tPA plus Plg (200 nM), or vehicle in the presence or absence of εACA (10 mM). Expression of the mRNAs encoding TNFα, IL-1β, IL-6 and CCL2 was determined (mean ± SEM; n = 3–4; *p<0.05, ***p<0.001; one-way ANOVA with Bonferroni’s post hoc test). (E) BMDMs were treated for 3 h with EA-tPA (12 nM) plus Plg (200 nM), aprotinin alone (33 Unit/ml), or EA-tPA, plasminogen plus aprotinin. RT-qPCR was performed to compare mRNA levels for TNFα (mean ± SEM; n = 4; ***p<0.001; one-way ANOVA with Bonferroni’s post hoc analysis). (F) BMDMs were treated for 3 h with the indicated concentrations of pre-activated plasmin (Pm) in the presence or absence of EI-tPA. TNFα mRNA was determined (n=4). The presented results show “fold increase” in mRNA expression compared with cultures treated with vehicle (*p<0.05, **p<0.01; statistical analysis is relative to the vehicle control).
Aprotinin is a 58-amino acid proteinase inhibitor from bovine pancreas and a naturally occurring plasmin inhibitor.33 Aprotinin blocked the increase in TNFα mRNA expression caused by active tPA plus 200 nM plasminogen (Fig. 5E). This result supports the hypothesis that plasmin enzyme activity is necessary for the pro-inflammatory response observed when tPA and plasminogen are added to cultures of BMDMs. To confirm this hypothesis, we examined the ability of pre-activated plasmin to induce TNFα expression in BMDMs in the absence tPA. The plasmin was generated using low molecular weight urokinase coupled to Sepharose and thus, not contaminated with plasminogen activators. Fig. 5F shows that pre-activated plasmin significantly increased TNFα mRNA expression and the response appeared plasmin concentration-dependent. Importantly, when cells were treated with EI-tPA together with pre-activated plasmin, the EI-tPA failed to block the increase in TNFα expression induced by pre-activated plasmin.
3.3 │. Involvement of cell-signaling receptors in the response of BMDMs to plasmin
We previously demonstrated that BMDMs express the NMDA-R, which serves as an essential receptor in the pathway by which tPA suppresses cytokine expression by BMDMs in response to LPS.6 We therefore tested whether the NMDA-R is involved in the pro-inflammatory response to plasmin by treating BMDMs with the non-competitive NMDA-R antagonist, Dizocilpine/MK-801 (1.0 μM). Fig. 6 shows that MK-801 failed to significantly inhibit the effects of enzymatically-active tPA plus plasminogen (200 nM) on expression of TNFα, IL-1β, IL-6, or CCL2.
Figure 6. The NMDA-R is not required for the pro-inflammatory response to plasmin.
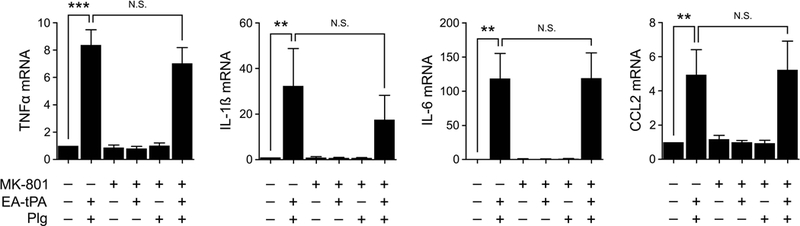
BMDMs were pre-treated with MK-801 (1.0 μM) or vehicle for 30 min and then for 3 h with 12 nM enzymatically-active tPA (EA-tPA), EA-tPA and plasminogen (Plg) (200 nM), Plg, or vehicle. Expression of the mRNAs encoding TNFα, IL-1β, IL-6 and CCL2 was determined (mean ± SEM; n = 6; **p<0.01, ***p<0.001, N.S., not statistically significant; one-way ANOVA with Bonferroni’s post hoc test). In separate control experiments (not shown), we demonstrated that the MK-801 blocked the ability of 12 nM EI-tPA to inhibit cytokine expression induced by LPS (0.1 μg/ml).
