

Abstract
Viruses drive carcinogenesis in human cancers through diverse mechanisms that have not been fully elucidated but include promoting immune escape. Here we investigated associations between virus-positivity and immune pathway alteration for 2009 tumors across six virus-related cancer types. Analysis revealed that for 3 of 72 human papillomavirus (HPV)-positive head and neck squamous cell carcinoma (HNSC) the HPV genome integrated in immune checkpoint genes PD-L1 or PD-L2, driving elevated expression in the corresponding gene. In addition to the previously described upregulation of the PD-1 immunosuppressive pathway in Epstein-Barr virus (EBV)-positive stomach tumors, we also observed upregulation of the PD-1 pathway in cytomegalovirus (CMV)-positive tumors. Furthermore, we found signatures of T-cell and B-cell response in HPV-positive HNSC and EBV-positive stomach tumors and HPV-positive HNSC patients were associated with better survival when T-cell signals were detected. Our work reveals that viral infection may recruit immune effector cells, and upregulate PD-1 and CTLA-4 immunosuppressive pathways.
Cao et al. show that human papillomavirus-positive, head and neck squamous cell carcinoma patients are associated with better survival when T-cells are activated. This study suggests that viral infection may recruit immune effector cells and that it may activate PD-1 and CTLA-4 immunosuppressive pathways.
Introduction
Since the sequencing of the first genomic DNA from a leukemia patient1, various studies have identified somatic and germline variants in key cancer genes2–5. These genomic biomarkers may aid in therapy selection6. Although large-scale sequencing projects such as The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC) continue to catalog variants across cancer types, only a minority of patients harbor tumors with genomic aberrations associated with sensitivity to targeted therapy.
Complementary to targeted therapy, cancer immunotherapy utilizes the host immune response to kill tumor cells7,8. PD-L1 and PD-L2 on tumor cells or antigen-presenting cells suppress T-cell immune response by binding to PD-1 on T-cells9,10. To escape attack by immune cells, tumor cells overexpress PD-L1 by gene amplification, utilization of an ectopic promoter, and disruption of 3′ untranslated regions (3′ UTRs)11, in addition to PTEN loss-of function12 and EGFR mutations13. Other studies indicate that EGFR mutations are not associated with an increased PD-L1 expression and a better clinical response of PD-L1 immune checkpoint inhibitors14,15. Elevated PD-L1 expression creates an immunosuppressive microenvironment that facilitates tumor progression16. Anti-PD-1 and anti-PD-L1 immune checkpoint blockades show favorable clinical outcome for treating patients with high PD-1 and PD-L1 expression17–20. Another important immune checkpoint pathway involves CTLA-4 and its ligands CD80 and CD86. CTLA-4 serves as a negative regulator of T-Cell activity. The anti-CTLA-4 blockade is also an effective therapeutic strategy to kill tumor cells21,22.
Immune infiltration of the tumor microenvironment correlates with improved survival in cancer patients23,24. Despite the importance of immune infiltrates and their theoretical associations with viral-positivity25, there is no systematic study of associations between virus-positive samples and the immune response except for some limited studies on human papillomavirus (HPV) in head and neck squamous cell carcinoma (HNSC)26–29. Our previous large-scale study demonstrated viral positivity in multiple cancer types30. Here, using TCGA RNA-Seq data for six virus-associated tumors, we systematically study the associations between virus-positivity and the tumor microenvironment, as measured by expression of PD-L1, PD-L2, PD-1, CD80, CD86, CTLA-4, Tim-3, LAG3, and 4-1BB and the prevalence of infiltrating immune cells across multiple types of human cancers. Specifically, we found the enrichment of a T-cell immune signature in HPV-positive and EBV-positive tumors compared to non-viral tumors. The increase of T-cell immune response is associated with a better prognosis in HPV-positive patients. In addition, we found HPV integrations at PD-L1 and PD-L2 are associated with high expression of these genes. Higher levels of PD-L1, PD-L2, PD-1, CD80, CD86, CTLA-4, Tim-3, LAG3, and 4-1BB expression were found in Epstein-Barr virus (EBV)-positive stomach adenocarcinoma (STAD) and cytomegalovirus (CMV)-positive colon and rectum adenocarcinoma (COADREAD) tumors compared to virus-negative tumors, providing the rationale for treating virus-positive tumors by anti-PD-1, and anti-CTLA-4 immune therapy. Besides PD-L1, PD-L2, PD-1, CD80, CD86, CTLA-4, Tim-3, LAG3, and 4-1BB, we found the increase of an inducible co-stimulator (ICOS) expression in both HPV-positive head and neck squamous cell carcinoma and EBV-positive stomach adenocarcinoma tumors. ICOS is the immune checkpoint protein, functionally and structurally related to CD2831. A positive ICOS signature may indicate a better clinical outcome of anti-CTLA-4 immune therapy in HPV-positive head and neck squamous cell carcinoma and EBV-positive stomach adenocarcinoma patients32.
