

Summary
The initiation and execution of cell death can be regulated by various lipids. How the levels of environmental (exogenous) lipids impact cell death sensitivity is not well understood. We find that exogenous monounsaturated fatty acids (MUFAs) potently inhibit the non-apoptotic, iron-dependent, oxidative cell death process of ferroptosis. This protective effect is associated with the suppression of toxic lipid reactive oxygen species (ROS) accumulation specifically at the plasma membrane and decreased levels of phospholipids containing oxidizable polyunsaturated fatty acids. We find that treatment with exogenous MUFAs reduces the sensitivity of plasma membrane lipids to lethal oxidation over several hours, and that this process requires MUFA activation by acyl-CoA synthetase long-chain family member 3 (ACSL3). Exogenous MUFAs also protect cells from apoptotic lipotoxicity caused by the accumulation of saturated fatty acids, but in an ACSL3-independent manner. Our work demonstrates that exogenous MUFAs and ACSL3 activity specifically promote a ferroptosis-resistant cell state.
Keywords: Ferroptosis, oleate, MUFAs, iron, lipid ROS, cell death, lipotoxicity
Graphical Abstract
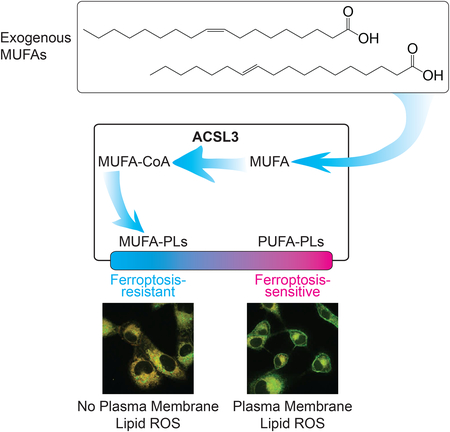
Exogenous lipids can modulate both apoptotic and non-apoptotic cell death. Here we show that exogenous monounsaturated fatty acids can suppress the non-apoptotic process of ferroptosis by promoting the displacement of polyunsaturated fatty acids from plasma membrane phospholipids in an ACSL3-dependent manner.
Introduction
Lipids are important regulators of cell death. In mammalian cells, both apoptotic and nonapoptotic cell death pathways can be induced, modulated or suppressed by different lipids, including free fatty acids and more complex lipids (Das, 1991; Green et al., 2014; Magtanong et al., 2016). For example, apoptosis can be triggered by increasing the ratio of intracellular saturated fatty acids (SFAs) to monounsaturated fatty acids (MUFAs), a process termed lipotoxicity (Garbarino and Sturley, 2009; Listenberger et al., 2003; Young et al., 2013). Necroptosis, a form of non-apoptotic cell death, is associated with increased very long chain fatty acid and ceramide levels that contribute to the initiation of cell death (Parisi et al., 2017). Ferroptosis, a distinct form of non-apoptotic cell death, is characterized by the iron-dependent accumulation of toxic lipid reactive oxygen species (ROS) (Dixon et al., 2012). The initiation and execution of cell death is therefore intimately linked to lipid metabolism.
Ferroptosis is an emerging form of cell death that can be selectively activated in certain cancer cells and that mediates a number of pathological cell death events in vivo (Stockwell et al., 2017). Unlike other forms of cell death, ferroptosis is not thought to require a specific protein effector, like a pore-forming protein. Rather, lipid oxidation and the resultant membrane damage are thought to be sufficient for the execution of this process (Stockwell et al., 2017). During ferroptosis, toxic lipid ROS arise from the reaction between iron and lipid peroxides, which are themselves generated by the oxidation of polyunsaturated fatty acid (PUFA)-containing phospholipids (PUFA-PLs) (Stockwell et al., 2017). Normally, lipid peroxides are reduced to non-reactive (i.e. non-toxic) lipid alcohols by the reduced glutathione (GSH)-dependent lipid hydroperoxidase glutathione peroxidase 4 (GPX4). However, GPX4 is inactivated under ferroptosis-inducing conditions, leading to lipid peroxide accumulation (Stockwell et al., 2017). Oxidizable PUFA-PLs are found in membranes throughout the cell, and mitochondria, lysosomes, and endoplasmic reticulum may all be important sites of lethal lipid ROS accumulation during ferroptosis (Gaschler et al., 2018; Kagan et al., 2016; Krainz et al., 2016; Torii et al., 2016). Whether lipid oxidation at one or more of these sites is sufficient for ferroptosis is presently unclear.
The execution of ferroptosis can be prevented by deletion of the acyl-CoA synthetase long chain family member 4 (ACSL4), one of five human ACSL enzymes (ACSL1, 3–6; ACSL2 does not exist) (Dixon et al., 2015; Doll et al., 2017; Kagan et al., 2017; Mashek et al., 2004). ACSL enzymes activate free fatty acids to fatty acyl-CoAs, which can then be incorporated into glycerophospholipids (Grevengoed et al., 2014). ACSL4 has a marked preference for activating PUFAs (Kang et al., 1997); therefore, deletion of ACSL4 prevents PUFAs from being incorporated into membrane PLs where they would become oxidized following GPX4 inactivation. Endogenous mechanisms that may act to inhibit ferroptosis by opposing PUFA activation and incorporation into membrane PLs have not been described.
Exogenous metabolites including lipids are potent modulators of cell function and fate (Cantor et al., 2017; Yao et al., 2016). Given the links between lipid metabolism and cell death, we examined how exogenous fatty acids impacted cell sensitivity to both ferroptotic and nonferroptotic lethal stimuli. Here we show that exogenous MUFAs potently suppress ferroptosis. Mechanistically, MUFAs inhibit the accumulation of lipid ROS specifically at the plasma membrane and displace PUFAs from this location in the cell. We find that ACSL3 is required for exogenous MUFAs to protect cells against ferroptosis, but not lipotoxicity induced by exogenous saturated fatty acids. ACSL3-dependent MUFA metabolism therefore emerges as key a regulator of ferroptotic cell death.
Results
A modulatory profile identifies MUFAs as suppressors of ferroptosis
In standard tissue culture medium supplemented with 10% fetal bovine serum (~1–10 μM) a representative SFA (palmitate, C16:0), MUFA (oleic acid, C18:1) and PUFA (linoleic acid, C18:2) were each an order of magnitude lower than those observed in human serum (Psychogios et al., 2011; Yao et al., 2016) (Figure 1A). We hypothesized that raising the concentration of different free fatty acid species to more physiological levels would impact cell death sensitivity. To test this, we examined how five different PUFA and MUFA species impacted cell death induced by seven mechanistically distinct lethal compounds. Each lethal compound was tested over a 10-point, 2-fold dilution series and cell death was quantified using scalable time-lapse analysis of cell death kinetics (STACK) (Forcina et al., 2017). This profiling experiment was performed in human HT-1080 cells stably expressing the live cell marker nuclear-localized mKate2 (i.e. HT-1080N cells) and incubated with the dead cell marker SYTOX Green (SG). Live (mKate2+) and dead (SG+) cells were counted every 2 h for a total of 72 h, resulting in a total of ~140,000 population cell death measurements across all conditions from three independent biological replicate experiments (Figure 1B).
Figure 1. Exogenous monounsaturated fatty acids suppress ferroptosis.

(A) Fatty acid levels reported in adult human serum (Serum, (Psychogios et al., 2011)) or measured in three independent samples of DMEM + 10% FBS tissue culture medium (Medium). (B) Overview of the lipid modulatory profiling experiment in HT-1080N cells. (C) A cell death lipid modulation map. LA: linoleic acid, α-LA: α-linolenic acid, γ-LA: γ-linolenic acid, POA: palmitoleic acid, OA: oleic acid, H2O2: hydrogen peroxide. (D-G) Cell death (lethal fraction) over time, extracted from (C), for erastin (D), thapsigargin (Thaps.) (E), H2O2 (F) and bortezomib (Bortez.) (G) ± OA or POA. (H) SYTOX Green positive (SG+) object (i.e. dead cell) counts in HT-1080, A549 and T98G cells treated ± erastin2 (era2) ± OA. Era2 = 1 μM (HT-1080, T98G) or 2 μM (A549). (I) Dead cell counts in IMR-90 cells. (J) Dead cell counts in HT-1080 cells treated as indicated ± different monounsaturated fatty acids (MUFAs). Data in (A,D-G) are mean ± SD. Each data point in (H-J) represents an independent biological replicate (n=3).
To summarize these data, the effect of each exogenous fatty acid on compound-induced cell death were first summarized across time, compound concentrations and biological replicates, normalized to control conditions, and plotted as a two-dimensional hierarchically clustered heat map (Figure 1C). Most strikingly, cell death induced by the pro-ferroptotic system xc− inhibitor erastin was suppressed by cotreatment with the MUFAs oleic acid (OA, 18:1 cis-9) and palmitoleic acid (POA, 16:1 cis-7) (Figure 1D, S1A). This protective effect was specific for erastin, as OA and POA had little or no ability to inhibit cell death induced by the ER stress-inducing agent thapsigargin (Thap), hydrogen peroxide (H2O2) or the proteasome inhibitor bortezomib (Btz) (Figure 1E–G). We confirmed that OA and POA maintained the viability of erastin-treated HT-1080 cells using a metabolic indicator (PrestoBlue), suggesting that our observations were not an artifact of our cell death imaging technique (Figure S1B).
Exogenous OA suppressed ferroptosis induced by a more potent erastin analog (erastin2) in HT-1080, T98G glioblastoma and A549 non-small cell lung carcinoma cells, as well as in non-transformed IMR-90 diploid fibroblasts, demonstrating that this protective effect was broadly generalizable (Figure 1H,I). Moreover, erastin and erastin2 were also the only compounds from a library of 261 bioactive compounds whose lethality was suppressed by OA in HT-1080N cells, further indicating that exogenous MUFAs do not broadly or non-specifically limit cell death sensitivity (Figure S1C). OA free fatty acid and OA conjugated to bovine serum albumin (i.e. OA-BSA) both prevented erastin2-induced ferroptosis, indicating that the mode of MUFA delivery to the cell was not crucial for this protective effect (Figure S1D). In addition to OA and POA, trans-vaccenic acid (t-VA, 18:1 trans-11) inhibited erastin2-induced ferroptosis, while the structurally related MUFAs cis-7-hexadecanoic acid (C16:1 cis-7), palmitelaidic acid (C16:1 trans-9) and elaidic acid (C18:1 trans-9) did not (Figure 1J). Thus, ferroptosis can be specifically inhibited in both transformed and non-transformed cells by exogenous MUFA in a structure-specific manner.
MUFAs inhibit ferroptosis downstream of or in parallel to GPX4
We next examined where in the ferroptosis pathway exogenous MUFAs acted to block this process. Erastin and related compounds inhibit the cystine/glutamate antiporter system xc−. This prevents cystine import and glutamate export, leads to transcriptional upregulation of the amino acid deprivation-responsive gene CHAC1, depletion of intracellular glutathione and inactivation of GPX4 (Stockwell et al., 2017) (Figure 2A). In the presence of exogenous OA, erastin retained the ability to inhibit system xc− function, deplete total glutathione (GSH+GSSG) and upregulate CHAC1 expression (Figure 2B–D). Moreover, OA and t-VA both potently suppressed ferroptosis that was induced by culturing cells directly in low cystine medium, and cells cultured under these conditions (i.e. low cystine + OA) for 48 h were subsequently able to resume proliferating if cystine was restored (Figure 2E, S2A,B). Both OA and a synthetic small molecule ferroptosis inhibitor, ferrostatin-1 (Fer-1) (Dixon et al., 2012), suppressed cell death in Pfa1 mouse embryonic fibroblasts, which harbor floxed Gpx4 alleles and express a 4-hydroxytamoxifen (Tam)-inducible Cre recombinase (MERCreMER), enabling Tam-dependent disruption of the Gpx4 locus and the induction of ferroptosis (Seiler et al., 2008) (Figure 2F,G).