Next, we tested whether cell-signaling receptors in the PAR family are involved in the BMDM response to plasmin. SCH 79797 is a selective PAR-1 antagonist.34 Fig. 7A shows that 2.0 μM SCH 79797 did not independently regulate pro-inflammatory cytokine expression by BMDMs. However, SCH 79797 blocked the increase in expression of TNFα, CCL2, IL-1β, and IL-6 caused by active tPA plus 200 nM plasminogen.
Figure 7. PAR inhibitors block plasmin-induced pro-inflammatory cytokine expression.
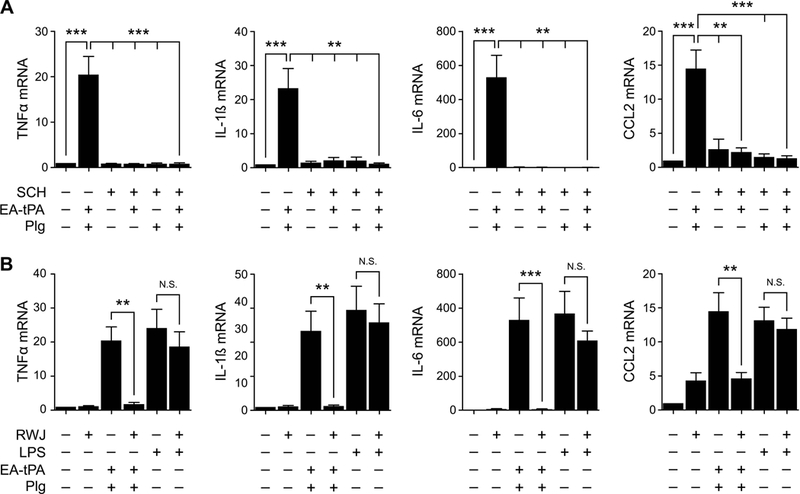
(A) BMDMs were treated with 12 nM enzymatically-active tPA (EA-tPA) plus 200 nM plasminogen (Plg), EA-tPA alone, or Plg alone, in the presence or absence of SCH-79797 (SCH) (2 μM) as indicated for 3 h. (B) BMDMs were pre-treated with RWJ-56110 (RWJ) (30 μM) or vehicle for 2 h and then with EA-tPA (12 nM) plus Plg (200 nM), LPS (0.1 μg/ml), or vehicle for 3 h. RT-qPCR was performed to compare mRNA levels for TNFα, IL-1β, IL-6 and CCL2 (mean ± SEM; n = 3–4; **p<0.01, ***p<0.001, N.S., not statistically significant; one-way ANOVA with Bonferroni’s post hoc analysis). The presented results show “fold increase” in mRNA expression compared with cultures treated with vehicle.
As a second approach for antagonizing PAR-1, we treated BMDMs with RWJ 56110 (30 μM). RWJ 56110 is a PAR-1 antagonist without activity against other PARs.35 Like SCH 79797, RWJ 56110 blocked the increase in expression of TNFα, IL-1β, IL-6, and CCL2 caused by active tPA plus 200 nM plasminogen (Fig. 7B). RWJ 56110 did not affect cytokine expression when added to BMDM cultures alone. RWJ 56110 also did not significantly affect cytokine expression by BMDMs treated with 0.1 μg/mL LPS.
3.4 │. tPA and plasmin have distinct effects on cell-signaling in BMDMs
We performed studies to determine how plasminogen activation affects the ability of tPA to regulate LPS-initiated cell-signaling in BMDMs. The BMDMs were transferred to SFM for 30 min and then treated with various proteins and reagents for 1 h. LPS (0.1 μg/mL) caused phosphorylation of IκBα and a decrease in the total abundance of IκBα (Fig. 8A) as anticipated; these changes report NFκB activation.36 In the absence of plasminogen, enzymatically-active tPA reversed the effects of LPS on IκBα phosphorylation and its total abundance, as previously reported.6 By contrast, when BMDMs were treated with active tPA and 200 nM plasminogen, in the presence of LPS or in its absence, IκBα was phosphorylated and the total abundance of IκBα was decreased. Thus, plasmin appears to independently promote IκBα phosphorylation in BMDMs even though tPA is present.
Figure 8. Plasmin activates cell-signaling in BMDMs.
