

SUMMARY
The spliceosome mediates precursor mRNA (pre-mRNA) splicing in eukaryotes, including the model organism Saccharomyces cerevisiae (yeast). Despite decades of study, no chemical inhibitors of yeast splicing in vivo are available. We have developed a system to efficiently inhibit splicing and block proliferation in living yeast cells using compounds that target the human spliceosome protein SF3B1. Potent inhibition is observed in yeast expressing a chimeric protein containing portions of human SF3B1. However, only a single point mutation in the yeast homolog of SF3B1 is needed for selective inhibition of splicing by pladienolide B (PB), herboxidiene (HB), or meayamycin (MAM) in liquid culture. Mutations which enable inhibition also improve splicing of branch sites containing mismatches between the intron and snRNA—suggesting a link between inhibitor sensitivity and usage of weak branch sites in humans. This approach provides powerful new tools for manipulating splicing in live yeast and studies of spliceosome inhibitors.
Keywords: splicing, pre-mRNA, spliceosome, SF3B1, yeast, pladienolide B, herboxidiene, meayamycin, inhibitor, RNA
Graphical Abstract
eTOC Blurb
No chemical approaches exist for inhibiting pre-mRNA splicing in budding yeast despite its widespread use in gene expression studies. Hansen et al. have discovered that mutation of the protein Hsh155 enables potent inhibition of yeast splicing in vivo by several drugs.
INTRODUCTION
In eukaryotic cells, pre-mRNA introns are removed and exons ligated together by the spliceosome to produce mature mRNA products (Figure 1A) (Hoskins and Moore, 2012; Wahl et al., 2009). Introns are recognized by conserved cis regulatory elements: the 5ʹ splice site (5ʹ SS), the branch site (BS), and the 3ʹ splice site (3ʹ SS). During spliceosome assembly, the BS forms a duplex with the U2 small nuclear RNA (snRNA) that is bound by the protein SF3B1 (Hsh155 in yeast) (Query et al., 1994; Wahl et al., 2009). The U2/BS duplex contains a bulged adenosine which serves as the nucleophile during 5ʹ SS cleavage and the branch point nucleotide (BP-A) for the intron lariat. The BP-A is accommodated within a binding pocket formed by HEAT repeats 15–16 (HR 15–16) of the HR domain of SF3B1/Hsh155 (Figure 1B) (Yan et al., 2016). Formation of the U2/BS duplex is likely coupled with other steps in expression of the nascent RNA (Alexander et al., 2010). However, research in this area has been hampered by the inability to chemically inhibit splicing in yeast despite its well-characterized gene expression machinery.
Figure 1. Small molecule inhibition of pre-mRNA splicing.
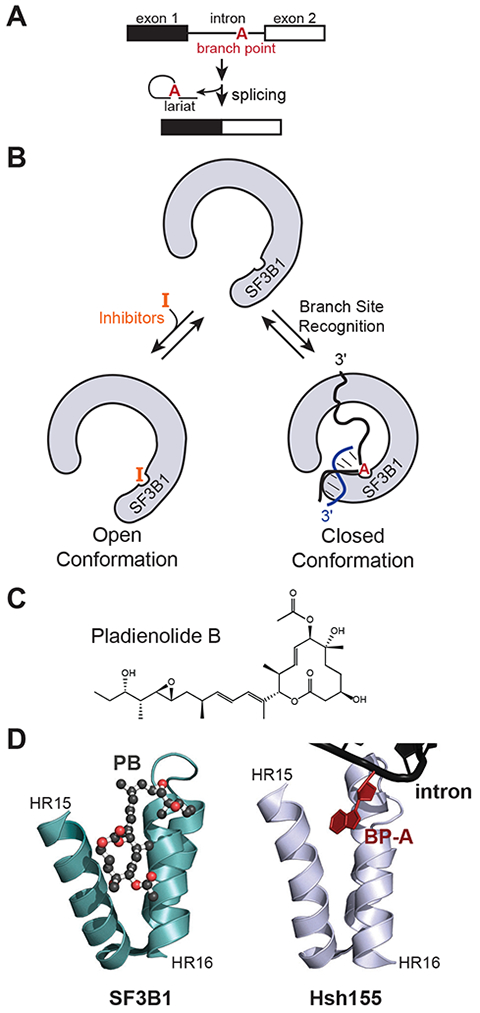
(A) Schematic of eukaryotic pre-mRNA splicing. (B) Splicing inhibitors that target SF3B1 are proposed to occupy the same site as the BP-A of the U2 snRNA/BS duplex. Inhibitor binding likely disrupts formation of a closed complex between SF3B1 and the RNA duplex. (C) Chemical structure of the splicing inhibitor PB. (D) Crystal structure of PB (ball and stick) bound to human SF3B1 (left, PDB ID: 6EN4) in comparison with the structure of Hsh155 bound to the U2/BS duplex including the bulged BP-A (right, PDB ID: 5GM6) (Cretu et al., 2018; Yan et al., 2016).
