

Abstract
Na+/H+ exchanger (NHE1) activation is required for multiple microglial functions. We investigated effects of selective deletion of microglial Nhe1 in Cx3cr1-CreER;Nhe1f/f mice on neuroinflammation and tissue repair after ischemic stroke. Infarct volume was similar in corn oil or tamoxifen (Tam)-treated mice at 48 hr and 14 days post-stroke. However, the Tam-treated mice showed significantly higher survival rate and faster neurological function recovery during day 1–14 post-stroke. Deletion of microglial Nhe1 prevented the elevation of CD11b+/CD45low-med microglia in the ischemic hemisphere at day 3 post-stroke, but stimulated expression of Ym1, CD68, TGF-β, IL-10, decreased expression of CD86 and IL-1β, and reduced GFAP+ reactive astrocytes. Moreover, at day 14 post-stroke, enhanced white matter myelination was detected in the microglial Nhe1 deleted mice. In comparison, neuronal Nhe1-null mice (the CamKII-Cre+/−;Nhe1f/f mice) showed a significant reduction in both acute and subacute infarct volume, along with increased survival rate and moderate neurological function recovery. However, these neuronal Nhe1-null mice did not exhibit reduced activation of CD11b+/CD45low-med microglia or CD11b+/CD45hi macrophages in the ischemic brains, and they exhibited no reductions in white matter lesions. Taken together, this study demonstrated that deletion of microglial and neuronal Nhe1 had differential effects on ischemic brain damage. Microglial NHE1 is involved in pro-inflammatory responses during post-stroke brain tissue repair. In contrast, neuronal NHE1 activation is directly associated with the acute ischemic neuronal injury but not inflammation. Our study reveals that NHE1 protein is a potential therapeutic target critical for differential regulation of ischemic neuronal injury, demyelination and tissue repair.
Keywords: inflammation, macrophages, microglia, phagocytosis, white matter tissue repair
1 |. INTRODUCTION
Microglia are resident macrophages of the central nervous system (CNS; Goldmann et al., 2016; Prinz & Priller, 2014), which structurally and functionally interact with both neuronal and non-neuronal cells in the healthy brain (Frost & Schafer, 2016; Kettenmann, Kirchhoff, & Verkhratsky, 2013; Schafer, Lehrman, & Stevens, 2013; Tremblay et al., 2011). Microglia actively survey the CNS to maintain normal CNS homoeostatic activity by phagocytosis of synaptic structures and newborn neurons during postnatal development and adult neurogenesis, or by remodeling of the peri-synaptic environment in mature and aging brains (Brown & Neher, 2014; Mosser, Baptista, Arnoux, & Audinat, 2017; Nayak, Roth, & McGavern, 2014; Paolicelli & Gross, 2011; Schafer et al., 2013; Tremblay, 2011; Wake, Moorhouse, Miyamoto, & Nabekura, 2013; Wu, Dissing-Olesen, MacVicar, & Stevens, 2015). Recently, microglia were found to regulate learning and learning-dependent synaptic remodeling by releasing brain-derived neurotrophic factor (BDNF) (Parkhurst et al., 2013). In addition, microglia mediate the main innate immune response in the brain and are activated following various pathological conditions including stroke (Gomez-Nicola & Perry, 2015). Following cerebral ischemia, microglia are rapidly activated, causing secondary injury by releasing inflammatory cytokines. However, alternatively activated microglia can secrete anti-inflammatory cytokines and neurotrophic factors that prevent neuronal apoptosis and promote tissue repair (Ma, Wang, Wang, & Yang, 2016; Michell-Robinson et al., 2015; Xiong, Liu, & Yang, 2016). However, the regulatory mechanisms in modulating the microglial inflammatory and adaptive functions remain unclear.
Na+/H+ exchanger isoform 1 (NHE1) belongs to a family of membrane transporter proteins that conduct the electroneutral transport of H+ efflux in exchange with Na+ influx (Jean, Frelin, Vigne, Barbry, & Lazdunski, 1985; Orlowski & Grinstein, 2004). NHE1 plays an important role in regulating microglial intracellular pH (pHi) homeostasis, their activation, and migration (Boscia et al., 2016; Liu et al., 2010; Shi et al., 2013; Zhao et al., 2016; Zhu et al., 2016). In particular, NHE1 maintains optimal pHi to support NADPH oxidase (NOX) function during microglial activation (Lam et al., 2013). Pharmacological inhibition of NHE1 reduces cultured microglial activation following various pro-inflammatory stimuli (Liu et al., 2010). Moreover, global genetic knockdown of NHE1 protein in mice or treatment with an NHE1 inhibitor (HOE642) inhibits microglial activation and pro-inflammatory responses in brain tissues after transient ischemic stroke (Shi, Chanana, Watters, Ferrazzano, & Sun, 2011). However, these studies targeting NHE1 in all cell types of the CNS cannot reveal the specific roles of NHE1 in microglial activation and function in ischemic brains.
In this study, we established an Nhe1f/f mouse line and crossed it with a microglia-specific chemokine fractalkine receptor Cx3cr1 genecontrolled Cre recombinase mouse line Cx3cr1-CreERT2-IRES-EYFP (Cx3cr1-CreER). We determined that specific deletion of Nhe1 in the Cx3cr1-CreER;Nhe1f/f mice did not reduce acute ischemic infarct; however, it prevented the elevation of CD11b+/CD45low-med microglia, reduced pro-inflammatory cytokine expression, and increased anti-inflammatory as well as phagocytic phenotypes after ischemic stroke. Microglial Nhe1-null mice showed accelerated tissue repair and faster recovery of post-stroke neurological function. In comparison, the CamKII-Cre+/−;Nhe1f/f mice (neuronal Nhe1-null) exhibited reduced acute infarct and improved functional recovery despite the sustained activation of microglia and macrophages during ischemic insult. These findings strongly suggest that neuronal and microglial NHE1 play different roles in the development of acute ischemic brain injury and post-stroke tissue repair.
2 |. MATERIALS AND METHODS
2.1 |. Materials
Tamoxifen and corn oil were from Sigma (St. Louis, MO, USA). Silicone-coated suture was from Doccol (Redlands, CA, USA). 2,3,5-triphenyltetrazolium chloride (TTC) was from Sigma. For flow cytometry, rat monoclonal anti-CD11b conjugated with APC (Invitrogen, Waltham, CA), rat monoclonal anti-CD45 conjugated with FITC (Invitrogen), rat monoclonal anti-CD16/32 conjugated with V450 (Affymetrix eBioscience, San Diego, CA, USA), rat monoclonal anti-CD206 conjugated with PE-Cy7 (Affymetrix eBioscience), rat monoclonal anti-CD86 conjugated with Axela 700 (BD Bioscience, San Jose, CA), rabbit monoclonal anti- Ym1 conjugated with PE (Abcam, Cambridge, MA), rat monoclonal anti-CD68 conjugated with PerCP-Cy5.5 (BioLegend, San Diego, CA), rat monoclonal anti-CD11b conjugated with PE (BD Biosciences, Franklin Lakes, NJ), rat monoclonal anti-TNF-α conjugated with BV605 (BioLegend), rat monoclonal anti-TGF-β conjugated with BV421 (BioLegend), rat monoclonal anti-IL-1β conjugated with FITC (Thermo Scientific, Waltham, MA) and rat monoclonal anti-IL-10 conjugated with PE-Cy7 (BioLegend) antibodies were used. Beads for compensation were from Affymetrix eBioscience. DMEM (Corning, NY), Anti-Anti (Catalog# 15240–062, Gibco, Gaithersburg, MD), GolgiPlug, GolgiStop, Cytofix/Cytoperm solution and Perm/Wash solution (BD Biosciences) were used for intracellular cytokine staining. For immunofluorescent staining, mouse monoclonal anti-glial fibrillary acidic protein (GFAP; Cell Signaling Technology, Danvers, MA), rabbit polyclonal anti-ionized calcium-binding adapter molecule 1 (IBA1; Wako, Richmond, VA), mouse monoclonal anti-adenomatous polyposis coli (APC; EMD Millipore, Billerica, MA), rabbit monoclonal anti-myelin basic protein (MBP; Abcam), mouse monoclonal anti-MAP2 (EMD Millipore) and rabbit monoclonal anti-NeuN (Abcam) primary antibodies and goat polyclonal anti-mouse IgG conjugated with Alexa Fluro 488 (Invitrogen), goat polyclonal anti-mouse IgG conjugated with Alexa Fluro 546 (Thermo Fisher Scientific), goat polyclonal anti-rabbit IgG conjugated with Alexa Fluro 546 (Invitrogen) and goat polyclonal anti-rabbit IgG conjugated with Alexa Fluro 488 (Thermo Fisher Scientific) secondary antibodies were used. To-Pro-3 was from Thermo Fisher Scientific. Vectashield mounting medium for fluorescence was from Vector Laboratories (Burlingame, CA).
2.2 |. Animals
All animal experiments and procedures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
2.2.1 |. Generation and maintenance of Nhe1flox/+ mice
Generation of heterozygous Nhe1flox/+ mice, with one LoxP site inserted in the intron upstream of exon 5 and the other LoxP site in the intron downstream of exon 5 was reported recently (Begum et al., 2017). Genotypes of the floxed mice were determined by PCR analysis of DNA from tail biopsies. For PCR analysis, forward (5′-CTG CTG CAT TCT CTA TCT TAC TC-3′, VS2513) and reverse (5′-GTT TCG AAG TGT AGG CTG TGA G-3′, VS2514) primers were used to amplify a 280-base pair product from the wild-type Nhe1+ gene or a 406-base pair product from the mutant Nhe1flox gene (Supporting Information Figure 1b). In parallel, forward (5′- GTC AAT CAG TAT ATG AAG TGA CG-3′, VS2511) and reverse (5′-GAA CTG CTC GAC ATA TGA TAA C-3′, VS25155) primers were used to amplify a 150-base pair product from the mutant to confirm the genotype. PCR amplification was performed using a thermocycler as follows: 94°C for 2 min, 30 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min, and 72°C for 3 min. PCR products were run and revealed on a 2% agarose gel.
2.2.2 |. Maintenance of Cx3cr1-CreERT2-IRES-EYFP mouse colony
Cx3cr1-CreERT2-IRES-EYFP (Cx3cr1-CreER) transgenic mouse line (B6.129P2 (Cg)-Cx3cr1tm2.1(cre/ERT2)Litt/WganJ, Stock# 021160, Jackson Laboratory, Bar Harbor, ME) was established as described previously (Parkhurst et al., 2013). For Cx3cr1-CreER PCR analysis, two pairs of primers were used. Forward (5′-GAA CTA CAA TCC TTT AAG GCT CAC G-3′) and reverse (5′-GCA GGA CCT CGG GGT AGT CAC-3) primers were used to amplify a 577-base pair product from the Cx3cr1 wild-type allele, and forward (5′-GAA CTA CAA TCC TTT AAG GCT CAC G-3′) and reverse (5′-CAC CAG AGA CGG AAA TCC ATC G-3′) primers were used to amplify a 757-base pair product from the CreER transgenic allele, as described previously (Parkhurst et al., 2013; Supporting Information Figure 1b).
2.2.3 |. Generation of conditional microglia-specific Nhe1 knockout in Cx3cr1-CreER;Nhe1f/f mice
To obtain microglia-specific Nhe1-null mice, Cx3cr1-CreER mice were bred with Nhe1f/f mice for two generations to obtain Cx3cr1-CreER;Nhe1f/f mice (Supporting Information Figure 1a). Mice (male or female) at postnatal day 30–40 (P30–40) were randomly assigned to receive either tamoxifen (Tam, Sigma; 75 mg/kg body weight/day at a concentration of 20 mg/ml in corn oil, intraperitoneally) for five consecutive days to induce nuclear translocation and expression of the Cre recombinase, or corn oil (Sigma; 3.75 ml/kg body weight/day, i.p.) as a vehicle control. A 30-day post-injection waiting period (Figure 1a) was used for clearance of Tam (Ajami, Bennett, Krieger, Tetzlaff, & Rossi, 2007; Fogg et al., 2006; Valny, Honsa, Kirdajova, Kamenik, & Anderova,2016) and for replenishing of Cx3cr1+ monocytes (Parkhurst et al., 2013) prior to induction of ischemic stroke.
FIGURE 1.