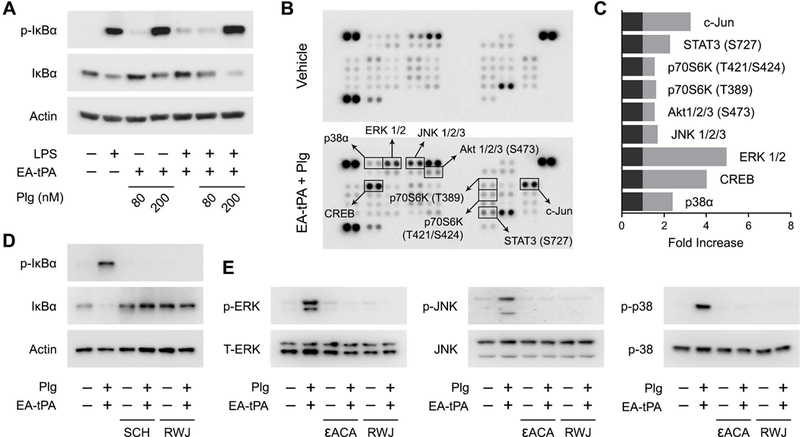
(A) BMDMs were treated for 1 h with LPS (0.1 μg/mL) or vehicle in the presence or absence of 12 nM enzymatically-active tPA (EA-tPA) and increasing concentrations of plasminogen (Plg). Equal amounts of cellular protein (20 μg) were subjected to immunoblot analysis to detect phospho-IκBα, total IκBα and β-actin. (B) BMDMs were treated with EA-tPA (12 nM) plus Plg (200 nM) or vehicle for 1 h. Protein phosphorylation was analyzed by array. Changes in phospho-epitopes, associated with the tPA/plasminogen treatment protocol, exceeding 1.6× are shown in boxes. (C) Densitometry analysis of the array shown in Panel B. The fold-increase in each phospho-epitope is shown. (D) BMDMs were pre-treated with RWJ-56110 (RWJ) (30 μM) for 2 h and then with EA-tPA (12 nM) plus Plg (200 nM) or with vehicle for 1 h. Separate cells were treated with EA-tPA plus plasminogen or with vehicle for 1 h in the presence and absence of SCH-79797 (SCH) (2 μM). Cell extracts were subjected to immunoblot analysis to detect phospho-IκBα, total IκBα, and β-actin. (E) BMDMs were treated with EA-tPA (12 nM) plus Plg (200 nM), εACA (10 mM), EA-tPA plus Plg and εACA, SCH (2 μM), EA-tPA plus Plg and SCH, or vehicle for 1 h. Cell extracts were subjected to immunoblot analysis to detect phospho-ERK1/2, total ERK1/2, phospho-c-Jun N-terminal kinase (p-JNK), total c-Jun N-terminal kinase (JNK), phospho-p38 MAP kinase, and total p38 MAP kinase.
Next, we examined cell-signaling in response to plasmin in BMDMs in an unbiased manner using an array platform that detects multiple distinct protein phosphorylation events. BMDMs were treated with enzymatically-active tPA (12 nM) and 200 nM plasminogen for 1 h. Protein extracts were then subjected to analysis (Fig. 8B). Phospho-epitopes that were increased at least 1.6-fold are enclosed in boxes. The results are summarized in Fig. 8C. The major MAP kinases, ERK1/2, c-Jun N-terminal kinase, and p38 MAP kinase were phosphorylated in plasmin-treated cells, together with the c-Jun N-terminal kinase substrate, c-Jun. CREB is a transcription factor phosphorylated downstream of diverse kinases including ERK1/2 and p38 MAP kinase.37 Phosphorylation of Akt at S473 is considered an mTORC2 event, required for full Akt activation.38 p70 S6 kinase is a downstream mediator of PI3K-Akt signaling involved in cell survival and growth.39 p70 S6 kinase activation also has been implicated in inflammatory cytokine production by macrophages.40,41
Because the array analysis is performed only once, validation studies were performed using conventional immunoblotting to examine phosphorylation of the three MAP kinases. These validation studies and separate experiments examining IκBα phosphorylation included incubations to test whether inhibitors that block cytokine expression in response to plasmin also block plasmin-initiated cell-signaling. Fig. 8D shows that plasmin-induced IκBα phosphorylation was blocked by SCH 79797 and RWJ 56110. Fig. 8E confirms that ERK1/2, c-Jun N-terminal kinase, and p38 MAP kinase are activated in BMDMs treated with active tPA and 200 nM plasminogen and also shows that RWJ 56110 and εACA block these cell-signaling events.