Results
Recurrent HPV Integrations at PD-L1 or PD-L2 in HNSC
We analyzed 498 TCGA HNSC tumors using VirusScan and identified 72 HPV-positive tumors (numbers of virus-supporting reads per hundred million reads mapped (RPKM) > 100) and 341 virus-negative tumors (RPKM < 5)30. The 413 HNSC tumors with clear HPV status were most common in two ethnicity groups: 364 Caucasians and 30 African Americans. There was no significant difference in HPV status between the two ethnicity groups. We additionally found that HPV-positive HNSC tumors were mostly from males (92%). See Supplementary Table 1. Of these, we identified three tumors with HPV integrations at PD-L1 or PD-L2 by using discordant read pair analysis (Methods, Fig. 1a–c). In tumor TCGA-CV-5443, HPV integration sites were localized to intron 4 of PD-L1. The same integration site in the same sample was also reported in a previous study33. Given a larger cohort size, we also found additional previously unidentified HPV integration sites at PD-L1 and PD-L2 in tumors TCGA-T2-A6X0 and TCGA-HL-7533, respectively. Inspection of the detailed discordant read pairs showed that the viral E7 gene integrates into the 5′ UTR region of PD-L1; see Fig. 1b. HPV integrations at PD-L2 appeared more complicated than at PD-L1, revealing multiple HPV integration sites in or after intron 3 of PD-L2. These three tumors originated in different anatomic sites (larynx, tonsil, and oral cavity). Although different integration patterns and anatomic sites were observed in the three tumors, HPV integrations at PD-L1 or PD-L2 were all accompanied by higher expression levels of these genes compared with those in virus-negative tumors (Fig. 1d), and PD-L1 or PD-L2 with HPV integrations are expression outliers (see Methods).
Fig. 1.

HPV integrations at PD-L1 and PD-L2. a–c Structural analysis of HPV integration events at PD-L1 or PD-L2 for three different HNSC tumors. The dots in the plot show the coordinates in HPV genome (X) and human chromosome (Y) of the breakpoints67. The vertical line represents a series of breakpoints clustered together by Pindel66. In the figure, e1 represents exon 1. HPV integrates intron 4 and 5′ UTR of PD-L1 in tumor TCGA-CV-5443 and TCGA-T2-A6X0, respectively. In tumor TCGA-HL-7533, HPV can integrate multiple locations in or after intron 3 of PD-L2. The bottom panel shows the number of mapped HPV reads (log2 ratio) along with HPV coordinates and E6, E7, E1, E2, E4, E5, L2, and L1 are different genes in HPV genome. d The expression of PD-L1 or PD-L2 for HPV-positive samples with and without HPV integrations and virus-negative tumors
To examine the prevalence of HPV integrations at PD-L1 or PD-L2 in other tumor types, we analyzed 229 TCGA HPV-positive cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC) tumors and found no putative HPV integrations at either gene. We also checked for integrations of viruses, including HPV, hepatitis B virus (HBV), EBV, and cytomegalovirus, in other cancer types and found no integration events at PD-L1 or PD-L2. This result indicates that high PD-L1 or PD-L2 expression induced by HPV integration is a phenomenon that is selectively associated with HPV in HNSC tumors, with PD-L1 or PD-L2 integrations occurring in 4.2% of HNSC HPV-positive tumors.
We further looked the relationship between HPV integration and expression in T-cell and B-cell genes. Supplementary Fig. 1 shows the distribution of expression for those genes with HPV integrations. Interestingly, we found that NR4A2, TBC1D1, BTNL9, DTX1, FOXP1, INPP4B, PDE4D, and STAT4 with HPV integration events are expression outliers (see Methods) and all these events are associated with the increase of expression.