In addition to playing an essential role in splicing, SF3B1 is a hotspot for mutations found in myelodysplastic syndrome, chronic lymphocytic leukemia, uveal melanoma, and breast cancer (Anczuków and Krainer, 2016; Lee and Abdel-Wahab, 2016). These mutations can alter BS usage and result in aberrant mRNA formation by activation of cryptic 3ʹ SS located near the new BS (Carrocci et al., 2017; Darman et al., 2015; Tang et al., 2016). Recently, several classes of natural products [e.g., pladienolide B (PB, Figure 1C), meayamycin (MAM), and herboxidiene (HB)] have been shown to modulate or inhibit splicing by targeting SF3B1 (Albert et al., 2007; Effenberger et al., 2017; Webb et al., 2013). Structures of PB and derivatives bound to SF3B1 show that these compounds occupy the BP-A binding pocket and suggest a mode of action where they can occlude binding of the U2/BS duplex (Figure 1B and 1D) (Cretu et al., 2018; Finci et al., 2018; Yan et al., 2016). Competition between these drugs and the U2/BS duplex for SF3B1 is supported by recent data in which purified spliceosomes containing SF3B1-bound RNA duplexes are insensitive to PB (Cretu et al., 2018). In addition, biochemical data indicates that these inhibitors also prevent stable formation of the U2/BS duplex itself (Folco et al., 2011). A derivative of PB (H3B-8800) is in clinical trials and has been shown to be effective at killing cancer cells expressing mutant alleles of SF3B1 or other splicing factors (Seiler et al., 2018). However, there is still a strong need for the discovery of new and selective SF3B1 ligands as well as for mechanistic elucidation of their mechanism of action (Webb et al., 2013).
We recently demonstrated that replacement of Hsh155 HR 5–16 with their counterparts from human SF3B1 could support yeast viability and splicing (Carrocci et al., 2018). Yeast were viable with the human/yeast chimeric protein (Hsh1555–16) as the only copy of HSH155 for the cell (Carrocci et al., 2018). When whole cell extracts were prepared from Hsh1555–16-containing strains, the in vitro splicing activity could be blocked by addition of PB. However, PB had little effect in extracts prepared from strains expressing the wild-type (WT) Hsh155 protein. The potency of PB as an in vitro inhibitor was similar in both human and yeast Hsh1555–16 extracts (IC50 ~25nM) (Effenberger et al., 2014). Herein, we report the chemical inhibition of yeast growth and splicing in cells using PB and other SF3B1-targeting compounds. A single point mutation in Hsh155 is sufficient to sensitize yeast to chemical inhibitors. These methods represent an innovative approach for blocking splicing in vivo in yeast and provide a new tool for identifying next-generation human splicing inhibitors.
RESULTS AND DISCUSSION
Our previous data showed that in vitro inhibition of pre-mRNA splicing was conserved between humans and yeast containing chimeric Hsh155; however, PB failed to significantly affect growth of those yeast strains (Figure S1) (Carrocci et al., 2018). Since yeast express several multidrug transporters, we reasoned that the loss of inhibitor potency between in vitro and in vivo assays was due to drug efflux (Jungwirth and Kuchler, 2006; Labunskyy et al., 2014; McMurray and Thorner, 2008). We deleted the genomic copy of HSH155 in a previously described transporter-deficient strain of yeast (pdr5Δ yor1Δ snq2Δ) while introducing a WT allele of HSH155 (Hsh155WT) on a URA3-marked plasmid (Key Resources Table, Table S1) (McMurray and Thorner, 2008). These yeast were then transformed with WT or humanized hsh155 on a TRP1-marked plasmid and loss of the URA3 plasmid selected for by growth on 5-fluoroorotic acid (5-FOA).
The resulting yeast strains were grown in liquid culture in rich media (YPD) in a 48-well microplate with varying concentrations of PB (Figure 2A). As expected from our in vitro analysis, growth of yeast containing either Hsh155WT or a chimeric protein containing human HR 1–12 (which contains humanized HR but not those involved in drug binding, Hsh1551–12) was not inhibited by addition of PB up to concentrations of 20 μM (Figure 2B, Figures S2A and S2B). However, growth of Hsh1555–16 yeast (which contains human HR involved in drug binding) was inhibited by PB in a concentration-dependent manner (Figures 2B and 2C; Table S2). To test if PB was causing cell death or stopping proliferation, we plated yeast containing Hsh1555–16 after exposure. Viable yeast were still present after PB treatment, and we conclude that PB causes an arrest of yeast growth but is not completely fungicidal even after 30 h of exposure at 10 μM PB (Figure S2D).
Figure 2. Microplate assay for yeast splicing inhibition.