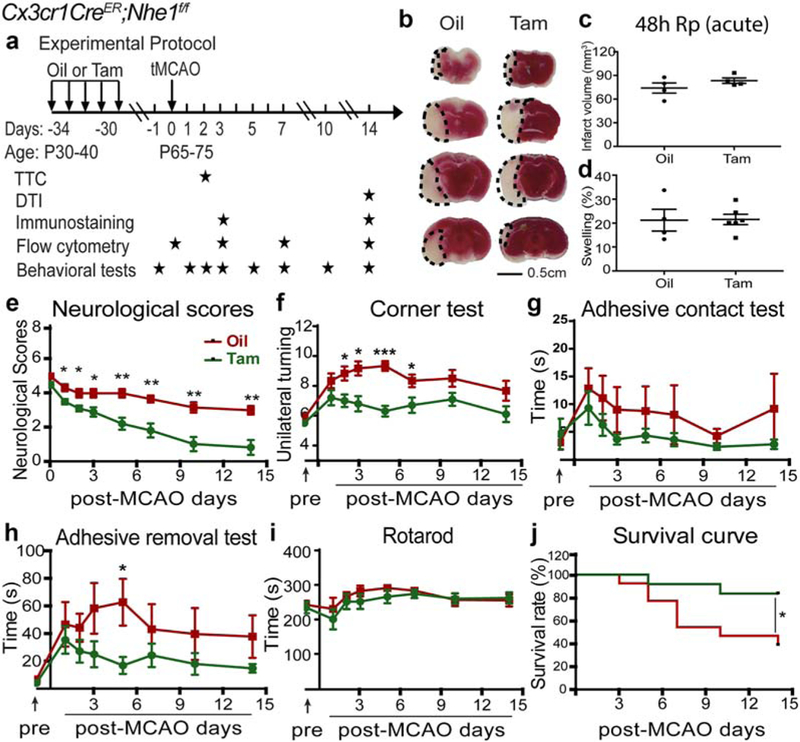
Specific deletion of Nhe1 in Cx3cr1-CreER;Nhe1f/f mice had no effects on acute ischemic infarct formation but significantly improved neurological functions after ischemic stroke. (a) Experimental protocol. Either corn oil (3.75 ml/kg) or tamoxifen (Tam, 75 mg/kg body weight, 20 mg/ml in corn oil, i.p.) was administered daily for consecutive 5 days in Cx3cr1-CreER;Nhe1f/f mice at postnatal day 30–40 (P30–40). A 30-day waiting period was given for complete clearance of Tam and peripheral Cx3cr1+ monocyte turnover. At P65–75, transient focal cerebral ischemia in mice was induced by 50 min of middle cerebral artery occlusion (tMCAO). 2,3,5-Triphenyltetrazolium chloride (TTC) staining was conducted at 48 hr reperfusion (Rp). Ex vivo DTI imaging was conducted at 14 days Rp. Immunostaining was conducted at 3 and 14 days Rp. Flow cytometry experiments were conducted at 0 (baseline), 3, 7, and 14 days Rp. Behavioral tests were conducted at 1 day prior to tMCAO, and at 1, 2, 3, 5, 7, 10, and 14 days Rp. (b) TTC staining of the oil- or Tam-treated Cx3cr1-CreER;Nhe1f/f mice at 48 hr Rp. (c and d) Infarct volume and brain swelling in mice at 48 hr Rp with TTC staining. Data are mean ± SEM. N = 4–6. (e) Neurological score of mice after tMCAO. Data are mean ± SEM. NTam = 10 (5 males, 5 females) and NOil = 6 (1 male, 5 females). *p < .05, **p < .01, oil- versus Tam-treated groups. (f-i) Corner test, adhesive contact test, adhesive removal test, and rotarod accelerating test of the same cohort of mice in e. Data are mean ± SEM. *p < .05, ***p < .001, oil- versus Tam-treated groups. (j) Survival curve of the same cohort of mice in e. Data are mean ± SEM. *p < .05, oil- versus Tam-treated groups [Color figure can be viewed at wileyonlinelibrary.com]
2.2.4 |. Generation of neuron-specific Nhe1-null mice
CamkIIa-Cre+/− transgenic mice (B6.Cg-Tg(Camk2a-cre)T29–1Stl/J, Stock# 005359, The Jackson Laboratory) were bred with Nhe1f/f mice for two generations to obtain CamKIIa-Cre+/−;Nhe1f/f (neuron-specific Nhe1-null) mice (Supporting Information Figure 1d), while CamKIIa-Cre+/− and/or Nhe1f/f mice were used as controls. For CamKIIa-Cre+/− PCR analysis, forward (5′-GCG GTC TGG CAG TAA AAA CTA TC-3′) and reverse (5′-GTG AAA CAG CAT TGC TGT CAC TT-3′) primers were used to amplify a 100-base pair product from the Cre+ transgenic allele (Orefice et al., 2016), and forward (5′-CTA GGC CAC AGA ATT GAA AGA TCT-3′) and reverse (5′-GTA GGT GGA AAT TCT AGC ATC ATC C-3′) primers from the IL-2 gene were used to amplify a 324-base pair product that served as an internal positive control (Cullen et al., 2011).
2.3 |. Transient focal ischemia model
Focal cerebral ischemia was induced by occlusion of the left middle cerebral artery (MCA) as described before (Shi et al., 2011). Briefly, mice were kept under 1.5% isoflurane anesthesia during the procedure and the core temperature (37.0°C) was maintained by a homoeother-mic blanket system. After midline skin incision, the left common carotid artery was exposed and the superior thyroid artery and occipital artery branches of the external carotid artery were isolated and coagulated. The animals were subjected to MCA occlusion (MCAO) by introduction of a silicone-coated suture (6–0 monofilament nylon, Doccol) inserted via the external carotid artery. Reperfusion was established by with-drawal of the filament after 60 min of transient MCAO (tMCAO). The incision was closed and the mice were allowed to recover under a heating lamp to maintain the core temperature (36°C-37°C) during a 30–60 min recovery period. After recovery, the animals were returned to their cages with free access to food and water. Sham controls under-went the same surgical procedures without introducing tMCAO.
2.4 |. Neurological function tests
Neurological functional deficits in mice were screened in a blinded manner with the following tests: neurological score, corner test, adhesive contact test, adhesive removal test, and rotarod test, all considered reliable for identifying and quantifying sensorimotor deficits and postural asymmetries. Animals that died during 14 days post-MCAO were excluded from the study.
2.4.1 |. Neurological score
A neurological deficit grading system (Xia et al., 2006; Zheng, Liu, Li, Xu, & Xu, 2009) was used to evaluate neurological deficit at 1, 2, 3, 5, 7, 10, and 14 days after tMCAO. The scores are as follows: 0: no neurological deficit; 1: forelimb flexion when suspended by the tail or failure to extend forepaw fully; 2: shoulder adduction when suspended by the tail; 3: reduced resistance to lateral push; 4: spontaneous movement in all directions with unilateral circling exhibited only if pulled by tail; 5: spontaneous unilateral circling; 6: walk only when stimulated; 7: no response to stimulation; 8: stroke-related death.
2.4.2 |. Corner test
Neurological functional deficits in mice were also determined by the corner test, as described before (Zhao et al., 2017). Corner tests were conducted prior to tMCAO and at 1, 2, 3, 5, 7, 10, and 14 days after tMCAO. The apparatus consisted of two cardboards (30 × 20 × 1 cm each) placed together at a 30° angle to form a narrow alley. The mouse was placed between the two angled cardboards facing the corner. When exiting the corner, uninjured mice would turn left or right with even probability. After tMCAO, animals with unilateral brain damage would exhibit unidirectional turning. The numbers of unilateral turning of each mouse during 10 trials were recorded, and turning movements that were not part of a rearing movement were not recorded. Pre-operative training was carried twice per day for consecutive three days.
2.4.3 |. Adhesive test
The adhesive contact test and the adhesive removal test were used to measure somatosensory deficits as described previously (Zhao et al., 2017). Animals were tested prior to tMCAO and at 1, 2,3, 5, 7,10, and 14 days after tMCAO. Two pieces of adhesive tape (4 × 3 mm) were attached to the forepaws in an alternating sequence and with equal pressure by the experimenter before each trial. The contact time was defined as the time at which the animal first made contact with the tape, and the removal time was defined as the time at which the animal removed the tape. The trial ended after the adhesive patch was removed or after 2 min had elapsed. Pre-operative training was carried twice per day for consecutive three days.
2.4.4 |. Rotarod test
The rotarod test was used to assess motor coordination and balance alterations, as described before (Zhao et al., 2017). Animals were tested prior to tMCAO and at 1, 2, 3, 5, 7, 10, and 14 days after tMCAO. The apparatus consisted of a black striated rod separated in four compartments (Model 755; IITC Life Science Inc., Woodland Hills, CA). Animals were first habituated to a stationary rod for 2 min and then placed on a rotating drum accelerating from 4 to 40 r/min over a 5-min period. The time the animal stayed on the drum was recorded. Three trials were performed with 15-min interval resting periods. The time at which the animal fell off the drum was recorded. Pre-operative training was carried twice per day for consecutive three days.
2.5 |. 2,3,5-Triphenyltetrazolium chloride (TTC) staining
At 48 hr post-tMCAO, mice were euthanized with 5% isoflurane vaporized in N2O and O2 (3:2) and decapitated. The brains were dissected and cut into four coronal slices of 2 mm thickness. Sections were stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC, Sigma) at 37°C for 15 min. Infarct volume was calculated using image analysis software Image J (NIH) with correction for edema as described by Swanson (Swanson et al., 1990). Briefly, the ischemic area for each slice was calculated by subtracting the non-infarct area in the ipsilateral (IL) hemisphere from the total area of the contralateral (CL) hemisphere. For swelling assessment during acute phase, percentage hemispheric swelling (%HSw) is determined by the difference in volume between two hemispheres and then divided by the CL hemispheric volume according to a formula: %HSw = (IL volume - CL volume)/CL volume × 100, as described before (Kim et al., 2015; Tan et al., 2013).
2.6 |. MRI and diffusion tensor imaging (DTI) of ex vivo brains
At 14 days post-tMCAO, mice were anesthetized with 5% isoflurane, transcardially perfused with 0.1 M PBS (pH 7.4), followed by ice-cold 4% paraformaldehyde (PFA) in 0.1 M PBS, and decapitated, as described before (Begum et al., 2017). Brains were maintained within the skull to avoid anatomical deformation. After post-fixation in 4% PFA overnight, heads were stored in PBS solution at 4°C. Magnetic Resonance Imaging (MRI) was performed at 500 MHz using a Bruker AV3HD 11.7 T/89 mm vertical bore small animal MRI scanner, equipped with a 20-mm quadrature radiofrequency (RF) coil and Paravision 6.0 software (Bruker Biospin, Billerica, MA). Following positioning and pilot scans, T2-weighted images (T2WI) were acquired using a Rapid Acquisition with Relaxation Enhancement (RARE) sequence, with the following parameters: Time of Echo/Time of Repetition (TE/TR) = 20/3,500 ms, averages = 2, 256 × 256 matrix, 25 slices with a 1 mm slice thickness, a RARE factor = 4, and a field of view (FOV) of 22 × 22 mm. A Diffusion Tensor Imaging (DTI) data set covering the entire brain was collected using a multislice spin echo sequence with three reference and 30 non-collinear diffusion-weighted images with the following parameters: TE/TR = 22/5,000 ms, four averages, matrix size = 192 × 192 reconstructed to 256 × 256, field of view = 22 × 22 mm, 25 axial slices, slice thickness = 1 mm, b-value = 1,200 s/mm2, and Δ/δ = 10/5 ms. DTI and T2 datasets were analyzed with DSI Studio (http://dsi-studio.labsolver.org/). In a blinded manner, region of interests (ROIs) were drawn segmenting corpus callosum (CC) and external capsule (EC) in both the CL and IL hemispheres. Values of fractional anisotropy (FA) were calculated for each ROI, as described before (Yin et al., 2017; Zhao et al., 2017).
2.7 |. Flow cytometry analysis of microglia/macrophage cells
Flow cytometry was conducted to investigate changes in CDllb+/CD45+ cell counts, polarization, and intracellular cytokines in brains without tMCAO or at 3, 7, or 14 days post-tMCAO. Animals were deeply anesthetized with 5% isoflurane and transcardially perfused with ice-cold saline. After removal of cerebellum and meninges, CL and IL hemispheric cortical tissues were cut into small pieces and dissociated into a single cell suspension by gentle physical disruption and enzymatic digestion using a tissue dissociation kit according to manufacturer’s instructions (Miltenyi Biotech Inc, Bergisch Gladbach, Germany). The single cell suspension sample was centrifuged through 30/70 Percoll (GE Healthcare, Little Chalfont, UK) gradient solution at 500g for 30 min to remove myelin. Brain cells were then collected from the middle layer and washed twice with HBSS containing 1% fetal bovine serum (FBS).
For CD11b+/CD45+ cell counts and polarization, cells (~106 cells) in 0.1 ml HBSS containing 1% FBS were incubated and labeled with APC-conjugated CD11b (1:400, Invitrogen), FITC-conjugated CD45 (1:400, Invitrogen), V450-conjugated CD16/32 (1:400, Affymetrix eBioscience), PE-Cy7-conjugated CD206 (1:400, Affymetrix eBioscience), Alexa 700-conjugated CD86 (1:400, BD Bioscience), PE-conjugated Ym1 (1:400, Abcam), and PerCP-Cy5.5-conjugated CD68 (1:400, BioLegend) antibodies for 30 min at 4°C in the dark. Cells were then rinsed with HBSS containing 1% FBS by centrifugation for 3 min at 2200 rpm and pellets were re-suspended in 300 μl of HBSS containing 1% FBS before flow cytometry analysis of the samples was acquired immediately with a LSR Fortessa flow cytometer (BD Biosciences) running FACS Diva software (BD Biosceiences) with the following settings: Forward scatter (FSC) V = 600, mode = Lin; Side scatter (SSC) V = 275, mode = Lin; APC V = 400, mode = Log; FITC V = 467, mode = Log; V450 V = 509, mode = Log; PE-Cy7 V = 358, mode = Log; Alexa 700 V = 623, mode = Log; PE V = 484, mode = Log; PerCP-Cy5.5 V = 574, mode = Log. In each experiment, at least 50,000 events were recorded from each hemispheric sample for analysis.
For intracellular cytokine staining, cells (~106 cells) in 0.1 ml HBSS containing 1% FBS were incubated and surface labeled with PE-conjugated CD11b (1:200, BD Biosciences) antibody for 30 min at 4°C in the dark. Cells were then washed twice and incubated in DMEM supplemented with 10% FBS, 1× Antibiotic-Antimycotic (Gibco), 2 μ/ml GolgiPlug and 1 μ/ml GolgiStop (BD Biosciences) for 4 hr at 37°C in the dark, allowing accumulation of intracellular cytokines by Golgi activity inhibition, as described before (Nikodemova & Watters, 2012). Cells were then permeablized and fixed with Cytofix/Cytoperm solution (BD Biosciences) for 20 min at 4°C and labeled with BV421-conjugated TGF-β (1:200, BioLegend), BV605-conjugated TNF-α (1:200, BioLegend), FITC-conjugated IL-1β (1:200, Thermo Scientific), and PE-Cy7-conjugated IL-10 (1:200, BioLegend) intracellular antibodies for 30 min at 4°C in the dark. Flow cytometry analysis of the samples was acquired with the following settings: Forward scatter (FSC) V = 600, mode = Lin; Side scatter (SSC) V = 275, mode = Lin; PE V = 450, mode = Log; BV421 V = 400, mode = Log; BV605 V = 450, mode = Log; FITC V = 500, mode = Log; PE-Cy7 V = 400, mode = Log. In each experiment, at least 10,000 events were recorded from each hemispheric sample for analysis.