Effect of viral infection on levels of immune checkpoint genes
Having correlated gene-specific viral integration with elevated PD-L1 and PD-L2 expression in HPV-positive HNSC, we next correlated any viral infection with PD-L1, PD-L2, PD-1, CD80, CD86, CTLA-4, Tim-3, LAG3, and 4-1BB expressions. In Fig. 2, we compare expression levels of these genes in four tumor types positive for different viruses: HPV in HNSC, EBV in STAD, HBV in liver hepatocellular carcinoma (LIHC), and cytomegalovirus in colon and rectum adenocarcinoma (COADREAD). For the three viruses, we only observed an association between the ethnicity groups for HBV status: 98% are from ASIAN group. We observed most of EBV and HBV tumor patients are Male; See Supplementary Table 1. In HNSC, we found that three tumors with HPV integrations had high expression of PD-L1 or PD-L2, i.e., TCGA-CV-5443 and TCGA-T2-A6X0 with 11.8 and 9.2 for PD-L1 and TCGA-HL-7533 with 10 for PD-L2 (RSEM in log2 scale). RSEM stands for RNA-Seq reads by expectation maximization, which is widely used for quantifying gene expression34. Overall, no significant difference in PD-L1, PD-L2, CD80, or CD86 expression between HPV-positive and virus-negative HNSC tumors was found. A similar observation was also made for HBV (Fig. 2c). However, we found a higher level of PD-L1, PD-L2, CD80, CD86, Tim-3, LAG3, and 4-1BB in EBV-positive STAD and cytomegalovirus-positive colon and rectum adenocarcinoma than in virus-negative tumor samples (Fig. 2b–d). To leverage the new findings of elevated immune escape pathways of cytomegalovirus-positive colon and rectum adenocarcinoma to other cytomegalovirus-positive tumors, we compared cytomegalovirus-positive and negative tumor samples from stomach and esophageal carcinoma (STES). Supplementary Fig. 2a shows a higher PD-L1, PD-L2, or CD80 expression in cytomegalovirus-positive stomach and esophageal carcinoma.
Fig. 2.

Expression of immune checkpoint genes in virus-positive and negative tumors. The comparison of the expressions of immune checkpoint genes PD-L1, PD-L2, PD-1, CD80, CD86, CTLA-4, Tim-3, LAG-3, and 4-1BB as well as T-cell CD4 and CD8 markers for a HPV-positive and virus-negative tumors in HNSC, b EBV-positive and virus-negative tumors in STAD, c HBV-positive and virus-negative tumors in liver hepatocellular carcinoma and d Cytomegalovirus-positive and virus-negative tumors in colon and rectum adenocarcinoma. The “*”, “**”, and “***” symbols indicate p-value in the regions [0.05, 0.01], (0.01,0.001], and <0.001, respectively
In our previous study, we found a high prevalence of EBV-positive and cytomegalovirus-positive esophageal cancers30. In the current study, we found upregulation of PD-L2 expression (p = 0.02), CD80 (p = 0.01), and CD86 (0.04) associated with EBV or cytomegalovirus positivity (Supplementary Fig. 2b). In this analysis, we combined the EBV and cytomegalovirus-positive tumor samples together to improve the statistical power. In esophageal cancers, we obtained 10 EBV or cytomegalovirus-positive and 89 virus-negative tumors for the statistical analysis. We also noted that PD-1, CTLA-4, CD4, and CD8 expressions were also higher in the EBV and cytomegalovirus-positive samples, suggesting a higher level of infiltrating T-cells compared with virus-negative tumors (Fig. 2). Although an elevated PD-L1 expression in EBV-positive samples has been reported in other studies35,36, our study shows that EBV or cytomegalovirus infection increases expression of genes encoding PD-L1, PD-L2, PD-1, CD80, CD86, CTLA-4, Tim-3, LAG3, and 4-1BB immune checkpoint genes together with other T-cell markers such as CD4 and CD8 in tumors along the gastrointestinal tract, including esophagus, stomach, and intestine.