Figure 2. MUFAs prevent ferroptosis downstream of GPX4 activity.

(A) Schematic of the ferroptosis pathway. (B) Glutamate release, a measure of system xc− activity, following 2 h compound treatment. Era: erastin, OA: oleic acid. (C) Total glutathione levels assayed using Ellman’s reagent at 11 h. Era2: erastin2. (D) RT-qPCR analysis of CHAC1 mRNA levels at 6 h. (E) Cell death (lethal fraction) of HT-1080N cells in low cystine (Cys2) medium. Fer-1: ferrostatin-1, 1 μM; MUFA: monounsaturated fatty acid; OA, 500 μM; t-VA: trans-vaccenic acid, 500 μM. (F) Schematic of inducible Gpx4 loss in Pfa1 mouse embryonic fibroblasts (MEFs). (G) Cell death of Pfa1 MEFs ± tamoxifen citrate (Tam, 1 μM). (H) Fold-change of HT-1080N live cells (mKate2+ counts). (I) Cell-free free radical scavenging tested using the 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay. (J,K) GPX4 (J) and ACSL4 (K) protein levels assessed by Western blotting. Compound treatments were for 10 h prior to cell pellet harvest. Era2, 1 μM; OA, 500 μM. Each data point represents an independent biological (B-H) or technical (I) replicate (n=3). Western blots in (J) and (K) were performed twice and results from one blot are shown.
When GPX4 is inactive, ferroptosis can be inhibited by iron chelators, such as deferoxamine, or radical trapping antioxidants like Fer-1 (Stockwell et al., 2017) (Figure 2A). Free intracellular iron promotes lipid ROS formation and is also required for cell proliferation. However, unlike deferoxamine, OA and t-VA did not inhibit HT-1080N proliferation (Figure 2H, S2C). OA and t-VA were likewise unable to directly scavenge free radicals in a cell-free assay (Figure 2I). Thus, exogenous MUFAs appeared unlikely to inhibit ferroptosis by acting like known iron chelators or radical trapping antioxidants. These experiments also confirmed that our MUFA stocks were free of such contaminants. Another explanation for our results was that exogenous MUFAs altered the expression of key proteins that regulate ferroptosis sensitivity. However, OA did not increase the expression of GPX4, which opposes ferroptosis, or decrease the expression of ACSL4, which promotes ferroptosis, either alone or in combination with erastin2 (Figure 2J,K). These results indicated that exogenous MUFAs most likely inhibited ferroptosis downstream of or in parallel to GPX4.
MUFAs block plasma membrane lipid ROS accumulation
Lipid ROS accumulation during ferroptosis can be detected using the ratiometric fluorescent lipid peroxidation sensor C11 BODIPY 581/591 (hereafter C11) in combination with flow cytometry (Dixon et al., 2012). We used this established approach to test the ability of MUFAs to prevent lipid ROS accumulation in erastin2-treated HT-1080 cells. As expected, erastin2 treatment increased the ratio of oxidized to total C11, and this increase was fully suppressed by DFO (Dixon et al., 2012) (Figure 3A). Unexpectedly, C11 oxidation was not detectably reduced in cells co-treated with OA (Figure 3A). It seemed unlikely that MUFAs suppressed ferroptosis downstream of lipid ROS accumulation, and so we hypothesized that MUFAs were acting to prevent the accumulation of a subset of lethal lipid ROS that could not be detected by flow cytometry.
Figure 3. OA preferentially decreases lipid reactive oxygen species (ROS) at the plasma membrane.

(A) Analysis of lipid ROS using C11 BODIPY 581/591 (C11) by flow cytometry in HT-1080 cells treated for 10 h. Each data point represents the ratio of oxidized (C11Ox) to total (non-oxidized C11 (C11Non-ox) + C11Ox) signal from an independent biological replicate (n=3). Era2: erastin2, DFO: deferoxamine, OA: oleic acid. (B) Confocal imaging of C11 in HT-1080 cells. After compound treatment for 10 h, cells were labeled with C11 (5 μM) and Concanavalin A, Alexa Fluor 350 (ConA-AlexaF, 25 μg/mL). Arrowheads indicate regions of highly oxidized C11 and/or ConA-AlexaF labeling. C11Non-ox: non-oxidized C11, C11Ox: oxidized C11. Scale bar = 20 μm. Images from one of four independent experiments are shown. (C) Schematic showing the two regions of the cell that were quantified in (D) and (E). (D,E) Quantification of (i) plasma membrane and (ii) perinuclear ratios of C11Ox. Fer-1: ferrostatin-1. Each data point represents an individual cell quantified in one of two (Fer-1-treated samples) or four (all other conditions) independent biological replicates. (F) Model summarizing the effects of ferroptosis inhibitors on lipid ROS accumulation.
To test this hypothesis we examined C11 oxidation using confocal microscopy, which we reasoned could allow for the detection in changes localized to a specific cellular region or structure. In vehicle-treated cells, overlapping oxidized and non-oxidized C11 signals were observed in the perinuclear region, consistent with previous reports (Drummen et al., 2002) (Figure 3B). Treatment with erastin2 (1 μM, 10 h) increased C11 oxidation in the perinuclear region and resulted in the appearance of a distinct ‘ring’ of C11 oxidation at the periphery of the cell that co-localized with the plasma membrane marker concanavalin A (Figure 3B). OA by itself had little effect on C11 oxidation, but in erastin2-treated cells the addition of OA reduced plasma membrane C11 oxidation to levels that were not significantly different from those observed in vehicle-treated controls (P < 0.05) (Figure 3C–D). By contrast, the levels of perinuclear C11 oxidation in cells co-treated with erastin2 and OA remained significantly higher than vehicle-treated controls (P < 0.05, two-way ANOVA with Tukey’s post-tests) (Figure 3B–D). Unlike OA, deferoxamine and Fer-1 effectively prevented C11 oxidation on both perinuclear membranes and the plasma membrane in erastin2-treated cells (Figure 3E, S3A). Thus, MUFAs preferentially inhibit plasma membrane C11 oxidation while DFO and Fer-1 do not show this same selectivity (Figure 3F).
Our results indicated that C11 oxidation at the plasma membrane was specifically associated with the execution of ferroptosis. We wondered if this was associated with unique morphological alterations at the plasma membrane. Using high-resolution, phase-contrast imaging we examined plasma membrane morphology in erastin2-treated HT-1080 cells over time. In response to erastin2 treatment cells appeared normal for many hours. However, roughly an hour prior to death, cells rounded up and then one or more ‘blisters’ appeared on the surface of the cell (median = 3, range = 1–7, n = 35) (Figure S3B,C). These balloon-shaped blisters formed rapidly without detaching from the cell and then appeared to deflate, coincident with apparent plasma membrane permeabilization. Erastin2-induced blister formation was suppressed in cells co-treated with OA (500 μM) or deferoxamine (50 μM) (Figure S3B,C). By contrast, OA did not prevent in HT-1080 cells the appearance of numerous smaller protrusions (i.e. apoptotic bodies) in response to the pro-apoptotic proteasome inhibitor bortezomib (100 nM) (Figure S3D). Thus, MUFAs suppress plasma membrane lipid ROS accumulation and membrane blistering phenotypes that are associated with the onset of ferroptosis.
Exogenous MUFAs reduce PUFA incorporation into phospholipids
During ferroptosis lipid ROS are generated from the oxidation of phospholipids (PLs) containing oxidizable PUFAs such as arachidonic acid (C20:4) (i.e. PUFA-PLs) (Stockwell et al., 2017). We hypothesized that exogenous MUFAs could block ferroptosis by perturbing PUFA-PL levels. In support of this hypothesis, treatment with OA (500 μM, 11 h) decreased the steady-state abundance of 7/8 detectable C18:0/C20:4 and C16:0/C20:4 PUFA-PLs, as determined using single reaction monitoring-based liquid chromatography coupled to mass spectrometry (Figure 4A). The effect of OA on PUFA-PL levels was not altered by erastin2 cotreatment (Figure 4A). These decreases were apparent for both less abundant PLs like phosphatidylethanolamines and highly abundant PLs like phosphatidylcholines, consistent with a substantial overall reduction in the steady-state abundance of oxidizable PUFA-PLs within the cell (Figure S4A). With the exception of C18:1 free fatty acid itself, the levels of other detectable free fatty acids (C20:4, C22:6, C16:0 and C18:0) and acyl carnitines (e.g. C12:0 acyl carnitine) were largely unaltered by OA or erastin2 treatment, suggesting that free fatty acid uptake, de novo synthesis, elongation and catabolism were largely normal (Figure 4B,C). With the exception of C18:0/C18:1 PI, the levels of seven other detectable C16:0/C18:1 and C18:0/C18:1 MUFA-PLs were not elevated by OA treatment above baseline levels (Figure 4D).
Figure 4. Exogenous OA reduces PUFA-PL levels.

(A-D) Lipid levels determined by mass spectrometry in HT-1080 cells treated for 10 h ± erastin2 (Era2, 1 μM) ± oleic acid (OA, 500 μM). PC: phosphatidylcholine, PI: phosphatidylinositol, PS: phosphatidylserine, PE: phosphatidylethanolamine. (E) Visualization of arachidonic acid (AA) localization. HT-1080 cells were incubated with arachidonic acid-alkyne (AA-alkyne, 20 μM) or vehicle control (ethanol, EtOH), and with either OA (conjugated to bovine serum albumin [BSA], 125 μM) or vehicle (BSA alone) for 2 h, then chased in medium containing only OA (125 μM) or vehicle for 10 h. AA-alkyne localization was visualized using copper-catalyzed click chemistry to azidefluor 488, to yield AA-488. The plasma membrane is identified by cadherin immunofluorescence. Scale bar = 20 μm. (F) Quantification of AA-488 fluorescence intensity within cadherin-positive (cad.+) regions. Each data point represents one field. The y-axis is in arbitrary units. Scale bar = 20 μm. (G) Cell death in HT-1080N cells either cotreated with OA and ML162 or pretreated for 10 h with OA prior to the addition of ML162. Data in (A-D) are mean ± SD of five independent biological replicates. AA-488 imaging and quantification (E) was performed on two independent biological replicates and results from one replicate are shown. Each data point in (G) represents an independent biological replicate (n = 3).
We next visualized the effect of exogenous OA on the incorporation of PUFAs into plasma membrane PLs using a ‘clickable’ arachidonic acid (AA) analog, AA-alkyne (Robichaud et al., 2016). HT-1080 cells were pulsed for 2 h with AA-alkyne (20 μM) in the presence or absence of exogenous OA (125 μM). Following a two-hour chase in regular medium, cells were fixed and AA-alkyne was detected by conjugation to azide-fluor 488 using copper(I)-catalyzed alkyne-azide cycloaddition, yielding AA-488. Immunofluorescence against the cell surface protein cadherin was used to visualize the plasma membrane. Significant overlap of AA-488 signal and cadherin staining was observed, and this overlap was unaffected by cotreatment OA (Figure S4B,C). Note that non-specific nuclear azide-fluor 488 signal made it difficult to confidently assess whether OA treatment altered AA-alkyne incorporation into perinuclear phospholipids. These results confirmed that exogenous OA did not impair PUFA (i.e. AA-alkyne) uptake or incorporation into plasma membrane PLs. However, this experiment left unresolved how exogenous MUFAs reduced PUFA-PL levels.