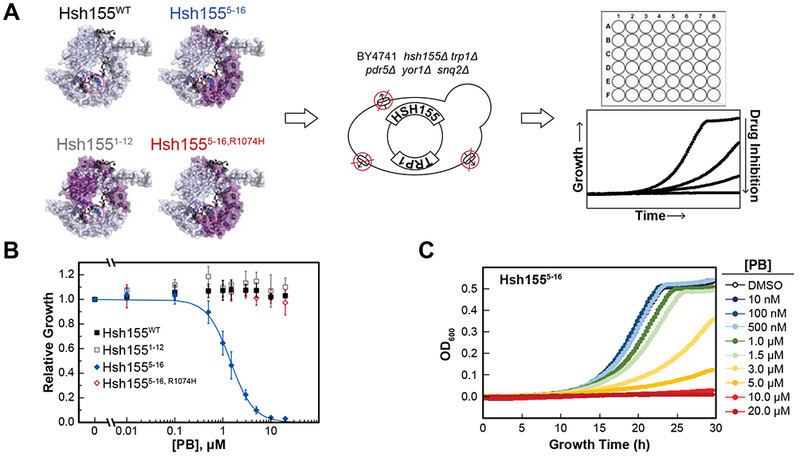
(A) Chimeric Hsh155 proteins were created by incorporating domains from human SF3B1 (purple). The BP-A and inhibitor binding pocket is circled (pink). The chimeric proteins were introduced into a transporter deletion strain of yeast by plasmid shuffle. Yeast growth was monitored in microplate wells containing varying concentrations of splicing inhibitors. (B) Impact of PB on yeast growth for strains expressing Hsh155WT (black) or humanized chimeras. Relative growth in the presence of the drug was determined after 20 h in comparison to DMSO alone. Each point represents the average from three replicates ± standard deviation (SD). (C) Microplate assay data showing growth inhibition of yeast expressing Hsh1555–16 at increasing concentrations of PB. See also Figures S1–S3 and Table S2.
A single point mutation in SF3B1 (R1074H) confers resistance to PB and other drugs both in human cells and in vitro (Teng et al., 2017; Yokoi et al., 2011). Mutation of the corresponding amino acid in the Hsh1555–16 chimera (Hsh1555–16,R1074H; note that the residue number refers to human SF3B1) also confers resistance to PB in yeast splicing assays (Carrocci et al., 2018). We next tested whether or not this mutation would confer resistance in vivo. Yeast containing Hsh1555–16,R1074H were not affected by PB and grew similarly to the Hsh155WT strain even at the highest drug concentrations (Figure 2B, Figure S2C). Thus, a PB resistance mutation found in human cell lines also confers resistance when introduced into the humanized, PB-sensitive yeast strain.
We also assayed sensitivity of Hsh1555–16 yeast towards other known splicing inhibitors. Both HB and MAM strongly inhibited growth of yeast containing the chimeric protein but not Hsh155WT (Figure S3, Table S2). Structurally diverse splicing inhibitors are therefore able to block yeast growth, presumably by binding to humanized Hsh155 and inhibiting splicing.
To gain insight into why human SF3B1 is sensitive to these inhibitors while Hsh155WT is not, we analyzed structures of human SF3B1 and yeast Hsh155 (Figure 3A) (Cretu et al., 2016; Yan et al., 2016). This analysis revealed two residues in the BP-A/drug binding pocket of HR 15 and 16 which are non-identical or non-similar between the two proteins (Figure 3A): yeast asparagine 747 is a valine in humans while leucine 777 is an asparagine. We tested whether or not mutation of just these amino acids in Hsh155 to their human counterparts would convey similar levels of sensitivity to inhibitors. We created yeast strains in the transporter-deficient background where one or both amino acids were mutated (Hsh155N747V, Hsh155L777N, and Hsh155NL/VN). Yeast containing Hsh155L777N were insensitive to PB or HB and grew similarly to Hsh155WT yeast (Figure 3B and 3C; Table S2). On the other hand, the Hsh155N747V mutation was sufficient to confer drug sensitivity to both PB and HB in vivo, albeit to a lesser extent than that observed when using the humanized Hsh1555–16 chimera (Figure 3B and 3C; Table S2). Although L777N alone does not impart drug sensitivity, this mutation significantly enhances growth inhibition when combined with N747V in the double mutant Hsh155NL/VN strain. In fact, the 50% growth-inhibitory concentration (GI50) values observed with Hsh155NL/VN approached those observed with chimeric Hsh1555–16 for all inhibitors tested (Table S2). These results highlight that only a limited number of amino acid substitutions in Hsh155 are needed for potent growth inhibition in the presence of human splicing inhibitors and that these mutations can be epistatic with one another.
Figure 3. A single point mutation in Hsh155 is sufficient for inhibition.