Effect of viral infection on host immune response
CD4+ and CD8+ T-cells and B-cells play important roles in fighting infection and cancer. Immune infiltration is frequently observed in solid tumors, and is associated with improved host survival23. Here, we evaluated associations between viral infection and immune cell infiltration in the tumor microenvironment. We collected a list of genes corresponding to T-cell and B-cell signatures (Supplementary Table 2) from previous publications37–39. We then identified 99 genes with significant differential expression (FDR < 0.05, see Methods) between HPV-positive and virus-negative HNSC tumors (Supplementary Data 1, Fig. 3). In the 99 selected genes, we also required that the difference of median values of gene expression (log2) is larger than 1 between the two cohorts. Overall, HPV-positive tumors displayed higher levels of T-cell signatures than virus-negative tumors (Fig. 3). In Fig. 3, we separated samples into four different groups based on supervised clustering results, i.e., Virus-/T-celllow, HPV+/T-celllow, Virus-/T-cellhigh, and HPV+/T-cellhigh. Overall, HPV-positive tumors displayed higher levels of T-cell signatures than virus-negative tumors (Fig. 3). The expressions of 99 T-cell genes in HPV-positive HNSC tumor are higher than the values in virus-negative samples (Supplementary Fig. 3). GSEA40,41 also shows the enrichment of T-cell gene set in HPV-positive HNSC tumors (Supplementary Fig. 4a). By using gene oncology (GO) database42,43, we found that most of the 99 genes are classified as gene sets related to immune response, lymphocyte and leukocyte, indicating infiltrated immune cells, and etc. No obviously different clusters were observed in term of GO annotation for these genes (Supplementary Fig. 5). We found that tumors with high T-cell signatures from our clustering method were associated with high PD-L1, PD-L2, PD-1, CD80, CD86, CTLA-4, Tim-3, LAG3, 4-1BB, CD8, and CD4 expression. These tumors also associated with high immune scores and lower tumor purity, indicative of a high level of immune infiltration (Supplementary Fig. 6a, c). Tumor purity and immune score were calculated based on the method used by Aran et al.44. In addition, HPV-positive HNSC tumors had elevated levels of PD-1, CTLA-4, CD8, and CD4 compared to virus-negative tumors (Fig. 2a). Similarly, we found an enrichment of B cell signatures in HPV-positive HNSC tumors (Supplementary Figs. 4b, 7).
Fig. 3.

The supervised clustering of T-cell genes in HPV-positive and virus-negative HNSC tumors. In the upper panel, we show the expression of T-ell markers such CD4, CD8 and immune checkpoint genes PD-L1, PD-L2, PD-1, CD80, CD86, CTLA-4, Tim-3, LAG-3, and 4-1BB. Tumors are clustered to four different groups based on the T-cell and virus statuses, i.e., Virus-/T-celllow, HPV+/T-celllow, Virus-/T-cellhigh, and HPV+/T-cellhigh from left to right
Next, we identified 78 T-cell genes with significant differential expression (FDR < 0.05, see Methods) between EBV-positive and virus-negative STAD tumors (Fig. 4 and Supplementary Data 1). In the 78 selected genes, we also required that the difference of median values of gene expression (log2) is larger than 1 between the two cohorts. GESA analysis indicates an enrichment of T-cell in EBV-positive STAD tumors (Supplementary Fig. 4c). GO database annotation shows that 77 differential expression are mostly related to immune response, lymphocyte, cell activation, etc. and we did not observe significant different clusters among these genes in term of GO annotation (Supplementary Fig. 8). Tumors with high T-cell signatures from our clustering analysis are concordant with high expression of PD-L1, PD-L2, PD-1, CD80, CD86, CTLA-4, Tim-3, LAG3, 4-1BB, CD8, and CD4, EBV-positive status (Fig. 5), and low tumor purity and high immune score (Supplementary Fig. 6b, d). B-cell response signatures were also observed in EBV-positive STAD tumors based on differential expression of 34 genes compared with non-viral tumors (Supplementary Fig. 9, 4d). This includes human leukocyte antigen (HLA) genes such as HLA-DQB1, HLA-DQA1, HLA-DMA, HLA-DMB, HLA-DRB5, and HLA-DOA. These genes play an important role for the formation of major histocompatibility complex (MHC) class II/peptide complex, which recognizes microbial antigens and cancer neoantigens45.
Fig. 4.

The supervised clustering of T-cell genes in EBV-positive and virus-negative STAD tumors. In the upper panel, we show the expression of T-cell markers such CD4, CD8 and immune checkpoint genes PD-L1, PD-L2, PD-1, CD80, CD86, CTLA-4, Tim-3, LAG-3, and 4-1BB. Tumors are clustered to four different groups based on the T-cell and virus statuses, i.e., Virus-/T-celllow, EBV+/T-celllow, Virus-/T-cellhigh, and EBV+/T-cellhigh from left to right
Fig. 5.