2.8 |. Immunofluorescent staining
Mice were deeply anesthetized with 5% isoflurane and transcardially perfused with 0.1 M PBS (pH 7.4), followed by ice-cold 4% PFA in 0.1 M PBS as described before (Begum et al., 2015). Brains were cryo-protected with 30% sucrose after an overnight post-fixation in 4% PFA (Begum et al., 2017). Coronal sections (30 μm, at the level of 0.26 mm posterior to Bregma) were selected and processed for immunofluorescent staining. The sections were incubated with blocking solution (10% normal goat serum and 0.3% Triton X-100 in PBS) for 1 hr at room temperature and were then incubated with mouse monoclonal anti-GFAP (1:200, Cell Signaling Technology) and rabbit polyclonal anti-IBA1 (1:200, Wako, Italy) antibodies, or mouse monoclonal anti-MAP2 (1:200, EMD Millipore) and rabbit monoclonal anti-NeuN (1:200, Abcam) antibodies, or mouse monoclonal anti-APC (1:100, EMD Millipore) and rabbit monoclonal anti-MBP (1:100, Abcam) antibodies in the blocking solution for overnight at 4°C. After washing in TBS-Triton X-100 (0.3%) for 3 × 10 min, the sections were incubated with goat anti-mouse Alexa 546-conjuagated IgG (1:200, Thermo Fisher Scientific) and goat anti-rabbit Alexa 488-conjugated IgG (1:200, Thermo Fisher Scientific), or goat anti-mouse Alexa 488-conjuagated IgG (1:200, Invitrogen) and goat anti-rabbit Alexa 546-conjugated IgG (1:200, Invitrogen) in the blocking solution for 1 hr. For negative controls, brain sections were stained with the secondary antibodies only. After washing for three times, nuclei were stained with To-pro-3 (1:500, Thermo Fisher Scientific) for 15 min at 37°C. Sections were mounted with Vectashield mounting medium (Vector Laboratories). Fluorescent images were captured under 40× lens using a Leica DMIRE2 inverted confocal laser-scanning microscope (Leica, Germany). Identical digital imaging acquisition parameters were used and images were obtained and analyzed in a blinded manner throughout the study.
For MAP2 staining used for measurement of infarct volume, brain sections at four different levels (0.62,0.14, −0.34, −0.82 mm posterior from Bregma) from each brain were selected and stained with mouse monoclonal anti-MAP2 (1:200, EMD Millipore) antibody, as described before (Dawson & Hallenbeck, 1996; Wang et al., 2015). Mouse Alexa 488-conjuagated IgG (1:200, Invitrogen) antibody was used as secondary antibody. Fluorescent images were captured under 4× lens using a Nikon Eclipse Ti epifluorescent microscope (Nikon, Tokyo, Japan) and processed with NIS-Elements Advance Research microscope imaging software (version 4.30.02, Nikon). The ischemic area for each slice was calculated by subtracting the non-infarct area in the IL hemisphere from the total area of the contralateral CL hemisphere (Swanson et al., 1990). For atrophy assessment during the subacute phase, percentage hemispheric shrinkage (%HSh) is determined by the difference in volume between two hemispheres and then divided by the CL hemispheric volume according to a formula: %HSh = (IL volume – CL volume)/CL volume × 100, as described before (Kim et al., 2015; Tan et al., 2013).
2.9 |. Statistical analysis
Data are expressed as mean ± SD or SEM. Statistical analysis was performed with Student’s t test in case of two-group comparisons and one-way or two-way ANOVA with post-hoc tests in case of multiple comparisons (Prism, GraphPad, La Jolla, CA). Non-parametric data were analyzed with Mann-Whitney Test (Prism, GraphPad). Correlation analysis was performed with Pearson correlation coefficient (Prism, GraphPad). p value < .05 was considered statistically significant.
3 |. RESULTS
3.1 |. Selective deletion of microglial Nhe1 in Cx3cr1-CreER;Nhe1f/f mice did not reduce acute ischemic infarction but improved survival and neurological function recovery
To obtain microglia-specific Nhe1-null mice, Cx3cr1-CreER;Nhe1f/f mice (male or female) at postnatal day 30–40 (P30–40) were randomly assigned to receive either tamoxifen (Tam) or oil treatment and transient middle cerebral artery occlusion (tMCAO) was induced at P65–75 (Figure 1a). The selective deletion of NHE1 protein in the IBA1+ cell populations was confirmed by immunofluorescent staining (Supporting Information Figure 1c). At 48 hr reperfusion (Rp) after tMCAO, oil-and Tam-treated mice exhibited similar infarct volume (82.8 ± 14.2 mm3 vs. 74.0 ± 6.4 mm3, p > .05) and hemispheric swelling (21.2% ± 4.5% vs. 21.5% ± 2.1%, p > .05; Figure 1b–d). Similar infarction and neuronal degeneration was detected in both oil- and Tam-treated mice at 48 hr post-MCAO using TTC and MAP2 staining (Supporting Information Figure 2). These data suggest that specific deletion of Nhe1 in microglia did not affect acute ischemic infarct formation. We then investigated whether the deletion of microglial Nhe1 in mice altered their post-stroke neurological functional recovery. An 8-scale neurological deficit grading system (Xia et al., 2006; Zheng et al., 2009) was used to evaluate neurological deficit during 1–14 days after tMCAO. The oil-treated mice showed poor neurological scores in the first week post-stroke and slightly improved to scores of 3–4 by day 7–14 post-stroke (Figure 1e). In contrast, the Tam-treated Cx3cr1-CreER;Nhe1f/f mice reduced the score to <2 (1.8 ± 0.4) by the end of the first week post-stroke, and to <1 by day 14 post-stroke (0.8 ± 0.4, p < .001).
We also conducted a panel of sensorimotor function tests in the same cohort of mice. In the corner test (Figure 1f), both oil-treated and Tam-treated mice showed no behavioral asymmetries prior to tMCAO and changed to unilateral turning at 1–14 days after tMCAO. However, the Tam-treated mice exhibited significantly less asymmetries than the oil-treated mice, especially during day 2–7 post-stroke (p < .001). In the adhesive tests (Figure 1g,h), there were no differences in baseline sensorimotor function between the oil-treated and Tam-treated mice prior to tMCAO. Both oil- and Tam-treated mice exhibited mild to moderate deficits in the contact test, without statistical differences between the two groups (Figure 1g). In contrast, ischemic stroke increased the tape removal time of the oil-treated mice at day 5 post-stroke by ~10-fold. The Tam-treated mice did not display such severe deficits throughout day 1–14 post-stroke (p < .05, Figure 1h). Both groups showed marked improvement by day 7–14 post-ischemia (Figure 1g,h). Interestingly, both oil- and Tam-treated mice displayed a relatively high latency to fall in the rotarod accelerating test and no significant differences in motor deficit was detected between the groups (Figure 1i). Figure 1jshows that the Tam-treated Cx3cr1-CreER;Nhe1f/f mice exhibited a significantly higher survival rate than the oil-treated mice at day 1–14 post-tMCAO (83.3% vs. 46.2%, p < .05). Taken together, microglial Nhe1-null mice showed robust improvement in sensorimotor function and survival after ischemic stroke, despite the large ischemic infarction.
3.2 |. Deletion of microglial Nhe1 blocked microglial CD11b+/CD45low-med stimulation and skewed pro-inflammatory responses to anti-inflammatory responses after ischemic stroke
Resident microglia express low-to-moderate levels of CD45 (Christensen, Woods, Carmody, Caughey, & Peterson, 2014; Greter, Lelios, & Croxford, 2015; Sedgwick et al., 1991). A very small population of cells expressing CD45high are present in the normal CNS, which normally represent as infiltrating macrophages (Sedgwick et al., 1991). As reported previously (Christensen etal., 2014; Martin, El-Behi, Fontaine, & Delarasse, 2017; Ritzel et al., 2015; Wong et al., 2017), CD11b+/ CD45low-med microglia and CD11b+/CD45hi macrophage populations were identified in a scatter plot (Figure 2a). Supporting Information Figure 3 shows that these microglial populations also expressed P2RY12, a special parenchymal microglial marker protein (Butovsky et al., 2014; Mildner, Huang, Radke, Stenzel, & Priller, 2017), with a strong positive correlation (a Pearson coefficient r of .9638) between CD11b+/CD45low-med and P2RY12+ cell counts (Supporting Information Figure 3b). Non-ischemic brains from the oil- and Tam-treated mice exhibited low numbers of CD11b+/CD45low-med microglia and low CD11b+/CD45hi macrophage counts in both contralateral (CL) and ipsilateral (IL) hemispheres (Figure 2b,c). But, at day 3 post-tMCAO, the oil-treated brains increased microglia by 2.6-fold and macrophage counts by 3.4-fold in the IL hemisphere (p < .01 and p < .05, respectively, Figure 2b, c). The microglial counts returned to the resting level by day 14 post-stroke, but the macrophage population remained elevated throughout day 1–14 post-stroke. In contrast, the Tam-treated brains showed an absence of elevation in the CD11b+/CD45low-med microglia population throughout 3–14 days after tMCAO (Figure 2b), but no decrease in the CD11b+/CD45hi macrophage counts (Figure 2c). The microglial activation was further evaluated by immunostaining for IBA1+ cells (Figure 2d,e). A few IBA1+ cells were detected in the CL hemispheres of either oil- or Tam-treated brains at day 3 post-stroke (data not shown). However, a significantly increased number of IBA1+ microglia were detected in the IL peri-lesion cortex and striatum areas of the oil-treated brains (Figure 2d,e). In contrast, the Tam-treated brains exhibited significantly less accumulation of IBA1+ microglia in the IL peri-lesion areas (Figure 2d,e). These data collectively demonstrate that microglial NHE1 protein is involved in the regulation of microglia activation in the ischemic brains.
FIGURE 2.
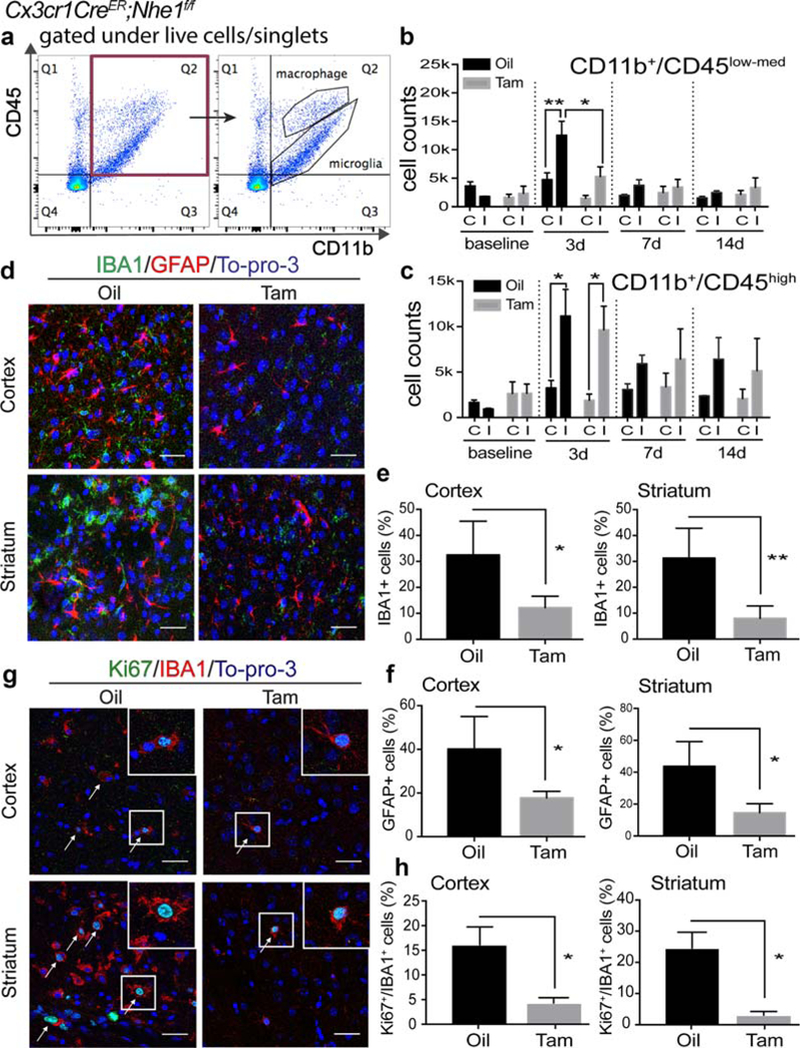
Tam-treated Cx3cr1-CreER;Nhe1f/f mice showed decreased number of microglia with less proliferation and reduced reactive astrocytes at the acute phase post-stroke. (a) Representative gating strategies of CD11b+/CD45low-med microglia and CD11b+/CD45hi macrophage populations. X-axis: number of CD11b+ cells. Y-axis: number of CD45+ cells. (b–c) Cell counts of CD11b+/CD45low-med microglia and CD11b+/CD45hi macrophages in the oil- and Tam-treated Cx3cr1-CreER;Nhe1f/f brains prior to tMCAO and at 3, 7, and 14 days Rp. A total cell count of 50,000 was recorded and analyzed. C, contralateral hemisphere; I, ipsilateral hemisphere. Data are mean ± SEM. At 0 day (baseline), 7 days and 14 days: N = 3; at 3 days: N = 9. *p < .05, **p < .01. (d) Representative immunofluorescent images of IBA1, GFAP, and To-pro-3 staining in the IL peri-lesion areas in the oil- and Tam-treated Cx3cr1-CreER;Nhe1f/f mice at 3 days post-tMCAO. (e) Quantitative analysis of IBA11 microglia in the IL peri-lesion areas in the cortex and striatum of the oil- and Tam-treated Cx3cr1-CreER;Nhe1f/f mice at 3 days post-tMCAO. Data are mean ± SEM. N = 4. *p < .05, **p < .01 oil- versus Tam-treated groups. (f) Quantitative analysis of GFAP1 astrocytes in the IL peri-lesion areas in the cortex and striatum of the oil- and Tam-treated Cx3cr1-CreER;Nhe1f/f mice at 3 days post-tMCAO. Data are mean ± SEM. N = 4. *p < .05, oil- versus Tam-treated groups. (g) Representative immunofluorescent images illustrate stroke-induced elevation of Ki67+/IBA1+ microglial cells in the IL hemispheres in the oi- but not in the Tam-treated Cx3cr1-CreER;Nhe1f/f mice at day 3 post-tMCAO. (h) Quantitative analysis of Ki67+/IBA1+ proliferative microglia populations. Data are mean ± SEM. N = 3. *p < .05, oil- versus Tam-treated groups [Color figure can be viewed at wileyonlinelibrary.com]
To determine that increased number of microglia in the IL hemispheres was due to proliferation, we assessed microglial proliferation by co-immunofluorescent staining for Ki67 expression in the IBA1 cells at day 3 post-stroke. Little expression of Ki67 was detected in the IBA1+ cells in the CL hemispheres of either oil- or Tam-treated brains (data not shown). However, the IBA1+/Ki67+ cell number was significantly increased in the oil-treated brains (Figure 2g,h). In contrast, no such an increase in the IBA1+/Ki67+ cell population was detected in the Tam-treated ischemic brains (Figure 2g,h). These data suggest that microglial NHE1 protein is involved in microglial activation and proliferation in the ischemic brains.