3.5 │. tPA fails to reverse cytokine expression caused by PAR-1 agonist peptide
The results presented thus far suggested that plasmin induces pro-inflammatory cytokine expression by activating a PAR, and most likely PAR-1. Because EI-tPA failed to inhibit the effects of pre-activated plasmin on cytokine expression, we tested whether tPA is ineffective at neutralizing pro-inflammatory responses mediated by PARs. BMDMs were treated with PAR-1 agonist peptide/TFLLR in the presence or absence of tPA. Fig. 9 shows that TFLLR induced expression of TNFα, IL-1β, IL-6, and CCL2 whereas the control peptide, RLLFT, did not. When either enzymatically-active tPA or EI-tPA was added together with TFLLR, cytokine expression was not inhibited. These results demonstrate that tPA is not effective in regulating PAR-induced cytokine expression in BMDMs and provide the first evidence for specificity in the anti-inflammatory activity of tPA.
Figure 9. tPA is not effective in regulating PAR-1-initiated cytokine expression in BMDMs.
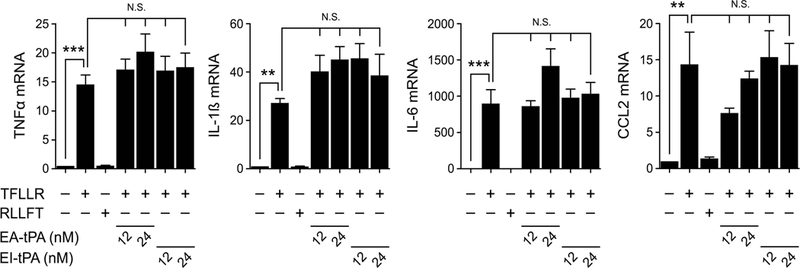
BMDMs were treated for 3 h with PAR-1 agonist peptide/TFLLR (10 μM) alone or simultaneously with enzymatically-active tPA (EA-tPA) or EI-tPA (12 or 24 nM). Separate cultures were treated with RLLFT (10 μM) as a negative control peptide. Expression of the mRNAs encoding TNFα, IL-1β, IL-6 and CCL2 was determined (mean ± SEM; n = 4; **p<0.01, ***p<0.001, N.S., not statistically significant; one-way ANOVA with Bonferroni’s post hoc test). The presented results show “fold increase” in mRNA expression compared with cultures treated with vehicle.
4. Discussion
tPA is an FDA-approved drug for stroke.42 The recently discovered activity of tPA as an inhibitor of innate immune system responses to LPS6 is thus of considerable interest. EI-tPA neutralized the toxicity of LPS when administered to mice at approximately two-times the dose of tPA recommended for patients with stroke.6 Because the effects of EI-tPA on hemostasis should be greatly attenuated compared with the active enzyme, higher doses of EI-tPA may be feasible in experiments testing the activity of EI-tPA in immunity.
Our research suggesting that tPA may antagonize the response of macrophages to stimuli that activate the innate immune system6,29 raises important questions regarding how tPA and its major substrate, plasminogen, interact in immunity. A robust body of literature indicates that plasmin regulates cell-signaling and gene expression in monocytes and macrophages and may contribute to a pro-inflammatory state.13–16 The major objective of this study was to consider the activities of tPA and plasminogen as regulators of innate immunity in concert. We applied a previously described mouse BMDM cell culture model system6,29 and demonstrated that, in the absence of plasminogen, tPA not only inhibits expression of pro-inflammatory cytokines by BMDMs in response to LPS, but also IL-10 and IL-1RA. Thus, tPA appears to inhibit the macrophage response to LPS comprehensively.