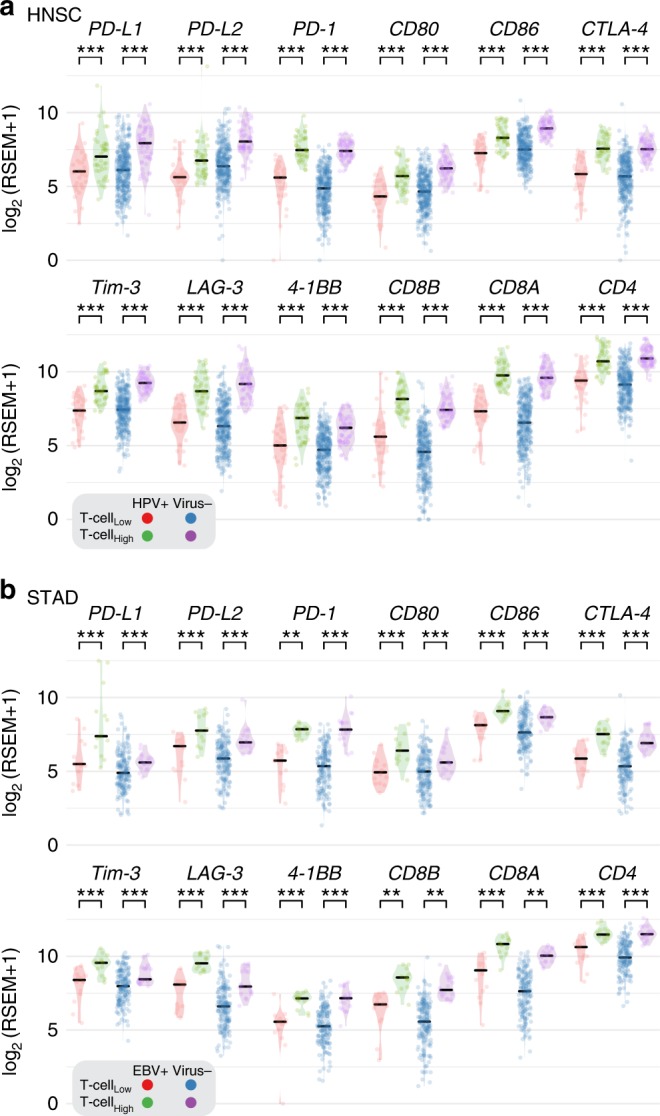
Expression of immune checkpoint genes in T-cellhigh and T-celllow tumors. The comparison of the expressions of immune checkpoint genes PD-L1, PD-L2, PD-1, CD80, CD86, CTLA-4, Tim-3, LAG-3, and 4-1BB as well as T-cell CD4 and CD8 markers between T-cellhigh and T-celllow tumors in HPV-positive HNSC, EBV-positive STAD and virus-negative cohorts
From our analyses, 78% of T-cell signature genes in EBV-positive tumors overlapped with those from HPV-positive HNSC, indicating that similar T-cell immune responses are associated with EBV and HPV. For instance, an ICOS signature was identified in EBV-positive STAD (Supplementary Table 2) as well as HPV-positive HNSC; The expression of ICOS increases about two-fold in EBV-positive STAD and HPV-positive HNSC compared to virus-negative samples. ICOS is an immune checkpoint gene in CD28 and CTLA-4 family, which plays important role in regulating the immune response and enhances the antitumor immune response in anti-CTLA-4 blockade31,32.
Furthermore, we selected genes, which showed at least four-fold difference in the relative expression between HPV-positive and EBV-positive samples compared to the corresponding virus-negative cohorts. In the T-cell gene list, six genes (PLAC8, CXCL9, SATB1, PDE9A, NPTXR, and NELL2) passed the cut-off. We note that for PLAC8, a gene that is associated with pancreatic cancer progression46, was highly elevated in HPV-positive samples with about 16-fold increase compared to the virus-negative HNSC samples. However, in EBV-positive STAD sample, the expression of PLAC8 was downregulated by about four-fold compared to virus-negative STAD sample (Supplementary Table 2). In the B-cell gene list, we identified an additional 18 genes fitting these criteria, such as STAG3, MET1, CDKN2A, COCH, BTNL9, SPIB, MS4A1, CD19, CR2, BLK, VPREB3, CXCR5, MIR600HG, BANK1, F5, BACH2, KYNU, and SLC22A3. CDKN2A showed distinct expression alteration in HPV-positive and EBV-positive samples. In HPV-positive samples, the expression of CDKN2A was 16-fold higher than virus-negative HNSC samples, but in EBV-positive samples, its expression was three-fold lower than the virus-negative samples. CDKN2A is a tumor suppressor, which is highly mutated in virus-negative HNSC samples47, but not in HPV-positive HNSC samples. The low expression of CDKN2A found in EBV-positive STAD and HPV-negative HNSC samples reflect two different mechanisms inactivating the gene function by EBV infection and somatic mutation. We note that CDKN2A is mostly involved in the regulation of cell cycle48, not necessarily related to the immune infiltration. The latter is mainly associated with a low tumor purity and increased overall expression of T-cell genes (Figs. 3, 4). In contrast to HPV-positive HNSC and EBV-positive STAD, we did not find significant enrichment of T-cell and B-cell signatures in HBV-positive liver hepatocellular carcinoma. For colon and rectum adenocarcinoma, we find higher levels of CD4, CD8A, and PD-1 in cytomegalovirus-positive tumors than in virus-negative tumors (Fig. 2), though other T-cell genes only showed modest differential expression in cytomegalovirus-positive and virus-negative tumors.
