

SUMMARY
Glioblastoma (GBM) is among the most common and malignant types of primary brain tumors in adult with a dismal prognosis. Although alkylating agents such as temozolomide are widely applied as the first-line treatment for GBM, they often cause chemoresistance and remain ineffective with recurrent GBM. Alternative therapeutics against GBM are urgently needed in the clinic. We report herein the discovery of a class of inhibitors (YZ129 and its derivatives) of the calcineurin-NFAT pathway that exhibited potent anti-tumor activity against glioblastoma. YZ129 induced GBM cell cycle arrest at the G2/M phase, promoted apoptosis and inhibited tumor cell proliferation and migration. At the molecular level, YZ129 directly engaged HSP90 to antagonize its chaperoning effect on calcineurin to abrogate NFAT nuclear translocation, and also suppressed other proto-oncogenic pathways including hypoxia, glycolysis and the PI3K/AKT/mTOR signaling axis. Our data highlight the potential for targeting the cancer-promoting HSP90 chaperone network to treat glioblastoma.
eTOC Blurb
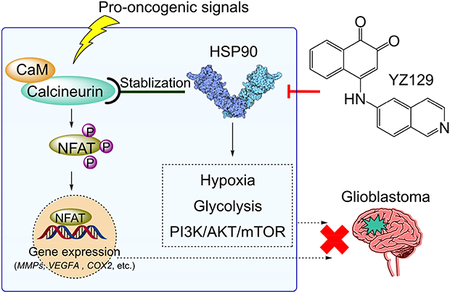
Liu et al. discovered a class of HSP90 inhibitors with strong therapeutic potential against glioblastoma. YZ129 directly interacted with HSP90 to antagonize its chaperoning effect on calcineurin to abrogate NFAT nuclear translocation, and also suppressed other proto-oncogenic pathways including hypoxia, glycolysis and the PI3K/AKT/mTOR axis.
INTRODUCTION
Glioblastoma (GBM) is among the most common and malignant primary brain cancer in adults, accounting for approximately 50% of all gliomas and up to 15% of all brain tumors (Preusser et al., 2015). The prognosis for GBM patients remains poor because the tumor cells can invade the surrounding brain tissues to cause secondary lethal brain disorders (Fritz et al., 2016). Even treated with surgical resection combined with radio-chemotherapy immediately after diagnosis, the median survival time of GBM is less than 17 months. Several FDA-approved alkylating drugs (e.g., lomustine, carmustine and temozolomide) have been used to treat GBM (Mittal et al., 2015), but tend to cause chemoresistance and are largely ineffective to recurrent glioblastoma (Simpson and Galanis, 2006). There remains an urgent clinical need for exploring the molecular basis of glioblastoma pathology (O’Duibhir et al., 2017) and discovering novel chemotherapeutic drugs (Bai et al., 2011).
The nuclear factor of activated T cells (NFAT) is a master transcription factor most well-characterized in the immune system and is critical for T cell activation (Müller and Rao, 2010). NFAT is found to be overexpressed or hyperactivated in multiple cancer types, including breast cancer, pancreatic cancer, leukemia, melanoma, colon cancer and glioblastoma (Mancini and Toker, 2009; Müller and Rao, 2010; Qin et al., 2014). In these cancer cells, dysregulation of the NFAT pathway elevates the expression of key cancer-associated genes (e.g., COX2 (cyclooxygenase-2), autotaxin, VEGF (vascular endothelial growth factor), and matrix metalloproteinases (MMPs)) to promote tumor growth and malignant transformation. In glioblastoma, malignant phenotypes are highly correlated with NFAT upregulation (Tie et al., 2013). Multiple upstream signals, such as aberrant activation of growth factor receptors, Ca2+ signaling and the p53-K120R mutant, can cooperate with and/or converge on NFAT to promote tumor progression in glioblastoma (Chigurupati et al., 2010; Monteiro et al., 2017; Pearson and Regad, 2017; Shinmen et al., 2009). These findings indicate that the NFAT pathway might represent a promising drug target for glioblastoma therapy.
It has been established that NFAT activation is regulated by the upstream Ca2+-calcineurin signaling (Müller and Rao, 2010). In mammalian cells, the binding of growth factors (e.g., FGF or VEGF) to their cognate receptors activates phospholipase C (PLC) with subsequent hydrolysis of phosphatidylinositol 4,5 bisphosphate (PIP2) to generate inositol-1,4,5-trisphosphate (IP3). IP3 binds to the ER-resident IP3 receptor and triggers the release of Ca2+ from the ER lumen into cytoplasm (Berridge, 1993). The decrease of free Ca2+ within ER lumen is sensed by the stromal interaction molecule 1 (STIM1) via its ER-luminal domain that contains a Ca2+-binding EF-hand motif (Huang et al., 2009; Liou et al., 2005; Roos et al., 2005; Zhang et al., 2005). Next, activated STIM1 forms oligomers and migrates toward ER-PM junctions, where it directly gates the ORAI1 Ca2+ channels to evoke Ca2+ influx (Gudlur et al., 2013; Hogan et al., 2010; Nguyen et al., 2018; Prakriya and Lewis, 2015; Soboloff et al., 2012; Zhou et al., 2010). The sustained elevation of cytosolic Ca2+ activates calcineurin, a Ca2+/calmodulin-dependent phosphatase that dephosphorylates NFAT. Upon dephosphorylation, NFAT translocates from the cytoplasm to the nucleus to regulate gene transcription. Conversely, the dephosphorylated NFAT can be rephosphorylated by kinases, such as glycogen synthase kinase 3 (GSK3), casein kinases 1 (CK1), and the dual specificity tyrosine phosphorylation-regulated kinases (DYRKs), which causes the nuclear export of NFAT (Müller and Rao, 2010). Targeting any node of this pathway can disturb the nucleocytoplasmic shuttling of NFAT.
In this study, we conducted a screening for inhibitors of the calcineurin-NFAT pathway and serendipitously identified a compound YZ129 that shows strong anti-tumor activity toward GBM. YZ129 directly binds to HSP90 to antagonize its chaperoning effect on calcineurin, a Ca2+-sensitive phosphatase that is required for NFAT nuclear translocation and subsequent gene expression. HSP90 is a key member of the heat shock protein (HSP) family that function as an ATP-dependent molecular chaperone (Schopf et al., 2017). In tumor cells, HSP90 plays a crucial role in maintaining tumor cell survival through chaperoning the degradation of oncogenic proteins, such as AKT, HIF, VEGF, MMP2, mutant p53, JAK, and RTKs. Our systematic proteomic and transcriptomic studies unveiled that YZ129 targets pro-oncogenic HSP90 clients to suppress cancer cell growth both in vitro and in vivo. Our study establishes that suppressing the HSP90-calcineurin-NFAT pathway constitutes a promising anti-cancer strategy to benefit GBM patients.
RESULTS
Identification of small molecule inhibitors for the calcineurin-NFAT pathway
We first set out to identify chemical modulators for the calcineurin-NFAT pathway by using Ca2+ store depletion-induced nuclear translocation of NFAT as readout. As NFAT1 was often overexpressed in cancer cells, we conducted this experiment in a HeLa cell line stably expressing NFAT1-GFP. Ca2+ store depletion was triggered by using thapsigargin (TG), a potent inhibitor of the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) that prevents the refilling of ER Ca2+ store to elicit Ca2+ influx and subsequent calcineurin activation and NFAT nuclear entry. Building upon this assay that was compatible with an automated high-content screening platform, we evaluated the inhibitory activity of over 50,000 compounds from SPECS database that spanned a wide range of chemical diversity. The initial screening results revealed that some of the compounds showed suppressive activity (normalized to Z’-score) on NFAT nuclear translocation (representative examples in Table S1). Among all the hits, a 1,2-naphthalenone compound, named YZ01, stood out as a promising candidate with an IC50 value of 60 μM (Figure 1A). In HeLa cells incubated with YZ01, we observed a robust suppression of TG-induced NFAT nuclear translocation (Figure 1B). Next, to further optimize the efficacy of YZ01, we designed and synthesized a series of YZ01 analogs by replacing the 4-SO3H-naphthalene group with other aromatic rings (Figure 1C and Table S2), with the anticipation that increased hydrophobicity might enhance the intracellular uptake to boost the blocking efficiency. Indeed, the inhibitory activities of many YZ01 analogs were markedly improved (Figure 1D and Table S2), with YZ129 exhibiting the highest potency (IC50 = 820 ± 130 nM; Figure 1D and Table S2). YZ129 also suppressed the expression of NFAT-regulated genes, such as COX2, MMP7, and MMP9, in U87 glioblastoma cells (Figure 1E). Together, through an unbiased high throughput screening, we identified YZ129 as a strong inhibitor for the calcineurin-NFAT pathway.
Figure 1. Identification of YZ129 as an inhibitor targeting NFAT.

A. Quantification of the inhibitory activity of screened YZ compounds. 200 μM of each compound was added for the initial screening. The raw score of the inhibitory activity (nucleus-over-total NFAT1-GFP signals) was normalized to Z’ score values. NC, negative control without addition of thapsigargin (TG) to induce Ca2+ store depletion. PC, addition of TG to induce maximal NFAT nuclear entry.
B. Confocal images of HeLa cells stably expressing NFAT1-GFP after treatment with DMSO (control) or YZ01 (200 μM). 1 μM TG was added to induce store depletion and subsequent NFAT nuclear entry. Scale bar, 20 μm.
C. Chemical structures of YZ01 and its derivatives. The 4-SO3H-naphthalene group in YZ01 was replaced by other aromatic rings to yield new analogs, with YZ129 (highlighted in yellow) exhibiting the highest potency.
D. IC50 curves for the indicated compounds. Compared to other compounds or the positive control SKF96365, YZ129 showed the most potent inhibitory effect on NFAT nuclear translocation. Data were shown as mean ± SD (n= 4).
E. Real-time PCR revealed the inhibitory effect of YZ129 on the expression of NFAT downstream genes, such as MMP7 and MMP9, and COX2. GAPDH was used as control. Data were shown as mean ± SD (n= 3). **P<0.01.
YZ129 promotes apoptosis and suppresses proliferation, migration and mobility of glioblastoma cells in vitro
Next, we moved on to evaluate the anti-tumor effects of YZ129 by using U87 glioblastoma cells as a model system. In U87 cells treated with 5 μM YZ129 for 24 h, we observed pronounced changes in cell morphology with the majority of cells becoming round with less protrusions, rather than the otherwise elongated shapes seen in untreated cells (Figure 2A). Subsequently, the inhibitory effect of YZ129 on U87 cell proliferation was assessed by using a WST-1 based colorimetric assay. Following incubation with U87 cells for 48 h, YZ129 pronouncedly impeded the growth of GBM cells, but not normal astrocytes, in a dose-dependent manner (Figure 2B), thereby attesting to the selectivity of YZ129 against normal non-cancerous cells. To probe the anti-proliferation mechanism, we further examined the influence of YZ129 on cell cycle by flow cytometry. As depicted in Figure 2C, YZ129 induced a prominent cell cycle arrest at the G2/M phase, but had little effect on the S phase. The percentage of cells in the G2/M phase was increased from 20% to 53% upon YZ129 treatment. In addition, some dead cells were noticed, likely arising from apoptosis. We subsequently analyzed cell apoptosis by flow cytometry and noted a dose-dependent increase in both early (marked by Annexin V-positive but PI-negative) and late apoptotic processes (marked by both Annexin V and PI positive; Figures 2D and 2E). We further validated these findings independently at the single-cell level by immunostaining Ki67 (marker for proliferation) and active caspase 3 (marker for apoptosis). In cells incubated with YZ129 for 24 h, we detected 2.5-fold reduction in Ki67-positive staining with a concomitant 2-fold increase in active caspase 3 staining (Figure S1A). Clearly, YZ129 exhibited anti-tumor activity by inhibiting cell proliferation via cell cycle arrest at the G2/M checkpoint and promoting cell apoptosis.