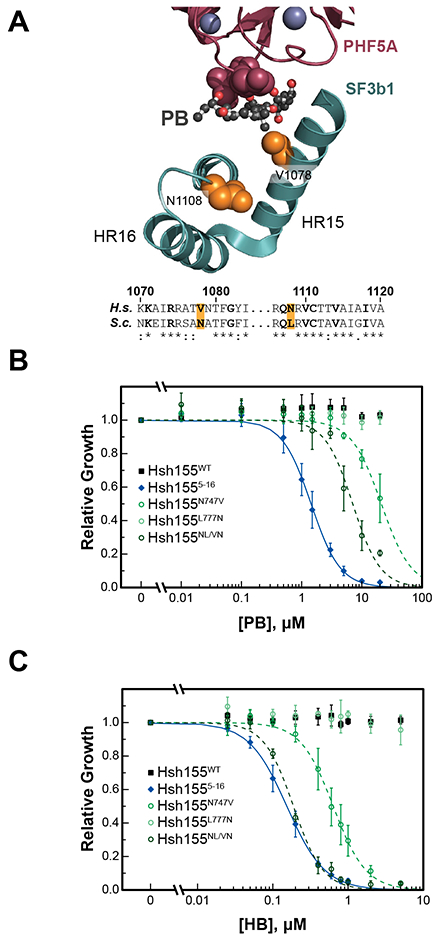
(A) Structure of the PB binding site of human SF3 (PDB ID: 6EN4) (Cretu et al., 2018). Shown are HR15 and 16 of SF3B1 (teal) and a fragment of PHF5A (purple). The PB binding site is composed, in part, of a narrow cleft between V1078 (yeast N747) and a loop from PHF5A (purple spheres). The non-conserved amino acids mutated in yeast Hsh155 are shown as orange spheres and highlighted in the sequence alignment below (residue numbers are for SF3B1). Amino acids lining the BP-A pocket of Hsh155 are noted in bold in the sequence alignment. (B and C) Impact of PB (B) and HB (C) on yeast growth for strains expressing Hsh155WT, the Hsh1555–16 (human-yeast chimera), or Hsh155 point mutants. Relative growth was determined after 20 h in comparison to a DMSO alone. Each point represents the average from three replicates ± SD. Hsh155WT and Hsh1555–16 data shown in (B) are reproduced from Figure 2B and included for comparison. See also Table S2.
Our growth assays strongly suggest that PB, HB, and MAM can target mutant versions of Hsh155 in yeast cells and that growth stasis is caused by splicing inhibition. To test this hypothesis, we quantified the amount of spliced product mRNA generated in Hsh155WT and Hsh155NL/VN strains after inhibitor exposure. These strains also contained a plasmid encoding an ACT1-CUP1 splicing reporter gene, which is easily quantified by primer extension (Figure 4A) (Lesser and Guthrie, 1993). To maintain this plasmid, yeast were grown in liquid drop-out media lacking leucine and tryptophan, conditions under which HB is a more potent growth inhibitor than in YPD (Figure S4A, Table S2). After 1 h of treatment with either DMSO or 0.1 μM HB, total cellular RNA was isolated from each strain and primer extension reactions were performed to quantify intron retention and splicing inhibition (Figure S4B). We observed an almost complete loss of spliced mRNA product and build-up of pre-mRNA in yeast containing Hsh155NL/VN treated with HB whereas Hsh155WT was not significantly affected (Figure 4B). These results are consistent with SF3B1 inhibitors stopping cell proliferation by blocking pre-mRNA splicing.
Figure 4. In vivo assays of splicing inhibition and branch site usage.
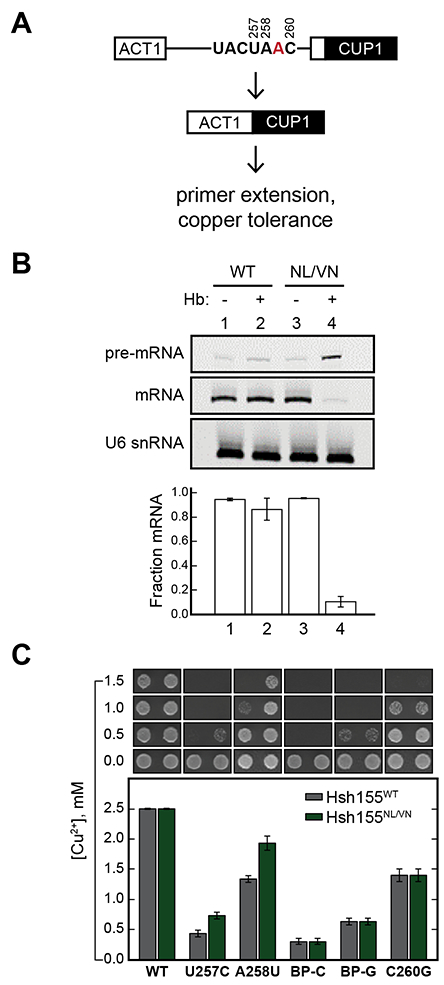
(A) Schematic of the ACT1-CUP1 reporter assay. (B) Primer extension analysis of splicing inhibition by HB in yeast expressing Hsh155WT or Hsh155NL/VN. U6 snRNA was used as a loading control. Quantified below the gel is the fraction mRNA (fraction spliced) plotted as the mean ± SD from three replicates. (C) Results of the ACT1-CUP1 Cu2+ tolerance assay. The maximum Cu2+ concentration on which a yeast strain survived is plotted below as the mean ± SD from three replicates. Representative images of yeast grown on Cu2+-containing plates are included above the bar plot. See also Figure S4.