When BMDMs were treated with enzymatically-active tPA and sufficiently high concentrations of plasminogen, plasmin was generated and a pro-inflammatory response was observed in the presence and absence of LPS. Plasmin induced expression of TNFα, IL-1β, IL-6, and CCL2 and the level of expression of these cytokines was similar to that detected with 0.1 μg/mL LPS. Plasmin also activated cell-signaling in BMDMs. IκBα was phosphorylated and ERK1/2, p38 MAP kinase, and c-Jun N-terminal kinase were activated. These responses occurred in the presence of 12 nM tPA. Plasmin generation, in cultures treated with tPA, 200–500 nM plasminogen, and LPS restored an LPS-like response, which was not observed in the absence of plasminogen. The inability of tPA to inhibit cytokine expression in response to plasmin was confirmed in experiments with EI-tPA and pre-activated plasmin. EI-tPA retained its anti-inflammatory activity when added to cultures together with LPS, even in the presence of the highest concentrations of plasminogen, a result that reflects the inability of EI-tPA to activate plasminogen. We conclude that EI-tPA is the preferred reagent, as opposed to enzymatically-active tPA, to assess the activity of tPA as a candidate anti-inflammatory agent in animal model systems.
The effects of tPA and plasmin on pro-inflammatory cytokine expression by BMDMs are mediated by distinct receptor systems. MK-801 blocks the activity of tPA, as a neutralizer of LPS;6 however, MK-801 had no effect on cytokine expression in response to plasmin. The NMDA-R, which plays an essential role in mediating many cellular activities of tPA,6,11,12 is not involved in the response of BMDMs to plasmin. By contrast, the effects of plasmin on BMDM gene expression were blocked by εACA, aprotinin, and two separate PAR-1-selective PAR antagonists. These results fit a model in which plasmin is generated by enzymatically-active tPA in association with cellular receptors in a facilitated reaction that is inhibited by εACA. Once plasmin is generated, it cleaves a PAR, activating cell-signaling downstream of this G protein-coupled receptor. If plasmin is inhibited by aprotinin or if PAR inhibitors are added, the response to plasmin is blocked. This model is supported by studies demonstrating that thrombin-activated cell-signaling through PAR-1 is pro-inflammatory,43,44 like the plasmin-initiated response described here.
The activity of PARs as plasmin response mediators is not straightforward because we and others have reported that plasmin may desensitize these receptors.45,46 At the biochemical level, this paradox is explained by the fact that plasmin cleaves PAR-1 at the same site as thrombin, in a reaction that activates cell-signaling, and at sites distal to the thrombin cleavage site. The latter reactions inactivate PAR-1.47 The predominant activity of plasmin, observed in different experimental model systems, may reflect the abundance of PARs on the cell surface and whether alternative PAR activators are present. It is also possible that activation of PARs by plasmin18,21−24 reflects selective presentation of PAR peptide bonds to plasmin that is bound to cell-surface plasminogen receptors.
Because of evidence implicating PARs in plasmin responses in monocytes and macrophages, the inability of EI-tPA to inhibit cytokine expression induced by pre-activated plasmin was examined in experiments with PAR-1 agonist peptide. TFLLR increased pro-inflammatory cytokine expression in BMDMs and both active tPA and EI-tPA failed to reverse this response. These data provide the first evidence for specificity in the anti-inflammatory activity of tPA. Whether tPA is effective in suppressing cytokine expression depends on the pro-inflammatory signal and the receptor system engaged.
The inability of BMDMs to mediate plasminogen activation in the absence of exogenously added tPA demonstrates that these cells do not produce large amounts of plasminogen activators in the absence of endogenous plasminogen activator inhibitors. When tPA and 80 nM plasminogen were added to BMDM cultures, plasmin generation was detected by chromogenic substrate assay; however, consistently, the amount of plasmin generated appeared insufficient to induce cytokine expression or reverse the activity of tPA as an inhibitor of LPS. These results suggest that there may be a threshold concentration of plasmin required for generating a response. This hypothesis merits further investigation.
The identification of distinct receptor systems for tPA and plasmin, which regulate macrophage activation and innate immunity, supports models linking hemostasis and inflammation. Understanding these receptor systems may be important not only in the response to injury but also in numerous diseases in which inflammatory pathways are inappropriately activated.
Acknowledgments
We thank Dr. Maria Kounnas for assistance with the purification of plasminogen. Support for this work was provided by National Institutes of Health grant R01 HL136395 to S.L.G. S.L.G. also was supported by NIH grant R01 NS097590. E.M. was supported by the Ministry of Education, Universities and Research, Italy, grant FIRB 2013 RBFR13RBK9.