We further compared the T-cell differentiation phenotype in virus-positive and negative tumors for different viruses by using markers such as CD28, CD27, CD45, CD103, perforin, GMP-17, and granzymeA49 (see Supplementary Fig. 10). High expression of CD28 and CD27 in HPV-positive HNSC and cytomegalovirus-positive colon and rectum adenocarcinoma tumors suggest an increase in the presence of CD28+CD27+ T-cells compared to virus-negative HNSC tumors and virus-negative colon and rectum adenocarcinoma tumors. CD28 and CD27 are markers for precursor or early differentiation T-cells49. The HPV-positive HNSC tumors and cytomegalovirus-positive colon and rectum adenocarcinoma tumors also had higher expression of NK T-cell markers (perforin, GMP-17, and granzymeA) and CD45 compared with virus-negative HNSC and colon and rectum adenocarcinoma tumors. Also, HPV-positive HNSC tumors and EBV-positive tumors had a higher expression of CD103, a marker for resident T-cells, compared to virus-negative samples, but not in cytomegalovirus-positive colon and rectum adenocarcinoma and HBV-positive liver hepatocellular carcinoma tumors.
Clinical relevance of virus-associated immune response
Based on our clustering analysis, HPV-positive tumors were more likely to have an elevated immune response (40 of 72 tumors, 55%) compared with virus-negative tumors (57 of 341 tumors, 16%) (p < 0.01, Fisher’s exact test, two-tailed) (Fig. 3). We first performed survival analysis to evaluate how immune response correlates with clinical outcome in HPV-positive and virus-negative cohorts (Fig. 6a, b, respectively). We found that immune response is associated with a positive prognosis in patients with HPV-positive HNSC, but not in those with virus-negative HNSC (Fig. 6a). We then examined mutational and gene expression patterns in two cohorts: HPV-positive HNSC with elevated immune response and virus-negative HNSC with elevated immune response (Fig. 6c); in total, we found 2695 genes with at least two-fold differential expression (FDR < 0.05). We identified 103 genes with 16-fold or higher differential expression (Fig. 6c). Notably, we found that the expression of FOXA1, a gene associated with better survival in breast cancer50, is about 16-fold higher in HPV-positive tumors. We also identified seven highly mutated genes (TP53, PIK3CA, CDKN2A, FAT1, NOTCH1, KMT2D, and NSD1) with frequency greater than 10% in HNSC tumors with elevated immune response. The HPV-positive cohort contained no variants in either TP53 or CDKN2A and only one in FAT1; in the HPV-negative cohort, these genes were frequently mutated. HNSC tumors with wild-type TP53 are more sensitive to radiation therapy than tumors with TP53 mutations51. In addition, we examined differentially expressed genes in the p53 signaling pathway between the two cohorts and found 4 of 16 p53 pathway genes showing substantial expression alteration (see Fig. 6d). Levels of B-cell CLL/lymphoma 2 (BCL2) and E2F transcription factor 1 (E2F1) are higher in HPV-positive tumors. Previous studies show favorable prognosis with a high expression of BCL252 and poor prognosis with overexpressed CCND153. The distinct expression pattern of p53 signaling pathway genes may also drive different clinical outcome of the two cohorts, though both cohorts are associated with an infiltrated immune cell microenvironment.
Fig. 6.