Figure 2. YZ129 suppresses glioblastoma cell growth, survival and migration in vitro.

A. Comparison of cell morphology after treating U87 GBM cells with DMSO or 5 μM YZ129 for 24 h. Scale bars, 100 μm.
B. Dose-dependent inhibition of the proliferation of U87 cells and normal astrocytes revealed by the WST-1 assay (mean ± SEM; n= 3).
C. DNA content and cell cycle distribution analysis of U87 cells after treatment with DMSO or 5 μM YZ129 for 24 h. Cells were stained with propidium iodide (PI) and analyzed by flow cytometry.
D. Assessment of cell apoptosis. U87 cells after treatment with DMSO or YZ129 for 24 h were stained with FITC-conjugated Annexin V and PI, and then analyzed by flow cytometry.
E. Quantification of PI-single positive (dead), Annexin V-single positive (early apoptosis) and Annexin V/PI-double positive (late apoptosis) cell populations (mean ± SEM, n=3).
F. Wound healing assays were performed to assess the rates of migration for control (DMSO) and YZ129-treated U87 cells. Scale bars, 100 μm. Bar graphs on the right showed percentage of migration cells (mean ± SD, n=5).
G. Effect of YZ129 (5 μM) on cell mobility evaluated by recording the movement trajectory of U87 cells over 1.5 h. Scale bars, 50 μm. Bar graph on the right showed the normalized cell movement distance (mean ± SD, n=5).
See also Figure S1
One of the most important hallmarks of malignant glioblastoma is their invasive behavior (Cheng et al., 2011; Rao, 2003). We therefore examined how YZ129 would impinge on cell migration and motility. As revealed by the wound healing assay results, YZ129 hindered the migration of U87 cells: the mean numbers of the cells in the denuded area were reduced by ~60% compared to the control group (Figure 2E). This finding was concordant with our earlier observation that YZ129 could suppress the expression of MMP-7 and MMP-9 (Figure 1E), two NFAT target genes that promote cancer cell migration (Tie et al., 2013). In parallel, we monitored the motility of U87 cells by continuously recording the cell movement trajectories under a confocal microscope (Memmel et al., 2017). As shown in Figure 2F, YZ129 pronouncedly impaired cell motility by restraining cells from traveling further away from their starting points: the average travelled distance per hour was reduced by 60% (Figure 2F, right panel). In aggregate, YZ129 could not only suppress cancer cell growth but also reduce cancer cell movement and migration.
YZ129 suppresses the growth of glioblastoma xenografts in vivo
Having confirmed that YZ129 exerted potent antitumor activity against GBM cells in vitro, we further evaluated its therapeutic potential in vivo by using a glioblastoma xenograft mouse model. Nude mice bearing U87-luciferase-expressing (U87-Luc) xenograft tumors were treated with YZ129 via intraperitoneal injection every day for 5 days. The bioluminescence imaging of the tumors was recorded every week. Compared to the control group, the tumor sizes, as reflected by bioluminescence, were substantially reduced in the YZ129-treated group (Figures 3A–C and Figure S2A). Three weeks after treatment, tumors were excised and the tumor volume was calculated. The tumor sizes were reduced by over 60% upon YZ129 treatment (Figures 3B and 3D). HE (hematoxylin and eosin) staining of xenograft tumors showed that, after treatment with YZ129, the tumor cell distribution became uneven with nuclei sizes varying greatly among different cells. In addition, karyopyknosis, which reflects the nuclear chromatin condensation in cells undergoing necrosis or apoptosis, was prominently noted in the xenograft tissues (Figure 3E). Consistent with the immunocytochemistry staining results, immunohistochemical (IHC) staining revealed that YZ129 also led to a decrease in Ki67-positive cell number but an increase in caspase-3 positive cells compared to control (Figure 3F), thereby validating the tumor suppressive and apoptosis-promoting effects of YZ129 in vivo.
Figure 3. YZ129 suppresses the growth of glioblastoma xenografts in vivo.

A. Representative bioluminescence images of U87-Luc xenografts. Nude mice were subcutaneously transplanted with U87-Luc GBM cells and treated with vehicle or YZ129 every day for 5 days at a dose of 2.5 μM/kg body weight. The tumor size was monitored each week using bioluminescence imaging.
B. Representative images of tumors dissected from vehicle-treated or YZ129-treated nude mice at week 3.
C. Tumor size was assessed by the weekly bioluminescence intensities in xenograft tumors and plotted relative to the bioluminescence at week 3 for the DMSO control group (mean ± SD, n = 6).
D. The volumes of tumor masses isolated from nude mice treated with DMSO or YZ129 (mean ± SD, n=6). Each dot represents the tumor volume from one mouse. ***P < 0.001 (Two-tailed Student’s t-test).
E. HE staining of the dissected subcutaneous xenograft tumor tissues from mice treated with vehicle (DMSO) or YZ129. Arrow, sign of karyopyknosis with nuclear chromatin condensation. Scale bars, 50 μm.
F. Representative images of immunohistochemical (IHC) staining of cell proliferation marker Ki67 and activated caspase 3 in GBM xenografts isolated from mice treated with vehicle or YZ129. Scale bars, 50 μm. Percentage of positive cells was scored from 4–6 random fields per tumor section. ***P < 0.001 (Two-tailed Student’s t-test).
See also Figure S2
To evaluate the potential toxicity of YZ129 in vivo, we further performed HE staining on major tissues obtained from YZ129-treated mice. YZ129 treatment did not elicit overt cellular and tissue destruction in the liver, kidney and small intestine (Figure S2C). In addition, during the course of xenograft experiments for up to three weeks, the body weight of the xenograft mice remained largely comparable to the control group (Figure S2D). Overall, YZ129 did not seem to induce significant toxic damages to cells and tissues in living mice.
To further assess the therapeutic potential of YZ129 on orthotopic GBM tumors, we implanted the U87-Luc glioblastoma cells in intracalvarium and monitored tumor growth by bioluminescence imaging. One week following YZ129 treatment, we observed a significant decrease in the bioluminescent intensities and areas, two parameters used to evaluate the intracranial tumor growth (Figure 1B). Given that we administrated YZ129 via intraperitoneal injection, it was highly possible that YZ129 could penetrate the blood-brain barrier to attack orthotopic GBM tumors, an ideal feature that is required for further drug development to treat brain tumors (Westphal and Lamszus, 2011).
YZ129 inhibits the calcineurin-NFAT pathway independent of Ca2+ influx
We next sought to probe the mechanism of action of YZ129 by assessing how it might impinge on each critical signaling node in the Ca2+/calcineurin-NFAT pathway. We first compared the TG-induced Ca2+ influx in cells treated with DMSO (as control) or YZ129 but did not observe a significant difference in both the initial and second phases of Ca2+ increase in the cytosol (Figure 4A), thereby ruling out the involvement of the upstream STIM/ORAI-mediated store-operated Ca2+ entry. We then assessed the effect of YZ129 on the next signaling component, the Ca2+-responsive phosphatase calcineurin. We expressed a mCherry (mCh)-tagged constitutively-active form of the catalytic subunit of calcineurin A (caCnA) (Clipstone and Crabtree, 1992) in NFAT1-GFP HeLa cells. Consequently, in the same imaging field, we observed spontaneous nuclear translocation of NFAT in mCherry-positive cells independent of store depletion, but not in those without expressing mCh-caCnA (Figure 4B, top panel). By contrast, in cells incubated with YZ129, we failed to observe spontaneous NFAT nuclear entry regardless of the expression of mCh-caCnA (Figure 4B, bottom panel). Finally, we examined whether YZ129 would further directly modulate NFAT or block the nuclear import machinery that is responsible for NFAT nucleocytoplasmic shuttling. We compared the subcellular localization of constitutively-active NFAT1 (caNFAT1) (Monticelli and Rao, 2002) in HeLa cells. In both cells treated with DMSO or YZ129, we noticed a clear nuclear localization of caNFAT1 (Figure 4C), thus ruling out a direct effect of YZ129 on the downstream NFAT or the nuclear import machinery. Taken together, our findings imply that calcineurin is one critical node in the signal transduction pathway that can be inhibited by YZ129.
Figure 4. YZ129 inhibits the calcineurin-NFAT pathway independent of Ca2+ influx A.

The effect of 5 μM YZ129 on store depletion-induced Ca2+ influx. Empty bar, 0 mM Ca2+ in the external medium; Black bar, 1 mM Ca2+. Blue bar, 1 μM TG used to passively induce store depletion.
B. The effect of YZ129 on NFAT nuclear translocation (green) induced by a constitutively active calcineurin (caCnA; red). mCherry (mCh)-tagged caCnA was transfected into an NFAT1-GFP stable HeLa cell line to induce nuclear entry of NFAT1-GFP (arrow). Transfected cells were either treated with DMSO or YZ129 (50 μM). Note that caCnA-induced NFAT nuclear entry was inhibited by YZ129. Scale bar, 20 μm.
C. YZ129 failed to suppress the nuclear localization of a constitutively active NFAT1 (caNFAT1). GFP-tagged caNFAT1 was transfected into HeLa cells, treated with DMSO (control) or YZ129 (50 μM). Scale bar, 10 μm.
YZ129 suppresses the calcineurin-NFAT pathway by targeting HSP90
To identify the direct molecular target of YZ129, we resorted to click chemistry and biotin-streptavidin binding to pull down its potential binding partners (Figure 5A). We synthesized three azide derivatives of YZ129 (YZ137–139; Table S2), in which the azido group was linked with 1,2-naphthoquinone by a carbon chain of varying lengths (4–6 carbon atoms). These derivatives largely preserved the activity on inhibiting NFAT nuclear translocation (Figures S3A and S3B and Table S2), with YZ137 containing five carbon atoms in the linker exhibiting the strongest IC50 (7.70 ± 2.4 μM). We subsequently immobilized YZ137 to streptavidin beads through a click reaction between YZ137 and biotin-alkyne as shown in Figure 5A. Immobilized YZ137 was used as a bait and incubated with cell lysates to pull down potential binders. Because the inhibitory activity of YZ129 against NFAT nuclear translocation was initially determined in HeLa cells, we first used this cell line for our proteomic study. Compared to controls, we indeed observed enrichment of proteins and a prominent band resolved at approximately 93 kDa, as revealed by the SDS-PAGE shown in Figure 5B. Through in-gel digestion coupled with mass spectrometry, we identified HSP90 as the molecular entity corresponding to this band (Figure S4). In parallel, we unbiasedly analyzed the whole eluted samples by mass spectrometry. Again, we detected HSP90 as the most enriched protein, as well as other significant hits with the majority being HSP90 clients or interactors, including tubulin, HSP27, and nucleolin (Figure 5C). To further validate this in vitro, we incubated the immobilized biotin-YZ137 conjugate with recombinant HSP90 and detected an enrichment of HSP90 on the beads (Figure 5D), clearly demonstrating a direct interaction between the YZ129 derivative and HSP90. We also repeated the pulldown experiment using U87 cell lysate. A distinct band resolved at ~93 kDa was also observed (Figure 5E). In the presence of soluble YZ129 as competitor, the band intensity was reduced while other bands remained largely unaffected (Figure 5E), implying the specificity and reversibility of the physical association between YZ129 and HSP90.
Figure 5. YZ129 directly targets HSP90 to destabilize calcineurin.

A. Design of a biotinylated YZ129-azide derivative (YZ137) as the bait to identify its cellular binding targets. YZ129 was immobilized to streptavidin (Stv) beads through a Click reaction between biotin-alkyne (immobilized on streptavidin beads) and YZ137 in the presence of THPTA, CuSO4 and ascorbic acid. Immobilized biotin-YZ137 was used as the bait to enrich its binding partners and subsequently subjected to mass spectrometry (MS) analysis.