Erastin analogs induce cell death beginning ~12 h after compound exposure (Figure 1D, S3B). We reasoned that MUFAs could act within this window to alter membrane PL composition and reduce ferroptosis sensitivity. We therefore examined the duration of exogenous OA exposure required for protection against ferroptosis. Notably, addition of OA (125 μM) to HT-1080N cells six hours after erastin2 (i.e. six hours before the onset of cell death within the population) was unable to prevent cell death (Figure S4D). Accordingly, we hypothesized that at least six hours would be required to see the effect of exogenous MUFAs on PUFA incorporation into membrane PLs. In support of this hypothesis, when HT-1080 cells were pulsed with AA-alkyne (20 μM) for 2 h, and then chased for a further 10 h in medium containing OA (125 μM), the intensity of AA-488 signal that overlapped with cadherin was suppressed to almost background levels (Figure 4E,F).
If exogenous MUFAs required at least six hours to alter PUFA-PL levels with the membrane, we predicted that this treatment would not protect against ferroptosis induced by an agent that can act in less than this amount of time. We tested this hypothesis using ML162, a covalent GPX4 inhibitor that initiates ferroptosis within six hours (Figure S4E) (Weiwer et al., 2012). As predicted, exogenous OA (125 μM) inhibited ferroptosis poorly when added at the same time as ML162, but potently suppressed ferroptosis when added 10 h before ML162 (Figure 4G). Collectively, these results suggest that exogenous MUFAs reduce PUFA-PL levels within the plasma membrane over the course of at least six hours, eventually resulting in a ferroptosis-resistant cellular state.
ACSL3 is required for the protective effects of exogenous MUFAs
To be incorporated into membrane PLs, free fatty acids must first be activated to fatty acyl-CoAs by specific fatty acid activating enzymes. We therefore examined whether a specific ACSL enzyme was required for exogenous MUFAs to protect against ferroptosis. We first tested a panel of genetically-modified human embryonic kidney (HEK)-293 cells lacking ACSL1, ACSL3 or ACSL4 protein expression (Figure S5A). The role of ACSL4 in the protective effects of exogenous MUFAs could not be determined, as deletion of this protein made cells insensitive to erastin2, consistent with recent reports (Dixon et al., 2015; Doll et al., 2017; Kagan et al., 2017) (Figure 5A). Wild-type (Control), ACSL1KO1/2 and ACSL3KO1/2 cells were sensitive to erastin2-induced ferroptosis, and exogenous OA (125 μM) inhibited this process in both Control and ACSL1KO1/2 cells (Figure 5A). The ability of exogenous OA to inhibit erastin2-induced ferroptosis was, however, lost in cells lacking ACSL3 (Figure 5A). Likewise, in A549N cells the ability of exogenous OA to protect from erastin2-induced ferroptosis was diminished when ACSL3 was disrupted using CRISPR/Cas9-mediated gene editing (Figure S5B,C). This effect was dose-dependent, as at higher concentrations of exogenous OA both A549N Control and ACSL3KO1/2 cells were protected equally from ferroptosis. This is consistent with the notion that ACSL enzymes other than ACSL3 can activate MUFAs when they are present at high concentrations (Soupene and Kuypers, 2008).
Figure 5. ACSL3 is required for OA-induced protection from ferroptosis.

(A) Dead cell counts (SG+ objects) in HEK-293 Control or ACSL1, 3, or 4 knockout (KO) cell lines treated ± erastin2 (Era2, 1 μM) ± oleic acid (OA, 125 μM). (B) HT-1080 Control and ACSL3LOF1 cells incubated ± oleic acid-alkyne (OA-alkyne, 20 μM) for 2 h, then chased in regular medium (no alkyne) for 2 h. Scale bar = 20 μm. (C) Quantification of AA-488 fluorescence intensity within cadherin-positive (cad.+) regions. Each data point represents one field. The y-axis is in arbitrary units. (D) Confocal imaging of C11 BODIPY 581/591 (C11) in HT-1080 ControlPM-mTq and ACSL3LOF1-PM-mTq cells after compound treatment for 24 h. Arrowheads indicate regions of highly oxidized C11 and/or plasma membrane-mTurquoise2 (PM-mTq) fluorescence. C11Non-ox: non-oxidized C11, C11Ox: oxidized C11. Scale bar = 20 m. (E) BODIPY 493/503 imaging of neutral lipid accumulation in HT-1080 Control and ACSL3LOF1/2 cells ± oleic acid (OA). Scale bar = 50 μm. (F) Schematic of triacylglycerol synthesis. DGATis is the combination of T863 (20 μM) and PF-06424439 (PF-064., 10 μM). FA: fatty acid. (G) Thin-layer chromatography of neutral lipid extracts from HT-1080 cells ± DGATis (6 h). TAG: triacylglycerol. (H) Dead cell counts in HT-1080 cells. Era2: 1 μM, OA: 500 μM. (I) Cell death ± erastin2 (Era2). (J) Significant correlations between low expression of ACSL genes and small molecule probe lethality from the Cancer Therapeutics Response Portal (CTRP) dataset. Each dot represent one compound. Ferroptosis-inducing compounds are highlighted in pink. Each data point in (A) and (H) represents an independent biological replicate (n ≥ 3). OA-488 imaging quantification (B), BODIPY 493/503 imaging (E) and TLC (G) were performed twice and results from one biological replicate are shown.
We next asked whether ACSL3 was required for incorporation of exogenous MUFAs into membrane PLs. For these studies, we generated HT-1080 Control and ACSL3 gene-disrupted cell lines using CRISPR/Cas9 technology. We obtained two independent strong loss of function (LOF) cell lines and confirmed that exogenous OA was less effective at protecting these cells against ferroptosis compared to unmodified Control cells (Figure S5D,E). We visualized OA localization in HT-1080 Control and ACSL3LOF1 cells using a clickable OA analog, OA-alkyne. Cells were pulsed with OA-alkyne (20 μM) for 2 h and then, following a 2 h chase period, this probe was conjugated to azide-fluor 488 using copper(I)-catalyzed alkyneazide cycloaddition, yielding OA-488. As in the AA-488 experiments above, immunofluorescence against the cell surface protein cadherin was used to visualize the plasma membrane. In Control cells we observed a strong increase in OA-488 signal over background at the plasma membrane and at perinuclear sites (Figure 5B,C). This increase was strongly attenuated in ACSL3LOF1 cells, demonstrating that incorporation of exogenous OA into membrane lipids is promoted by ACSL3 (Figure 5B,C).
Next, we examined whether ACSL3 was necessary for exogenous OA to prevent lipid ROS accumulation at the plasma membrane. For these experiments, we specifically visualized the plasma membrane of HT-1080 Control and ACSL3LOF1 cells lines by stably expressing a plasma membrane-localized mTurquoise2 (PM-mTq) reporter. In HT-1080 ControlPM-mTq cells, erastin2 treatment induced substantial oxidation of the C11 probe at the plasma membrane that was fully suppressed by co-treatment with exogenous OA (250 μM) (Figure 5D). By contrast, exogenous OA did not suppress C11 oxidation at the plasma membrane in erastin2-treated HT-1080 ACSL3LOF1-PM-mTq cells (Figure 5D). Therefore, ACSL3 is required for exogenous MUFAs to prevent lipid ROS accumulation at the plasma membrane during ferroptosis.
Lipid droplet formation is not required for exogenous MUFAs to inhibit ferroptosis
In some systems, lipid droplet formation protects from oxidative cell death by sequestering PUFAs away from membrane PLs (Bailey et al., 2015; Li et al., 2018). In HEK-293 cells, the ability of exogenous OA to inhibit ferroptosis correlated with the stimulation of neutral lipid (i.e. triacylglycerol) synthesis (Figure S5F). Furthermore, HT-1080 ACSL3LOF1/2 cells accumulated lower levels of neutral lipids in response to exogenous OA compared to Control cells, as determined using BODIPY 493/503 staining (Figure 5E). We therefore asked whether triacylglycerol synthesis and lipid droplet formation were necessary for exogenous OA to inhibit ferroptosis. Using the acyl-CoA:diacylglycerol acyltransferases (DGAT) 1 and 2-specific inhibitors T863 and PF-06424439 (Nguyen et al., 2017) we specifically blocked neutral lipid synthesis and lipid droplet formation in HT-1080 cells (Figure 5F,G, S5G). However, exogenous OA was still capable of suppressing erastin2-induced ferroptosis in the presence of DGATis across all tested concentrations of OA (Figure 5H, S5H). Similar results were obtained using the natural product ACSL inhibitor triacsin C which, for unknown reasons, inhibits de novo glycerophospholipid (e.g. triacylglycerol) synthesis more potently than lysophospholipid reacylation (Igal et al., 1997). Triacsin C (10 μM) blocked OA-stimulated neutral lipids synthesis but had no effect on the ability of OA or t-VA to suppress erastin2-induced ferroptosis (Figure S5I,J). Thus, triacsin C-insensitive ACSL3 activity is sufficient for exogenous MUFAs to suppress ferroptosis in the absence of neutral lipid synthesis or lipid droplet formation.
Basal ACSL3 expression correlates with sensitivity to ferroptosis-inducing agents
We noted that A549N cells lacking ACSL3 were more sensitive to erastin2-induced ferroptosis than Control cells, with both a higher maximal cell death and a substantially earlier onset of cell death (i.e. DO, (Forcina et al., 2017), Control DO = 11.1 h, ACSL3KO1 DO = 6.6 h) (Figure 5I). To investigate whether the relationship between low ACSL3 expression and enhanced ferroptosis sensitivity was more broadly generalizable, we examined data from the Cancer Therapeutics Response Portal (CTRP) (Rees et al., 2015), which reports associations between basal gene expression and sensitivity to 481 small molecule probes across as many as 823 different human cancer cell lines. Remarkably, across all cancer cell lines low ACSL3 expression was strongly associated with sensitivity to covalent GPX4 inhibitors (1S,3R-RSL3, ML162 and ML210) as well as erastin (Figure 5J). By contrast, low expression of ACSL1, ACSL5, and ACSL6 were not associated with sensitivity to ferroptosis-inducing compounds (Figure 5J, note: data for ACSL4 is not available). Thus, across cancer cell types low ACSL3 expression correlates with increased ferroptosis sensitivity.
MUFAs inhibit apoptotic lipotoxicity and ferroptosis through distinct mechanisms
Exogenous MUFAs were previously shown to inhibit apoptotic lipotoxicity caused by the accumulation of saturated fatty acids (SFAs) (Listenberger et al., 2003; Young et al., 2013), potentially positioning MUFAs as regulators of both apoptosis and ferroptosis. Indeed, in HT-1080 cells, exogenous OA blocked cell death induced by either erastin2 or treatment with the exogenous SFA palmitic acid (C16:0), while Fer-1 was only effective at suppressing erastin2-induced cell death (Figure 6A). We confirmed the caspase-dependent nature of the lipotoxic process in these cells, showing that the pan-caspase inhibitor Q-VD-OPh (20 μM), but not Fer-1, blocked cell death induced by growing cells in 2% serum medium and blocking the de novo oleoyl-CoA synthesis using small molecule inhibitors of Δ−9 stearoyl-CoA desaturase 1 (SCD1) (Kamphorst et al., 2013; Young et al., 2013) (Figure 6B). Thus, lipotoxicity and ferroptosis are mechanistically distinct, but both blocked by exogenous MUFAs (Figure S6A).