Survival and gene expression analyses in HPV-positive and negative tumors. The comparison of overall survival in T-cellhigh and T-celllow tumors between two different cohorts: a HPV-positive and b virus-negative. c The mutational status for seven mutated genes in HNSC and the supervised clustering based on 103 genes which demonstrate strong significantly differential expression among HPV-positive and virus-negative HNSC tumors (FDR < 0.05, see Methods). d The differentially expressed genes involved in p53 signaling pathway between HPV-positive and virus-negative tumors
Discussion
In this study, we systematically investigated associations between virus infection or integration and alteration of the tumor microenvironment. We found a significant difference (p = 0.01, Fisher’s exact test) between HPV integration status at PD-L1 or PD-L2 in HPV-positive HNSC (N = 72) and CESC (N = 229) tumors. Specifically, we found three integrations among the HNSC samples with high expression of PD-L1 or PD-L2 and no integrations among the CESC samples. It is likely that HPV has co-evolved to target PD-L1 or PD-L2 to create an immunosuppressive tumor microenvironment in some head and neck cancers. Further investigation with a large sample set is important for leveraging the observed relationship between virus integration at PD-L1 or PD-L2 in HNSC and the increased expression. We also found samples with HPV integrations in other immune-related genes (NR4A2, TBC1D1, BTNL9, DTX1, FOXP1, INPP4B, PDE4D, and STAT4) have an increased expression of these genes. Previous studies show that high expression of NR4A2, BTNL9, FOXP1, or PDE4D can antagonize immune response or is associated with tumor progression54–57. In EBV-positive STAD, cytomegalovirus-positive colon and rectum adenocarcinoma and EBV or cytomegalovirus positive stomach and esophageal carcinoma, we found that viral infection associates with high expression of PD-L1 or PD-L2, CD80, CD86, PD-1, CTLA-4, Tim-3, LAG3, and 4-1BB without integrating into the human genome. Moreover, our study indicates that EBV and cytomegalovirus elevate PD-L1 or PD-L2, CD80, CD86, PD-1, CTLA-4, Tim-3, LAG3, and 4-1BB expression in multiple cancers along the gastrointestinal tract including stomach and esophageal carcinoma and colon and rectum adenocarcinoma.
A previous study58 demonstrated elevated PD-L1 expression in both tumor and immune cells across a large number of tumor samples by using immunohistochemistry (IHC) assays. For example, in 101 head-neck tumors, they found 28 and 19% of immune cell and tumor cell, respectively, were positive for PD-L1 in their samples. The anti-PD-L1 antibody works well on PD-L1 positive tumors to neutralize PD-L1 and make the tumor susceptible to attack by the immune system58. Our studies show an elevated PD-L1 expression in EBV and cytomegalovirus-positive samples, suggesting clinical trials of PD-L1 immunotherapy in these patients may be beneficial. Also, in a subset of HPV-positive HNSC, PD-L1 is highly expressed when HPV integrates into the PD-L1, suggesting these patients may have responded to anti-PD-L1 therapies. The previous clinical trial on HNSC has shown longer overall survival in both HPV-positive and PD-L1-positive tumors when treating with ant-PD-1 monoclonal antibody than the standard single-agent therapy59. Furthermore, we found that the expression of PD-L2, CD80, CD86, and CTLA-4 are also elevated in cytomegalovirus and EBV-positive tumor patients, suggesting anti-PD-L2 and anti-CTLA4 immunotherapy may be effective in patients with these types of tumors.
In contrast to the clear association between EBV and stomach adenocarcinoma, some controversy exists about the association between cytomegalovirus and colorectal cancer60–62. In our previous large-scale study, we found a higher abundance of cytomegalovirus in tumors than in adjacent normal samples30. In the current study, we discovered a high level of PD-L1/PD-L2 in cytomegalovirus-positive tumors across the gastrointestinal tract suggesting that cytomegalovirus mediates the tumor microenvironment, which helps tumor cells to avoid the attack of immune cells.
In addition, we found distinct immune responses for different viruses in different cancer types. A high level of immune response was observed in HPV-positive HNSC and EBV-positive STAD samples but not in HBV-positive liver hepatocellular carcinoma. One explanation is that HBV promotes cancer in a different way than EBV/HPV, which are directly oncogenic, HBV promotes cancer by making the liver cirrhotic/inflamed chronically. The immune response was measured by gene expression of characteristic T-cell and B-cell markers, including CD4, CD8, and PD-1 T-cell markers, and tumor purity, which when low indicates high immune cell infiltration into the tumor microenvironment. We also observed high expression of ICOS and CTLA-4 in both HPV-positive HNSC and EBV-positive STAD, suggesting these tumors may have had an effective clinical response to anti-CTLA-4 immune therapy. Survival analysis shows high immune response is associated with favorable survival in HPV-positive but not HPV-negative HNSC samples. The different mutational status and expression patterns may lead to different clinical outcomes of the immune response. For instance, we found different expression alteration in key genes involved p53 signaling pathway in two cohorts. The complete retention of wild-type TP53 in HPV-positive HNSC tumors is another key factor driving the difference, as previous studies show better radiotherapy sensitivity in HNSC patients with wild-type TP5351. For cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), we separated tumors into low and elevated immune infiltration cohorts according to HPV-positive T-cell signatures (Supplementary Fig. 11). Although tumors with elevated immune response show a better survival rate before eight years, there is no significant difference in overall survival rate based on immune response (Supplementary Fig. 12). Further clustering samples based on a CD8+ T-cell gene list show improved association survival rate and CD8+ T-cell status and patients with CD8+ T-cell status have a higher chance of a tumor-free status (Supplementary Figs. 13, 14).