Moreover, GFAP+ astrocyte number was low in the CL hemispheres of either oil- or Tam-treated brains (data not shown). However, a significantly increased number of GFAP+ reactive astrocytes accumulated in the IL peri-lesion cortex and striatum areas of the oil-treated brains (Figure 2d,f). In contrast, the Tam-treated brains exhibited significantly less accumulation of GFAP+ reactive astrocytes in the IL peri-lesion areas (Figure 2d,f). These data suggest that deletion of Nhe1 in microglial cells reduces astrogliosis in the ischemic brains.
In the case of the CD11b+/CD45+ microglia/macrophage subpopulation analysis, the oil-treated brains showed a significant increase in the pro-inflammatory marker CD86 expression at day 3 post-stroke (3784.9 ± 1282.8, p < .05, Figure 3a,b), which remained high by day 7 post-stroke. In contrast, no significant elevation of CD86 expression was detected in the CD11b+/CD45+ microglia/macrophage populations of the Tam-treated brains (Figure 3a,b). In addition, the Tam-treated brains exhibited a 5.1-fold increase in expression of the anti-inflammatory marker Ym1 in the CD11b+/CD45+ microglia/macrophage at day 3 post-stroke, which was not detected in the oil-treated brains (p < .05, Figure 3a,b). For the phagocytic marker CD68, the Tam-treated brains showed a significant increase at day 3 postischemia (11162.7 ± 2480.3 vs. 4326.3 ± 777.5, p < .01), but no such change was detected in the oil-treated brains (p < .05, Figure 3a,b). However, similar increases in CD16/32 expression were detected in the CD11b+/CD45+ microglia/macrophage from both oil- and Tam-treated brains (Figure 3b). No changes in CD206 expression were found in either oil- or Tam-treated brains after ischemia (Figure 3b).
FIGURE 3.
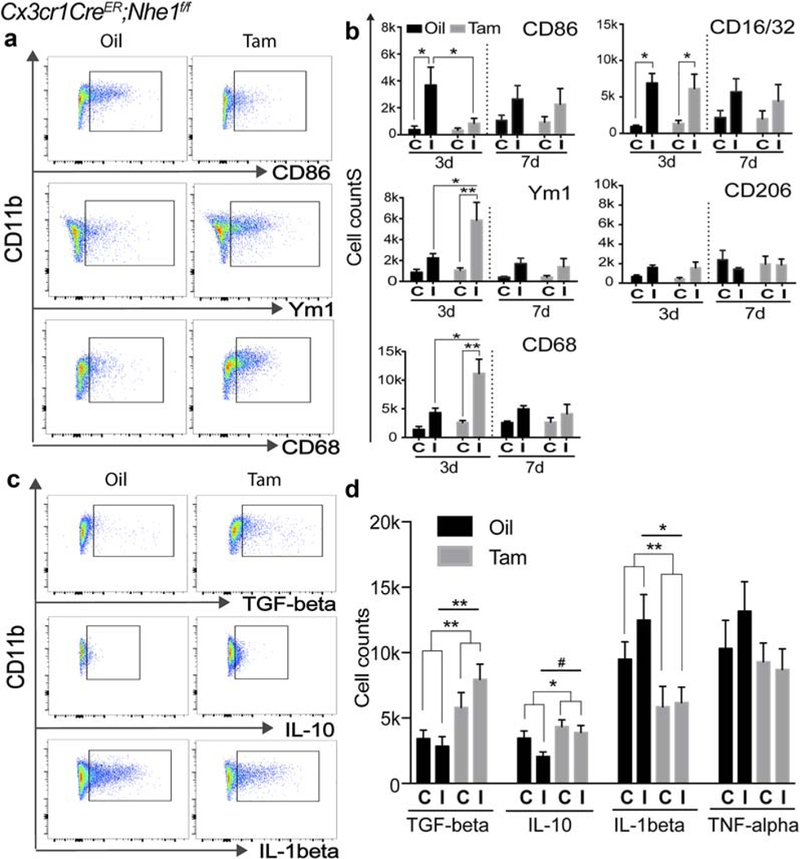
Tam-treated Cx3cr1-CreER;Nhe1f/f mice decreased pro-inflammatory responses and increased anti-inflammatory responses of microglia/macrophages in the acute stage after ischemic stroke. (a) Representative gating strategies for CD86+, Ym1+, and CD68+ cells within CD11b+ microglia/macrophage populations in oil- or Tam-treated Cx3cr1-CreER;Nhe1f/f brains. (b–f) Cell counts of CD86+, CD16/32+, Ym1+, CD206+ and CD68+ cells gated under CD11b+ cells in oil- and Tam-treated Cx3cr1-CreER;Nhe1f/f brains at 3 and 7 days Rp, respectively. A total cell count of 50,000 was recorded and analyzed. C, contralateral hemisphere; I, ipsilateral hemisphere. Data are mean ± SEM. At 3 days: N = 5; at 7 days: N = 3. *p < .05, **p < .01. (g) Representative gating strategies for TGF-β+, IL-10+, and IL-1β+ cells within CD11b+ microglia/macrophage populations in oil- or Tam-treated Cx3cr1-CreER;Nhe1f/f brains. (h) Cell counts of TGF-β+, IL-10+, IL-1β+, and TNF-α+ cells gated under CD11b+ cells in oil- and Tam-treated Cx3cr1-CreER;Nhe1f/f brains at 3 days Rp, respectively. At least a total cell count of 10,000 was recorded and analyzed. C, contralateral hemisphere; I, ipsilateral hemisphere. Data are mean ± SEM. N = 3. *p < .05, **p < .01, #p < .10 [Color figure can be viewed at wileyonlinelibrary.com]
We further investigated whether the decreased pro-inflammatory and increased anti-inflammatory profiles were associated with changes of cytokine release in CD11b+/CD45+ microglia/macrophages. Figure 3c,d illustrate that compared with the oil-treated brains at day 3 post-ischemia, the Tam-treated brains exhibited higher release of restorative TGF-beta (7957.5 ± 1168.7 vs. 2844.6 ± 734.8, p < .01), IL-10 (3896.5 ± 534.5 vs. 2050.3 ± 366.3, p < .10), and less detrimental IL-1beta (6166.7 ± 1184.7 vs. 12485.5 ± 1958.8, p < .05). The changes of TNF-alpha were not statistically significant. Similar trends of the changes were also detected in the CD11b+/CD45low-med/P2RY12+ cell population (Supporting Information Figure S4). These data strongly suggest that deletion of microglia Nhe1 selectively suppresses the pro-inflammatory responses and concurrently increases the anti-inflammatory responses in the microglial cells in ischemic brains.
3.3 |. Deletion of microglial Nhe1 promoted neuronal recovery and white matter repair during the subacute phase after ischemic stroke
Our ex vivo T2-Weighted Imaging (T2WI) study reveals that the Tam-treated brains showed similar infarction and brain atrophy as the oil-treated brains at day 14 post-stroke (Figure 4a,b). This finding is consistent with the MAP2 staining data obtained from the same cohort of ex vivo brains (Figure 4c,d). High intensity of MAP2 staining was detected in the non-ischemic CL hemispheres of both groups (Figure 4e). In the oil-treated brains, a drastic decrease in MAP2 intensity occurred in the IL ischemic core and peri-lesion cortex and striatum areas (p < .01 and p < .001, respectively, Figure 4e). In contrast, although the Tam-treated brains exhibited significantly decreased MAP2 intensity in the ischemic core region, the cortex and striatum peri-lesion areas showed significantly higher MAP2 protein levels than the oil-treated brains (p < .01 and p < .05, respectively, Figure 4e). These data strongly suggest that deletion of the microglial Nhe1 affects neuronal repair in ischemic brains.
FIGURE 4.
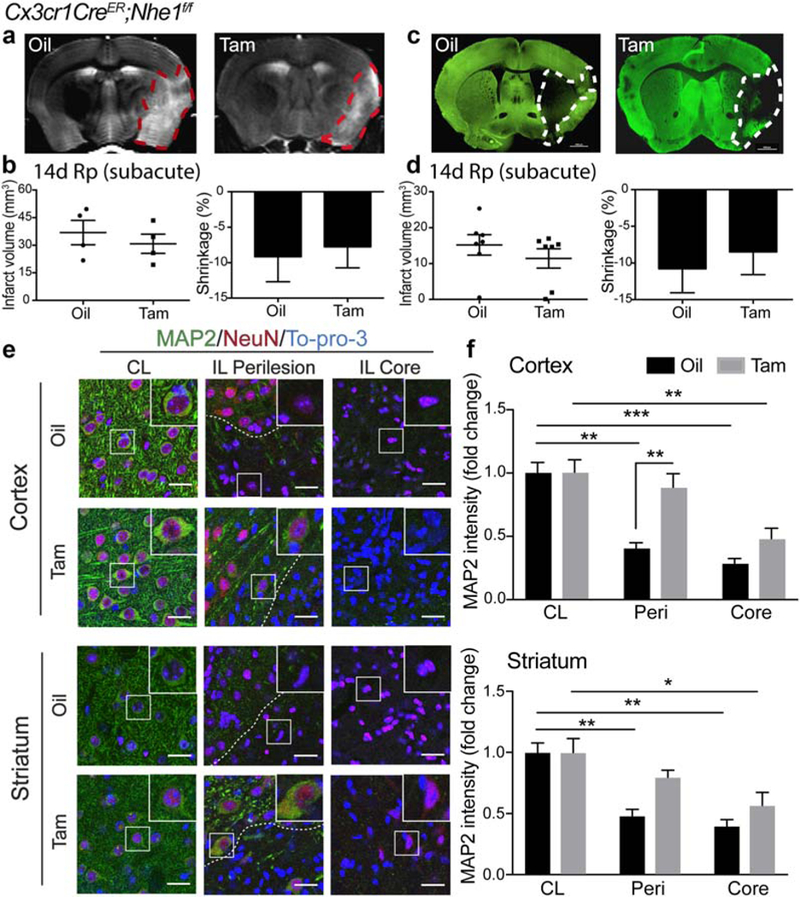
Tam-treated Cx3cr1-CreER;Nhe1f/f mice did not affect subacute infarction but reduced neuronal injury at the subacute phase postischemia. (a) Representative T2-weighted imaging (T2WI) of ex vivo oil- and Tam-treated Cx3cr1-CreER;Nhe1f/f brains at 14 days Rp. (b) Infarct volume and brain atrophy in oil- or Tam-treated Cx3cr1-CreER;NHE1f/f brains at 14 days Rp with T2WI. Data are mean ± SEM. N = 4. (c) Representative MAP2 staining of oil- and Tam-treated Cx3cr1-CreER;Nhe1f/f brains at 14 days Rp. (d) Infarct volume and brain atrophy in oil- or Tam-treated Cx3cr1-CreER; NHE1f/f brains at 14 days Rp with MAP2 staining. Data are mean ± SEM. N = 7. (e) Representative immunofluorescent images of MAP2, NeuN, and To-pro-3 staining in the CL, IL perilesion and IL core areas in the cortex and striatum of the oil- and Tam-treated Cx3cr1-CreER;Nhe1f/f mice at 14 days post-tMCAO. (f) Quantitative analysis of MAP2 intensity in the CL, IL perilesion and IL core areas in the cortex and striatum of the oil- and Tam-treated Cx3cr1-CreER;Nhe1f/f mice at 14 days post-tMCAO. N = 3. *p < .05, **p < .01, ***p < .001 [Color figure can be viewed at wileyonlinelibrary.com]
Diffused Tensor Imaging (DTI) of the ex vivo brains showed that ischemic stroke did not cause persistent reduction in the fractional anisotropy (FA) in the corpus callosum (CC) or external capsule (EC) in the oil-treated brains by day 14 post-stroke (p > .05, Figure 6a,b). Interestingly, we detected a small but significant increase in the FA values of EC in both non-ischemic and ischemic hemispheres in the Tam-treated brains (p < .01, Figure 6a,b). The EC DTI FA values were significantly higher in the Tam-treated brains than the oil-treated brains (0.35 ± 0.02 vs. 0.29 ± 0.01, p < .05, Figure 6a,b). We further determined myelination status by measuring expression of myelin basic protein (MBP) and mature oligodendrocyte (APC+) counts in the CC and EC white matter tracts at day 3 post-stroke (Figure 5). Ischemic stroke triggered a similar loss of MBP intensity in the ischemic CC and EC of both oil- and Tam-treated brains (Figure 5a,b). However, there was a differential change in APC+ counts between the two groups. The oil-treated ischemic brains displayed a significant decrease in APC+ counts in the EC (Figure 5c). No such a change was detected in the EC of the Tam-treated brains (Figure 5c). By day 14 post-stroke, no significant recovery of the APC+ mature oligodendrocyte numbers or MBP intensity were seen in the IL hemisphere CC and EC of the oil-treated brains (Figure 6c–e). In contrast, the Tam-treated brains showed a significant increase in both the APC+ cell count and MBP intensity (Figure 6c–e). Taken together, these data clearly demonstrate that microglial NHE1 protein contributes to early oligodendrocyte damage and sustains white matter injury after ischemic stroke.