B. Silver staining of SDS-PAGE showing a typical pull-down result of immobilized YZ137 after incubation with HeLa cell lysate. The band framed in the box was identified as HSP90 (see Figure S4).
C. Mass spectrometry analysis of the total mixtures eluted from the YZ129-immobilized or control beads (the same as panel B). Candidates that were only present in the YZ29 group (>150 spectra counts) or highly enriched after YZ129 treatment (> 4 fold counts when compared to control) were considered as significant hits. HSP90 clients or its known interactors were highlighted in red.
D. SDS-PAEG visualized by Silver staining after pulldown of recombinant HSP90 by immobilized YZ137.
E. The silver staining of SDS-PAGE showing the pull-down result of immobilized YZ137 after incubation with U87 cell lysate. In the presence of competitive YZ129, the intensity of the ~93kDa band was reduced.
F. Representative IC50 curves showing the competitive binding of YZ129, YZ126, or 17-AAG (positive control) to the HSP90/geldanamycin (GA)-FITC complex (left) or the inhibitory effect on NFAT nuclear entry (right). GA is a known HSP90 inhibitor that binds to the APT-binding pocket.
G: Confocal images showing the effect of 50 μM 17-AAG on NFAT nuclear entry (mean ± SEM, n= 4). Scale bar, 40 μm.
H. Immunoblot analysis of the expression of calcineurin in U87 cells treated with DMSO or YZ129.
I. Measurement of mRNA levels of calcineurin in U87 cells treated with DMSO or YZ129. The quantification was shown on the right (mean ± SEM, n= 3).
See also Figures S3–S5 and Tables S3 and S4
Subsequently, we used a competitive fluorescent polarization assay to determine the binding affinity of YZ129 with HSP90. In this assay, geldanamycin-FITC, a fluorescent probe that binds specifically to the HSP90 ATP-binding pocket, was used. The well-established HSP90 inhibitor, 17-AAG (Li et al., 2014), was used as positive control. We obtained an IC50 value of 29.5 nM for YZ129, which was comparable to 17-AAG (Figure 5F). Meanwhile, because YZ129 emitted blue fluorescence (Figure S5A), we were able to conduct a similar competitive fluorescence polarization assay by titrating 17-AAG into the pre-incubated HSP90-YZ129 mixture. As anticipated for a typical reversible protein-ligand binding event, we observed a dose-dependent decrease in the fluorescence polarization signals when high doses of 17-AAG were gradually added to outcompete YZ129 from the complex (Figure S5B). Moreover, 17-AAG as a classical HSP90 inhibitor could also inhibit TG-induced NFAT nuclear entry with an IC50 value of 5.90 μM (Table S3, and Figures 5F and 5G). Similar to YZ129, 17-AAG inhibited caCnA induced NFAT nuclear translocation, but not the nuclear accumulation of caNFAT1 (Figures S3C and S3D).
Given that calcineurin is one of the client protein of HSP90 (Singh et al., 2009) and YZ129 directly inhibited HSP90, we reasoned that YZ129 might block the chaperon activity of HSP90 to render calcineurin more unstable with concomitant loss of its catalytic activity, thereby leading to the apparent suppression on NFAT nuclear translocation. Indeed, our immunoblotting analysis results confirmed our speculation: we noticed a gradual reduction in the protein levels of calcineurin (Figure 5H) in U87 cells upon YZ129 treatment; whereas the mRNA levels remained insignificantly altered (Figure 5I). These results establish that YZ129 inhibits NFAT nuclear translocation likely by disrupting HSP90-mediated chaperone activity on calcineurin.
Global effects of YZ129 assessed by transcriptomic and proteomic profiling in glioblastoma cells
To unbiasedly delineate how YZ129 might impinge on the cancer signaling networks, we performed integrative omics studies in U87 cells treated with DMSO (control) or YZ129. We first evaluated the impact of YZ129 on gene expression at mRNA levels using RNA-seq. In proliferation and migration assays, when U87 cells were exposed to 5 μM YZ129 for 24 h, it could generate significant pharmacological effects without inducing massive cell death. We therefore used this condition throughout our transcriptomic profiling experiments. We detected a total of 407 differentially expressed genes (DEGs; fold-change ≥ 2 and FDR < 0.05), with 226 showing upregulation and 181 downregulated (Figure 6A and Figure S6). The functional enrichment analysis of DEGs was performed using the GSEA software in the hallmark gene sets category (Liberzon et al., 2015). The downregulated genes were largely associated with oncogenic pathways, including hypoxia, glycolysis, the PI3K/AKT/mTOR pathway, epithelial-mesenchymal transition, and G2/M checkpoint (Figure 6B and Figure S6). Conversely, the functions of the upregulated genes were largely found to be tumor suppressive, including genes involved in the TNFα signal pathway, apoptosis, p53 pathways, and tumor suppressors.
Figure 6. Global assessment of molecular responses in U87 GBM cells treated with YZ129.

Gene expression profiled by RNA-seq (GEO ID: GSE108749), whereas changes in protein expression/phosphorylation levels of key cancer-associated proteins were evaluated with RPPA.
A. Volcano plot of RNA-seq results. Data for all genes were plotted as log2 fold change versus the −log10 of the adjusted p-value. Genes with significant changes in expression (changed ≥2-fold) after treatment with YZ129 were presented as blue (181 downregulated genes) or red dots (226 upregulated genes), respectively.
B. Functional enrichment analysis of the significantly changed genes highlighted in the volcano plot.
C. Heatmap showing the differential expression and/or phosphorylation levels of key cancer-associated proteins in U87 cells treated with DMSO or YZ129. The RPPA experiments were carried out with two biological replicates.
D. Immunoblot analysis of the expression of AKT and its phosphorylated forms (T308) in U87 cells treated with DMSO or YZ129.
E. Summary of major cancer-associated pathways other than NFAT that are affected by YZ129 in U87 GBM cells. The RTK/PI3K/AKT/mTORC, TGF-β/Smad3, hypoxia (HIF1) and glycolysis (LDHA) pathways were most affected, thereby suppressing cancer cell proliferation, migration, and survival to exert anti-tumor effects.
See also Figures S1 and S6
To further capture the molecular signature of YZ129’s effects on GBMs, especially at the protein expression level and posttranslational modifications, we profiled the alterations of protein expression before and after YZ129 treatment in glioblastoma cells by using the Reverse Phase Protein Array (RPPA) assay, in which the expression or phosphorylation states of 295 key cancer-associated proteins are probed by specific antibodies (Wachter et al., 2015). We found that a large fraction of downregulated proteins (>50% shown in Figure 6C) were among known or predicted HSP90 chaperoning clients (http://www.picard.ch/downloads/Hsp90interactors.pdf). Most notably, many signaling proteins in the proto-oncogenic RTKs/PI3K/AKT/mTOR pathway (Liberzon et al., 2015) was significantly inhibited by YZ129 (Figure 6C), including PDGFR-β, IGF1R, VEGFR, AKT, mTOR, Raptor, Rictor, and TSC1/2 (Figure 6C). The downregulation of AKT and its phosphorylated form (at residue T308) were further confirmed by immunoblotting (Figure 6D). It is also worth noting that the downregulation of vimentin was most prominent (4-fold; Figure 6C). Vimentin belongs to the intermediate filament family of cytoskeletal proteins, and also acts as a maker of epithelial-mesenchymal transition (Satelli and Li, 2011). Downregulation of vimentin may result in cytoskeleton destruction to impede cell migration and invasion. Moreover, the cytoskeletal destruction arising from vimentin downregulation could contribute to the cell morphology changes observed in Figure 2A. It is interesting to note that that two negative immune regulators, B7–H3 and PD-L1, were also downregulated, which may reduce immune evasion during tumor progression (Kataoka et al., 2016; Liu et al., 2011). In parallel, the expression levels of a series of pro-oncogenic proteins, including fibronectin, BRAF, Notch3, FAK, YB1, β-catenin, Sox2, c-kit, Gys1, were also reduced (Figure 6C), which collectively suppress tumor growth, invasion and metastasis.
The upregulated proteins were mostly associated with G2/M cell cycle checkpoint regulators and apoptosis, including cyclinB1, PLK1, Chk2, MAPK14 (p38), 14–3–3-zeta/epsilon (Su et al., 2001), Smac (Du et al., 2000) and cyclophilin-F (Tavecchio et al., 2013) (Figure 6C). The G2/M checkpoint was activated by inhibiting the CDK1 (Cdc2)/cyclinB1 complex, which prevents cells entering mitosis and results in cell cycle arrest at G2/M phase(Kastan and Bartek, 2004; Wang et al., 2009). This finding dovetailed with our cell cycle analysis results shown in Figure 2C. Dephosphorylation of CDK1 by Cdc25B and Cdc25C is required for activation of CDK1 (Donzelli and Draetta, 2003; Wang et al., 2009). The upregulation of Chk2 and MAPK14 could phosphorylate Cdc25B at S309 site and Cdc25C at the S216 site, unmasking the binding sites of Cdc25B and Cdc25C protein to 14–3–3 protein (Bulavin and Fornace, 2004; Bulavin et al., 2001; Tsvetkov et al., 2003). The Cdc25B/C-14–3–3 complex can sequestrate Cdc25B/C in the cytoplasm and inactivate CDK1 activity (Bulavin et al., 2003). Therefore, it can be speculated that the cell cycle G2/M arrest induced by YZ129 may result from inactivation of Cdc25B/C via Chk2, MAPK14, and 14–3–3.
Discussions
Glioblastoma is a lethal disease featured with high aggressiveness, poor prognosis, and low survival rates. Presently, limited drugs have been approved to treat glioblastoma. NFAT is an attractive antitumor target, particularly for highly aggressive tumors. NFAT controls the expression of key cancer-associated genes, including COX2, VEGF, autotaxin and MMPs. In addition, NFAT can promote the exhaustion of tumor-infiltrating T cells by enhancing the expression of negative regulators of anti-tumor immunity, such as TIM3 and the inhibitory immune checkpoint protein PD-1 (Martinez et al., 2015). NFAT can thus be exploited as an alternative therapeutic target for cancer treatment.
In this study, we report the discovery of a previously-unrecognized class of small molecules, YZ129 and its derivatives, that share a common core structure of 1,2-naphthalenone. YZ129 potently inhibits the HSP90-calcineurin-NFAT signaling axis to effectively suppress GBM growth and survival. YZ129 suppresses NFAT nuclear translocation and its downstream gene expression by disrupting HSP90-mediated chaperone activity on calcineurin. Furthermore, YZ129 can induce cell cycle arrest at the G2/M phase, promote cancer cell apoptosis and inhibit cell proliferation and migration (Figure 2). In addition, we have demonstrated that YZ129 can suppress the GBM xenografts growth in vivo while imposing limited toxicity to healthy tissues in living animals (Figure 3 and Figure S2). Most notably, intraperitoneal administration of YZ129 could further inhibit GBM cell growth in an orthotopic xenograft model, strongly indicating that YZ129 can travel across the blood-brain barrier (BBB), which is of great importance for CNS disease therapy.
To yield an integrated view on the mechanisms of action of YZ129 on glioblastoma, we have performed comparative transcriptomic and proteomic profiling in GBM cells with or without YZ129 treatment. These unbiased omics studies could facilitate understanding the molecular mechanisms of drugs, help identify the pharmacodynamic markers of drug response, and aid the discovery of gene expression patterns that account for drug resistance and sensitivity (Clarke etal., 2004). As anticipated, we detected the downregulation of a set of NFAT downstream genes, such as VEGFA, VEGFR-2, MMP2, TMEM45A, PFKFB3, and Smad3. Both RNA-seq and RPPA assay results converge to support the conclusion that YZ129 also inhibits other key cancer-associated pathways, including hypoxia, glycolysis and the RTK/PI3K/AKT/mTOR axis (Figure 6E and Figure S6). Particularly, the hypoxia regulator HIFα and the glycolysis catalytic enzyme LDHA are both downregulated, which may cut off the energy supply to tumor cells while improving the low-oxygen acidic tumor microenvironment. In addition, cytoskeleton protein vimentin and myosin-IIa were the most downregulated proteins, which may contribute to the cell morphology changes observed in GBM cells after YZ129 treatment (Figure 2A). Furthermore, YZ129 can induce cell cycle arrest at the G2/M phase in GBM cells. From our bioinformatic analyses on the RNA-seq and RPPA datasets, we speculate that the molecular basis underlying this phenomenon probably involves the downregulation of UBE2C, Chk2, MAPK14, 14–3–3-zeta/epsilon at the mRNA level and Chk2 and MAPK14 at the protein level.