Figure 6. OA protects from apoptotic and ferroptotic cell death through distinct mechanisms.

(A) Dead cell counts in HT-1080 cells treated ± palmitic acid (200 μM) or erastin2 (1 μM). OA: oleic acid, Fer-1: ferrostatin-1. (B) Dead cell counts in HT-1080 cells treated in 2% serum-containing medium with CAY10566 (200 nM), A939572 (200 nM) or erastin2 (1 μM) ± Fer-1 or the pan-caspase inhibitor Q-VD-OPh. (C) Neutral lipid staining in HT-1080N cells treated for 6 h ± palmitic acid (PA, 200 μM) ± oleic acid (OA, 500 μM) ± DGATis (T863 [20 μM] and PF-06424439 [10 μM]). Scale bar = 50 μm. (D) Cell death in HT-1080N cells at 48 h treated ± oleic acid ± palmitic acid (200 μM) ± DGATis, as in panel C. (E) Cell death at 24 h with two different concentrations of OA ± palmitic acid (200 μM) or erastin2 (1 μM). (F) Model for how ACSL3-dependent MUFA activation generates a ferroptosis-resistant cell state by increasing the phospholipid MUFA to PUFA ratio. Each data point in (A,B,D,E) represents an independent biological replicate (n = 3). Imaging in (C) was performed twice and results from one experiment are shown.
We next investigated whether MUFAs inhibited lipotoxicity and ferroptosis through a similar molecular mechanism. In MEFs, exogenous MUFAs can suppress lipotoxicity by channeling SFAs into neutral lipids for storage in lipid droplets (Listenberger et al., 2003). In HT-1080 cells exogenous OA stimulated neutral lipid synthesis and this process was further enhanced by exogenous palmitic acid (Figure 6C). However, as with ferroptosis (Figure 5G,H), inhibition of lipid droplet formation using DGATis did not block palmitic acid-induced lipotoxicity (Figure 6C,D). We next examined whether ACSL3 was required for exogenous OA to inhibit both ferroptosis and lipotoxicity. While exogenous OA (125 or 500 μM) fully suppressed palmitic acid-induced lipotoxicity in both HT-1080 Control and ACSL3LOF1 cells these same concentrations of OA did not fully protect ACSL3LOF1 cells from erastin2-induced ferroptosis (Figure 6E). CRISPR/Cas9-mediated disruption of ACSL4 likewise did not inhibit palmitic acid-induced cell death or prevent exogenous OA from protecting against lipotoxicity, further distinguishing lipotoxicity from ferroptosis, which requires ACSL4 (Stockwell et al., 2017) (Figure S6B,C). Finally, as shown above, exogenous OA could not protect HEK-293 ACSL3KO1/2 cells from erastin2-induced ferroptosis (Figure 5A). However, exogenous OA fully protected both HEK-293 Control and ACSL3KO1 cells from lipotoxicity induced by growing these cells in 2% serum in the presence of SCD1 inhibitors (Figure S6D). Thus, ACSL3 is more important for exogenous MUFAs to inhibit ferroptosis than lipotoxicity (Figure 6F).
Discussion
Our studies highlight MUFAs as key lipid modulators of both non-apoptotic and apoptotic cell death. Recent studies show that a more mesenchymal phenotype, de-differentiation, and acquired resistance to targeted inhibitors can all promote a ferroptosis-sensitive cell state (Hangauer et al., 2017; Tsoi et al., 2018; Viswanathan et al., 2017). Here we show that exogenous MUFAs can drive cells into a state that makes them resistant to ferroptosis induced by system xc− inhibition, cystine deprivation or GPX4 inactivation. These results are consistent with an earlier observation that exogenous OA could inhibit cell death in response to the covalent GPX4 inhibitor RSL3 (Yang et al., 2016), and now provide mechanistic insight into this protective mechanism. We propose that over time certain exogenous MUFAs, once activated by ACSL3, can displace PUFAs from PLs located at the plasma membrane and possibly other sites, reducing the sensitivity of these membranes to oxidation and lethal ROS accumulation following GPX4 inactivation (Figure 6F). By contrast, MUFAs inhibit SFA-induced apoptotic lipotoxicity through a distinct mechanism that appears independent of ACSL3 and likely unrelated to effects on membrane lipid ROS accumulation per se (Young et al., 2013).
It has been suggested that lysosomes, endoplasmic reticulum or mitochondria could be key sites of lethal ROS accumulation during ferroptosis (Gaschler et al., 2018; Kagan et al., 2016; Krainz et al., 2016; Torii et al., 2016). We confirm widespread lipid ROS accumulation on internal membranes, most likely including these organelles. However, the strong correlation between the ability of exogenous MUFAs to inhibit ferroptosis and preferentially block lipid ROS accumulation at the plasma membrane suggests that the accumulation of lipid ROS at this site is essential for ferroptosis. One intriguing possibility is that triacsin C-insensitive ACSL3 activity generates MUFA-CoAs that directly compete with PUFA-CoAs for insertion into lysophospholipids during plasma membrane remodeling. This could explain why exogenous MUFAs appear to inhibit lipid ROS accumulation at the plasma membrane more effectively than at intracellular sites, in contrast to classic small molecule inhibitors of ferroptosis (ferrostatin-1 and deferoxamine) that block lipid ROS accumulation throughout the cell. Lipid ROS accumulation at the plasma membrane may explain the blistering of the membrane just prior to frank membrane permeabilization, and provides a simple model linking lethal ROS accumulation to membrane permeabilization and cell death during ferroptosis.
The ability of exogenous MUFAs to inhibit ferroptosis could conceivably be relevant in several contexts in vivo, especially for cells that can extract exogenous MUFAs directly from the bloodstream. In healthy individuals, raising serum MUFA levels could provide a means to prevent ferroptosis in various pathological cell death contexts (e.g. neurodegeneration or kidney failure, (Stockwell et al., 2017)). However, increased levels of exogenous MUFAs could also enable cancer cells to evade ferroptosis. Intriguingly, KRAS-driven lung tumors express high levels of ACSL3 (Padanad et al., 2016). RAS pathway mutations also enable cancer cells to scavenge MUFA-containing lysophospholipids from the environment (Kamphorst et al., 2013). RAS-driven ACSL3 expression and increased lipid scavenging could together drive cancer cells into a more ferroptosis-resistant state. Treatments that inhibit MUFA uptake, activation or de novo synthesis could potentially help overcome this state and improve the lethality of existing pro-ferroptotic agents. The observation that low ACSL3 expression is strongly correlated with sensitivity to ferroptosis-inducing agents across hundreds of cancer cell lines supports the potential utility of such an approach.
STAR◆METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to the Lead Contact, Scott Dixon (sjdixon@stanford.edu). Pfa-1 cells were obtained from Dr. Marcus Conrad (Helmholtz Zentrum München) under the terms of a materials transfer agreement.
Experimental Model and Subject Details
Cell Lines and Culture Conditions
HT-1080 (gender: male), A549 (gender: male), T98G (gender: male) and IMR-90 (gender: female) cells were obtained from ATCC, expanded for one passage then frozen down in multiple aliquots used for all subsequent experiments. Pfa-1 cells are mouse embryonic fibroblasts (gender: not specified). Polyclonal Nuc::mKate2-expressing HT-1080 (HT-1080N) and A549N cells were described previously (Forcina et al., 2017). HEK-293 (gender: female) Cas9 control cells and ACSL1KO, ACSL3KO and ACSL4KO cells were generated as described below. HT-1080 cells were grown in Dulbecco’s modified Eagle medium (DMEM, Cat# MT-10–013-CV, Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (FBS, Cat# 26140–079, Gibco), 1x non-essential amino acids (NEAAs, Cat# 11140–050, Gibco) and 0.5 U/mL Pen/Strep (P/S, Cat# 15070–063, Gibco). A549, T98G, IMR90, Pfa1 and HEK-293 cells were grown in DMEM, 10% FBS and 0.5 U/mL P/S. HBSS (Cat# 14025–134) and trypsin (Cat# 25200114) are from Gibco. Low cystine medium was constituted using DMEM (Cat# 17–204-CI, Thermo Fisher Scientific) supplemented with 10% FBS, 0.5 U/mL P/S, 201 μM L-methionine (Cat# M9625, Sigma Aldrich, St. Louis, MO) and 3.9962 μM L-glutamine (Cat# G3126, Sigma Aldrich). For all experiments, cells were trypsinized and counted using a Cellometer Auto T4 cell counter (Nexcelom, Lawrence, MA, USA).
Method Details
Chemicals
Erastin and erastin2 (compound 35MEW28 in (Dixon et al., 2014)) were described in (Forcina et al., 2017). ML162 was synthesized by Acme (Palo Alto, CA). SYTOX Green (Cat# S7020) was from Life Technologies. Dimethyl sulfoxide (Cat# 276855), thapsigargin (Cat# T9033), vinblastine (Cat# V1377), staurosporine (Cat# S6942), hydrogen peroxide (Cat# 216763), bovine serum albumin (BSA, Cat# A4612), BSA-oleic acid (Cat# O3008), ferrostatin-1 (Cat# SML0583), T863 (Cat# SML0539), PF-06424439 (Cat# PZ0233) and etomoxir (Cat# E1905) were from Sigma-Aldrich. Ethanol (Cat# AX0441–3) was from EMD Millipore (Billerica, MA). Methanol (Cat# A935), bortezomib (Cat# NC0587961) and camptothecin (Cat# AC276721000) were from Fisher Scientific. BODIPY 493/503 (Cat# D3922) and C11 BODIPY 581/591 (Cat# D3861) were from Molecular Probes (Thermo Fisher Scientific) and dissolved in anhydrous ethanol or methanol, respectively. Oleic acid (Cat# 90260), α-linolenic acid (Cat# 90150), γ-linolenic acid (Cat# 90220), linoleic acid (Cat# 90150), arachidonic acid (Cat# 90010), palmitic acid (Cat# 10006627), elaidic acid (Cat# 90250), trans-vaccenic acid (Cat# 15301), cis-7-hexadecenoic acid (Cat# 10007290), palmitelaidic acid (Cat# 9001798), oleic acid-alkyne (Cat# 9002078), CAY10566 (Cat# 10012562), triacsin C (Cat# 10007448) and deferoxamine mesylate (Cat# 14595) were from Cayman Chemical (Ann Arbor, MI). A939572 (Cat# B3607) was from ApexBio (Houston, TX). Tamoxifen (Cat# S1972) was from Selleck Chemicals (Houston, TX). Concanavalin A-Alexa Fluor 350 (Cat# C11254, Molecular Probes) was a gift from R. Sala (Tim Stearns Lab, Dept. of Biology, Stanford University, Stanford, CA). Triolein (thin-layer chromatography TAG standard) was a gift from C. Pataki (Ron Kopito lab, Dept. of Biology, Stanford University, Stanford, CA). All chemicals were stored at −20°C until use.