FIGURE 6.
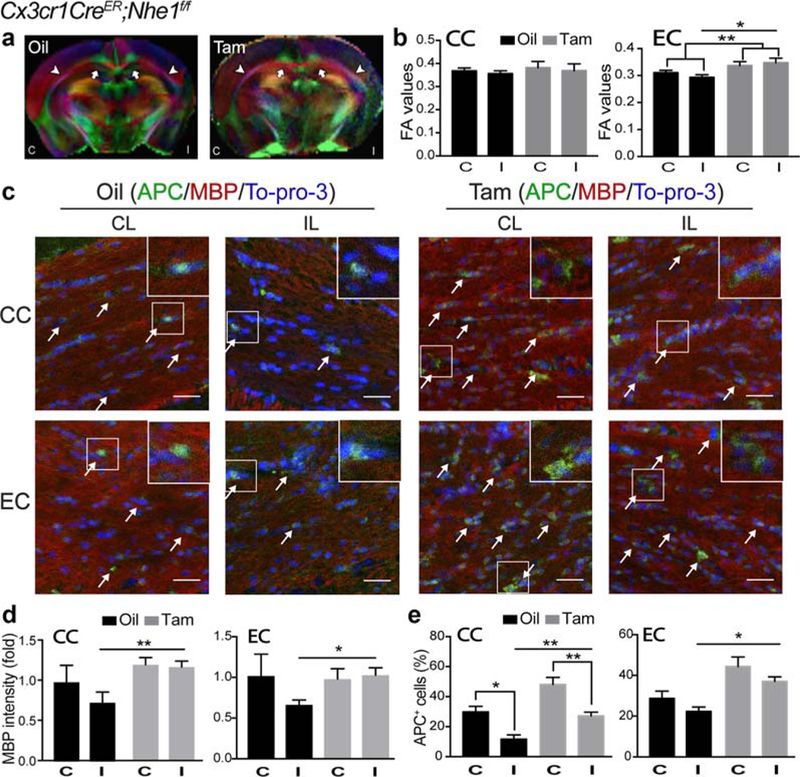
Tam-treated Cx3cr1-CreER;Nhe1f/f mice enhanced white matter repair at the subacute phase post-ischemia. (a) Representative Directionally Encoded Color (DEC) maps of ex vivo oil- and Tam-treated Cx3cr1-CreER;Nhe1f/f brains at 14 days Rp. C, contralateral hemisphere; I, ipsi-lateral hemisphere. Arrow: corpus callosum (CC). Arrowhead: external capsule (EC). (b) Quantitative analysis of fractional anisotropy (FA) values in CC and EC of oil- or Tam-treated Cx3cr1-CreER;Nhe1f/f brains at 14 days Rp. C, contralateral hemisphere; I, ipsilateral hemisphere. Data are mean ± SEM. N = 4. *p < .05, **p < .01. (c) Representative immunofluorescent images of APC, MBP, and To-pro-3 staining in CC and EC areas in the CL and IL of oil- and Tam-treated Cx3cr1-CreER;Nhe1f/f brains at 14 days Rp. (d) Quantitative analysis of MBP intensity (fold change) in CC and EC areas in the CL (C) and IL (I) of oil- and Tam-treated Cx3cr1-CreER;Nhe1f/f brains at 14 days Rp. Data are mean ± SEM. N = 4. *p < .05, **p < .01. (e) Quantitative analysis of APC+ cell percentages in CC and EC areas in the CL (C) and IL (I) of oil- and Tam-treated Cx3cr1-CreER; Nhe1f/f brains at 14 days Rp. Data are mean ± SEM. N = 4. *p < .05, **p < .01 [Color figure can be viewed at wileyonlinelibrary.com]
FIGURE 5.
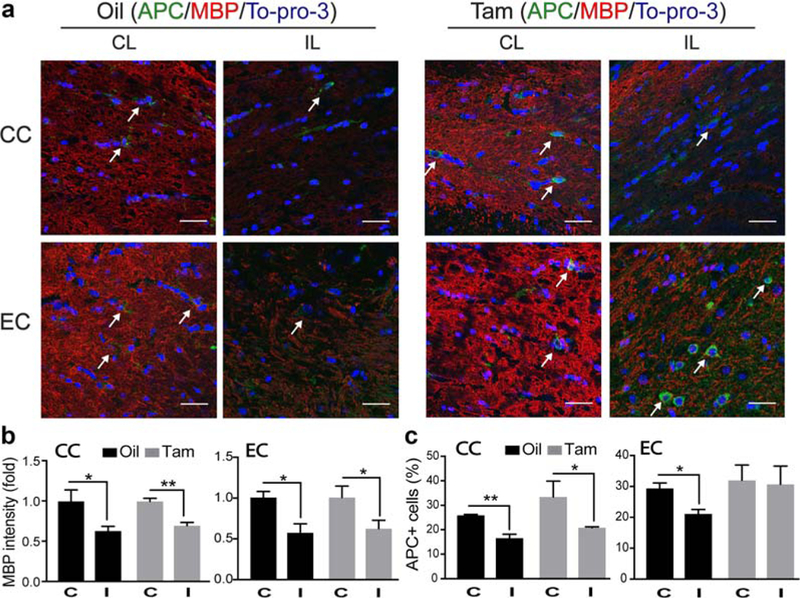
Tam-treated Cx3cr1-CreER;Nhe1f/f mice exhibited similar demyelination but preserved more mature oligodendrocytes at the acute phase post-stroke. (a) Representative immunofluorescent images of APC, MBP, and To-pro-3 staining in CC and EC areas in the CL and IL of oil- and Tam-treated Cx3cr1-CreER;Nhe1f/f brains at day 3 post-tMCAO. (b) Quantitative analysis of MBP intensity (fold change) in CC and EC areas the oil- and Tam-treated Cx3cr1-CreER;Nhe1f/f brains at day 3 post-tMCAO. Data are mean ± SEM. N = 3. *p < .05, **p < .01. (c) Quantitative analysis of APC+ cells in CC and EC areas in the oil- and Tam-treated Cx3cr1-CreER;Nhe1f/f brains at day 3 post-tMCAO. Data are mean ± SEM. N = 3. *p < .05, **p < .01 [Color figure can be viewed at wileyonlinelibrary.com]
3.4 |. Selective deletion of neuronal Nhe1 in CamKIIa-Cre+/−;Nhe1f/f mice reduces acute infarct and improves sensorimotor function recovery after ischemic stroke
CamKIIa-Cre+/−;Nhe1f/f mice (neuron-specific Nhe1-null) were created by crossing the Nhe1f/f mice with CamKIIa-Cre+/− mice (Supporting Information Figure S1d). The selective deletion of neuronal NHE1 protein in the CamKIIa-Cre+/−;Nhe1f/f mice was confirmed by a lack of immunofluorescent staining for NHE1 protein in the NeuN+ cell populations (Supporting Information Figure S1e). tMCAO was induced in both CamKIIa-Cre+/− control mice and CamKIIa-Cre+/−;Nhe1f/f neuronal Nhe1-null mice at P65–75 (Figure 7a). The control mice exhibited 79.6 ± 9.8 mm3 infarct at 48 hr post-tMCAO with severe neurological deficits on day 1–5 post-stroke, which improved by day 7–14 post-stroke. In contrast, the neuronal Nhe1-null mice showed significantly smaller infarction (33.0 ± 9.2 mm3, p < .05) and hemispheric swelling (14.4% ± 1.7% vs. 26.8% ± 4.4%, p < .05, Figure 7b–d), as well as faster neurological function recovery with the score reduced to <2 as early as day 5 after stroke (p < .05, Figure 7e), and reached the nearly normal neurological scores of 0–1 at day 7–14 post-stroke (Figure 7e).
FIGURE 7.
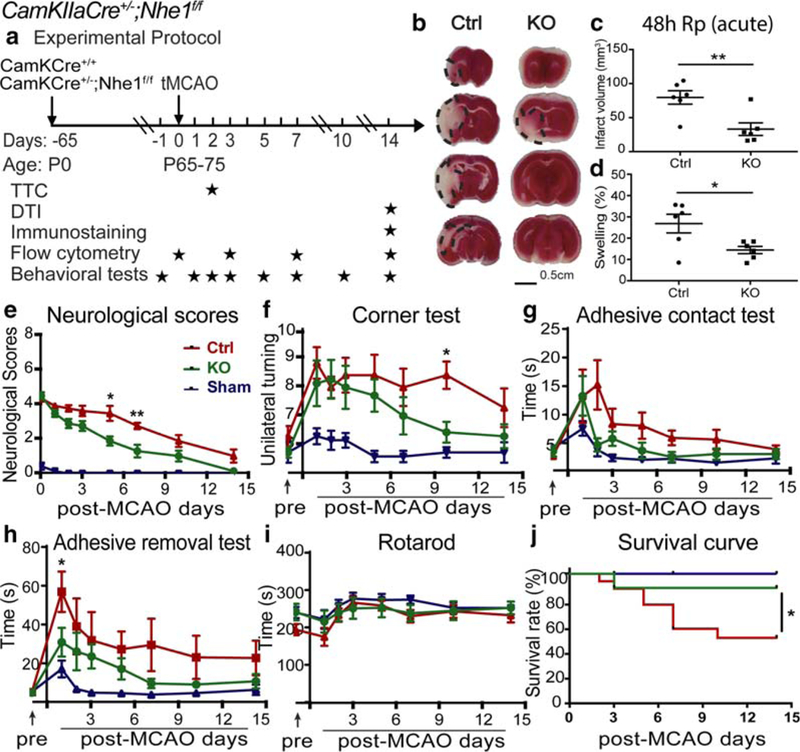
Neuron-specific deletion of Nhe1 in CamKIIa-Cre+/−;Nhe1f/f mice significantly reduced acute infarct volume and improved survival and neurological function recovery after ischemic stroke. (a) Experimental protocol. Focal cerebral ischemia was induced by tMCAO in of CamKIIa-Cre+/− (Ctrl) and CamKIIa-Cre+/−;Nhe1f/f (KO) mice at P65–75. TTC staining was conducted at 48 hr Rp. Ex vivo DTI imaging and immunostaining were conducted at 14 days Rp. Flow cytometry experiments were conducted at 0 (baseline), 3, 7, and 14 days Rp. Behavioral tests were conducted at 1 day prior to tMCAO, and at 1, 2, 3, 5, 7, 10, and 14 days Rp. (b-d) Infarct volume and brain swelling of Ctrl and KO mice at 48 h Rp with TTC staining. Data are mean ± SEM. N = 6. *p < .05, **p < .01. (e) Neurological score of Ctrl, KO, or sham mice after 50 min tMCAO. Data are mean ± SEM. NKO = 7 (5 males and 2 females), NCtrl = 7 (4 males and 3 females), NSham = 9 (5 males and 4 female). *p < .05, **p < .01, Ctrl versus KO mice. (f-i) Corner test, adhesive contact test, adhesive removal test, and rotarod accelerating test of the same cohort of mice in e. Data are mean ± SEM. *p < .05, Ctrl versus KO mice. (j) Survival curve of the same cohort of mice in e. *p < .05, Ctrl versus KO mice [Color figure can be viewed at wileyonlinelibrary.com]
The panel of sensorimotor function tests showed that no neurological deficits were observed in the sham group during the 14-day post-procedure period (Figure 7f–i), and no differences in baseline sensorimotor function between the control mice and the neuronal Nhe1-null mice prior to tMCAO (Figure 7f–i). In the corner test, both the control group and the neuronal Nhe1-null mice exhibited increased asymmetries after stroke and showed no differences between the groups. However, the neuronal Nhe1-null mice exhibited faster recovery on day 10 post-stroke (6.4 ± 0.3 vs. 8.4 ± 0.5, p < .05, Figure 7f). In the adhesive tests, both the control mice and the neuronal Nhe1-null mice exhibited mild deficits in the contact test, and showed no differences between the groups (Figure 7g). In contrast, ischemic stroke increased the time in the removal test of the control mice by ~11-fold on day 2 post-stroke and did not display such severe deficit in the neuronal Nhe1-null mice throughout day 1–14 post-stroke (p < .05, Figure 7h). No significant motor deficits were detected in either group of mice in the rotarod accelerating test (Figure 7i). Lastly, in the survival study, all sham animals survived during the 14-day recovery period (100%, Figure 7j). Compared with the control mice (either CamKIIa-Cre+/− or Nhe1f/f), the CamKIIa-Cre+/−;Nhe1f/f mice exhibited a higher survival rate at day 14 post-tMCAO (88.9% vs. 52.9%, p < .05, Figure 7j). These data clearly show that neuronal Nhe1 plays an indispensable role in ischemic brain damage and the sensorimotor dysfunction after ischemic stroke.