Findings from the in cellulo and in vivo studies consistently point to an apoptosis-promoting effect of YZ129 on cancer cells. Apoptotic cell death is controlled by the coordinated actions of a set of apoptosis promoters and inhibitors (Nagata, 2018), such as caspases, the Bcl-2 family, and the inhibitor of apoptosis protein (IAP) family. Some of them have been established as HSP90 clients, such as Bcl-2, Bcl-xL, and Beclin 1. Indeed, anti-apoptotic Bcl-xL, Bcl-2 and XIAP proteins were downregulated following YZ129 treatment in glioblastoma cells (Figure S1), as one would expect for inhibition on HSP90 chaperoning. In addition, pro-apoptotic protein Bax and cleaved caspase-7 were upregulated. These changes at molecular levels might explain the overall pro-apoptotic role of YZ129 (Figures 2D and 2E; and Figure 3F).
Aside from these changes, we also observed reciprocal changes in the expression of some tumor-promoting genes (downregulation of fibronectin, BRAF, Notch3, FAK, YB1, β-catenin, Sox2, c-kit, and Gys1) and tumor suppressive genes (upregulation of BTG2, ATF3, BMP2, NR4A3, TAGLN, DKK1, PRAS40, and MEG3), with all contributing to the suppression of tumor growth, survival and invasion. However, it should be noted that some tumor-promoting genes (e.g., FOSB, INHBA, EDN1, IRS2, KDM6B, TGM2, PREX1, PAI-1, CyclinB1, PLK1, NEAT1 and MALAT) are also upregulated, which might ultimately render tumor cells resistant to treatment. Interestingly, some non-coding RNAs, including NEAT1, MALAT1 (NEAT2) and MEG3, were upregulated. It has been reported that NEAT1 and MALAT1 can generate tumor-promoting effects and may account for chemoresistance, while MEG3 has tumor suppressive function (Adriaens et al., 2016; Gutschner et al., 2013; Zhou et al., 2012). These omics data could provide valuable pharmacological information for HSP90 inhibitor action in glioblastoma cells.
As a chaperone, HSP90 assists other proteins to fold and assemble properly, stabilizes proteins against aggregation, and facilitates protein degradation (Whitesell and Lindquist, 2005). Till now, over one hundred proteins, ranging from receptors, kinases, E3 ubiquitin ligases to transcription factors, have been identified as the client proteins of HSP90, many of which are involved in tumorigenesis, such as AKT, HIF, JAK, and RTKs (http://www.picard.ch/downloads/Hsp90interactors.pdf). Moreover, overexpression of HSP90 is correlated with poor prognosis of cancer patients (Chatterjee and Burns, 2017). Therefore, HSP90 is an actively pursed anticancer target in the pharmaceutical industry with 17 drug candidates undergoing clinical trials (Jhaveri et al., 2012; Khandelwal et al., 2018; Shrestha et al., 2016). While most ongoing studies center on the HIF, AKT/mTOR, and Ras-Raf-ERK pathways (Mahalingam et al., 2009; Whitesell and Lindquist, 2005), our study unveils the importance of a previously neglected pathway that involves calcineurin and NFAT. Our study fills a critical gap in the systematic dissection of crosstalk between HSP90 and the calcineurin-NFAT pathway in the context of cancer. It is our immediate future plan to further evaluate whether targeting the HSP90/calcineurin/NFAT axis can overcome the chemoresistance encountered in the treatment of glioblastoma.
SIGNIFICANCE
Glioblastoma (GBM) is among the most common and malignant primary brain tumors with a dismissal prognosis. Due to its aggressiveness and tendency to widely metastasize within the brain, it is almost impossible to remove the tumor lesion completely through surgical resection without affecting the normal brain function. Therefore, effective chemotherapeutic drugs are urgently needed in the clinic for improving patient survival. Here we report the discovery of a previously-unrecognized class of HSP90 inhibitors (YZ129 as a lead compound) that share the core structure of 1,2-naphthalenone. YZ129 disrupted the chaperone network of HSP90 to deregulate the calcineurin-NFAT pathway and other cancer-associated signaling events. As a result, YZ129 curtailed glioblastoma growth and promoted cancer cell apoptosis both in vitro and in xenograft GBM mouse models. Capitalizing on unbiased integrative omics approaches, we further characterized the global impact of YZ129 on cancer-associated signaling pathways both at the mRNA and protein levels. Collectively, our study has established the preclinical feasibility of targeting HSP90 client network to treat GBM. The anti-cancer HSP90 inhibitors developed in this study can furnish an alternative therapeutic strategy for the clinical management of GBM in the near future, and might hold promise to overcome chemoresistance encountered in the current GBM therapies.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yubin Zhou (yzhou@ibt.tamhsc.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Line Authentication
The human GBM cell lines U87-MG (male glioblastoma, ATCC, # HTB-14) and HeLa (taken from a female with cervical cancer, ATCC, # CRM-CCL-2) were purchased from American Type Culture Collection (ATCC). The cell line was validated by short tandem repeat profiling analysis by ATCC. The NFAT1(1–460)-GFP stably transfected HeLa cell line was a gift from Anjana Rao at La Jolla Institute. U87 cell line that stably expresses luciferase (U87-Luc) was established through lentiviral transduction. Transduced colonies (bioluminescent U87 cells, or U87-Luc) were selected with 5 mg/mL blasticidin and examined for luciferase expression by detecting its activity with D-Luciferin.
Cell Culture Conditions
U87, HeLa, NFAT1(1–460)-GFP HeLa and U87-Luc cells were all cultured in Dulbecco’s modified eagle’s medium (DMEM) supplemented with 10% FBS in 5% CO2 at 37 °C.
Mice
All animal experiments were approved by the Ethics Committee and IACUC of the Third Military University and were conducted according to the guidelines for laboratory animals care and use. BALB/c nude mice used in this study were 6–8 weeks of age with either sex and were purchased from a pathogen-free Animal Center of the Third Military University (Chongqing, China).
METHOD DETAILS
NFAT Nuclear Translocation Assay
The stable NFAT1-GFP HeLa cells were cultured in Dulbecco’s modified eagle’s medium (DMEM) supplemented with 10% FBS in 5% CO2 at 37 °C. When it reached 90% confluency, a cell suspension with a concentration of 80,000/mL was prepared, and an amount of 2000 cells per well (25 μL/well) was seeded in a 384-well plate. After overnight culture, cells were used for high-content screening. A series of compounds with a final concentration of 200 μM (diluted from 20–50 mM stock dissolved in DMSO) in DMEM were prepared in a a 96 well plate and then added to 384-well plate using a Beckman Counter automatic liquid handling system. Each condition was repeated for four times. SKF96365 was used as the positive control. 1 μM TG and 1 mM Ca2+ were added to each well and incubated with the cells for 20 min. Subsequently, the 384-well plate was washed with PBS twice gently, and the cells were fixed with 4% PFA (25 μL per well) for 15 min at room temperature, washed with PBS, treated with 0.5% Triton X-100 (25 μL per well) for 5 min at room temperature. Permeabilized cells were subsequently stained with DAPI (1 μg/mL) for 5~10 min at room temperature and washed with PBS for three times to remove residual dyes. Prior to imaging, 30 μL PBS was added to each well and the 384-well plate was mounted for high-content imaging using an IN Cell Analyzer 6000 cell imaging system (objective lens: 10×. FITC and UV; GE Healthcare). Using the Pipeline Plot software (with a user customized NFAT nuclear translocation counting protocol), the intensities of green fluorescence in the nuclei and cytosol of each cell were counted to calculate the nucleus-to-cytosol ratio. For normalization, the ratio in cells without treatment of TG was set as 0 while the ratio in cells treated with TG and DMSO as 1. Finally, the data were exported to GraphPad Prism 5 for calculation of IC50 values. For each plate, the Z’-score was calculated as: Z’= (RatioTest – RatioMean) / σStd, where RatioTest represents the normalization ratio of each compound, RatioMean represents the mean normalization ratio value of all the tested compounds, and σStd represents standard deviation of all the compounds’ ratios.
Ca2+ Influx Assay
Intracellular [Ca2+] was measured with the green calcium indicator, Fluo-4 AM (5 μM, Invitrogen). In brief, HeLa cells were seeded in a glass-bottom dish (MatTek) at least 1 day before imaging. Before Ca2+ measurements, the culture dishes were washed with Ca2+-free extracellular solution buffer (107 mM NaCl, 1.2 mM KCl, 1.2 mM MgCl2, 1.2 mM glucose, 20 mM HEPES-NaOH, PH 7.2). Cells were incubated with medium alone and 5 μM YZ129 for 20–30 min before the addition of thapsigargin (TG), a non-competitive inhibitor of the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA). Cells were first kept in Ca2+-free imaging solution and then in 1 mM CaCl2 imaging solution. Fluorescence intensities were recorded using a Nikon confocal microscope. At least 20–30 cells from 3–4 biological replicates were selected for each condition.
Plasmids
The full-length cDNA of calcineurin was purchased from Addgene (#11785). The fragment encoding the N-terminal catalytic domain of calcineurin (CnA; residues 1–381) was amplified by PCR. Then, a constitutively active calcineurin (caCnA) construct was produced by inserting this amplified fragment into pCDNA3.1-mCherry vector between the EcoRI and XhoI sites. Primers were shown in Table S4. Constitutively active NFAT1 (caNFAT1) was obtained from Addgene (#11792). The sequence encoding caNFAT1 (aa 1–460) was amplified by standard PCR using the KOD hot start DNA polymerase (EMD Millipore) and then inserted into the pEGFP-N1 vector between HindIII and AgeI sites.
Confocal Imaging
Cells were grown in DMEM medium supplemented with 10% (v/v) fetal bovine serum at 37 °C with 5% CO2 on 35-mm glass-bottom dishes. To measure the inhibitory activity of TG-induced NFAT nuclear translocation, YZ01 (200 μM), HSP27 inhibitor Quercetin (Heowns, 200 μM), and 17-AAG (50 μM), YZ129 azide analogues (YZ136–139, 200 μM) were incubated with NFAT1-GFP stably transfected HeLa for 30 min, and then 1 μM TG and 1 mM Ca2+ were added. Fluorescence imaging was performed on a Nikon A1 confocal microscope. To measure the inhibitory activity of caCnA induced NFAT translocation, 2.5 μg caCnA was transfected into NFAT1-GFP stable expressing HeLa cells using TransIT transfection reagents (Mirus). 24 h posttransfection, YZ129 (50 μM), or 17-AAG (50 μM) were incubated with the transfected cells for 1–2 h. The method used for evaluating the inhibitory activity of YZ129 (50 μM) and 17-AAG (50 μM) on caNFAT1 was similar, except that NFAT1-GFP HeLa cells were replaced with HeLa cells transiently expressing caNFAT1.