While both yeast Hsh155 and human SF3B1 bind the U2/BS duplex, the BS consensus sequence is quite different between species. Yeast almost exclusively use a strong UACUAAC (BP-A underlined) BS, which has perfect complementarity to the U2 snRNA pairing region with the exception of the bulged BP-A. In contrast, human introns are defined by highly divergent BS sequences with limited predicted pairing to U2 (i.e., weak BS) (Gould et al., 2016; Harris and Senapathy, 1990; Mercer et al., 2015; Qin et al., 2016). We previously reported that the Hsh1555–16 chimera permitted both in vitro inhibition of splicing by PB and increased usage of BS containing mismatches to U2 snRNA in yeast (Carrocci et al., 2018). Furthermore, mutation of the N747 position was sufficient to alter BS usage. We therefore tested the Hsh155NL/VN mutant for changes in BS usage in vivo. We again used the ACT1-CUP1 reporter gene, which allows for changes in splicing in cells to be correlated with a Cu2+-resistant growth phenotype (Lesser and Guthrie, 1993). We detected changes in growth on plates containing varying [Cu2+] for yeast strains harboring ACT1-CUP1 reporters with consensus or weak BS.
Hsh155NL/VN does not alter Cu2+-resistance with an ACT1-CUP1 reporter containing a consensus BS, consistent with past studies of other mutations within this region (Figure 4C) (Carrocci et al., 2018; Carrocci et al., 2017). However, we observed increased Cu2+ tolerance of Hsh155NL/VN yeast expressing reporters with mismatches at −1 and −2 relative to the BP-A (Figure 4C). Together our data suggest that some features of SF3B1 which enhance usage of weak BS also confer sensitivity to small molecule inhibitors that target the BP-A binding pocket.
The exact mechanisms by which Hsh155NL/VN causes drug sensitivity or alters BS usage are not yet clear. We note that the N747V mutation introduces a less bulky amino acid at this position, while L777N introduces a similarly sized but more hydrophilic amino acid. It is tempting to speculate that together these amino acid substitutions allow for better steric accommodation of either small molecule inhibitors or U2/BS duplexes containing mismatches. However, the human SF3B1 mutation V1078A (equivalent to N747A in yeast Hsh155) also incorporates a smaller amino acid at this position but instead results in resistance to a PB derivative (E7107) and HB (Teng et al., 2017). A recent crystal structure of human SF3B1 bound to PB noted that V1078 is located in a binding pocket with shape complementarity to the PB macrolide moiety and that SF3B1 undergoes significant conformational changes within this same region (Cretu et al., 2018). It is possible that sensitivity to inhibitors depends on both complementarity of the binding pocket to the drug as well as SF3B1 being in a conformation susceptible to drug binding. Amino acids N747 and L777 in Hsh155 likely are influencing these attributes in yeast.
Small molecule inhibition is a powerful tool for exploring conserved mechanisms of gene expression including DNA replication (hydroxyurea), RNA transcription (α-amanitin), and translation initiation (hippuristanol) and elongation (cycloheximide) in yeast. Yet, chemical approaches for inhibiting yeast pre-mRNA splicing have been lacking. We have developed a novel and practical method for inhibiting splicing in live yeast. We believe that this will be an attractive tool for molecular biologists studying splicing as well as those interested in decoupling splicing from other steps in gene expression including transcription, chromatin modification, 3’ end formation, and nuclear export (Alexander and Beggs, 2010; Herzel et al., 2017; Kim and Dreyfuss, 2001; Reed and Hurt, 2002). Additionally, the ability to easily screen human splicing inhibitors in yeast has potential in drug discovery, both for rapidly testing derivatives of current lead candidates and screening libraries of novel compounds. Yeast containing humanized spicing factors such as the Hsh1555–16 chimera could even be used as a basis for screening inhibitors targeting specific SF3B1 disease alleles or resistance mutations.
SIGNIFICANCE
Budding yeast is an important model organism for studying eukaryotic gene expression. While methods have long existed for chemically inhibiting replication, transcription, and translation in yeast cells, there has been no complementary approach for inhibition of pre-mRNA splicing by the spliceosome. Here, we report that mutation of the Hsh155 protein (the yeast homolog of human SF3B1) enables selective inhibition of yeast proliferation and pre-mRNA splicing in vivo by multiple different human splicing modulators. These inhibitors are potent in liquid culture with 50% growth inhibition values ranging from 10−8-10−5 M. Only a single amino acid substitution is needed to confer drug sensitivity to yeast; however, inhibition is enhanced by also including a secondary mutation that does not change sensitivity on its own. These same mutations increase usage of weak branch sites by the splicing machinery. This suggests that features of the human spliceosome which facilitate the use of weak branch sites also confer susceptibility to SF3B1 inhibitors. Chemical inhibition of splicing in yeast will likely prove useful for elucidating connections between splicing and other steps in gene expression, understanding mechanisms of SF3B1 inhibition, and screening novel compounds for splicing inhibitory activity.