Abbreviations:
- tPA
tissue-type plasminogen activator
- LPS
lipopolysaccharide
- BMDMs
bone marrow-derived macrophages
- NMDA-R
N-methyl-D-aspartate receptor
- EI
enzymatically-inactive
- PAR
protease-activated receptor
- IL-1RA
interleukin-1 receptor-antagonist
- PA
plasminogen activator
- Plg
plasminogen
- Pm
plasmin
Footnotes
Disclosures
The authors report no conflicts of interest.
References
- 1.Diegelmann RF, Evans MC. Wound healing: An overview of acute, fibrotic and delayed healing. Front Biosci. 2004;9:283–289 [DOI] [PubMed] [Google Scholar]
- 2.Laurens N, Koolwijk P, de Maat MP. Fibrin structure and wound healing. J Thromb Haemost. 2006;4:932–939 [DOI] [PubMed] [Google Scholar]
- 3.Reinke JM, Sorg H. Wound repair and regeneration. European surgical research. 2012;49:35–43 [DOI] [PubMed] [Google Scholar]
- 4.Castellino FJ, Ploplis VA. Structure and function of the plasminogen/plasmin system. Thromb Haemost. 2005;93:647–654 [DOI] [PubMed] [Google Scholar]
- 5.Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, De Vos R, van den Oord JJ, Collen D, Mulligan RC. Physiological consequences of loss of plasminogen activator gene function in mice. Nature. 1994;368:419–424 [DOI] [PubMed] [Google Scholar]
- 6.Mantuano E, Azmoon P, Brifault C, Banki MA, Gilder AS, Campana WM, Gonias SL. Tissue-type plasminogen activator regulates macrophage activation and innate immunity. Blood. 2017;130:1364–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicole O, Docagne F, Ali C, Margaill I, Carmeliet P, MacKenzie ET, Vivien D, Buisson A. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med. 2001;7:59–64 [DOI] [PubMed] [Google Scholar]
- 8.Yepes M Tissue-type plasminogen activator is a neuroprotectant in the central nervous system. Frontiers in Cellular Neuroscience. 2015;9:304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lesept F, Chevilley A, Jezequel J, Ladepeche L, Macrez R, Aimable M, Lenoir S, Bertrand T, Rubrecht L, Galea P, Lebouvier L, Petersen KU, Hommet Y, Maubert E, Ali C, Groc L, Vivien D. Tissue-type plasminogen activator controls neuronal death by raising surface dynamics of extrasynaptic NMDA receptors. Cell Death & Disease. 2016;7:e2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeanneret V, Yepes M. Tissue-type plasminogen activator is a homeostatic regulator of synaptic function in the central nervous system. Neural Regeneration Research. 2017;12:362–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mantuano E, Lam MS, Shibayama M, Campana WM, Gonias SL. The NMDA receptor functions independently and as an LRP1 co-receptor to promote schwann cell survival and migration. J Cell Sci. 2015; 128:3478–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mantuano E, Lam MS, Gonias SL. LRP1 assembles unique co-receptor systems to initiate cell signaling in response to tissue-type plasminogen activator and myelin-associated glycoprotein. J Biol Chem. 2013; 288:34009–34018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Syrovets T, Jendrach M, Rohwedder A, Schule A, Simmet T. Plasmin-induced expression of cytokines and tissue factor in human monocytes involves AP-1 and IKKβ-mediated NF-κB activation. Blood. 2001;97:3941–3950 [DOI] [PubMed] [Google Scholar]
- 14.Burysek L, Syrovets T, Simmet T. The serine protease plasmin triggers expression of MCP-1 and CD40 in human primary monocytes via activation of p38 MAPK and janus kinase (jak)/stat signaling pathways. J Biol Chem. 2002;277:33509–33517 [DOI] [PubMed] [Google Scholar]
- 15.Li Q, Laumonnier Y, Syrovets T, Simmet T. Plasmin triggers cytokine induction in human monocyte-derived macrophages. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:1383–1389 [DOI] [PubMed] [Google Scholar]