Pull-down Experiments
Firstly, M-280 streptavidin magnetic beads were washed with buffer A (pH=7.3) that contained 20 mM Tris-HCl, 150 mM NaCl and then Biotin-PEG4-Alkyne was added. After incubation at 4 °C for 2 h, the beads were washed with buffer A three times, with each time for 10 min at 4 °C. Then, YZ137 (1 eq) was added to react with the immobilized alkyne by incubation with THPTA (0.2 eq), CuSO4 (0.1 eq) and ascorbic acid (1 eq) for 2 h at room temperature in a solution of 1:3 glycerol and water. As a result, the YZ129 azide analog was immobilized to streptavidin beads through a click chemistry reaction between the alkyne and azide (Schulke et al., 2014; Sibbersen et al., 2014; Tashiro and Imoto, 2012). Unreacted free YZ137 was washed away by using buffer A. Subsequently, the conjugated beads was incubated with cell lysates (2–5 mg/ml) extracted from HeLa cells or U87 cells for 4 h at 4 °C and then rinsed with buffer B (pH=7.3) that contained 20 mM Tris-HCl, 150 mM NaCl, 5% glycerol, protease inhibitor for three times, with each time for 10 min at 4 °C. Finally, the enriched proteins were eluted using 8 M urea (pH=1.5) and then subjected to SDS-PAGE. A differential band corresponding to ~93kDa was cut out and digested with trypsin for further MALDI-MS analysis. Alternatively, the whole eluted samples were subjected to mass spectrometry analysis using LC-MS/MS at the in-house MS core facility. For competetive pull-down experiment, 10 μM YZ129 was added in the cell lysate. The procedure for the U87 cell lysis and recombinant HSP90α protein (ab80369, Abcam, USA) pull-down experiment was similar as procedures done with the HeLa cell lysate.
HSP90 Competitive Binding Assay
The binding affinity of the synthesized compounds against HSP90 was determined using a competitive fluorescent polarization (FP) assay based on a FITC-conjugated geldanamycin (GA-FITC) that binds specifically to the HSP90 ATP-binding pocket. In brief, 50 μL of 100 nM recombinant HSP90α protein (ab80369, Abcam, USA), 25 μL of 10 nM GA-FITC and 25 μL of different concentrations of compounds in the FP assay buffer (20 mM HEPES, 50 mM KCl, 2mM DTT, 5mM MgCl2, 20 mM Na2MoO4, 0.01% Triton X-100 with 0.1 mg/mL BSA) were added to a microplate. After incubating at 4 °C for 4 h, plates were mounted on a Beckman Coulter DTX80 multi-mode microplate reader with excitation at 470/5 nm and emission at 515/5 nm. The polarization value was calculated using the following equation mP=1000× (Iparallel – G*Iperpedicular)/(Iparallel + G*Iperpedicular). The correction factor G (G=1.12) was introduced to eliminate differences in the transmission of vertically and horizontally polarized light. The negative control contained GA-FITC and HSP90 protein, while the blank control only contained GA-FITC. The inhibition curves were fitted using the GraphPad Prism 5 software. 17-AAG, a highly selective HSP90 inhibitor, was used as the positive control in the FP assay.
Immunoblotting
U87 cells were treated with 0.25 μM, 5 μM YZ129 or DMSO (as control) for 24 h. Cells were washed with PBS three times and lysed on ice with RIPA buffer containing protease inhibitors cocktails (Roche) for 30 min. The lysate was centrifuged at 12,000×g for 10 min at 4 °C and the protein content of the supernatant was quantified by using the BCA kit (Thermo Scientific). The lysate was separated on a 4–15% gel (Bio-Rad), transferred to PVDF membranes and probed with antibodies against different proteins (anti-AKT, Cell Signaling Technology, Cat No. #9272; anti-pAKT-T308, Cell Signaling Technology, #4056; anti-calcineurin, Abcam, #ab109412).
RT-PCR
U87 cells were treated with YZ129 (5 μM; unless otherwise noted) or DMSO (control) for 24 h. RNA was extracted from harvested cells using TRIzol. The quality of extracted RNA was confirmed by a spectrophotometer, and agarose gel electrophoresis to ensure that samples had no obvious degradation. The total RNA was reverse transcribed to cDNA by using the amfiRivert Platinum One cDNA Synthesis Master Mix Kit (GenDEPOT, #R6100) according to manufacturer’s instructions. GAPDH was used as a normalization control. Primers were listed in Table S4.
Cell Proliferation Assay
Cell proliferation was measured using the WST-1 proliferation colorimetric assay kit. Briefly, 5–6×103 cells per well were seeded in 96-well plates with a total volume of 100 μL. After culturing overnight, cells were treated with varying concentrations of YZ129 for 24 h, 48 h at 37 °C with 5% CO2. Then, 10 μL of WST-1 reagents were added to each well, and incubated with cells for another 2 h. The absorbance at 460 nm of each well was measured on a microplate reader. The inhibitory rate was calculated with the following equation: inhibition rate = 100%× (ODcontrol well − ODtreated well)/(ODcontrol well −ODblank well). The inhibition curves were fitted using GraphPad Prism 5 software, and IC50 values (defined as the drug concentration that required for inhibiting growth by 50% relative to controls) were calculated. A
Cell Cycle Analysis
U87 cells were treated with or without 5 μM YZ129 at 37 °C with 5% CO2. After incubation for 24 h, cells were collected by trypsinization and fixed with ice-cold 95% ethanol for 30 min. Subsequently, the cell suspension was centrifuged at 1000 rpm for 5 min, and washed with PBS once. The cell pellets were resuspended in 0.4 mL PBS and treated with RNAase A (0.1 mg/mL) at 37 °C for 30 min. Then, propidium iodide (50 μg/mL, Sigma) was added, and the cells were stained for 30 min at 37 °C in the dark. Analysis of cellular DNA content was performed by a BD FACSAria flow cytometer.
Cell Apoptosis Analysis
Cell apoptosis was measured using Annexin V-FITC Apoptosis Detection kit (sigma Aldrich). Briefly, U87 cells were treated with or without 5 μM YZ129 for 24 h at 37 °C with 5% CO2. Then, cells were collected, washed twice with ice-cold PBS and resuspended in 100 μL 1× binding buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4). Subsequently, the cell suspension was incubated with 5 μL FITC Annexin V and Propidium Iodide or 15 min at 25 °C in the dark. After that, 400 μL 1× binding buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4) was added, and the sample was analyzed by flow cytometry within 1 h.
Wound Healing Assay and Real-time Monitoring of Cell Motility
The effect of YZ129 on inhibiting cell migration was evaluated by using the wound healing assay. Briefly, U87 cells were seeded in a six-well plate, and a wound was generated by scratching the cell monolayer with a 200 μL pipette tip. Then, cells were continued to be cultured in the presence or absence of 5 μM YZ129 and the photographic recording was performed at 0 h and 24 h respectively. The number of the migrated cells were calculated with the ImageJ software (NIH). In addition, the cell motility in the presence or absence of 5 μM YZ129 was monitored by a Nikon confocal microscopy, with the movement trajectory analyzed using ImageJ.
Immunofluorescence
U87 cells were seeded on glass coverslips and cultured overnight. Then, cells were treated with 5 μM YZ129 at 37 °C with 5% CO2. After 24 h, cells were fixed with 4 % PFA, rinsed with PBS, incubated with 0.3 % TritonX-100 for 5 minutes and blocked with 3 % BSA for 60 min. The glass coverslips were incubated with primary Ki67 (Abcam; cat #ab8191), and Caspase3 (Novus; cat #AF-605-SP) antibodies overnight at 4 °C, washed with PBS, and incubated with fluorescenceconjugated secondary antibodies (Alexa 568-labeled donkey anti-mouse, or Alexa 488-labeled donkey anti-goat, 1:1000, Invitrogen; cat #A10037 and A-11055) at 4 °C overnight. The immunoreactivity was determined using a ×40 objective oil lens on a Nikon confocal laser microscope with an excitation wavelength appropriate for 488 or 568 nm. Cell nuclei were stained with DAPI (Sigma, 0.1 μg/mL in PBS) at room temperature for 15 min. Cell counting was conducted on 5 randomly chosen fields for each coverslip by using the densitometer Image-Pro Plus image analysis system.
Xenograft tumor models
All animal experiments were approved by the Ethics Committee and IACUC of the Third Military University and were conducted according to the guidelines for laboratory animals care and use. A U87 cell line that stably expresses luciferase was established by lentiviral transduction. U87 cells were transduced with a lentiviral vector encoding the luciferase gene. Transduced colonies (bioluminescence U87 cells, U87-Luc) were selected with 5 mg/mL Blasticidin and examined for luciferase expression by detecting its activity with D-Luciferin. The selected cell line stably expressing luciferase was cultured in DMEM medium supplemented with 10% (v/v) fetal bovine serum (Sigma) in an atmosphere of 5% CO2, 95% air. Balb/c nude mice, 6–8 weeks of age with either sex, were purchased from Animal Center of the Third Military University. To generate xenografts tumor model, 100 μL U87-Luc cells with a density of 5×104/μL were subcutaneously injected under the bilateral posterior limbs of a nude mouse. Mice were randomized into vehicle control (n=6) and YZ129 treatment groups (n=6). YZ129 was administered via intraperitoneal injection every day for 5 days at a dose of 2.5 μM/kg body weight, and the control group received an equal volume of saline solution containing the same amount of DMSO.
The tumor size was monitored each week using bioluminescence imaging method as follows: mice were anesthetized with isoflurane and injected with D-luciferin (120 mg/kg, i.p), and after ten minutes, bioluminescent signals in tumors were recorded on a Xenogen IVIS System. After three weeks, mice were sacrificed, and the solid tumors were isolated. The tumors were measured with a vernier caliper and the volume was calculated with the following equation: TV(cm3) = L×W2 ×1/2, where L represents the largest tumor diameter and W represents the perpendicular tumor diameter.
To generate the orthotopic GBM model within the brain, 10 μL (5×104/μL) U87-Luc cells suspensions were implanted in the brain with a Hamilton syringe (180 μM needle) using a micropump system.(LIBERZON ET AL., 2015; SAUVAGEOT ET AL., 2009; WU ET AL., 2017) Then, animals with established tumors were assigned into two groups: the YZ129 treatment group (n=3, 2.5 μM, 6 μL/g/kg per day, intraperitoneal administration), and the control group (n=3, saline solution containing the same amount of DMSO, intraperitoneal administration). One week later, tumor growth after implantation was verified and assessed by luciferase bioluminescence imaging as described above.
Immunohistochemistry (IHC)
The tumor tissue was isolated from the animals as described above and then fixed with 10% PFA for 4 h at room temperature. Then, the tumor tissue was embedded in paraffin using standard procedures as follows: 70% ethanol for 3 h, 85% ethanol for 1 h, 95% ethanol for 1 h or overnight, 100% ethanol for 1 h × 2, 1/2 ethanol + 1/2 xylene 20 min, xylene 30 min × 2, paraffin I 1 h, paraffin II 1 h, and paraffin III 1 h. The 5 μM sections were processed (deparaffinization, rehydration, target retrieval) and stained with the hematoxylin and eosin (HE) dye or antibodies directed against cell proliferation marker Ki67 (Abcam; Cat # ab833) or activated caspase 3 (Abcam; Cat # ab2302) (Huang et al., 2016).
Reverse Phase Protein Array (RPPA)
U87 cells were treated with or without 5 μM YZ129 for 24 h at 37 °C with 5% CO2. Then, cells were collected using a cell scraper. The cell pellet was washed twice with ice-cold PBS and lysed on ice with a lysis buffer that contains 1% Triton X-100, 50 mM HEPES, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 100 mM NaF, 10 mM sodium pyrophosphate, 1 mM Na3VO4, 10% glycerol (freshly added protease and phosphatase inhibitors from Roche Applied Science Cat. # 05056489001 and 04906837001, respectively). The cell lysate was centrifuged at 18,000 × g for 10 min at 4°C and the supernatant was carefully collected with the protein concentration determined using a BCA kit (adjusted to a final concentration of 1–1.5 μg/μL using lysis buffer). The cell lysate was mixed with 4 × SDS sample buffer without bromophenol blue and boiled for 5 minutes. The samples were then subjected to RPPA as described previously at the MD Anderson RPPA Core Facility (Iadevaia et al., 2010; Tibes et al., 2006).