Free fatty acid quantification
The concentrations of the FFAs C16:0, C18:2, and C18:1 were quantified in three independent samples of tissue culture medium DMEM containing 10% FBS (see above) by LC/MS as described previously (Yao et al., 2016). 50 μL aliquots of media were extracted by using 100 μL of a 1:1 methanol:acetonitrile solution containing 5 μM U-13C palmitate as an internal standard. Samples were then vortexed for 30 sec, bath sonicated for 10 min, and stored at −20°C for 2 h. After centrifuging at 3000 × g for 15 min, sample supernatants were transferred to LC vials for LC/MS analysis. No plastics were used to minimize palmitate contamination. An aliquot of each extracted sample (1 μL) was injected onto a Luna Aminopropyl column (150 mm long × 1mm inner diameter or 100 mm long × 2 mm diameter packed with 3-μm particles; Phenomenex) coupled to an Agilent 1200 series high performance liquid chromatography (HPLC) system or a 1200 series ultra-high performance liquid chromatography (UHPLC) system. The columns were used in hydrophilic interaction liquid chromatography (HILIC) mode with the following buffers and gradients: A = 95% water, 5% acetonitrile (ACN), 10 mM ammonium hydroxide, 10 mM ammonium acetate; B = 95% ACN, 5% water; 95% to 0 % B from 0 – 12 min, 0% B from 12 – 15 min at 0.2 mL/min as a flow rate (UHPLC) and 100% to 0% B from 0 – 40 min, 0 % B from 40 – 45 min at 50 μL/min as a flow rate (HPLC). MS detection was carried out on an Agilent 6530 and 6540 Q-TOF in negative ESI mode with a mass range of 65–1,500 Da. Serum lipid concentrations for palmitic acid (HMDB0000220), linoleic acid (HMDB0000673), oleic acid (HMDB0000207) were reported by (Psychogios et al., 2011) and accessed from The Human Metabolome Database (http://www.hmdb.ca/) on January 17th, 2018.
Cell death assessment using STACK
In the lipid modulatory profiling and other experiments cell death was assessed using scalable time-lapse analysis of cell death kinetics (STACK) (Forcina et al., 2017). Cell lines stably expressing nuclear-localized mKate2 (Nuc::mKate2) were incubated in medium containing SYTOX Green (Cat# S7020, Life Technologies) at a final concentration of 20 nM. Counts of live (mKate2+) and dead (SG+) objects were obtained from images obtained every 2 or 4 h. The following image extraction parameter values were used to count HT-1080N mKate2+ objects: Parameter adaptive, threshold adjustment 1; Edge split on; Edge sensitivity 50; Filter area min 20 μm2, maximum 8100 μm2; Eccentricity max 1.0; and SG+ objects: Parameter adaptive, threshold adjustment 10; Edge split on; Edge sensitivity −5; Filter area min 20 μm2, maximum 750 μm2; Eccentricity max 0.9. Counts were exported to Excel (Microsoft Corp.) and lethal fraction (LF) scores were computed from mKate2+ and SG+ counts exactly as described (Forcina et al., 2017). For non-mKate2+-marked cells (HT-1080, A549, T98G), the following image extraction parameter values were used to count SG+ objects: Parameter adaptive, threshold adjustment 1; Edge split on; Edge sensitivity −10; Filter area min 5 μm2, maximum 800 μm2; Eccentricity max 0.9.
Lipid modulatory profile
The day before the start of the experiment, 1,500 HT-1080N cells/well were seeded into 384-well plates (Cat#3712, Corning). The next day, the medium was removed by flicking and immediately replaced using a multichannel pipette with 20 μL medium containing SYTOX Green (40 nM, 20 nM final) and the fatty acid vehicle control (ethanol) or one following fatty acids, each at a concentration of 500 μM (250 μM final): linoleic acid (LA, 18:2n-6), α-linolenic acid (α-LA, 18:3n-3), γ-linolenic acid (γ-LA, 18:3n-6), oleic acid (OA, 18:1n-9) and palmitoleic acid (POA, 16:1n-7). Immediately following the addition of fatty acids, 20 μL of control (DMSO) or 2x lethal compound solutions were added to the cells. The seven lethal compounds each tested in a 10-point, 2-fold dilution series, generated by serial pipetting using a multichannel pipette. The lethal compounds (and final 1x high concentration) tested were: bortezomib (200 nM), camptothecin (10 μM), erastin (20 μM), H2O2 (10 mM), staurosporine (500 nM), thapsigargin (250 nM) and vinblastine (100 nM). Following addition of lethal compounds, plates were spun briefly (250 rpm, 3 sec) to remove air bubbles and then transferred into an Essen IncuCyte Zoom live cell imaging station (Essen BioScience, Ann Arbor, MI, USA) housed within a standard tissue culture incubator.
Counts of live and dead cells were obtained automatically every 2 h for 72 h using time-lapse, live-cell imaging as described (Forcina et al., 2017). The lethal fraction (LF) was computed for each population over time, then area under the curve (AUC) values from each LF profile of each compound-fatty acid combination were used to summarize cell death over the entire timecourse as a single value. We then computed mean AUC values across all concentrations of each lethal compound and the fold-difference AUC value between the vehicle control (ethanol) and each exogenous FA was determined and plotted as a heatmap using R (https://www.r-project.org/).
PrestoBlue cell viability assay
At the end of the lipid modulatory profiling experiment, cell death was assessed using PrestoBlue (Cat# A13261, ThermoFisher). Following 72 h compound treatment, 10 μL of a 5x PrestoBlue/fresh medium solution was pipetted into the plates and mixed using a multichannel pipettor (final 10% v/v). Cells were incubated for 2 h at 37°C in a humidified tissue culture incubator and fluorescence was measured using a multimode fluorescence plate reader (Cytation3, BioTek Instruments, Winooski, VT, USA) at ex/em 530/590 nm. Data was normalized to the control treatment (DMSO), which was set to 100% cell viability.
Small molecule library screen
A library of 261 bioactive compounds (Cat# L2000) was obtained from Selleck Chemicals (Houston, TX) and stored at −80°C. The library was re-formatted from 96-well to 384-well format using a Versette automated liquid handler configured with a 96-channel pipetting head, and diluted to 2 mM in DMSO. The day before screen, 1,500 HT-1080N cells/well were seeded into two 384-well plates in 40 μL medium. The next day, the medium was removed and replaced with medium containing SYTOX Green (20 nM final) and compounds from a freshly thawed library master stock plate (1 compound/well) were added to a final concentration of 5 μM, using a Versette liquid handler equipped with a 384-channel pipetting head. One plate was co-treated with EtOH and the other with oleic acid (125 μM). Plates were imaged immediately and every 4 h thereafter for a total of 120 h using the Essen IncuCyte Zoom. Counts of SYTOX Green and mKate2 objects per mm2 were obtained and the lethal fraction calculated as described in (Forcina et al., 2017). The area under the curve (AUC) of lethal fraction scores across the full 120 h were calculated using the trapezoid rule in Prism 7.0.
Glutathione measurements
The day before the experiment, 150,000 HT-1080 cells/well were seeded into 6-well dishes (Cat# 07–200–83, Corning Life Sciences, Tewksbury, MA). The next day, cells were treated with ethanol (vehicle) or oleic acid (500 μM) and DMSO (vehicle) or erastin2 (1 μM) for 11 h. After 11 h, cells were harvested and prepared for measurement of total intracellular glutathione (GSH+GSSG) using a glutathione assay kit based on Ellman’s reagent according to the manufacturer’s protocol (Cat# 703002, Cayman Chemical, Ann Arbor, USA). The concentration of glutathione was determined using a glutathione standard curve and normalized to total protein level in each sample. Three independent biological replicates were performed for each condition.
RT-qPCR
The day before the experiment, 150,000 HT-1080 cells/well were seeded into 6-well dishes. The next day, cells were treated with ethanol (vehicle) or oleic acid (500 μM) and DMSO (vehicle) or erastin2 (1 μM) for 6 hours. After 6 hours, treatment medium was removed and 1 mL cold HBSS was applied to the cells. Cells were harvested by scraping into a 1.5 mL tube and frozen at −80°C until RNA extraction. RNA extra ction was performed using the QIAshredder and RNeasy Mini kits (Cat# 79654 and Cat# 74134, respectively, QIAGEN) according to the manufacturer’s protocol. 1–2 μg of mRNA was used as a template for each RT reaction, performed using the TaqMan RT kit (Cat# N8080234, Applied Biosystems/Thermo Fisher Scientific, Foster City, CA). Primers used to amplify CHAC1 and ACTB, the internal reference control, are listed in Table S1. qPCR reactions were performed using the Power SYBR Green PCR Master Mix (Cat# 4367659, Applied Biosystems). Triplicate samples per treatment were analyzed on a QuantStudio 3 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using absolute quantification settings. Differences in CHAC1 mRNA levels compared to internal reference ACTB mRNA levels between control and experimental conditions were calculated using the ΔΔCt method. Three independent biological replicates were performed for each condition.
Cystine deprivation
The day before the experiment, 5,000 HT-1080N cells/well were seeded into a 96-well plate (Cat# 3904, Corning). The next day, cells were washed 3 times with 1x HBSS and then low cys2 medium plus the appropriate vehicle or compound was applied to the cells. Cells were imaged on the Essen IncuCyte Zoom and analyzed as described (Forcina et al., 2017). For the transient cystine deprivation experiment, cystine-replete medium was added back after 24 or 48 h. Three independent biological replicates were performed for each condition.
Pfa1 cell viability analysis
The day before the experiment, 4,000 Pfa1 cells/well were seeded into a 96-well plate. The next day, the cell culture medium was replaced with DMSO (vehicle) or tamoxifen (1 μM) and vehicle, Fer-1 (1 μM) or OA (125 μM) was applied to the cells. Cells were imaged on the Essen IncuCyte Zoom and analyzed as described (Forcina et al., 2017). Three independent biological replicates were performed for each condition.
DPPH assay
The stable radical DPPH (Cat# D9132, Sigma-Aldrich) was dissolved in methanol to a final concentration of 50 μM. Each test compound (or vehicle) was added to 1 mL of DPPH solution so that the final compound concentration was 50 μM. Samples were inverted 5–6 times and allowed to incubate at room temperature for 10 min. Samples were then aliquoted in triplicate to 96-well black-side clear-bottom dishes (Cat# 3904, Corning) and absorbance at 517 nm was measuring using a multimode fluorescence plate reader (Cytation3). Samples were subtracted for background (methanol only) and normalized to starting DPPH absorbance. The experiment was repeated three times.