3.5 |. Neuronal Nhe1-null mice did not suppress pro-inflammatory responses of CD11b+/CD45+ microglia/macrophage after stroke
Both the control and the neuronal Nhe1-null brains exhibited low numbers of CD11b+/CD45low-med microglia and CD11b+/CD45hi macrophages in both CL and IL hemispheres prior to tMCAO (Figure 8a,b). The CD11b+/CD45low-med microglia counts initially increased at day 3 post-stroke and peaked by day 7 post-stroke (Figure 8a). Elevation of CD11b+/CD45hi macrophages also peaked at day 7 post-ischemia in both groups (Figure 8b). Pro-inflammatory markers CD86 and CD16/32, anti-inflammatory markers Ym1 and CD206, as well as the phagocytic marker CD68 were all expressed at similar levels in the control and the neuronal Nhe1-null brains (Figure 8c-g). Therefore, selective deletion of neuronal Nhe1 has no effect on brain inflammation (Supporting Information Figure S6b).
FIGURE 8.
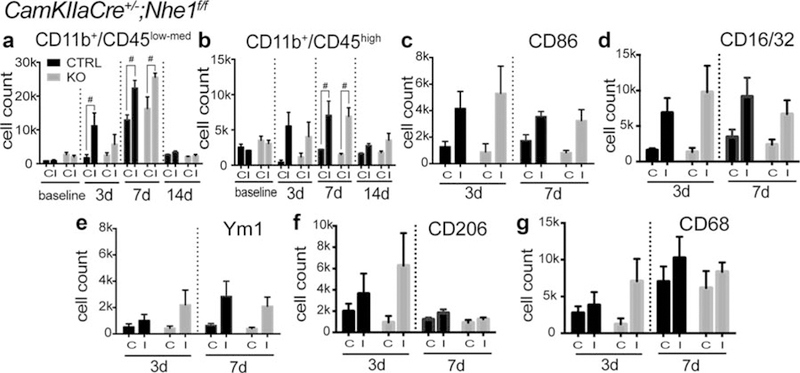
CamKIIa-Cre+/−;Nhe1f/f mice showed delayed activation but no significant effects on polarizations of microglia/macrophages after ischemic stroke. (a) Cell counts of CD11b+/CD45low-med microglia in Ctrl and KO brains before tMCAO and at 3, 7, and 14 days Rp. A total cell count of 50,000 was recorded and analyzed. In all panels C, contralateral; I, ipsilateral. Data are mean ± SEM. N = 3–5. #p < .10 control versus KO groups. (b) Cell counts of CD11b+/CD45hi macrophages in Ctrl and KO brains before tMCAO and at 3, 7, and 14 days Rp. A total cell count of 50,000 was recorded and analyzed. Data are mean ± SEM. N = 3–5. #p < .10 control versus KO groups. (c–g) Cell counts of CD86+, CD16/32+, Ym1+, CD206+, and CD68+ cells gated under CD11b+ cells in Ctrl and KO brains at 3 and 7 days Rp. A total cell count of 50,000 was recorded and analyzed. Data are mean ± SEM. N = 5
3.6 |. Neuronal Nhe1-null mice did not show increased white matter recovery after ischemic stroke
Compared with the control mice, T2WI of ex vivo brains from the neuronal Nhe1-null mice illustrated significantly smaller infarction (17.8 ± 3.0 mm3 vs. 34.2 ± 3.8 mm3, p < .01, Figure 9A-B) and brain atrophy (−2.8% ± 1.0% vs. −5.5% ± 0.5%, p < .05, Figure 9a,b) at day 14 post-stroke. Concurrently, MAP2 staining showed that subacute infarction and brain atrophy were both reduced in the neuronal Nhe1-null brains (p < .01 and p < .05, respectively, Figure 9c,d). Ex vivo DTI did not show significant differences in the FA values of the CC or EC between control and neuronal Nhe1-null mice at day 14 post-stroke (Figure 9e,f). These data demonstrate that deletion of neuronal Nhe1 reduces subacute neuronal damage and brain atrophy, but has no effect on white matter repair.
FIGURE 9.
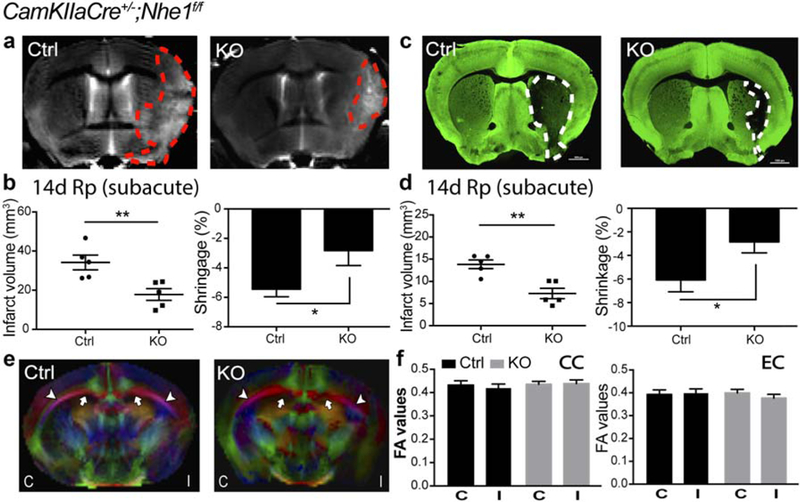
CamKIIa-Cre+/−;Nhe1f/f mice exhibited smaller infarction and atrophy but no improvements in white matter injury at the subacute phase post-ischemia. (a) Representative T2WI of ex vivo Ctrl and KO brains at 14 days Rp. (b) Infarct volume and brain atrophy of Ctrl and KO brains at 14 days Rp with T2WI. Data are mean ± SEM. N = 5. *p < .05, **p < .01, Ctrl versus KO groups. (c) Representative MAP2 staining of Ctrl and KO brains at 14 days Rp. (d) Infarct volume and brain atrophy of Ctrl and KO brains at 14 days Rp with MAP2 staining. Data are mean ± SEM. N = 5. *p < .05, **p < .01, Ctrl versus KO groups. (e) Representative DEC maps of ex vivo Ctrl and KO brains at 14 days Rp. C, contralateral; I, ipsilateral. Arrow: CC. Arrowhead: EC. (f) Quantitative analysis of FA values in CC and EC of Ctrl and KO brains at 14 days Rp. C, contralateral; I, ipsilateral. Data are mean ± SEM. N = 5 [Color figure can be viewed at wileyonlinelibrary.com]
4 |. DISCUSSION
4.1 |. Selective deletion of Nhe1 in Cx3cr1-CreER;Nhe1f/f mice did not affect ischemic infarction but improved neurological function after ischemic stroke
In this study, we used the newly established Nhe1f/f mouse line (Begum et al., 2017) to generate a microglia-specific Nhe1-null mouse line Cx3cr1-CreER;NHE1f/f and selectively ablated NHE1 protein in microglial cells. Selective deletion of Nhe1 in Cx3cr1+ microglia did not reduce acute ischemic infarction at 48 hr nor subacute infarct lesion volume at day 14 post-stroke. However, the microglial Nhe1-null mice exhibited a significantly higher survival rate and faster neurological function recovery over 1–14 days after ischemia. These findings suggest that microglial NHE1 protein is not involved in acute ischemic brain cell death but affects brain tissue repair in response to ischemic stroke. This study reveals novel roles of microglial NHE1 in ischemic brain injury and repair.
4.2 |. Selective deletion of Nhe1 in Cx3cr1+ microglia suppressed acute inflammatory responses and skewed them towards adaptive phenotypes in the ischemic brains
Our flow cytometry data sheds light on the underlying mechanisms on microglial Nhe1 in microglial activation and inflammation. Ischemic stroke triggered a significant elevation of CD11b+/CD45low-med microglial cell counts in the control mice at day 3 post-stroke, a peak time of microglial activation as reported previously (Kim & Cho, 2016; Schilling et al., 2005; Tang et al., 2014). In contrast, no such elevation was detected in the microglial Nhe1-null mice. We further demonstrated that the low CD11b+/CD45low-med microglial cell counts resulted from inhibition of microglial proliferation in the microglial Nhe1-null mice. It has been well documented that NHE1-mediated H+ efflux maintains alkaline pH, and supports cell proliferation by stimulating glycolysis and promoting protein, pyruvate and ATP production (Pedersen, 2006; Putney & Barber, 2004). NHE1 protein is also engaged in cell proliferation through cell cycle modulation (Putney & Barber, 2003). Our data demonstrate that microglial NHE1 protein plays an important role in the regulation of microglia activation and proliferation in ischemic brains. Additional studies are warranted to investigate mechanisms involved in microglial proliferation in the Nhe1-null mice after ischemic stroke.
Selective deletion of Nhe1 in microglial cells altered function of CD11b+/CD45low-med microglial populations, which were characterized by reduced pro-inflammatory CD86+ expression but increased antiinflammatory Ym1+ subpopulation, along with significantly decreased expression of IL-1β and elevation of TGF-β and IL-10. These reduced pro-inflammatory responses are consistent with our previous in vitro findings that pharmacological blockage of NHE1 protein in cultured microglia abolished production of pro-inflammatory cytokines (IL-6, IL-1β, and TNFα; Liu et al., 2010). Moreover, in this study, microglial Nhe1-null brains showed a significant increase in CD68+ phagocytic microglial population, which is important for phagocytosis of damaged tissue. Lastly, a reduced number of GFAP+ reactive astrocytes were detected in the microglial Nhe1-null mice. It is reported that neurotoxic-reactive astrocytes can be induced by pro-inflammatory microglia, lead to death of neurons and oligodendrocytes, and are detrimental to neuronal outgrowth and synaptogenesis (Liddelow et al., 2017). TGF-β and IL-10 play roles against inflammation by suppressing GFAP+ reactive astrocytes (Balasingam & Yong, 1996; Cekanaviciute et al., 2014; Makwana et al., 2007). Taken together, these data suggest that microglial NHE1 protein is involved in inflammatory responses, and that deletion of microglial Nhe1 causes a switch from the pro-inflammatory responses towards restorative functions in ischemic brains.
4.3 |. Impact of specific deletion of microglial Nhe1 on white matter myelination and ischemic tissue repair
The microglial Nhe1-null mice exhibited accelerated improvements in sensorimotor functional recovery, increased preservation of mature oligodendrocytes, and promoted white matter remyelination despite the initial large infarct lesion. Specifically, as shown in Supporting Information Figure S6a, increased DTI FA values in both non-ischemic and ischemic hemispheres and elevated MBP protein expression in microglial Nhe1-null mice strongly suggest that microglial NHE1 protein plays an important role in white matter damage and can be targeted for white matter tissue repair. It has been reported that suppression of classically-activated microglia ameliorated axonal damage, neuronal degeneration, and demyelination through increased microglial phagocytic capacity in animal models of multiple sclerosis (Rinaldi et al., 2016; Starossom et al., 2012). In consistence with these reports, we showed here that improved white matter myelination and elevated CD68+ phagocytic microglia/macrophage populations were concurrently detected in the microglial Nhe1-null brains. Phagocytic microglia function as scavengers for debris in damaged brain tissues, and are involved in modulation of synapse function and pruning, as well as axonal growth (Fu, Shen, Xu, Luo, & Tang, 2014; Paolicelli et al., 2011; Rajbhandari et al., 2014; Schafer et al., 2012). Secondly, the microglial Nhe1-null mice also exhibited increased TGF-β and IL-10, as well as decreased IL-1β. It has been well established that neurotrophic factors such as TGF-β and insulin-like growth factor 1 (IGF1) promote neuronal survival and axonal growth (Bialas & Stevens, 2013; Constam et al., 1992; Luo et al., 2016; Ueno et al., 2013). It has been recently discovered that TGF-β mediates synaptic pruning by regulation of neuronal expression of complement protein C1q in a microglia-dependent manner (Bialas & Stevens, 2013; Derecki & Kipnis, 2013). IL-10 released from microglia is sufficient for dendritic spine outgrowth and synaptic formation in hippocampal neuron culture (Lim et al., 2013). IL-10 is also suggested to recover neurite outgrowth by suppressing microglia-regulated nitric oxide and glutamate production under oxidative stress (Silva et al., 2012). IL-1β, on the other hand, plays an antagonizing role against IL-10 and suppresses long-term potentiation in the hippocampus (Kelly et al., 2001). It is reported that IL-13 produced by microglia reduces cortical synapses and mature oligodendrocytes, damages node of Ranvier, and suppresses axonal development after lipopolysaccharide injection (Han et al., 2017; Xie et al., 2016). Therefore, increased TGF-β, IL-10, and decreased IL-1β in microglial Nhe1-null mice could collectively enhance white matter tissue repair and improve neurological functions after ischemic stroke. Additional studies are needed to examine changes of synapse plasticity and oligodendrocyte maturation and function in microglial Nhe1-null mice.