RNA–seq and Data Analysis
U87 cells were treated with 5 μM YZ129 for 24 h at 37 °C with 5% CO2. Then, cells were collected, and RNA was extracted. Total RNA was isolated with RNeasy kit (Qiagen). One microgram of total RNA was isolated and selected twice with Ambion MicroPoly(A)Purist Kit (Thermo Fisher Scientific). cDNA conversion and library preparation were performed by using NEB RNA-Seq library prep kit (New England BioLabs). RNA-Seq was performed on Illumina NextSeq500. Each sample contains 2 biological replicates. The pair-ended reads were mapped against mm10 using STAR with default parameters, and the RefSeq gene annotation was obtained from the UCSC’s genome database (September 7, 2015). RNA-Seq data were aligned to hg19 human reference genome using Tophat2 v2.0.12. The abundance of genes was quantified by Cufflinks v2.2.1(Trapnell et al., 2010).
Raw sequence data are deposited in the Gene Expression Omnibus (GEO) database under the accession number GSE108749 with token as odebcomyprkfdip. We used a paired Student’s t-test to assess the statistical difference in the gene expression level between DMSO control group and YZ129 drug treatment group, and considered |fold change| ≥ 2 and FDR < 0.05 to be significant. The functional enrichment analyses were performed using GSEA software in hallmark gene sets category online. For the heatmaps, row-wise scaled RPKM values across all samples were plotted using the function heatmap.2 in the R package gplots (www.r-project.org).
Chemicals
Compounds YZ01–75 described in the high-content screening were purchased from Specs Inc. Compounds YZ126–135 were synthesized with the synthetic procedures shown in Scheme S1 below. The resources of other agents were shown in KEY SOURCES TABLE.
Scheme S1.

The synthesis route of compounds YZ126–135
Chemical Synthesis of Compounds YZ126-YZ139
4-((4-chlorophenyl)amino)naphthalene-1,2-dione (YZ126)
A mixture of 1,2-naphthoquinone-4-sulfonic sodium (52 mg, 0.2 mmol) and K2CO3 (43 mg, 0.2 mmol) in 50 mL water was stirred to get a clear solution. Then, 4-chloroaniline (51 mg, 0.4 mmol) in 5mL ethanol was slowly added. The reaction mass was stirred for 1 h, filtered and washed with water several times to generate YZ126 with a 83.2% yield, m.p: 243–265°C.1H-NMR (300 MHz, CDCl3): δ=8.33 (d, J=7.8Hz, 1H), 8.07 (dd, J=7.5Hz, J=1.2Hz, 1H), 7.89 (t, J=7.8Hz, 1H), 8.07 (dt, J=7.8Hz, J=0.9Hz, 1H), 7.55 (d, J=11.4Hz, 2H), 7.24 (br.s, 2H), 5.94(br.s, 1H); ESI-HRMS: m/z calcd for C16H10ClNO2 [(M+H)+], 284.0478; found, 284.0546
4-(naphthalen-2-ylamino)naphthalene-1,2-dione (YZ127)
YZ127 was prepared following the method as described for the preparation of YZ126. 1,2-naphthoquinone-4-sulfonic sodium (52 mg, 0.2 mmol) and naphthalen-2-amine (58 mg, 0.4 mmol) were used to obtain YZ128 with a yield of 82.6%, m.p: 285–292 °C. 1H-NMR (300 MHz, CDCl3): δ=8.41 (d, J=7.8Hz, 1H), 8.10–8.06 (m, 3H), 8.00–7.93 (m, 2H), 7.90–7.88 (m, 1H), 7.80–7.75 (m, 1H), 7.59–7.53 (m, 2H), 7.52(br.s, 1H), 6.20 (br.s, 1H); ESI-HRMS: m/z calcd for C20H13NO2 [(M+H)+], 300.1025; found, 300.0989
4-(naphthalen-1-ylamino)naphthalene-1,2-dione (YZ128)
YZ128 was prepared following the method described for the preparation of YZ126, employing 1,2-naphthoquinone-4-sulfonic sodium (52 mg, 0.2 mmol) and naphthalen-1-amine (58 mg, 0.4 mmol) to obtain YZ128 with a yield of 70.0%, m.p: 213–222 °C. 1H-NMR (300 MHz, CDCl3): δ=8.56 (d, J=7.8Hz, 1H), 8.12 (dd, J=7.8Hz, J=1.2Hz, 1H), 8.04 (d, J=7.8Hz, 1H), 7.95 (dt, J=7.5Hz, J=1.5Hz, 2H), 7.83–7.77 (m, 2H), 7.64–7.51 (m, 3H), 7.23(br.s, 1H), 5.71(br.s, 1H); ESI-HRMS: m/z calcd for C20H13NO2 [(M+H)+], 300.1025; found, 300.0990
4-(isoquinolin-6-ylamino)naphthalene-1,2-dione (YZ129)
YZ130 was prepared following the method described for the preparation of YZ126, employing 1,2-naphthoquinone-4-sulfonic sodium (52 mg, 0.2 mmol) and isoquinolin-6-amine (58 mg, 0.4 mmol) to obtain YZ129 with a yield of 91.6%, m.p: 183–215 °C. 1H-NMR (300 MHz, CDCl3): δ=9.72 (s, 1H), 8.62 (d, J=6.3Hz, 1H), 8.53 (d, J=8.7Hz, 1H), 8.38 (d, J=7.8Hz, 1H), 8.31 (d, J=6.6Hz, 1H), 8.12 (dd, J=7.8Hz, J=4.2Hz, 1H), 7.93 (dt, J=7.5Hz, J=1.5Hz, 1H), 7.85–7.83 (m, 2H), 7.76–7.69 (m, 1H), 6.20 (s, 1H); ESI-HRMS: m/z calcd for C19H12N2O2 [(M+H)+], 301.0977; found, 301.0968
6-((3,4-dioxo-3,4-dihydronaphthalen-1-yl)amino)naphthalene-1-sulfonic acid (YZ130)
YZ130 was prepared following the method as described for the preparation of YZ126, employing 1,2-naphthoquinone-4-sulfonic sodium (52 mg, 0.2 mmol) and 6-aminonaphthalene-1-sulfonic acid (89 mg, 0.4 mmol) to obtain YZ130 with a yield of 68.4%, m.p: 280–295 °C. 1H-NMR (300 MHz, CDCl3): δ=8.89 (d, J=9Hz, 1H), 8.46 (d, J=7.5Hz, 1H), 8.05 (dd, J=7.5Hz, J=1.2Hz, 1H), 7.91–7.79 (m, 3H), 7.73 (dt, J=7.5Hz, J=0.9Hz, 1H), 7.54 (s, 1H), 7.46–7.41 (m, 1H), 7.34 (dd, J=8.7Hz, J=1.2Hz, 1H), 5.86 (s, 1H); ESI-HRMS: m/z calcd for C20H13NO5S [(M+H)−], 378.0436; found, 378.0434
5-((3,4-dioxo-3,4-dihydronaphthalen-1-yl)amino)naphthalene-2-sulfonic acid (YZ131)
YZ128 was prepared following the method as described for the preparation of YZ126, employing 1,2-naphthoquinone-4-sulfonic sodium (52 mg, 0.2 mmol) and 5-aminonaphthalene-2-sulfonic acid (89 mg, 0.4 mmol) to obtain YZ131 with a yield of 56.6%, m.p: >300 °C. 1H-NMR (300 MHz, CDCl3): δ=10.70 (br.s, 1H), 8.59 (d, J=7.8Hz, 1H), 8.25 (s, 1H), 8.11 (d, J=7.2Hz, 1H), 7.95 (t, J=7.5Hz, 1H), 7.82–7.72 (m, 3H), 7.65 (t, J=7.5Hz, 1H), 7.20 (br.s, 1H), 5.70 (br.s, 1H); ESI-HRMS: m/z calcd for C20H13NO5S [(M+H)−], 378.0436; found, 378.0433
4-((3,4-dioxo-3,4-dihydronaphthalen-1-yl)amino)benzenesulfonic acid (YZ132)
YZ132 was prepared following the method as described for the preparation of YZ126, employing 1,2-naphthoquinone-4-sulfonic sodium (52 mg, 0.2 mmol) and 4-aminobenzenesulfonic acid (70 mg, 0.4 mmol) to get YZ127 with a yield of 68.1%. m.p: 189–208 °C。1H-NMR (300 MHz, CDCl3): δ=11.67 (brs, 1H), 8.02 (dt, J=6.9Hz, J=0.9Hz, 2H), 7.96 (d, J=1.5Hz, 1H), 7.94 (t, J=1.2Hz, 1H), 7.87–7.78 (m, 4H), 6.18 (s, 1H); ESI-HRMS: m/z calcd for C16H11NO5S [(M+H)−], 328. 0280; found, 328.0318
4-((6-bromonaphthalen-2-yl)amino)naphthalene-1,2-dione (YZ133)
YZ133 was prepared following the method as described for the preparation of YZ126, employing 1,2-naphthoquinone-4-sulfonic sodium (52 mg, 0.2 mmol) and 6-bromonaphthalen-2-amine (90 mg, 0.4 mmol) to get YZ133 as a red solid in 83.7% yield, m.p: 273–290 °C. 1H-NMR (300 MHz, CDCl3): δ=9.88 (s, 1H), 8.40 (d, J=6.3Hz, 1H), 8.26 (s, 1H), 8.09–8.01 (m, 3H), 7.92–7.87 (m, 2H), 7.79 (t, J=7.5Hz, 1H), 7.47–7.46 (m, 1H), 5.50 (s, 1H); ESI-HRMS: m/z calcd for C20H12BrNO2 [(M+H)+], 378.0130, 380.0109; found, 378.0163, 380.0148
6-((3,4-dioxo-3,4-dihydronaphthalen-1-yl)amino)-2-ethyl-1H-benzo[de]isoquinoline-1,3(2H)-dione (YZ134)
YZ134 was prepared following the method as described for the preparation of YZ126, employing 1,2-naphthoquinone-4-sulfonic sodium (52 mg, 0.2 mmol) and N-ethyl-4-amino-1,8-Naphthalimide t (104 mg, 0.4 mmol) to get YZ134 as a red solid in 65.8% yield, m.p: 242–268 °C, 1H-NMR (300 MHz, CDCl3): δ=11.07 (s, 1H), 8.62–8.52 (m, 3H), 8.44(t, J=8.4Hz, 1H), 8.14 (dd, J=7.5Hz, J=0.9Hz, 1H), 7.95 (dt, J=7.5Hz, J=1.2Hz, 1H), 7.85–7.78 (m, 2H), 7.30 (d, J=7.5Hz, 1H), 6.04 (s, 1H), 4.16–4.03 (m, 2H), 1.27–1.03 (m, 3H); ESI-HRMS: m/z calcd for C24H16N2O4 [(M+H)+], 397.1188; found, 397.1166
4-((2-oxo-2H-chromen-6-yl)amino)naphthalene-1,2-dione (YZ135)
A mixture of 6-nitrocoumarin (64 mg, 0.4 mmol) and zinc powder (260 mg, 4 mmol) in 20mL acetic acid was stirred for 24 h at room temperature. Then, the reaction solution was filtered, washed with CH2Cl2 (30 mL×3), and concentrated under vacuum to get 6-aminocoumarin as a yellow oil, which was directly used for the next step reaction. YZ135 was prepared following the method as described for the preparation of YZ126, employing 1,2-naphthoquinone-4-sulfonic sodium (52 mg, 0.2 mmol) and 6-aminocoumarin which was prepared as described above to get YZ135 as a red solid in 41% yield, m.p: 242–268 °C, m.p: 281–295 °C. 1H-NMR (300 MHz, CDCl3): δ=8.37 (d, J=7.5Hz, 1H), 8.15–8.02 (m, 3H), 7.93–7.82 (m, 2H), 7.79–7.70 (m, 2H), 7.54–7.47 (m, 2H), 6.59 (d, J=9.3Hz, 1H); ESI-HRMS: m/z calcd for C19H11NO4 [(M+H)+], 318.0766; found, 318.0774
(E)-2-(4-azidobutoxy)-4-(isoquinolin-6-ylimino)naphthalen-1(4H)-one (YZ136)
YZ129 (90 mg, 0.3 mmol) in 3 mL DMF, K2CO3 (41 mg, 0.3 mmol) and 1,4-dibromobutane (0.12 mL, 1.0 mmol) were mixed. After stirring for 6 h at 40 °C, the reaction mixture was diluted with water and extracted with ethyl acetate. The organic layers were washed with water and brine, dried over sodium sulfate, and filtered. The filtrate was concentrated and purified by column chromatography on silica gel with CH2Cl2/CH3OH (200:1) as eluant to afford 70 mg red oil, which was not stable and therefore was immediately dissolved in 3 mL DMF directly for next step. Next, sodium azide (42 mg, 0.65 mmol) was added. After stirring for 8 h at 80 °C, the usual workup and purification described above afforded 54 mg orange solid. The total yield for two steps was 45%. 1H NMR (400 MHz, CDCl3): δ=9.24 (s, 1H), 8.64–8.49 (m, 2H), 8.22 (d, J=8.0 Hz, 1H), 8.02 (d, J=8.0 Hz, 1H), 7.76 (t, J=8.0 Hz, 1H), 7.69 (t, J=8.0 Hz, 1H), 7.62 (d, J=5.6 Hz, 1H), 7.24–7.22 (m, 2H), 6.19 (s, 1H), 3.73 (t, J=6.4 Hz, 2H), 3.30 (t, J=6.4 Hz, 2H), 1.87–1.81 (m, 2H), 1.74–1.67 (m, 2H); 13C NMR (100 MHz, CDCl3): δ=179.9, 156.2, 155.6, 152.3, 152.2, 143.8, 136.7, 134.2, 133.4, 131.4, 131.1, 129.0, 126.6, 126.2, 125.4, 122.2, 120.0, 114.0, 102.1, 68.1, 50.9, 25.6, 25.5; ESI-HRMS: m/z calcd for C23H20N5O2 [(M+H)+], 398.1612; found, 398.1608.