Western blot analysis
The day before the experiment, 2 × 106 HT-1080 cells/plate (one plate per condition) were seeded into 10 cm dishes (Cat# CC7682–3394, USA Scientific, Ocala, FL). The next day, cells were treated with ethanol (vehicle) or oleic acid (500 μM) and DMSO (vehicle) or erastin2 (1 μM) for 11 h. After 11 h, the media was removed and cells were washed once with 3 mL 1x PBS. After washing, 2 mL of 1x PBS was added to the cells. Cells were scraped off the plate and 0.5 mL of cell suspension was transferred into a 1.5 mL microfuge tube. Cells were spun down (2,000 rpm, 2 min), the supernatant removed and discarded. The cell pellet was immediately stored at −80 °C until needed. Cell pel lets were thawed at room temperature and immediately lysed with 150–175 μL 9 M urea or RIPA/SDS. Lysates were sonicated [(1 s on, 1 s off, 100 % amplitude) × 10 cycles] and spun down to remove any debris (13,300 rpm, 15 min, room temperature or 4°C). Clarified lysates were tr ansferred to a new 1.5 mL tube. Lysates from cells treated with scrambled or GPX4 siRNA were used as controls to demonstrate antibody specificity. All lysates were quantified using a Bradford protein assay (Bradford Protein Assay Dye Reagent Concentrate, Cat# 5000006, Bio-Rad, Hercules, CA) with a standard BSA curve. Equal amounts of protein were combined with 4x Bolt LDS Sample Buffer (Cat# B0007) and 10x Bolt Sample Reducing Agent (Cat# B0009) (Life Technologies), heated to 95°C for 5 min and loaded onto a Bolt 4–12% Bis-Tris Plus Gel (Cat# NW04120BOX) (Life Technologies). Protein was transferred to a nitrocellulose membrane using an iBlot2 transfer stack (Life Technologies). The membrane was blocked using Odyssey Blocking Buffer (Cat# 927–50000, LI-COR Biotechnology, Lincoln, NE) (1 h, room temperature) and then incubated in primary antibody mixture (24 h, 4°C). Primary antib odies used were α-GPX4 (Cat# ab125066, Abcam, Cambridge, MA; 1:250 dilution), α-actin (Cat# sc-1616, Santa Cruz Biotechnology, Dallas, TX; 1:2000 dilution), α-ACSL4 (Cat# SAB2701949, Sigma-Aldrich; 1:2500 dilution) and α-tubulin (Cat# MS581P1, Millipore Sigma, Billerica, MA; 1:2000 dilution). The membrane was washed 3x in TBST and then incubated in secondary antibody mixture (1 h, room temperature). Secondary antibodies used were donkey α-rabbit (Cat# 925–32213, LI-COR Biotechnology, Lincoln, NE, 1:15,000 dilution) and donkey α-goat (Cat# 925–32214, LI-COR, 1:15,000 dilution) or donkey α-mouse (Cat# 926–68022, LI-COR, 1:15,000 dilution), and the secondary antibody buffer was 0.5:25:25 10% SDS:Odyssey Blocking Buffer (TBS):TBST or Odyssey Blocking Buffer. The membrane was washed 3 times in TBST and then scanned on an Odyssey CLx Imaging System (LI-COR). Western blots were performed on two independent biological replicates for each condition.
C11 BODIPY 581/591 Analysis
For flow cytometry experiments, the day before the experiment, 200,000 HT-1080 cells/well were seeded into 6-well dishes. The next day, cells were treated with ethanol (vehicle), oleic acid (500 μM) or DFO (50 μM), and DMSO (vehicle) or erastin2 (1 μM) for 11 h. After 11 h, cells were harvested by trypsinization and 200,000 cells were transferred to a 1.5 mL microfuge tube, pelleted (3000 rpm, 5 min), and then resuspended in C11 BODIPY 581/591 (5 μM) dissolved in HBSS. Cell suspensions were incubated at 37°C for 10 min. After 10 min, the cells were pelleted (3000 rpm, 5 min), and then resuspended in 0.2 mL fresh HBSS. Samples were strained through a cell strainer prior to flow cytometry analysis. Flow cytometry analysis was performed on a custom Stanford and Cytek upgraded FACScan analyzer (Scanford, BluFL1 and YelFL1 for detecting green and red fluorescence, respectively, Stanford Shared FACS Facility). Oxidation of C11 BODIPY 581/591 was calculated as the ratio of the green fluorescence (which indicates oxidized probe) to total (green + red, which indicates total reduced plus oxidized probe) fluorescence (Pap et al., 1999). Three independent biological replicates were performed for each condition.
For confocal imaging experiments, the day before the experiment, 120,000 HT-1080 cells/well were seeded into 6-well dishes that had one 22 mm2 glass coverslip in each well. The next day, cells were treated with ethanol (vehicle), oleic acid (500 μM) or DFO (50 μM) and DMSO (vehicle) or erastin2 (1 μM). After 10 h, the treatment media was removed and cells were washed once with HBSS. HT-1080 cells were then treated with 1 mL Concanavalin AAlexa Fluor 350 (25 μg/mL) and incubated at 37°C for 20 min. After 20 mi n, 1 mL C11 BODIPY 581/591 (final concentration = 5 μM) dissolved in HBSS was added to each sample. Cells were incubated at 37 °C for an additional 10 min. After 10 min, the label was removed and 1 mL of fresh HBSS was added to the cells. The cover slip was removed from the well and inverted onto a glass microscope slide onto which 25 μL of fresh HBSS had been applied. Cells were imaged using a Zeiss Axio Observer microscope with a confocal spinning-disk head (Yokogawa, Tokyo, Japan), PlanApoChromat 63×/1.4 NA oil immersion objective, and a Cascade II:512 electron-multiplying (EM) CCD camera (Photometrics, Tucson, AZ). Imaging was performed on four (DMSO/erastin2 ± EtOH/OA or ± DMSO/DFO) and two (DMSO/erastin2 ± DMSO/Fer-1) independent biological replicates per treatment. Images were processed in ImageJ 1.48v and quantified as follows. Two regions of interest (ROI) were defined for each nucleus: one perinuclear and one plasma membrane-localized. Red and green fluorescence values were background-corrected by quantifying the red or green fluorescence in cell-free areas from at least four images and subtracting to determine the final fluorescence values. Oxidation of C11 BODIPY 581/591 was calculated as described above. A similar approach was used to treat and image HT-1080 ControlPM-mTq and ACSL3LOF1-PM-mTq (see below for cell line details) cells with the following modifications. The day before the experiment, 150,000 cells/well were seeded. The next days, cells were treated with ethanol (vehicle) or oleic acid (250 μM) and DMSO (vehicle) or erastin2 (1 μM) for 24 h before C11 labeling.
Phase contrast time-lapse imaging
The day before the experiment, 60,000 HT-1080 cells/well were seeded into 4-well chamber slides (Cat# 177399, Thermo Scientific). The next day, the growth medium was removed and cells were treated with the appropriate compound(s) and imaged every 5 min using Micro-Manager (Edelstein et al., 2010) on a Nikon Diaphot microscope (Nikon Instruments, Melville, NY) equipped with an environmental chamber allowing incubation at 37°C, a Nikon 20x Ph2 DL phase contrast objective and a Hamamatsu ORCA-ER B/W CCD digital camera (Model# C4742–80, Hamamatsu Photonics K.K., Hamamatsu City, Japan). Compounds and concentrations (in parentheses) used were: DMSO (vehicle), erastin2 (1 μM), bortezomib (100 nM), DFO (50 μM), ethanol (vehicle), OA (500 μM).
Single reaction monitoring-based LC-MS
The day before the experiment, 2 × 106 HT-1080 cells/plate were seeded into 10 cm dishes (Cat# CC7682–3394, USA Scientific, Ocala, FL). The next day, cells were treated with ethanol (vehicle) or oleic acid (500 μM) and DMSO (vehicle) or erastin2 (1 μM) for 11 h. After 11 h, the media was removed and cells were washed once with 3 ml 1x PBS. After washing, 2 mL of 1x PBS was added to the cells. Cells were scraped off the plate and 1 mL of cell suspension was transferred into a 1.5 mL microfuge tube. Cells were spun down (2000 rpm, 2 min), the supernatant removed and discarded. The cell pellet was then immediately stored at −80 °C, prior to further analysis. Lipids were extracted using 4 mL of a 2:1:1 solution of chloroform:methanol:phosphate-buffered saline that included dodecylglycerol (10 nM) and pentadecanoic acid (10 nM) as internal standards. The organic layer was separated and the aqueous layer was acidified with 0.1% formic acid and re-extracted in chloroform. The organic layers were combined and dried down under a nitrogen stream. Dried extracts were resolubilized in chloroform and 10 l was injected onto an Agilent 6430 triple quadrupole (QQQ)-liquid chromatography-mass spectrometry (LC-MS) instrument for single-reaction monitoring (SRM)-based analysis as described (Benjamin et al., 2015; Louie et al., 2016). Five independent biological replicates were performed for each condition.
Alkyne labeling, imaging and quantification
The day before the experiment, 50,000 HT-1080 cells/well were seeded into a 12-well plate (Cat# 3513, Corning) that had one 12 mm2 coverslip in each well. On the day of the experiment, cells were washed once with HBSS, and then treated in DMEM + 10% FBS ± arachidonic acid-alkyne (AA-alk, 20 μM), and bovine serum albumin (BSA) or BSA-OA for 2 h. After 2 h, the treatment medium was removed and replaced with fresh HT-1080 growth medium (no treatment) and BSA or BSA-OA, and the cells were allowed to recover for 10 h. After 10 h, cells were washed 3 times with 1x PBS, then fixed in 4% PFA in 1x PBS (10 min, 60 rpm, RT). Cells were washed 3 times with 1x PBS, then permeabilized with 0.1% Triton-X in PBS (2 min, RT). Cells were washed 3 times with 1x PBS, then treated with the click reaction: 0.1 mM Azide-fluor 488 (Cat# 760765, Sigma-Aldrich), 1 mM cupric sulfate (CuSO4, Cat# S25285, Fisher Scientific), 1 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Cat# C4706, Sigma Aldrich) in 1x PBS (1 h, RT) in a light-impermeable humid chamber. Cells were washed 5 times with 1x PBS, then blocked with PBS-BT (1x PBS, 3% BSA, 0.1% Triton X-100, 0.02% NaN3) (45 min, RT). PBS-BT was removed and replaced with primary antibody mixture: α-pan-cadherin (1:100) (Cat# ab6528, Abcam) in 1x PBS-BT (1 h, RT). Cells were washed 3 times with 1x PBS-BT, then treated with secondary antibody mixture: donkey-α-mouse Alexa Fluor 568 (1:1000) (Cat# A10037, Invitrogen) in 1x PBS-BT (45 min, RT). Cells were washed 3 times with 1x PBS-BT, then treated with 100 ng/ml 4’,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI, Cat# D1306, Thermo Fisher) in 1x PBS (15 min, RT). Cells were washed 3 times with 1x PBS, then the coverslips were mounted onto a glass slide with 5 μL of ProLong Gold Antifade Mountant (Cat# P10144, Thermo Fisher). Cells were imaged by confocal microscopy (described above). A similar protocol was used for OA-alkyne labeling except that cells were treated ± oleic acid-alkyne and then allowed to recover for 2 h prior to fixation. Two independent biological experiments were performed for each condition. Images acquired for the alkyne experiments were processed in ImageJ 1.48v and quantified as follows. A region of interest (ROI) was defined for each image based on red fluorescence (i.e. Alexa Fluor 568) intensity falling within an empirically defined range. Then, the green fluorescence (i.e. azide-fluor 488) intensity within the ROI was quantified. Images were background corrected by quantifying the green fluorescence in cell-free areas from at least four images and subtracting to determine the final green fluorescence values.