- 16.Syrovets T, Lunov O, Simmet T. Plasmin as a proinflammatory cell activator. J Leukoc Biol. 2012;92:509–519 [DOI] [PubMed] [Google Scholar]
- 17.Sugimoto MA, Ribeiro ALC, Costa BRC, Vago JP, Lima KM, Carneiro FS, Ortiz MMO, Lima GLN, Carmo AAF, Rocha RM, Perez DA, Reis AC, Pinho V, Miles LA, Garcia CC, Teixeira MM, Sousa LP. Plasmin and plasminogen induce macrophage reprogramming and regulate key steps of inflammation resolution via annexin A1. Blood. 2017;129:2896–2907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bock A, Tucker N, Kelher MR, Khan SY, Gonzalez E, Wohlauer M, Hansen K, Dzieciatkowska M, Sauaia A, Banerjee A, Moore EE, Silliman CC. Alpha-enolase causes proinflammatory activation of pulmonary microvascular endothelial cells and primes neutrophils through plasmin activation of protease-activated receptor 2. Shock. 2015;44:137–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schuliga M, Langenbach S, Xia YC, Qin C, Mok JS, Harris T, Mackay GA, Medcalf RL, Stewart AG. Plasminogen-stimulated inflammatory cytokine production by airway smooth muscle cells is regulated by annexin A2. American Journal of Respiratory Cell and Molecular Biology. 2013;49:751–758 [DOI] [PubMed] [Google Scholar]
- 20.Miles LA, Baik N, Lighvani S, Khaldoyanidi S, Varki NM, Bai H, Mueller BM, Parmer RJ. Deficiency of plasminogen receptor, Plg-rkt, causes defects in plasminogen binding and inflammatory macrophage recruitment in vivo. J Thromb Haemost. 2017;15:155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagai T, Ito M, Nakamichi N, Mizoguchi H, Kamei H, Fukakusa A, Nabeshima T, Takuma K, Yamada K. The rewards of nicotine: Regulation by tissue plasminogen activator-plasmin system through protease activated receptor-1. J Neurosci. 2006;26:12374–12383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kamio N, Hashizume H, Nakao S, Matsushima K, Sugiya H. Plasmin is involved in inflammation via protease-activated receptor-1 activation in human dental pulp. Biochemical Pharmacology. 2008;75:1974–1980 [DOI] [PubMed] [Google Scholar]
- 23.Mannaioni G, Orr AG, Hamill CE, Yuan H, Pedone KH, McCoy KL, Berlinguer Palmini R, Junge CE, Lee CJ, Yepes M, Hepler JR, Traynelis SF. Plasmin potentiates synaptic N-methyl-D-aspartate receptor function in hippocampal neurons through activation of protease-activated receptor-1. J Biol Chem. 2008;283:20600–20611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carmo AA, Costa BR, Vago JP, de Oliveira LC, Tavares LP, Nogueira CR, Ribeiro AL, Garcia CC, Barbosa AS, Brasil BS, Dusse LM, Barcelos LS, Bonjardim CA, Teixeira MM, Sousa LP. Plasmin induces in vivo monocyte recruitment through protease-activated receptor-1-, MEK/ERK-, and CCR2-mediated signaling. J Immunol. 2014;193:3654–3663 [DOI] [PubMed] [Google Scholar]
- 25.Sheehan JJ, Zhou C, Gravanis I, Rogove AD, Wu YP, Bogenhagen DF, Tsirka SE. Proteolytic activation of monocyte chemoattractant protein-1 by plasmin underlies excitotoxic neurodegeneration in mice. J Neurosci. 2007;27:1738–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annual Review of Immunology. 2001;19:683–765 [DOI] [PubMed] [Google Scholar]
- 27.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–2147 [PubMed] [Google Scholar]
- 28.Gonias SL, Braud LL, Geary WA, VandenBerg SR. Plasminogen binding to rat hepatocytes in primary culture and to thin slices of rat liver. Blood. 1989;74:729–736 [PubMed] [Google Scholar]
- 29.Mantuano E, Brifault C, Lam MS, Azmoon P, Gilder AS, Gonias SL. LDL receptor-related protein-1 regulates NFκB and microRNA-155 in macrophages to control the inflammatory response. Proc Natl Acad Sci U S A. 2016;113:1369–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campana WM, Mantuano E, Azmoon P, Henry K, Banki M, Kim JH, Pizzo DP, Gonias SL. Ionotropic glutamate receptors activate cell signaling in response to glutamate in Schwann cells. FASEB J. 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eder C Mechanisms of interleukin-1β release. Immunobiology. 2009;214:543–553 [DOI] [PubMed] [Google Scholar]