(E)-2-((5-azidopentyl)oxy)-4-(isoquinolin-6-ylimino)naphthalen-1(4H)-one (YZ137)
YZ137 was prepared following the method as described for the preparation of YZ136, employing YZ129 (90 mg, 0.3 mmol), 1,5-dibromopentane (0.14 mL, 1.0 mmol) and sodium azide (46 mg, 0.71 mmol) to get YZ137 as a red solid in 41.0% yield. 1H NMR (400 MHz, CDCl3): δ=9.24 (s, 1H), 8.54–8.49 (m, 2H), 8.22 (d, J=8.0 Hz, 1H), 8.02 (d, J=8.0 Hz, 1H), 7.76 (t, J=8.0 Hz, 1H), 7.69 (t, J=8.0 Hz, 1H), 7.61 (d, J=5.6 Hz, 1H), 7.24–7.21 (m, 2H), 6.17 (s, 1H), 3.70 (t, J=6.4 Hz, 2H), 3.23 (t, J=6.4 Hz, 2H), 1.81–1.75 (m, 2H), 1.61–1.54 (m, 2H), 1.50–1.42 (m, 2H). 13C NMR (100 MHz, CDCl3): δ=180.0, 156.3, 155.7, 152.4, 152.2, 143.8, 136.7, 134.2, 133.4, 131.4, 131.2, 129.0, 126.6, 126.2, 125.4, 122.3, 120.1, 114.0, 102.0, 68.5, 51.1, 28.4, 27.8, 23.2. ESI-HRMS: m/z calcd for C23H20N5O2 [(M+H)+], 412.1768; found, 412.1764.
(E)-2-((6-azidohexyl)oxy)-4-(isoquinolin-6-ylimino)naphthalen-1(4H)-one (YZ138)
YZ138 was prepared following the method as described for the preparation of YZ136, employing YZ129 (90 mg, 0.3 mmol), 1,6-dibromohexane (0.15 mL, 1.0 mmol) and sodium azide (18 mg, 0.39 mmol) to get YZ138 as a red solid in 22.0% yield. 1H NMR (400 MHz, CDCl3): δ=9.24 (s, 1H), 8.53–8.49 (m, 2H), 8.22 (d, J=8.0 Hz, 1H), 8.02 (d, J=8.0 Hz, 1H), 7.76 (t, J=8.0 Hz, 1H), 7.69 (t, J=8.0 Hz, 1H), 7.61 (d, J=5.6 Hz, 1H), 7.25–7.21 (m, 2H), 6.17 (s, 1H), 3.70 (t, J=6.4 Hz, 2H), 3.22 (t, J=6.4 Hz, 2H), 1.79–1.72 (m, 2H), 1.58–1.51 (m, 2H), 1.43–1.30 (m, 4H). 13C NMR (100 MHz, CDCl3): δ=180.0, 156.4, 155.7, 152.4, 152.2, 143.8, 136.7, 134.2, 133.4, 131.4, 131.2, 129.0, 126.6, 126.2, 125.4, 122.3, 120.1, 114.0, 102.0, 68.6, 51.2, 28.6, 28.1, 26.3, 25.5. ESI-HRMS: m/z calcd for C25H23N5O2 [(M+H)+], 426.1925; found, 426.1920.
(E)-2-((8-azidooctyl)oxy)-4-(isoquinolin-6-ylimino)naphthalen-1(4H)-one (YZ139)
YZ139 was prepared following the method as described for the preparation of YZ136, employing YZ129 (90 mg, 0.3 mmol), 1,8-dibromooctane (0.18 mL, 1.0 mmol) and sodium azide (32 mg, 0.49 mmol) to get YZ139 as a red solid in 29.0% yield. 1H NMR (400 MHz, CDCl3): δ=9.24 (s, 1H), 8.53–8.49 (m, 2H), 8.23 (d, J=8.0 Hz, 1H), 8.02 (d, J=8.0 Hz, 1H), 7.76 (t, J=8.0 Hz, 1H), 7.69 (t, J=8.0 Hz, 1H), 7.61 (d, J=6.0 Hz, 1H), 7.25–7.22 (m, 2H), 6.17 (s, 1H), 3.69 (t, J=6.4 Hz, 2H), 3.23 (t, J=6.4 Hz, 2H), 1.77–1.71 (m, 2H), 1.59–1.51 (m, 2H), 1.34–1.26 (m, 8H). 13C NMR (100 MHz, CDCl3): δ=180.0, 156.4, 155.8, 152.4, 152.2, 143.8, 136.7, 134.2, 133.4, 131.4, 131.2, 129.0, 126.6, 126.2, 125.4, 122.3, 120.1, 114.0, 102.0, 68.8, 51.4, 29.0, 28.9, 28.7, 28.2, 26.5, 25.7. ESI-HRMS: m/z calcd for C27H27N5O2 [(M+H)+], 454.2238; found, 454.2233.
QUANTIFICATION AND STATISTICAL ANALYSIS
To determine the significance between groups, comparison was made with Student’s t-test. For all statistical tests, the 0.05 confidence level was considered statistically significant. In all figures, * denotes p < 0.05 and ** denotes p < 0.01 in an unpaired Student’s t-test.
DATA AND SOFTWARE AVAILABILITY
The software used in this study is listed in the Key Resources Table. Additional experimental data are provided as Supplemental Information and are available from the corresponding author upon request. RNA-Seq raw data are available at Gene Expression Omnibus (GEO) database (accession number GSE108749): https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108749
Supplementary Material
Highlights.
A class of HSP90 inhibitors with a core structure of 1,2-naphthalenone
Azide-alkyne Click chemistry to identify unknown binding targets in living cells
Omics approaches to unbiasedly evaluate anti-cancer mechanisms of HSP90 inhibitors
Targeting the calcineurin-NFAT signaling and HSP90 client network for GBM therapy
ACKNOWLEDGEMENTS
This work was supported by grants from the National Institutes of Health (R01GM112003, R21GM126532 and R01HL134780), the Cancer Prevention and Research Institute of Texas (RP170660, RR140053 and RR150085), the National Natural Science foundation of China (No. 81673393 and 81728019), the John S. Dunn Foundation Collaborative Research Award, the Welch Foundation (BE-1913) and the American Cancer Society (RSG-16–215–01-TBE and RSG-18–043–01-LIB), the Taishan Scholar Program at Shandong Province, the Qilu/Tang Scholar Program at Shandong University, and the China Postdoctoral Science Foundation (No. 2016M592204, 2017M622225), Postdoctoral innovation foundation of Shandong province (201703009) and the Texas A&M University President’s Excellence Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes 6 Supplementary Figures (S1–S6) and 4 Supplementary Tables (S1–S4). In addition, the structural spectra (1H NMR, ESI-HRMS) for major compounds used in the study were also shown in the Supplemental Information.