Thin-layer chromatography
The day before the experiment, 100,000 HT-1080 cells or 500,000 HEK-293 cells/well were seeded into 6-well dishes. The next day, cells were treated with ethanol (vehicle) or oleic acid (various concentrations) [and DMSO (vehicle) or T863 (20 μM) + PF-06424439 (10 μM) where indicated] for 6 hours. Cells were trypsinized and washed twice in 1x PBS, and cell pellets were frozen at −20°C until use. Cell pellets were l ysed in 200 μL 1x PBS + 0.5 % Nonidet P 40 substitute (Sigma-Aldrich). Protein concentration was assayed using the Pierce Microplate BCA Protein Assay Kit (Cat# 23252, Thermo Scientific) and the same amount of protein was used from each sample for lipid extraction. For neutral lipid extraction, 300 μL of water and 1 mL of CHCl3:MeOH (2:1) was added to each sample. Samples were vortexed for 20 seconds, and then spun at 3000 rpm, 5 min. The organic phase was transferred to a clean 1.5 mL tube and dried down in a speed vacuum at 60°C, 30 min. The dried extract was resuspended in 5 μL CHCl3:MeOH (2:1) and immediately applied to a TLC plate (Cat# 70644, Sigma-Aldrich). The TLC plate was developed using hexane:ethyl ether:acetic acid (80:20:1). After air-drying, the plate was dipped in a solution of cerium ammonium molybdate (CAM, for the Sigma plate). Excess solution was blotted off using Whatman paper. The plate was placed in a 95°C oven until bands were visible. Bands were compared to standards applied and developed on the same TLC plate. Two independent biological replicates were performed for each condition.
Neutral lipid analysis using BODIPY 493/593
The day before the experiment, 200,000 HT-1080 cells/well were seeded into 6-well dishes. The next day, cells were treated with ethanol (vehicle) or fatty acid (500 μM) [and DMSO (vehicle) or T863 (20 μM) + PF-06424439 (10 μM) or Triacsin C (10 μM), where relevant] for 6 hours. After 6 hours, the media was removed and cells were washed twice with HBSS. Cells were incubated with BODIPY 493/503 (1 μM) in HBSS at 37°C for 30 min. After 30 min, cells were washed three times with HBSS. After the final wash, 1 mL of HBSS was added and cells were imaged (10x objective, Essen IncuCyte Zoom). At least three independent biological replicates were performed for each condition.
CRISPR/Cas9 genome editing
HEK-293 cells lacking ACSL1, ACSL3 and ACSL4 were generated using the single-guide (sg) RNA sequences listed in Table S1, which were cloned into px459 (Addgene; plasmid #48139) Oligonucleotide pairs were phosphorylated and annealed at a concentration of 10 μM using T4 polynucleotide kinase (Cat# M0201, New England Biolabs) at 37°C for 30 min, followed by 95°C for 5 min, and cooled to 25°C at a rate of 5°C per minute. The oligo duplex was then diluted 1:250 in ddH2O and used for ligation into the plasmid px459. For the ligation, 2 μL of the diluted duplex was added to 1000 ng of px459, 2 μL of 10x FastDigest buffer (Thermo Scientific), 1 μL FastDigest BpiI (Cat# D1014, Thermo Scientific), 0.5 μL T4 DNA ligase (Cat# M0202, New England Biolabs), and completed to 20 μL with ddH2O. The ligation reaction was incubated for 1 hour [(37°C, 5 min; 21°C, 5 min) x 6 cycles]. 2 μL of the ligation product was used to transform competent NEB5α (Cat# C2987, New England Biolabs). Plasmid DNA was extracted using a QIAGEN spin column (Cat# 27106, QIAGEN) and validated by sequencing. 5,000,000 HEK-293 cells were seeded into 10 cm dishes. The following day, 10 μg plasmid was transfected using X-tremeGENE HP (Cat# 06366546001, Roche) at a 1:2 DNA:XtremeGENE HP ratio in 1 mL Opti-MEM (Cat# 31985–070, Invitrogen). After 24 h cells were trypsinized and replated in media supplemented with 1 μg/mL puromycin. Media was then replenished every 3 days until the untransfected controls were dead and then puromycin was removed from the growth medium. Clonal cells were isolated by limited dilution, single colonies were expanded, and knockout confirmed by western blotting using the following antibodies: α-ACSL1 (Cat# 13989–1-AP, Proteintech), α-ACSL3 (Wu et al., 2009), α-ACSL4 (Sigma Aldrich), and α-GAPDH (Cat# MAB374, Millipore).
HT-1080 and A549N cells lacking ACSL3 were generated as follows. The sgRNA primer set ACSL3 sg2 (Table S1) was used. sgRNAs were cloned as described (Ran et al., 2013). Briefly, for ACSL3, a mixture of 1 μL of prSD482 (100 μM) and 1 μL of prSD483 (100 μM) (for ACSL4, prSD484 and prSD485 were used), plus 8 μL of ddH2O was prepared, heated to 95°C, then cooled at a rate of 2.5°C/minute to 25°C. The oligo duplex was then diluted 1:200 in ddH2O and used for cloning into pSD224, the pSpCas9(BB) plasmid. The ligation reaction was: 100 ng pSD224, 2 μL of the diluted oligo duplex, 2 μL of 10x FastDigest buffer (Thermo Scientific), 1 μL 10 mM DTT, 1 μL 10 mM ATP, 1 μL FastDigest BpiI (Cat# D1014, Thermo Scientific), 0.5 μL T4 DNA ligase (Cat# EL0014, Thermo Scientific), and completed to 20 μL with ddH2O. The ligation reaction was incubated for 1 hour [(37°C, 5 min; 21°C, 5 min) × 6 cycles]. 2 μL of the ligation product was used to transform competent DH5α. Transformants were selected on LB+amp (100 μg/mL). Plasmid DNA was extracted using a QIAGEN spin column (Cat# 27106, QIAGEN, Hilden, Germany) and sent for sequencing. Cells were transfected using Opti-MEM I Reduced Serum Medium (Cat# 31985–062, Life Technologies) and Lipofectamine LTX Reagent (Cat# 15338–100, Life Technologies), according to the manufacturer’s instructions. The following morning post-transfection, the transfection medium was removed and replaced with fresh medium. Cells were allowed to recover for 24 h, and the following day, GFP+ cells were single-sorted using a BD FACSJazz Cell Sorter (BD Biosciences) (Stanford Shared FACS Facility) into a 96-well plate containing DMEM + 30% FBS, 1x NEAAs and 0.5 U/mL P/S. Cells were incubated at 37°C until distinct clones appeared. Individual clones were amplified in normal (10% FBS) HT-1080 medium to produce enough cells for gDNA extraction and protein lysis. gDNA extraction was performed using a NucleoSpin Tissue column (Cat# 740952, Takara Bio). 50–100 ng of gDNA was used as PCR template to amplify a region of DNA flanking the sgRNA recognition site. An ACSL3 amplicon was generated using primers listed in Table S1. The PCR product was purified using a QIAGEN PCR Purification column (Cat# 28106) and sent for sequencing. The ACSL3 amplicon was sequenced using the sequencing primer listed in Table S1. Protein levels were examined by Western blot (described above, except that the lysis was done in RIPA/SDS plus protease inhibitor cocktail (Cat# P8340, Sigma Aldrich)) using α-ACSL3 ((Wu et al., 2009); 1:1000 dilution) as the primary antibody. HT-1080 cells lacking ACSL4 were generated as described for HT-1080 cells lacking ACSL3 except that primers to target (ACSL4 sg2), amplify and sequence ACSL4 were used as listed in Table S1.
Lentiviral cloning, production and infection
The sequence of plasma membrane-targeted pPalmitoyl-mTurquoise2 was reported (Goedhart et al., 2012). A gBlock (Integrated DNA Technologies, Coralville, IA, USA) of this sequence was synthesized containing a KOZAK-FLAG tag sequence inserted in-frame in between the pPalmitoyl and the mTurquoise2 sequences (collectively called PM-mTurq2). This gBlock was cloned via Gibson Assembly (Cat# E5510S, New England Biolabs, Ipswich, MA, USA) into pSD291 (pLenti-CMV-DEST), previously linearized with EcoRV (Cat# FD0304, Thermo Scientific), to generate pSD427 (pLenti-CMV-PM-mTq). pSD427 was co-transfected with 3rd generation lentiviral packaging plasmids (pMDLg/pRRE, pRSV-Rev and pMD2.G, which were gifts from Didier Trono, Addgene plasmids #12251, #12253 and #12259, respectively) into 293T cells using PolyJet according to the manufacturer’s instructions (Cat# SL100688, SignaGen Laboratories, Rockville, MD, USA). The viral supernatant was harvested, filtered through a 0.45 μm PVDF filter (Cat# SLHV033RS, Merck Millipore, Burlington, MA, USA) and stored in aliquots at −80°C until use.
The day before the infection, 90,000 HT-1080 Control or ACSL3LOF1 cells/well were seeded into 12-well plates. The next day, the medium was removed and replaced with 0.95 mL of 1.05x Polybrene (Cat# H9268, Sigma Aldrich, final concentration = 8 μg/mL) in HT-1080 medium and 0.05 mL filtered viral supernatant. After 2 days, the medium was removed and replaced with HT-1080 medium containing 1 μg/mL puromycin (Cat# A11138–03, Life Technologies). After 4 days, the medium was removed and replaced with HT-1080 medium without puromycin. Cells were amplified and frozen down in aliquots at −140°C. The cell lines were named HT-1080 ControlPM-mTq and ACSL3LOF1-PM-mTq.
DNA Sequencing
Sanger sequencing of HT-1080 ACSL3LOF1 identified different mutations on each DNA strand that were resolved by TOPO-TA cloning (Cat# 450071, Thermo Fisher Scientific). One strand had a nine nucleotide deletion that resulted in a single amino acid change at the 5’ end of the deletion. The other strand had a two nucleotide deletion that resulted in a frameshift mutation. Sanger sequencing of HT-1080 ACSL3LOF2 identified a six nucleotide deletion that resulted in a 2 amino acid deletion. By Western blot both clones may retain a small amount of ACSL3 protein. Sanger sequencing of A549N ACSL3KO1 identified a four nucleotide deletion that resulted in a frameshift mutation. Sanger sequencing of A549N ACSL3KO2 identified different mutations on each DNA strand that were resolved by Tracking of Indels by Decomposition (TIDE, https://tide.deskgen.com/). One strand had a three nucleotide deletion and the other strand had a 1 nucleotide insertion that resulted in a frameshift mutation. By Western blot both A549 clones are protein nulls. Sanger sequencing of HT-1080 ACSL4KO1 identified different mutations on each DNA strand that were resolved by TIDE. One strand had a 2 bp deletion and the other strand had a 5 bp deletion, both of which resulted in frameshift mutations. Sanger sequencing of HT-1080 ACSL4KO2 identified different mutations on each DNA strand that were resolved by TIDE. One strand had a 11 bp deletion and the other strand had a 2 bp insertion, both of which resulted in frameshift mutations. By Western blot both clones are protein nulls.
Analysis of CTRP dataset
We analyzed data from Cancer Therapeutics Response Portal (CTRP) v2.1 dataset available from https://ocg.cancer.gov/ctd2-data-project/broad-institute-screening-dependencies-cancer-cell-lines-using-small-molecules-0 on October 6th, 2018. The data for statistically significant Pearson correlations between basal gene expression for ACSL3, ACSL1, ACSL5 and ACSL6 and small-molecule sensitivity, from all cancer cell lines only, were extracted from the v21.data.gex_global_analysis.txt table and plotted using Prism 6.0h.