- 32.Miles LA, Levin EG, Plescia J, Collen D, Plow EF. Plasminogen receptors, urokinase receptors, and their modulation on human endothelial cells. Blood. 1988;72:628–635 [PubMed] [Google Scholar]
- 33.Wiman B On the reaction of plasmin or plasmin-streptokinase complex with aprotinin or alpha 2-antiplasmin. Thrombosis research. 1980;17:143–152 [DOI] [PubMed] [Google Scholar]
- 34.Ahn HS, Foster C, Boykow G, Stamford A, Manna M, Graziano M. Inhibition of cellular action of thrombin by n3-cyclopropyl-7-[[4-(1-methylethyl)phenyl]methyl]-7h-pyrrolo[3, 2-f]quinazoline-1,3-diamine (SCH 79797), a nonpeptide thrombin receptor antagonist. Biochemical Pharmacology. 2000;60:1425–1434 [DOI] [PubMed] [Google Scholar]
- 35.Andrade-Gordon P, Maryanoff BE, Derian CK, Zhang HC, Addo MF, Darrow AL, Eckardt AJ, Hoekstra WJ, McComsey DF, Oksenberg D, Reynolds EE, Santulli RJ, Scarborough RM, Smith CE, White KB. Design, synthesis, and biological characterization of a peptide-mimetic antagonist for a tethered-ligand receptor. Proc Natl Acad Sci U S A. 1999;96:12257–12262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Henkel T, Machleidt T, Alkalay I, Kronke M, Ben-Neriah Y, Baeuerle PA. Rapid proteolysis of i kappa b-alpha is necessary for activation of transcription factor NF-kappa b. Nature. 1993;365:182–185 [DOI] [PubMed] [Google Scholar]
- 37.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609 [DOI] [PubMed] [Google Scholar]
- 38.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biology. 2009;7:e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harada H, Andersen JS, Mann M, Terada N, Korsmeyer SJ. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule bad. Proc Natl Acad Sci U S A. 2001;98:9666–9670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lim HK, Choi YA, Park W, Lee T, Ryu SH, Kim SY, Kim JR, Kim JH, Baek SH. Phosphatidic acid regulates systemic inflammatory responses by modulating the Akt-mammalian target of rapamycin-p70 S6 kinase 1 pathway. J Biol Chem. 2003;278:45117–45127 [DOI] [PubMed] [Google Scholar]
- 41.Xie S, Chen M, Yan B, He X, Chen X, Li D. Identification of a role for the pi3k/akt/mtor signaling pathway in innate immune cells. PLoS One. 2014;9:e94496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.National Institute of Neurological D, Stroke rt PASSG. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995;333:1581–1587 [DOI] [PubMed] [Google Scholar]
- 43.Cunningham MA, Rondeau E, Chen X, Coughlin SR, Holdsworth SR, Tipping PG. Protease-activated receptor 1 mediates thrombin-dependent, cell-mediated renal inflammation in crescentic glomerulonephritis. J Exp Med. 2000;191:455–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cirino G, Cicala C, Bucci MR, Sorrentino L, Maraganore JM, Stone SR. Thrombin functions as an inflammatory mediator through activation of its receptor. J Exp Med. 1996;183:821–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Penny WF, Ware JA. Platelet activation and subsequent inhibition by plasmin and recombinant tissue-type plasminogen activator. Blood. 1992;79:91–98 [PubMed] [Google Scholar]
- 46.Turner JS, Redpath GT, Humphries JE, Gonias SL, Vandenberg SR. Plasmin modulates the thrombin-evoked calcium response in C6 glioma cells. Biochem J. 1994;297:175–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuliopulos A, Covic L, Seeley SK, Sheridan PJ, Helin J, Costello CE. Plasmin desensitization of the PAR1 thrombin receptor: Kinetics, sites of truncation, and implications for thrombolytic therapy. Biochemistry. 1999;38:4572–4585 [DOI] [PubMed] [Google Scholar]