STAR METHODS TEXT
KEY RESOURCES TABLE
Submitted as a separate Word document.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Aaron Hoskins (ahoskins@wisc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Plasmid and Yeast Strain Construction
Descriptions and sources for recombinant DNA and Saccharomyces cerevisiae strains can be found in the KEY RESOURCES TABLE and Table S1. Plasmids encoding WT or humanized Hsh155 have been previously described (Carrocci et al., 2018). Point mutations in HSH155 were introduced by inverse polymerase chain reaction (PCR) and confirmed by DNA sequencing. Standard yeast growth media and conditions were used unless otherwise specified. The transporter deficient yeast strain (BY4741 pdr5Δ0::kanMX snq2Δ0:kanMX yor1Δ0::kanMX) was a gift from Jeremey Thorner (UC-Berkeley) (McMurray and Thorner, 2008). This haploid strain was transformed with a URA3/CEN plasmid encoding HSH155 and then the chromosomal copy of HSH155 was deleted using a hygromycin B resistance cassette (Goldstein and McCusker, 1999). To facilitate shuffling of plasmids containing HSH155, the TRP1 gene from this strain was also deleted using a nourseothricin resistance cassette to create strain yAAH1912, the parental strain used for growth assays unless otherwise noted (Goldstein and McCusker, 1999). Plasmids containing mutant hsh155 were shuffled into yAAH1912 and loss of the URA3/CEN plasmid selected for by plating onto 5-FOA (1 g/L) (Amberg and Burke, 2016).
Plasmids containing ACT1-CUP1 splicing reporter genes were obtained from the laboratories of Dr. Charles Query (Albert Einstein College of Medicine) and David Brow (U. Wisconsin-Madison) and have been previously described (Lesser and Guthrie, 1993). These reporter plasmids were introduced either into the transporter deficient strains described above or a Cu2+-sensitive HSH155 shuffle strain that has been previously described (Carrocci et al., 2017).
METHOD DETAILS
Splicing Inhibitors
The sources of the splicing inhibitors are listed in the KEY RESOURCES TABLE. MAM was a gift from Kazunori Koide (University of Pittsburgh). The inhibitors were dissolved at a high concentration in DMSO, aliquoted, and stored at −80°C.
Microplate Assay of Yeast Growth Inhibition
Yeast strains were first grown in yeast synthetic drop-out media lacking tryptophan overnight in a shaking incubator at 30°C (220 rpm). Immediately prior to the assay, cultures were diluted to OD600=0.06 in YPD. A portion of the resulting culture (100 μL) was then combined in a Corning Costar 48-well Flat Bottom Cell Culture plate with 100 μL 2xYPD, 92.5 μL sterile MilliQ H2O, and 7.5 μL DMSO (±inhibitor) to yield a final OD600=0.02 and 2.5% v/v DMSO in 300 μL rich media (1xYPD). Once filled, the plate was then covered with a Breathe-Easy plate sealing membrane to limit evaporation.
Cultures contained in the microplate were then incubated in a Tecan Infinite 200Pro plate reader at 30°C with orbital shaking an d OD600 measurements were taken every 15 min for 30 h. Absorbance values were corrected using blank measurements from wells where YPD was added instead of yeast culture.
Primer Extension Assays
Yeast transformed with ACT1-CUP1 reporters were grown in yeast synthetic drop-out media lacking tryptophan and leucine in a shaking incubator at 30°C at 220 rpm. Once the cultures reached an OD600=0.9–1.0, they were split into two 4.9 mL cultures. DMSO (125 μL) or herboxidiene (4 μM in DMSO, 125 μL) were then added to the culture and yeast incubated at 30°C for 60 min with shaking at 220 rpm. Cells were collected by centrifugation, resuspended with MilliQ H2O, and transferred to a 1.5 mL microfuge tube. Cells were then pelleted again by centrifugation. RNA was isolated from the yeast using a MasterPure RNA isolation kit according to the manufacturer’s protocol. Primer extension was carried out using superscript reverse transcriptase III according to manufacturer directions. The reactions simultaneously contained primers complementary to the ACT1-CUP1 RNA and the U6 snRNA, which was used as a loading control. Primers were purchased from Integrated DNA Technologies with a near-infrared fluorophore at the 5ʹ end (see KEY RESOURCES TABLE). Reactions were quenched with deionized formamide RNA gel loading buffer (lacking loading dye) and the products were separated on a 20cm by 32cm 7% w/v 19:1 acrylamide:bisacrylamide/8M urea/1x TBE gel at 35 W for 80 min at room temperature. Fluorescent primer extension products were visualized using an Amersham Typhoon NIR imager and quantified using ImageQuant.