4.4 |. Neuronal NHE1 protein in neurodegeneration after ischemic stroke
In the parallel study, neuron-specific Nhe1-null CamKIIa-Cre+/−;Nhe1f/f mice showed decreased acute infarct and persistently less neuronal degeneration in post-stroke brains, which was accompanied with improved survival and faster neurological function recovery. However, deletion of the neuronal Nhe1 had no effects on elevation of either CD11b+/CD45low-med microglial cells or CD11b+/CD45hi macrophages in ischemic brains (Supporting Information Figure S6b). Therefore, the beneficial effects exhibited in neuronal Nhe1-null mice did not result from reduced microglia-mediated neuroinflammation. We have previously reported that overstimulation of NHE1 activity leads to neuronal death in part by increasing intracellular Na+ and Ca2+ overload (Luo, Chen, Kintner, Shull, & Sun, 2005) and superoxide production mediated by NOX2 (Lam et al., 2013). Therefore, deletion of neuronal NHE1 reduces ischemic damage by direct protection of neurons without affecting microglia-mediated inflammation.
4.5 |. Selectivity of tamoxifen for Cx3cr1+ microglial cells and its potential neuroprotective effects
Specific gene deletion in microglia can be achieved using Cx3cr1-CreER expressing animals (Goldmann et al., 2013; Parkhurst et al., 2013; Wieghofer, Knobeloch, & Prinz, 2015). The effects on peripheral circulating Cx3cr1+ monocytes (Geissmann, Jung, & Littman, 2003; Jung et al., 2000) can be eliminated by a 30-day waiting period for replenishment of these blood-borne monocytes with a 21-day self-renewing cycle (Hashimoto et al., 2013). In addition, the 30-day post-Tam injection period is needed for clearance of Tam-mediated neuroprotection effects. Tam has neuroprotective effects against neurodegenerative diseases via possible mechanisms such as stimulating MAPK/ERK signaling pathways (Wakade et al., 2008; Zou et al., 2015), preventing mitochondrial reactive oxygen species generation (Khan, Wakade, de Sevilla, & Brann, 2015; Wakade et al., 2008), and generating antioxidant enzymes (Zhang, Milatovic, Aschner, Feustel, & Kimelberg, 2007). Despite complete clearance of Tam and its metabolites in the brain by 8 days post-injection (Valny et al., 2016), we detected prolonged neuroprotective effects of Tam (75 mg/kg for consecutive 5 days) in C57/B6J, Cx3cr1-CreER or Nhe1f/f, as well as Cx3cr1-CreER;Nhe1f/f mice when tMCAO was induced at 1, 12, and 20 days after Tam injection, compared with the vehicle oil control mice (Supporting Information Figure S5). The 30-day post-Tam injection waiting period completely eliminated the Tam-mediated neuroprotective effects since no differences in infarction were detected between the oil-treated and Tam-treated mice after tMCAO (Figure 1B).
5 |. CONCLUSION
Our inducible, microglia-specific Nhe1 knockout mouse model provides the first line of evidence that microglial NHE1 protein plays a role in neuroinflammation, white matter demyelination, and brain tissue repair after ischemic stroke (Figure 10). In contrast, neuronal NHE1 protein activation is directly associated with the acute ischemic neuronal injury but not related to inflammation (Figure 10). Our novel findings reveal that NHE1 protein has unique functions in microglia and neurons. NHE1 protein presents a therapeutic target for differential regulation of ischemic brain injury and tissue repair via modulating inflammation, white matter demyelination, and neurodegeneration.
FIGURE 10.
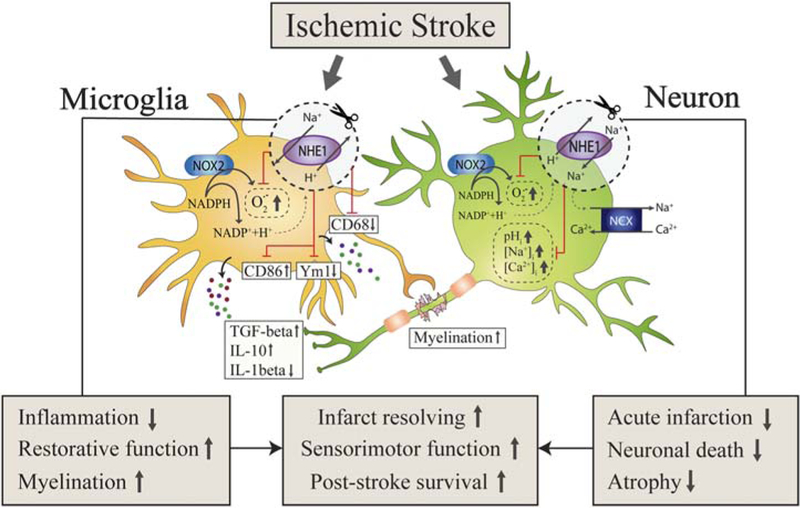
Schematic illustration of microglial and neuronal NHE1 activation in ischemic brain damage and tissue repair. Ischemic stroke triggers NHE1 activation in both microglia and neurons. Specific deletion of Nhe1 in microglia has no effects on acute infarct formation but significantly ameliorates inflammatory microenvironment, promotes restorative microglial function, and enhances myelination. In contrast, specific deletion of neuronal Nhe1 shows no involvement in inflammatory responses, but reduces acute infarction, neuronal damage, and brain atrophy. Therefore, microglial and neuronal Nhe1 deletion contribute differentially to post-ischemic recovery, but both lead to improved infarct resolving, promoted sensorimotor functions, and increased survival rate after ischemic stroke [Color figure can be viewed at wileyonlinelibrary.com]
Supplementary Material
ACKNOWLEDGMENT
We would like to thank Dr. Wenbiao Gan from the New York University for his generosity of providing us with the transgenic Cx3cr1-CreER mouse line. This work was supported by NIH grants R01NS 48216 and R01NS 38118 (D. Sun), and American Heart Association Postdoctoral Fellowship 17POST32440002 (S. Song). The authors have no conflicts of interest to declare.
Funding information NIH, Grant Numbers: R01NS 48216 and R01NS 38118; AHA Postdoctoral Fellowship, Grant Number: 17POST32440002
Footnotes
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
REFERENCES
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, & Rossi FM (2007). Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nature Neuroscience, 10(12), 1538–1543. 10.1038/nn2014 [DOI] [PubMed] [Google Scholar]
- Balasingam V, & Yong VW (1996). Attenuation of astroglial reactivity by interleukin-10. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 16(9), 2945–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum G, Song S, Wang S, Zhao H, Bhuiyan MI, Li E, ... Sun D (2017). Selective knockout of astrocytic Na+/H+ exchanger isoform 1 reduces astrogliosis, BBB damage, infarction, and improves neurological function after ischemic stroke. Glia, 10.1002/glia.23232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum G, Yuan H, Kahle KT, Li L, Wang S, Shi Y, ... Sun D (2015). Inhibition of WNK3 kinase signaling reduces brain damage and accelerates neurological recovery after stroke. Stroke, 46(7), 1956–1965. 10.1161/STR0KEAHA.115.008939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialas AR, & Stevens B (2013). TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nature Neuroscience, 16(12), 1773–1782. 10.1038/nn.3560 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Boscia F, Begum G, Pignataro G, Sirabella R, Cuomo O, Casamassa A, Annunziato L (2016). Glial Na(+) -dependent ion transporters in pathophysiological conditions. Glia, 64(10), 1677–1697. 10.1002/glia.23030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC, & Neher JJ (2014). Microglial phagocytosis of live neurons. Nature Reviews Neuroscience, 15(4), 209–216. 10.1038/nrn3710 [DOI] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, ... Weiner HL (2014). Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nature Neuroscience, 17(1), 131–143. 10.1038/nn.3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cekanaviciute E, Fathali N, Doyle KP, Williams AM, Han J, & Buckwalter MS (2014). Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia, 62(8), 1227–1240. 10.1002/glia.22675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen LB, Woods TA, Carmody AB, Caughey B, & Peterson KE (2014). Age-related differences in neuroinflammatory responses associated with a distinct profile of regulatory markers on neonatal microglia. Journal of Neuroinflammation, 11(1), 70 10.1186/1742-2094-11-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constam DB, Philipp J, Malipiero UV, ten Dijke P, Schachner M, & Fontana A (1992). Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. Journal of Immunology, 148(5), 1404–1410. [PubMed] [Google Scholar]
- Cullen M, Elzarrad MK, Seaman S, Zudaire E, Stevens J, Yang MY, ... St. Croix B (2011). GPR124, an orphan G protein-coupled receptor, is required for CNS-specific vascularization and establishment of the blood-brain barrier. Proceedings of the National Academy of Sciences of the United States of America, 108(14), 5759–5764. 10.1073/pnas.1017192108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson DA, & Hallenbeck JM (1996). Acute focal ischemia-induced alterations in MAP2 immunostaining: Description of temporal changes and utilization as a marker for volumetric assessment of acute brain injury. Journal of Cerebral Blood Flow & Metabolism, 16(1), 170–174. 10.1097/00004647-199601000-00020 [DOI] [PubMed] [Google Scholar]
- Derecki NC, & Kipnis J (2013). From neurons to microglia, with complements. Nature Neuroscience, 16(12), 1712–1713. 10.1038/nn.3579 [DOI] [PubMed] [Google Scholar]
- Fogg DK, Sibon C, Miled C, Jung S, Aucouturier P, Littman DR, ... Geissmann F (2006). A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science, 311(5757), 83–87. 10.1126/science.1117729 [DOI] [PubMed] [Google Scholar]
- Frost JL, & Schafer DP (2016). Microglia: Architects of the developing nervous system. Trends in Cell Biology, 26(8), 587–597. 10.1016/j.tcb.2016.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu R, Shen Q, Xu P, Luo JJ, & Tang Y (2014). Phagocytosis of microglia in the central nervous system diseases. Molecular Neurobiology, 49(3), 1422–1434. 10.1007/s12035-013-8620-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Jung S, & Littman DR (2003). Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity, 19(1), 71–82. [DOI] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Jordão MJC, Prutek F, Hagemeyer N, Frenzel K, Prinz M (2016). Origin, fate and dynamics of macrophages at central nervous system interfaces. Nature Immunology, 17 (7), 797–805. 10.1038/ni.3423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Müller PF, Wolf Y, Varol D, Yona S, ... Prinz M (2013). A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nature Neuroscience, 16(11), 1618–1626. 10.1038/nn.3531 [DOI] [PubMed] [Google Scholar]
- Gomez-Nicola D, & Perry VH (2015). Microglial dynamics and role in the healthy and diseased brain: A paradigm of functional plasticity. The Neuroscientist, 21(2), 169–184. 10.1177/1073858414530512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greter M, Lelios I, & Croxford AL (2015). Microglia versus myeloid cell nomenclature during brain inflammation. Frontiers in Immunology, 6, 249 10.3389/fimmu.2015.00249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Q, Lin Q, Huang P, Chen M, Hu X, Fu H, ... Deng Y (2017). Microglia-derived IL-1beta contributes to axon development disorders and synaptic deficit through p38-MAPK signal pathway in septic neonatal rats. Journal of Neuroinflammation, 14(1), 52 10.1186/s12974-017-0805-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, ... Merad M (2013). Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity, 38(4), 792–804. 10.1016/j.immuni.2013.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean T, Frelin C, Vigne P, Barbry P, & Lazdunski M (1985). Biochemical properties of the Na+/H+ exchange system in rat brain synaptosomes. Interdependence of internal and external pH control of the exchange activity. Journal of Biological Chemistry, 260(17), 9678–9684. [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, & Littman DR (2000). Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Molecular Cell Biology, 20(11), 4106–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly Á, Lynch A, Vereker E, Nolan Y, Queenan P, Whittaker E,.Lynch MA (2001). The anti-inflammatory cytokine, interleukin (IL)-10, blocks the inhibitory effect of IL-1 beta on long term potentiation. A role for JNK. Journal of Biological Chemistry, 276(49), 45564–45572. 10.1074/jbc.M108757200 [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Kirchhoff F, & Verkhratsky A (2013). Microglia: New roles for the synaptic stripper. Neuron, 77(1), 10–18. 10.1016/j.neuron.2012.12.023 [DOI] [PubMed] [Google Scholar]
- Khan MM, Wakade C, de Sevilla L, & Brann DW (2015). Selective estrogen receptor modulators (SERMs) enhance neurogenesis and spine density following focal cerebral ischemia. Journal of Steroid Biochemistry and Molecular Biology, 146, 38–47. 10.1016/j.jsbmb.2014.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, & Cho S (2016). Microglia and monocyte-derived macrophages in stroke. Neurotherapeutics, 13(4), 702–718. 10.1007/s13311-016-0463-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Woo M-S, Qin L, Ma T, Beltran CD, Bao Y, Cho S (2015). Daidzein augments cholesterol homeostasis via ApoE to promote functional recovery in chronic stroke. Journal of Neuroscience, 35(45), 15113–15126. 10.1523/JNEUROSCI.2890-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam TI, Brennan-Minnella AM, Won SJ, Shen Y, Hefner C, Shi Y, ... Swanson RA (2013). Intracellular pH reduction prevents excitotoxic and ischemic neuronal death by inhibiting NADPH oxidase. Proceedings of the National Academy of Sciences of the United States of America, 110(46), E4362–E4368. 10.1073/pnas.1313029110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, ... Barres BA (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541(7638), 481–487. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SH, Park E, You B, Jung Y, Park AR, Park SG, & Lee JR (2013). Neuronal synapse formation induced by microglia and interleukin 10. PLoS One, 8(11), e81218 10.1371/journal.pone.0081218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Kintner DB, Chanana V, Algharabli J, Chen X, Gao Y, ... Sun D (2010). Activation of microglia depends on Na+/H+ exchange-mediated H+ homeostasis. Journal of Neuroscience, 30 (45), 15210–15220. 10.1523/JNEUROSCI.3950-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Chen H, Kintner DB, Shull GE, & Sun D (2005). Decreased neuronal death in Na+/H+ exchanger isoform 1-null mice after in vitro and in vivo ischemia. Journal of Neuroscience, 25(49), 11256–11268. 10.1523/JNEUROSCI.3271-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo SX, Timbang L, Kim JC, Shang Y, Sandoval K, Tang AA, ... Huang EJ (2016). TGF-beta signaling in dopaminergic neurons regulates dendritic growth, excitatory-inhibitory synaptic balance, and reversal learning. Cell Reports, 17(12), 3233–3245. 10.1016/j.celrep.2016.11.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Wang J, Wang Y, & Yang GY (2016). The biphasic function of microglia in ischemic stroke. Progress in Neurobiology, 10.1016/j.pneurobio.2016.01.005 [DOI] [PubMed] [Google Scholar]