Data File 1
Data S1. Structural spectra of compounds YZ126-YZ139 (Related to the STAR methods and Results sections)
REFERENCES
- Adriaens C, Standaert L, Barra J, Latil M, Verfaillie A, Kalev P, Boeckx B, Wijnhoven PW, Radaelli E, Vermi W, et al. (2016). p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nature medicine 22, 861–868. [DOI] [PubMed] [Google Scholar]
- Bai RY, Staedtke V, and Riggins GJ (2011). Molecular targeting of glioblastoma: Drug discovery and therapies. Trends in molecular medicine 17, 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ (1993). Inositol trisphosphate and calcium signalling. Nature 361, 315–325. [DOI] [PubMed] [Google Scholar]
- Bulavin DV, and Fornace AJ Jr. (2004). p38 MAP kinase’s emerging role as a tumor suppressor. Advances in cancer research 92, 95–118. [DOI] [PubMed] [Google Scholar]
- Bulavin DV, Higashimoto Y, Demidenko ZN, Meek S, Graves P, Phillips C, Zhao H, Moody SA, Appella E, Piwnica-Worms H, et al. (2003). Dual phosphorylation controls Cdc25 phosphatases and mitotic entry. Nature cell biology 5, 545–551. [DOI] [PubMed] [Google Scholar]
- Bulavin DV, Higashimoto Y, Popoff IJ, Gaarde WA, Basrur V, Potapova O, Appella E, and Fornace AJ (2001). Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 411, 102–107. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, and Burns TF (2017). Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. International journal of molecular sciences 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Wu Q, Guryanova OA, Huang Z, Huang Q, Rich JN, and Bao S (2011). Elevated invasive potential of glioblastoma stem cells. Biochemical and biophysical research communications 406, 643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chigurupati S, Venkataraman R, Barrera D, Naganathan A, Madan M, Paul L, Pattisapu JV, Kyriazis GA, Sugaya K, Bushnev S, et al. (2010). Receptor channel TRPC6 is a key mediator of Notch-driven glioblastoma growth and invasiveness. Cancer research 70, 418–427. [DOI] [PubMed] [Google Scholar]
- Clarke PA, te Poele R, and Workman P (2004). Gene expression microarray technologies in the development of new therapeutic agents. European journal of cancer 40, 2560–2591. [DOI] [PubMed] [Google Scholar]
- Clipstone NA, and Crabtree GR (1992). Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature 357, 695–697. [DOI] [PubMed] [Google Scholar]
- Donzelli M, and Draetta GF (2003). Regulating mammalian checkpoints through Cdc25 inactivation. EMBO reports 4, 671–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Fang M, Li Y, Li L, and Wang X (2000). Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102, 33–42. [DOI] [PubMed] [Google Scholar]
- Fritz L, Dirven L, Reijneveld JC, Koekkoek JA, Stiggelbout AM, Pasman HR, and Taphoorn MJ (2016). Advance Care Planning in Glioblastoma Patients. Cancers 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudlur A, Zhou Y, and Hogan PG (2013). STIM-ORAI interactions that control the CRAC channel. Current topics in membranes 71, 33–58. [DOI] [PubMed] [Google Scholar]
- Gutschner T, Hammerle M, and Diederichs S (2013). MALAT1 -- a paradigm for long noncoding RNA function in cancer. Journal of molecular medicine 91, 791–801. [DOI] [PubMed] [Google Scholar]
- Hogan PG, Lewis RS, and Rao A (2010). Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annual review of immunology 28, 491–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Dong Z, Chen Y, Wang F, Wang CJ, Peng H, He Y, Hangoc G, Pollok K, Sandusky G, et al. (2016). Small-molecule inhibitors targeting the DNA-binding domain of STAT3 suppress tumor growth, metastasis and STAT3 target gene expression in vivo. Oncogene 35, 783–792. [DOI] [PubMed] [Google Scholar]
- Huang Y, Zhou Y, Wong HC, Chen Y, Chen Y, Wang S, Castiblanco A, Liu A, and Yang JJ (2009). A single EF-hand isolated from STIM1 forms dimer in the absence and presence of Ca2+. The FEBS journal 276, 5589–5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadevaia S, Lu Y, Morales FC, Mills GB, and Ram PT (2010). Identification of optimal drug combinations targeting cellular networks: integrating phospho-proteomics and computational network analysis. Cancer research 70, 6704–6714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri K, Taldone T, Modi S, and Chiosis G (2012). Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochimica et biophysica acta 1823, 742–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, and Bartek J (2004). Cell-cycle checkpoints and cancer. Nature 432, 316–323. [DOI] [PubMed] [Google Scholar]
- Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, Maeda T, Nagata Y, Kitanaka A, Mizuno S, et al. (2016). Aberrant PD-L1 expression through 3’-UTR disruption in multiple cancers. Nature 534, 402–406. [DOI] [PubMed] [Google Scholar]
- Khandelwal A, Kent CN, Balch M, Peng S, Mishra SJ, Deng J, Day VW, Liu W, Subramanian C, Cohen M, et al. (2018). Structure-guided design of an Hsp90beta N-terminal isoform-selective inhibitor. Nature communications 9, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Jia L, Wang J, Wu X, Hao H, Wu Y, Xu H, Wang Z, Shi G, Lu C, et al. (2014). Discovery of diamine-linked 17-aroylamido-17-demethoxygeldanamycins as potent Hsp90 inhibitors. European journal of medicinal chemistry 87, 346–363. [DOI] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell systems 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr., and Meyer T (2005). STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Current biology: CB 15, 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Tekle C, Chen YW, Kristian A, Zhao Y, Zhou M, Liu Z, Ding Y, Wang B, Maelandsmo GM, et al. (2011). B7–H3 silencing increases paclitaxel sensitivity by abrogating Jak2/Stat3 phosphorylation. Molecular cancer therapeutics 10, 960–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalingam D, Swords R, Carew JS, Nawrocki ST, Bhalla K, and Giles FJ (2009). Targeting HSP90 for cancer therapy. British Journal Of Cancer 100, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini M, and Toker A (2009). NFAT proteins: emerging roles in cancer progression. Nat Rev Cancer 9, 810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez GJ, Pereira RM, Aijo T, Kim EY, Marangoni F, Pipkin ME, Togher S, Heissmeyer V, Zhang YC, Crotty S, et al. (2015). The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity 42, 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memmel S, Sisario D, Zoller C, Fiedler V, Katzer A, Heiden R, Becker N, Eing L, Ferreira FLR, Zimmermann H, et al. (2017). Migration pattern, actin cytoskeleton organization and response to PI3K-, mTOR-, and Hsp90-inhibition of glioblastoma cells with different invasive capacities. Oncotarget 8, 45298–45310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal S, Pradhan S, and Srivastava T (2015). Recent advances in targeted therapy for glioblastoma. Expert review of neurotherapeutics 15, 935–946. [DOI] [PubMed] [Google Scholar]
- Monteiro AR, Hill R, Pilkington GJ, and Madureira PA (2017). The Role of Hypoxia in Glioblastoma Invasion. Cells 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monticelli S, and Rao A (2002). NFAT1 and NFAT2 are positive regulators of IL-4 gene transcription. European journal of immunology 32, 2971–2978. [DOI] [PubMed] [Google Scholar]
- Müller MR, and Rao A (2010). NFAT, immunity and cancer: a transcription factor comes of age. Nature Reviews Immunology 10, 645–656. [DOI] [PubMed] [Google Scholar]
- Nagata S (2018). Apoptosis and Clearance of Apoptotic Cells. Annual review of immunology 36, 489–517. [DOI] [PubMed] [Google Scholar]
- Nguyen NT, Han W, Cao WM, Wang Y, Wen S, Huang Y, Li M, Du L, and Zhou Y (2018). Store-Operated Calcium Entry Mediated by ORAI and STIM. Comprehensive Physiology 8, 981–1002. [DOI] [PubMed] [Google Scholar]
- O’Duibhir E, Carragher NO, and Pollard SM (2017). Accelerating glioblastoma drug discovery: Convergence of patient-derived models, genome editing and phenotypic screening. Molecular and Cellular Neuroscience 80, 198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson JRD, and Regad T (2017). Targeting cellular pathways in glioblastoma multiforme. Signal transduction and targeted therapy 2, 17040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, and Lewis RS (2015). Store-Operated Calcium Channels. Physiological reviews 95, 1383–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preusser M, Lim M, Hafler DA, Reardon DA, and Sampson JH (2015). Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nature Reviews Neurology 11, 504–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin JJ, Nag S, Wang W, Zhou J, Zhang WD, Wang H, and Zhang R (2014). NFAT as cancer target: mission possible? Biochimica et biophysica acta 1846, 297–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao JS (2003). Molecular mechanisms of glioma invasiveness: the role of proteases. Nat Rev Cancer 3, 489–501. [DOI] [PubMed] [Google Scholar]
- Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, et al. (2005). STIM1, an essential and conserved component of store-operated Ca2+ channel function. The Journal of cell biology 169, 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satelli A, and Li S (2011). Vimentin in cancer and its potential as a molecular target for cancer therapy. Cellular and molecular life sciences: CMLS 68, 3033–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvageot CM, Weatherbee JL, Kesari S, Winters SE, Barnes J, Dellagatta J, Ramakrishna NR, Stiles CD, Kung AL, Kieran MW, et al. (2009). Efficacy of the HSP90 inhibitor 17-AAG in human glioma cell lines and tumorigenic glioma stem cells. Neuro-oncology 11, 109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopf FH, Biebl MM, and Buchner J (2017). The HSP90 chaperone machinery. Nature reviews Molecular cell biology 18, 345–360. [DOI] [PubMed] [Google Scholar]
- Schulke JP, McAllister LA, Geoghegan KF, Parikh V, Chappie TA, Verhoest PR, Schmidt CJ, Johnson DS, and Brandon NJ (2014). Chemoproteomics demonstrates target engagement and exquisite selectivity of the clinical phosphodiesterase 10A inhibitor MP-10 in its native environment. ACS chemical biology 9, 2823–2832. [DOI] [PubMed] [Google Scholar]
- Shinmen N, Koshida T, Kumazawa T, Sato K, Shimada H, Matsutani T, Iwadate Y, Takiguchi M, and Hiwasa T (2009). Activation of NFAT signal by p53-K120R mutant. FEBS letters 583, 1916–1922. [DOI] [PubMed] [Google Scholar]
- Shrestha L, Bolaender A, Patel HJ, and Taldone T (2016). Heat Shock Protein (HSP) Drug Discovery and Development: Targeting Heat Shock Proteins in Disease. Current topics in medicinal chemistry 16, 2753–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibbersen C, Lykke L, Gregersen N, Jorgensen KA, and Johannsen M (2014). A cleavable azide resin for direct click chemistry mediated enrichment of alkyne-labeled proteins. Chemical communications 50, 12098–12100. [DOI] [PubMed] [Google Scholar]
- Simpson L, and Galanis E (2006). Recurrent glioblastoma multiforme: advances in treatment and promising drug candidates. Expert review of anticancer therapy 6, 1593–1607. [DOI] [PubMed] [Google Scholar]
- Singh SD, Robbins N, Zaas AK, Schell WA, Perfect JR, and Cowen LE (2009). Hsp90 governs echinocandin resistance in the pathogenic yeast Candida albicans via calcineurin. PLoS pathogens 5, e1000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboloff J, Rothberg BS, Madesh M, and Gill DL (2012). STIM proteins: dynamic calcium signal transducers. Nature reviews Molecular cell biology 13, 549–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su TT, Parry DH, Donahoe B, Chien CT, O’Farrell PH, and Purdy A (2001). Cell cycle roles for two 14–3–3 proteins during Drosophila development. Journal of cell science 114, 3445–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro E, and Imoto M (2012). Target identification of bioactive compounds. Bioorganic & medicinal chemistry 20, 1910–1921. [DOI] [PubMed] [Google Scholar]
- Tavecchio M, Lisanti S, Lam A, Ghosh JC, Martin NM, O’Connell M, Weeraratna AT, Kossenkov AV, Showe LC, and Altieri DC (2013). Cyclophilin D extramitochondrial signaling controls cell cycle progression and chemokine-directed cell motility. The Journal of biological chemistry 288, 5553–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, and Kornblau SM (2006). Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Molecular cancer therapeutics 5, 2512–2521. [DOI] [PubMed] [Google Scholar]
- Tie X, Han S, Meng L, Wang Y, and Wu A (2013). NFAT1 is highly expressed in, and regulates the invasion of, glioblastoma multiforme cells. PloS one 8, e66008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov L, Xu X, Li J, and Stern DF (2003). Polo-like kinase 1 and Chk2 interact and co-localize to centrosomes and the midbody. The Journal of biological chemistry 278, 8468–8475. [DOI] [PubMed] [Google Scholar]
- Wachter A, Bernhardt S, Beissbarth T, and Korf U (2015). Analysis of Reverse Phase Protein Array Data: From Experimental Design towards Targeted Biomarker Discovery. Microarrays 4, 520–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ji P, Liu J, Broaddus RR, Xue F, and Zhang W (2009). Centrosome-associated regulators of the G(2)/M checkpoint as targets for cancer therapy. Molecular Cancer 8, 8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal M, and Lamszus K (2011). The neurobiology of gliomas: from cell biology to the development of therapeutic approaches. Nature Reviews Neuroscience 12, 495–508. [DOI] [PubMed] [Google Scholar]
- Whitesell L, and Lindquist SL (2005). HSP90 and the chaperoning of cancer. Nat Rev Cancer 5, 761–772. [DOI] [PubMed] [Google Scholar]
- Wu W, Su J, Tang C, Bai H, Ma Z, Zhang T, Yuan Z, Li Z, Zhou W, Zhang H, et al. (2017). cybLuc: An Effective Aminoluciferin Derivative for Deep Bioluminescence Imaging. Analytical Chemistry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, and Cahalan MD (2005). STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Meraner P, Kwon HT, Machnes D, Oh-hora M, Zimmer J, Huang Y, Stura A, Rao A, and Hogan PG (2010). STIM1 gates the store-operated calcium channel ORAI1 in vitro. Nature structural & molecular biology 17, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhang X, and Klibanski A (2012). MEG3 noncoding RNA: a tumor suppressor. Journal of molecular endocrinology 48, R45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The software used in this study is listed in the Key Resources Table. Additional experimental data are provided as Supplemental Information and are available from the corresponding author upon request. RNA-Seq raw data are available at Gene Expression Omnibus (GEO) database (accession number GSE108749): https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108749