ACT1-CUP1 Assays of Cu2+ Tolerance
Strains expressing Hsh155WT or mutants and ACT1-CUP1 reporters were grown to mid-log phase in drop-out media, adjusted to OD600 = 0.5 and equal volumes were spotted onto drop-out/agar plates containing 0, 0.025, 0.05, 0.075, 0.1, 0.15, 0.2, 0.25, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9, 2.0, 2.25, or 2.5 mM CuSO4. Plates were scored after 3 days growth at 30°C fo r the presence of yeast growth.
QUANTIFICATION AND STATISTICAL ANALYSIS
All growth and in vitro experiments (Figures 2, 3, and 4; Figures S1, S2, S3, and S4) were performed in triplicate. For the growth inhibition assays the growth (blank-adjusted OD600) relative to the DMSO control was measured at the 20 hour time point. The mean ± SD was plotted for each concentration of PB, HB, or MAM tested. These results were fit and the 50% growth-inhibitory concentrations (GI50) were determined using a four parameters logistic regression in MATLAB (R2018a) (Cardillo, 2012). Errors in the fit used to obtain GI50 values represent the 95% confidence interval and are reported in Table S2 directly from the MATLAB output.
Supplementary Material
Highlights.
Mutation of Hsh155 enables chemical inhibition of pre-mRNA splicing in living yeast
Only a single point mutation in Hsh155 is required for inhibition in cells
Mutations in Hsh155 can synergize with one another to enhance inhibitor sensitivity
Mutations that enable inhibition increase splicing of introns with weak branch sites
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (R01 GM112735 to AAH), Shaw Scientist and Beckman Young Investigator Awards, and startup funding from the University of Wisconsin-Madison, Wisconsin Alumni Research Foundation (WARF), and the Department of Biochemistry. S.R.H. was supported in part by the NIH Chemistry–Biology Interface Training Grant (T32 GM008505). T.J.C. was supported in part by the NIH Biotechnology Training Grant (T32 GM08349) as well as a William H. Peterson Fellowship. We thank Kaz Koide (University of Pittsburgh) for comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Albert BJ, Sivaramakrishnan A, Naka T, Czaicki NL, and Koide K (2007). Total syntheses, fragmentation studies, and antitumor/antiproliferative activities of FR901464 and its low picomolar analogue. J. Am. Chem. Soc 129, 2648–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander R, and Beggs JD (2010). Cross-talk in transcription, splicing and chromatin: who makes the first call? Biochem. Soc. Trans 38, 1251–1256. [DOI] [PubMed] [Google Scholar]
- Alexander RD, Innocente SA, Barrass JD, and Beggs JD (2010). Splicing-dependent RNA polymerase pausing in yeast. Mol. Cell 40, 582–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberg DC, and Burke DJ (2016). Classical Genetics with Saccharomyces cerevisiae. Cold Spring Harb. Protoc. 2016. [DOI] [PubMed] [Google Scholar]
- Anczuków O, and Krainer AR (2016). Splicing-factor alterations in cancers. RNA 22, 1285–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardillo G (2012). Four parameters logistic regression - There and back again (MathWorks File Exchange). [Google Scholar]
- Carrocci TJ, Paulson JC, and Hoskins AA (2018). Functional analysis of Hsh155/SF3b1 interactions with the U2 snRNA/branch site duplex. RNA 24, 1028–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrocci TJ, Zoerner DM, Paulson JC, and Hoskins AA (2017). SF3b1 mutations associated with myelodysplastic syndromes alter the fidelity of branchsite selection in yeast. Nucleic Acids Res. 45, 4837–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cretu C, Agrawal AA, Cook A, Will CL, Fekkes P, Smith PG, Lührmann R, Larsen N, Buonamici S, and Pena V (2018). Structural Basis of Splicing Modulation by Antitumor Macrolide Compounds. Mol. Cell 70, 265–273.e268. [DOI] [PubMed] [Google Scholar]
- Cretu C, Schmitzova J, Ponce-Salvatierra A, Dybkov O, De Laurentiis EI, Sharma K, Will CL, Urlaub H, Lührmann R, and Pena V (2016). Molecular Architecture of SF3b and Structural Consequences of Its Cancer-Related Mutations. Mol. Cell 64, 307–319. [DOI] [PubMed] [Google Scholar]
- Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. (2015). Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3’ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 13, 1033–1045. [DOI] [PubMed] [Google Scholar]
- Effenberger KA, Anderson DD, Bray WM, Prichard BE, Ma N, Adams MS, Ghosh AK, and Jurica MS (2014). Coherence between cellular responses and in vitro splicing inhibition for the anti-tumor drug pladienolide B and its analogs. J. Biol. Chem 289, 1938–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effenberger KA, Urabe VK, and Jurica MS (2017). Modulating splicing with small molecular inhibitors of the spliceosome. Wiley interdisciplinary reviews RNA 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finci LI, Zhang X, Huang X, Zhou Q, Tsai J, Teng T, Agrawal A, Chan B, Irwin S, Karr C, et al. (2018). The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action. Genes Dev. 32, 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folco EG, Coil KE, and Reed R (2011). The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev. 25, 440–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein AL, and McCusker JH (1999). Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast (Chichester, England) 15, 1541–1553. [DOI] [PubMed] [Google Scholar]