- Makwana M, Jones LL, Cuthill D, Heuer H, Bohatschek M, Hristova M, Raivich G (2007). Endogenous transforming growth factor beta 1 suppresses inflammation and promotes survival in adult CNS. Journal of Neuroscience, 27(42), 11201–11213. 10.1523/JNEUROSCI.2255-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, El-Behi M, Fontaine B, & Delarasse C (2017). Analysis of microglia and monocyte-derived macrophages from the central nervous system by flow cytometry. Journal of Visualized Experiments, 124 10.3791/55781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michell-Robinson MA, Touil HE, Healy LM, Owen DR, Dura-fourt BA, Bar-Or A, ... Moore CS (2015). Roles of microglia in brain development, tissue maintenance and repair. Brain, 138(5), 1138–1159. 10.1093/brain/awv066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Huang H, Radke J, Stenzel W, & Priller J (2017). P2Y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia, 65(2), 375–387. 10.1002/glia.23097 [DOI] [PubMed] [Google Scholar]
- Mosser CA, Baptista S, Arnoux I, & Audinat E (2017). Microglia in CNS development: Shaping the brain for the future. Progress in Neurobiology, 149-150, 1–20. https://doi.org/10.1016Z.pneurobio.2017.01.002 [DOI] [PubMed] [Google Scholar]
- Nayak D, Roth TL, & McGavern DB (2014). Microglia development and function. Annual Review of Immunology, 32(1), 367–402. 10.1146/annurev-immunol-032713-120240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikodemova M, & Watters JJ (2012). Efficient isolation of live microglia with preserved phenotypes from adult mouse brain. Journal of Neuroinflammation, 9(1), 147 10.1186/1742-2094-9-147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orefice LL, Zimmerman AL, Chirila AM, Sleboda SJ, Head JP, & Ginty DD (2016). Peripheral mechanosensory neuron dysfunction underlies tactile and behavioral deficits in mouse models of ASDs. Cell, 166(2), 299–313. 10.1016/j.cell.2016.05.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlowski J, & Grinstein S (2004). Diversity of the mammalian sodium/proton exchanger SLC9 gene family. Pflugers Archives, 447(5), 549–565. 10.1007/s00424-003-1110-3 [DOI] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Gross CT (2011). Synaptic pruning by microglia is necessary for normal brain development. Science, 333(6048), 1456–1458. 10.1126/science.1202529 [DOI] [PubMed] [Google Scholar]
- Paolicelli RC, & Gross CT (2011). Microglia in development: Linking brain wiring to brain environment. Neuron Glia Biology, 7(01), 77–83. 10.1017/S1740925X12000105 [DOI] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, Lafaille JJ, ... Gan WI (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell, 155(7), 1596–1609. 10.1016/j.cell.2013.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen SF (2006). The Na+/H+ exchanger NHE1 in stress-induced signal transduction: Implications for cell proliferation and cell death. Pflugers Archiv European Journal of Physiology, 452(3), 249–259. 10.1007/s00424-006-0044-y [DOI] [PubMed] [Google Scholar]
- Prinz M, & Priller J (2014). Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nature Reviews Neuroscience, 15(5), 300–312. 10.1038/nrn3722 [DOI] [PubMed] [Google Scholar]
- Putney LK, & Barber DL (2003). Na-H exchange-dependent increase in intracellular pH times G2/M entry and transition. Journal of Biological Chemistry, 278(45), 44645–44649. 10.1074/jbc.M308099200 [DOI] [PubMed] [Google Scholar]
- Putney LK, & Barber DL (2004). Expression profile of genes regulated by activity of the Na-H exchanger NHE1. BMC Genomics, 5(1), 46 10.1186/1471-2164-5-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajbhandari L, Tegenge MA, Shrestha S, Ganesh Kumar N., Malik A, Mithal A, Venkatesan A (2014). Toll-like receptor 4 deficiency impairs microglial phagocytosis of degenerating axons. Glia, 62 (12), 1982–1991. 10.1002/glia.22719 [DOI] [PubMed] [Google Scholar]
- Rinaldi M, Thomas L, Mathieu P, Carabias P, Troncoso MF, Pas-quini JM, Pasquini LA (2016). Galectin-1 circumvents lysolecithin-induced demyelination through the modulation of microglial polarization/phagocytosis and oligodendroglial differentiation. Neurobiology Disorders, 96, 127–143. 10.1016/j.nbd.2016.09.003 [DOI] [PubMed] [Google Scholar]
- Ritzel RM, Patel AR, Grenier JM, Crapser J, Verma R, Jellison ER, & McCullough LD (2015). Functional differences between microglia and monocytes after ischemic stroke. Journal of Neuroinflammation, 12(1), 106 10.1186/s12974-015-0329-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Stevens B (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron, 74(4), 691–705. https://doi.org/10.1016Z.neuron.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, & Stevens B (2013). The “quad-partite” synapse: Microglia-synapse interactions in the developing and mature CNS. Glia, 61(1), 24–36. 10.1002/glia.22389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling M, Besselmann M, Muller M, Strecker JK, Ringelstein EB, & Kiefer R (2005). Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: An investigation using green fluorescent protein transgenic bone marrow chimeric mice. Experimental Neurology, 196 (2), 290–297. 10.1016/j.expneurol.2005.08.004 [DOI] [PubMed] [Google Scholar]
- Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, & ter Meulen V (1991). Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proceedings of the National Academy of Sciences of the United States of America, 88(16), 7438–7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Chanana V, Watters JJ, Ferrazzano P, & Sun D (2011). Role of sodium/hydrogen exchanger isoform 1 in microglial activation and proinflammatory responses in ischemic brains. Journal of Neurochemistry, 119(1), 124–135. 10.1111/j.1471-4159.2011.07403.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yuan H, Kim D, Chanana V, Baba A, Matsuda T, ... Sun D (2013). Stimulation of Na(+)/H(+) exchanger isoform 1 promotes microglial migration. PLoS One, 8(8), e74201 10.1371/journal.pone.0074201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva SL, Vaz AR, Diógenes MJ, van Rooijen N, Sebastião AM, Fernandes A, ... Brites D (2012). Neuritic growth impairment and cell death by unconjugated bilirubin is mediated by NO and glutamate, modulated by microglia, and prevented by glycoursodeoxy-cholic acid and interleukin-10. Neuropharmacology, 62(7), 2398–2408. 10.1016/j.neuropharm.2012.02.002 [DOI] [PubMed] [Google Scholar]
- Starossom SC, Mascanfroni ID, Imitola J, Cao LI, Raddassi K, Hernandez SF, ... Rabinovich GA (2012). Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity, 37(2), 249–263. 10.1016/j.immuni.2012.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, & Sharp FR (1990). A semiautomated method for measuring brain infarct volume. Journal of Cerebral Blood Flow & Metabolism, 10(2), 290–293. 10.1038/jcbfm.1990.47 [DOI] [PubMed] [Google Scholar]
- Tan Z, Turner RC, Leon RL, Li X, Hongpaisan J, Zheng W, ... Huber JD (2013). Bryostatin improves survival and reduces ischemic brain injury in aged rats after acute ischemic stroke. Stroke, 44(12), 3490–3497. 10.1161/STROKEAHA.113.002411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Gan Y, Liu Q, Yin JX, Liu Q, Shi J, & Shi FD (2014). CX3CR1 deficiency suppresses activation and neurotoxicity of microglia/macrophage in experimental ischemic stroke. Journal of Neuroinflammation, 11(1), 26 10.1186/1742-2094-11-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay ME (2011). The role of microglia at synapses in the healthy CNS: Novel insights from recent imaging studies. Neuron Glia Biology, 7(01), 67–76. 10.1017/S1740925X12000038 [DOI] [PubMed] [Google Scholar]
- Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, & Nimmerjahn A (2011). The role of microglia in the healthy brain. Journal of Neuroscience, 31(45), 16064–16069. 10.1523/JNEUR-OSCI.4158-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M, & Yamashita T (2013). Layer V cortical neurons require microglial support for survival during postnatal development. Nature Neuroscience, 16(5), 543–551. 10.1038/nn.3358 [DOI] [PubMed] [Google Scholar]
- Valny M, Honsa P, Kirdajova D, Kamenik Z, & Anderova M (2016). Tamoxifen in the mouse brain: Implications for fate-mapping studies using the tamoxifen-inducible Cre-loxP system. Frontiers in Cellular Neuroscience, 10, 243 10.3389/fncel.2016.00243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakade C, Khan MM, De Sevilla LM, Zhang QG, Mahesh VB, & Brann DW (2008). Tamoxifen neuroprotection in cerebral ischemia involves attenuation of kinase activation and superoxide production and potentiation of mitochondrial superoxide dismutase. Endocrinology, 149(1), 367–379. 10.1210/en.2007-0899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Miyamoto A, & Nabekura J (2013). Microglia: Actively surveying and shaping neuronal circuit structure and function. Trends in Neuroscience, 36(4), 209–217. 10.1016/j.tins.2012.11.007 [DOI] [PubMed] [Google Scholar]
- Wang J, Xia J, Zhang F, Shi Y, Wu Y, Pu H, ... Chen J (2015). Galectin-1-secreting neural stem cells elicit long-term neuroprotection against ischemic brain injury. Scientific Reports, 5(1), 9621 10.1038/srep09621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieghofer P, Knobeloch KP, & Prinz M (2015). Genetic targeting of microglia. Glia, 63(1), 1–22. 10.1002/glia.22727 [DOI] [PubMed] [Google Scholar]
- Wong K, Noubade R, Manzanillo P, Ota N, Foreman O, Hackney JA, Ouyang W (2017). Mice deficient in NRROS show abnormal microglial development and neurological disorders. Nature Immunology, 18(6), 633–641. 10.1038/ni.3743 [DOI] [PubMed] [Google Scholar]
- Wu Y, Dissing-Olesen L, MacVicar BA, & Stevens B (2015). Microglia: Dynamic mediators of synapse development and plasticity. Trends in Immunology, 36(10), 605–613. 10.1016/j.it.2015.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia CF, Smith RS Jr., Shen B, Yang ZR, Borlongan CV, Chao L, & Chao J (2006). Postischemic brain injury is exacerbated in mice lacking the kinin B2 receptor. Hypertension, 47(4), 752–761. 10.1161/01.HYP.0000214867.35632.0e [DOI] [PubMed] [Google Scholar]
- Xie DI, Shen F, He S, Chen M, Han Q, Fang M, ... Deng Y (2016). IL-1beta induces hypomyelination in the periventricular white matter through inhibition of oligodendrocyte progenitor cell maturation via FYN/MEK/ERK signaling pathway in septic neonatal rats. Glia, 64(4), 583–602. 10.1002/glia.22950 [DOI] [PubMed] [Google Scholar]
- Xiong XY, Liu L, & Yang QW (2016). Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Progress in Neurobiology, 142, 23–44. 10.1016/j.pneurobio.2016.05.001 [DOI] [PubMed] [Google Scholar]
- Yin Y, Li E, Sun G, Yan HQ, Foley LM, Andrzejczuk LA, ... Sun D (2017). Effects of DHA on hippocampal autophagy and lysosome function after traumatic brain injury. Molecular Neurobiology, 10.1007/s12035-017-0504-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Milatovic D, Aschner M, Feustel PJ, & Kimelberg HK (2007). Neuroprotection by tamoxifen in focal cerebral ischemia is not mediated by an agonist action at estrogen receptors but is associated with antioxidant activity. Experimental Neurology, 204(2), 819–827. 10.1016/j.expneurol.2007.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Carney KE, Falgoust L, Pan JW, Sun D, & Zhang Z (2016). Emerging roles of Na(+)/H(+) exchangers in epilepsy and developmental brain disorders. Progress in Neurobiology, 138–140, 19–35. 10.1016/j.pneurobio.2016.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Nepomuceno R, Gao X, Foley LM, Wang S, Begum G, ... Sun D (2017). Deletion of the WNK3-SPAK kinase complex in mice improves radiographic and clinical outcomes in malignant cerebral edema after ischemic stroke. Journal of Cerebral Blood Flow & Metabolism,37(2),550–563. 10.1177/0271678X16631561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YQ, Liu JX, Li XZ, Xu L, & Xu YG (2009). RNA interference-mediated downregulation of Beclin1 attenuates cerebral ischemic injury in rats. Acta Pharmacologica Sinica, 30(7), 9l9–927. 10.1038/aps.2009.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Carney KE, Pigott VM, Falgoust LM, Clark PA, Kuo JS, & Sun D (2016). Glioma-mediated microglial activation promotes glioma proliferation and migration: Roles of Na+/H+ exchanger isoform 1. Carcinogenesis, 37(9), 839–85l. 10.1093/car-cin/bgw068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Fang C, Ji X, Liang X, Liu Y, Han C, ... Liu J (20l5). Estrogen receptor (ER)-alpha36 is involved in estrogen- and tamoxifen-induced neuroprotective effects in ischemic stroke models. PLoS One, 10(10), e0140660 10.1371/journal.pone.0140660 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.