Quantification and statistical analysis
Lethal fraction scoring was performed using Microsoft Excel 14.6.0 (Microsoft Corporation, Redmond, WA). LED curve fitting was performed using Prism 6.0h (GraphPad Software, La Jolla, CA). Flow cytometry data were processed using FlowJo 10.1r5 (FlowJo LLC, Ashland, OR). Confocal images were processed in ImageJ 1.48v (U.S. National Institutes of Health, Bethesda, MD). Graphing and statistical analyses were performed using Prism 6.0h. Figures were assembled using Adobe Illustrator (Adobe Systems, San Jose, CA). Statistical details of experiments and statistical tests used can be found in the main text, figure legends, and STAR Methods.
Data Availability
For the 261-member bioactive compound screen in HT-1080N cells (both ethanol only and oleic acid (OA)-treated) the calculated area under the curve of lethal fraction scores are available online via the Mendeley Data repository https://data.mendeley.com/datasets/g2xhxht7cw/1.
Supplementary Material
Highlights:
Exogenous MUFAs inhibit ferroptosis in a structure-specific manner
MUFAs block lipid ROS accumulation specifically at the plasma membrane
Exogenous MUFAs inhibit ferroptosis in an ACSL3-dependent manner
Exogenous MUFAs inhibit apoptotic lipotoxicity in an ACSL3-independent manner
Significance.
Metabolites present in the extracellular environment can potently modulate cellular phenotypes and potentially serve as therapeutics for various diseases. Ferroptosis is a non-apoptotic cell death process characterized by the generation of toxic lipid reactive oxygen species (ROS). In this study we report that exogenous monounsaturated fatty acids (MUFAs) can promote a ferroptosis-resistant cellular state characterized by reduced total levels of polyunsaturated fatty acid-containing phospholipids (PUFA-PLs) and reduced sensitivity to oxidation of the lipid ROS probe C11 BODIPY 581/591 at the plasma membrane. The ability of exogenous MUFAs to inhibit ferroptosis and suppress lipid ROS accumulation at the plasma membrane requires the free fatty acid activating enzyme, ACSL3. We propose that non-oxidizable MUFAs displace oxidizable PUFAs from membrane phospholipids, thereby limiting the sensitivity of the membrane to PUFA-dependent oxidation. Exogenous MUFAs also suppress apoptotic lipotoxicity triggered by the accumulation of saturated fatty acids (SFAs), but this protective effect is less dependent on ACSL3. Thus, exogenous MUFAs can inhibit both ferroptosis and lipotoxicity but through distinct mechanisms. This study suggests that variation in environmental lipid levels and the inherent competition between individual PUFA, MUFA and SFA species for incorporation into membrane lipids shapes the cell state and sensitivity to both non-apoptotic and apoptotic cell death.
ACKNOWLEDGEMENTS
We thank Marcus Conrad (Helmholtz Zentrum München) for Pfa-1 cells, Jingwen Liu (Palo Alto VA hospital) for the anti-ACSL3 antibody, Tim Stearns for concanavalin A-Alexa Fluor 350 and assistance with microscopy, and Roberta Sala for experimental assistance. This work was supported by NIH awards to P-J.K. (T32 GM007276), G.J.P. (R35ES028365), D.N.M. (R01CA172667), J.A.O. (5R01GM112948), and S.J.D. (1R01GM122923). J.A.O. is supported by the American Heart Association (16GRNT30870005). S.J.D. is supported by a Damon Runyon-Rachleff Innovator award and the Hellman Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental information including six supplemental figures can be found with this article online at LINK.
Declaration of interests
S.J.D. is a member of the scientific advisory board of Ferro Therapeutics.
References
- Bailey AP, Koster G, Guillermier C, Hirst EMA, MacRae JI, Lechene CP, Postle AD, and Gould AP (2015). Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell 163, 340–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin DI, Li DS, Lowe W, Heuer T, Kemble G, and Nomura DK (2015). Diacylglycerol Metabolism and Signaling Is a Driving Force Underlying FASN Inhibitor Sensitivity in Cancer Cells. ACS Chem. Biol 10, 1616–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor JR, Abu-Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A, Lewis CA, and Sabatini DM (2017). Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 169, 258–272.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das UN (1991). Tumoricidal action of cis-unsaturated fatty acids and their relationship to free radicals and lipid peroxidation. Cancer Lett. 56, 235–243. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, et al. (2014). Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3, e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G, and Stockwell BR (2015). Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol 10, 1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol 13, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummen GPC, van Liebergen LCM, Op den Kamp JAF, and Post JA (2002). C11-BODIPY(581/591), an oxidation-sensitive fluorescent lipid peroxidation probe: (micro)spectroscopic characterization and validation of methodology. Free Radical Biology and Medicine 33, 473–490. [DOI] [PubMed] [Google Scholar]
- Edelstein A, Amodaj N, Hoover K, Vale R, and Stuurman N (2010). Computer control of microscopes using μManager. Curr Protoc Mol Biol Chapter 14, Unit14.20–14.20.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcina GC, Conlon M, Wells A, Cao JY, and Dixon SJ (2017). Systematic Quantification of Population Cell Death Kinetics in Mammalian Cells. Cell Syst 4, 600–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbarino J, and Sturley SL (2009). Saturated with fat: new perspectives on lipotoxicity. Curr Opin Clin Nutr Metab Care 12, 110–116. [DOI] [PubMed] [Google Scholar]
- Gaschler MM, Hu F, Feng H, Linkermann A, Min W, and Stockwell BR (2018). Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. ACS Chem. Biol 13, 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedhart J, von Stetten D, Noirclerc-Savoye M, Lelimousin M, Joosen L, Hink MA, Weeren L, Gadella TWJ, and Royant A (2012). Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat Commun 3, 751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Galluzzi L, and Kroemer G (2014). Cell biology. Metabolic control of cell death. Science 345, 1250256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grevengoed TJ, Klett EL, and Coleman RA (2014). Acyl-CoA metabolism and partitioning. Annu. Rev. Nutr 34, 1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL, et al. (2017). Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igal RA, Wang P, and Coleman RA (1997). Triacsin C blocks de novo synthesis of glycerolipids and cholesterol esters but not recycling of fatty acid into phospholipid: evidence for functionally separate pools of acyl-CoA. Biochem. J 324 (Pt 2), 529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al. (2016). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol 13, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol 13, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, Thompson CB, and Rabinowitz JD (2013). Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. U.S.a 110, 8882–8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MJ, Fujino T, Sasano H, Minekura H, Yabuki N, Nagura H, lijima H, and Yamamoto TT (1997). A novel arachidonate-preferring acyl-CoA synthetase is present in steroidogenic cells of the rat adrenal, ovary, and testis. Proc. Natl. Acad. Sci. U.S.a 94, 2880–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krainz T, Gaschler MM, Lim C, Sacher JR, Stockwell BR, and Wipf P (2016). A Mitochondrial-Targeted Nitroxide Is a Potent Inhibitor of Ferroptosis. ACS Cent Sci 2, 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Sancak Y, Frasor J, and Atilla-Gokcumen GE (2018). A Protective Role for Triacylglycerols during Apoptosis. Biochemistry 57, 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Ory DS, and Schaffer JE (2003). Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. U.S.a 100, 3077–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie SM, Grossman EA, Crawford LA, Ding L, Camarda R, Huffman TR, Miyamoto DK, Goga A, Weerapana E, and Nomura DK (2016). GSTP1 Is a Driver of Triple-Negative Breast Cancer Cell Metabolism and Pathogenicity. Cell Chem Biol 23, 567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magtanong L, Ko PJ, and Dixon SJ (2016). Emerging roles for lipids in non-apoptotic cell death. Cell Death Differ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashek DG, Bornfeldt KE, Coleman RA, Berger J, Bernlohr DA, Black P, DiRusso CC, Farber SA, Guo W, Hashimoto N, et al. (2004). Revised nomenclature for the mammalian long-chain acyl-CoA synthetase gene family. J. Lipid Res 45, 1958–1961. [DOI] [PubMed] [Google Scholar]
- Nguyen TB, Louie SM, Daniele JR, Tran Q, Dillin A, Zoncu R, Nomura DK, and Olzmann JA (2017). DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev. Cell 42, 9–21. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padanad MS, Konstantinidou G, Venkateswaran N, Melegari M, Rindhe S, Mitsche M, Yang C, Batten K, Huffman KE, Liu J, et al. (2016). Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep 16, 1614–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pap EH, Drummen GP, Winter VJ, Kooij TW, Rijken P, Wirtz KW, Op den Kamp JA, Hage WJ, and Post JA (1999). Ratio-fluorescence microscopy of lipid oxidation in living cells using C11-BODIPY(581/591). FEBS Lett. 453, 278–282. [DOI] [PubMed] [Google Scholar]
- Parisi LR, Li N, and Atilla-Gokcumen GE (2017). Very Long Chain Fatty Acids Are Functionally Involved in Necroptosis. Cell Chem Biol 24, 1445–1454. e1448. [DOI] [PubMed] [Google Scholar]
- Psychogios N, Hau DD, Peng J, Guo AC, Mandai R, Bouatra S, Sinelnikov I, Krishnamurthy R, Eisner R, Gautam B, et al. (2011). The human serum metabolome. PLoS ONE 6, e16957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S, Javaid S, Coletti ME, Jones VL, Bodycombe NE, et al. (2015). Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol 12, 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaud PP, Poirier SJ, Boudreau LH, Doiron JA, Barnett DA, Boilard E, and Surette ME (2016). On the cellular metabolism of the click chemistry probe 19-alkyne arachidonic acid. J. Lipid Res 57, 1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Râdmark O, Wurst W, et al. (2008). Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 8, 237–248. [DOI] [PubMed] [Google Scholar]
- Soupene E, and Kuypers FA (2008). Mammalian long-chain acy-CoA synthetases. Exp. Biol. Med. (Maywood) 233, 507–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK, Kagan VE, et al. (2017). Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torii S, Shintoku R, Kubota C, Yaegashi M, Torii R, Sasaki M, Suzuki T, Mori M, Yoshimoto Y, Takeuchi T, et al. (2016). An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J 473, 769–777. [DOI] [PubMed] [Google Scholar]
- Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X, et al. (2018). Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 33, 890–904. e895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiwer M, Bittker J, Lewis TA, Shimada K, Yang WS, MacPherson L, Dandapani S, Palmer M, Stockwell BR, Schreiber SL, et al. (2012). Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg. Med. Chem. Lett 22, 1822–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Liu H, Chen W, Fujimoto Y, and Liu J (2009). Hepatic expression of long-chain acyl-CoA synthetase 3 is upregulated in hyperlipidemic hamsters. Lipids 44, 989–998. [DOI] [PubMed] [Google Scholar]
- Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, and Stockwell BR (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. U.S.a 113, E4966–E4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao C-H, Fowle-Grider R, Mahieu NG, Liu G-Y, Chen Y-J, Wang R, Singh M, Potter GS, Gross RW, Schaefer J, et al. (2016). Exogenous Fatty Acids Are the Preferred Source of Membrane Lipids in Proliferating Fibroblasts. Cell Chem Biol 23, 483–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RM, Ackerman D, Quinn ZL, Mancuso A, Gruber M, Liu L, Giannoukos DN, Bobrovnikova-Marjon E, Diehl JA, Keith B, et al. (2013). Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev. 27, 1115–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For the 261-member bioactive compound screen in HT-1080N cells (both ethanol only and oleic acid (OA)-treated) the calculated area under the curve of lethal fraction scores are available online via the Mendeley Data repository https://data.mendeley.com/datasets/g2xhxht7cw/1.