- Gould GM, Paggi JM, Guo Y, Phizicky DV, Zinshteyn B, Wang ET, Gilbert WV, Gifford DK, and Burge CB (2016). Identification of new branch points and unconventional introns in Saccharomyces cerevisiae. RNA 22, 1522–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris NL, and Senapathy P (1990). Distribution and consensus of branch point signals in eukaryotic genes: a computerized statistical analysis. Nucleic Acids Res. 18, 3015–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzel L, Ottoz DSM, Alpert T, and Neugebauer KM (2017). Splicing and transcription touch base: co-transcriptional spliceosome assembly and function. Nat. Rev. Mol. Cell Biol 18, 637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins AA, and Moore MJ (2012). The spliceosome: a flexible, reversible macromolecular machine. Trends Biochem. Sci 37, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungwirth H, and Kuchler K (2006). Yeast ABC transporters-- a tale of sex, stress, drugs and aging. FEBS Lett. 580, 1131–1138. [DOI] [PubMed] [Google Scholar]
- Kim VN, and Dreyfuss G (2001). Nuclear mRNA binding proteins couple pre-mRNA splicing and post-splicing events. Mol. Cells 12, 1–10. [PubMed] [Google Scholar]
- Labunskyy VM, Suzuki Y, Hanly TJ, Murao A, Roth FP, and Gladyshev VN (2014). The insertion Green Monster (iGM) method for expression of multiple exogenous genes in yeast. G3 (Bethesda, Md) 4, 1183–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, and Abdel-Wahab O (2016). Therapeutic targeting of splicing in cancer. Nat. Med 22, 976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesser CF, and Guthrie C (1993). Mutational analysis of pre-mRNA splicing in Saccharomyces cerevisiae using a sensitive new reporter gene, CUP1. Genetics 133, 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray MA, and Thorner J (2008). Septin stability and recycling during dynamic structural transitions in cell division and development. Curr. Biol 18, 1203–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Clark MB, Andersen SB, Brunck ME, Haerty W, Crawford J, Taft RJ, Nielsen LK, Dinger ME, and Mattick JS (2015). Genome-wide discovery of human splicing branchpoints. Genome Res. 25, 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin D, Huang L, Wlodaver A, Andrade J, and Staley JP (2016). Sequencing of lariat termini in S. cerevisiae reveals 5’ splice sites, branch points, and novel splicing events. RNA 22, 237–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Query CC, Moore MJ, and Sharp PA (1994). Branch nucleophile selection in pre-mRNA splicing: evidence for the bulged duplex model. Genes Dev. 8, 587–597. [DOI] [PubMed] [Google Scholar]
- Reed R, and Hurt E (2002). A conserved mRNA export machinery coupled to pre-mRNA splicing. Cell 108, 523–531. [DOI] [PubMed] [Google Scholar]
- Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, Agrawal AA, Caleb B, Csibi A, Sean E, et al. (2018). H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nature Med. 24, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Rodriguez-Santiago S, Wang J, Pu J, Yuste A, Gupta V, Moldon A, Xu YZ, and Query CC (2016). SF3B1/Hsh155 HEAT motif mutations affect interaction with the spliceosomal ATPase Prp5, resulting in altered branch site selectivity in pre-mRNA splicing. Genes Dev. 30, 2710–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng T, Tsai JH, Puyang X, Seiler M, Peng S, Prajapati S, Aird D, Buonamici S, Caleb B, Chan B, et al. (2017). Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A-SF3b complex. Nat. Commun 8, 15522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, and Lührmann R (2009). The spliceosome: design principles of a dynamic RNP machine. Cell 136, 701–718. [DOI] [PubMed] [Google Scholar]
- Webb TR, Joyner AS, and Potter PM (2013). The development and application of small molecule modulators of SF3b as therapeutic agents for cancer. Drug Discov. Today 18, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Wan R, Bai R, Huang G, and Shi Y (2016). Structure of a yeast activated spliceosome at 3.5 A resolution. Science 353, 904–911. [DOI] [PubMed] [Google Scholar]
- Yokoi A, Kotake Y, Takahashi K, Kadowaki T, Matsumoto Y, Minoshima Y, Sugi NH, Sagane K, Hamaguchi M, Iwata M, et al. (2011). Biological validation that SF3b is a target of the antitumor macrolide pladienolide. FEBS J. 278, 4870–4880. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.