Our study highlights the importance of viral integration and infection in shaping tumor microenvironments. The current study is necessarily based on gene expression data from RNA-Seq. Proteomics data from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) will enable us to investigate the virus-mediated tumor microenvironment at the protein level63. The highly immunogenic property of HPV16 virus, the dominant HPV subtype affecting HNSC patients, can help to explain the increased immune response in HPV-positive HNSC samples (Supplementary Fig. 15a). There is no significant difference in terms of mutational burden in HPV-positive and virus-negative samples (Supplementary Fig. 15b). However, why different patients have different responses to different viral presentation requires more detailed work in the future. In addition, though the current work does not identify clear mechanisms by which virus infection affects PD-L1 and PD-L2 expression, it nonetheless suggests that viruses may aid tumors in evading the PD-1 immune checkpoint pathway across multiple cancer types. Our analysis of elevated expression in both PD-1, CTLA-4, Tim-3, LAG3, and 4-1BB checkpoint genes and immune response in virus-positive tumors may contribute to therapy selection in these patients.
Methods
Virus integration
We discovered viruses in the tumor samples by using the VirusScan pipeline30, which is available from Github64. For the identification of virus integration sites in human genome, we first used BWA65 to align RNA-Seq data to the human plus viral reference. From the re-aligned bam file, we extracted the discordant read pairs, where one read of a read pair maps to human, the other to virus. Pindel66 was used to identify exact breakpoints for all samples with ten or more human-virus discordant reads. The breakpoints in Fig. 1 were visualized by using BreakPointSurveyor67.
Statistical analysis
Survival analysis was implemented by using R package survival. We used the Student’s t-test to extract differentially expressed genes in virus-positive and negative samples, using FDR = 0.05 as the cut-off. The FDR value was obtained by p.adjust with Benjamini and Hochberg correction from R package. The heatmap figure was generated by using Heatmap.3R package with default parameters.
Expression outlier analysis
To investigate if genes with virus integrations are expression outliers, we used the Tukey’s standard formula to quantify the outlier score:
Outlier score = (x − Q3)/IQR for upper tail and (x − Q1)/IQR for low tail,
where IQR is the interquartile range, Q1 and Q3 are the first and third quartiles, respectively and x is the RSEM value in a log2 scale. In the current study, genes with an outlier score greater than 1.0 or less than −1.0 are considered to be expression outliers.
Neoantigen prediction
Different lengths of epitopes (8mer, 9mer, 10mer, and 11mer) are constructed from HPV16 protein sequences. We use NetMHC3pan68 to predict the binding affinity between epitopes and MHC based on the HLA type in each tumor. The HLA type was adopted from ref. 69. Epitopes with binding affinity ≤500 nM which are also not present in Ensembl 70.37 database are extracted for the following neoantigen analysis.
Reporting Summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
This work was supported by the National Cancer Institute grant R01CA178383 and National Human Genome Research Institute grant U01HG006517 to L.D. The Cancer Genome Atlas (cancergenome.nih.gov) was the source of primary data.
Author contributions
L.D. designed and supervised research. S.C., K.W., M.A.W., A.K., J.L., S.S., and R.J.M. analyzed the data. S.C. performed statistical analysis. S.C., M.A.W., A.K., and W.L. prepared figures and tables. X.W., K.J., J.F.D., H.G., L.R., F.C., D.R.A., and L.D. provided disease specific analysis and guidance. S.C. and L.D. wrote the manuscript. K.W., R.J.M., K.J., J.F.D., H.G., F.C., D.R.A., and L.D. revised the manuscript.
Data availability
We collected gene expression (RSEM), and clinical data from Broad firehose70 across six cancer types including cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), colon adenocarcinoma and rectal adenocarcinoma (COADREAD), esophageal cancer (ESCA), head/neck squamous cell carcinoma (HNSC), stomach adenocarcinomas (STAD), and liver hepatocellular carcinoma (LIHC) from The Cancer Genome Atlas (TCGA). The aligned TCGA RNA-Seq bams included in this study can be downloaded from the NCI’s Genomic Data Commons (GDC). The source gene expression data are available in the Supplementary Data 1.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s42003-019-0352-3.
References
- 1.Ley TJ, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lawrence MS, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kandoth C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang JH, et al. Germline mutations in predisposition genes in pediatric cancer. New Engl. J. Med. 2015;373:2336–2346. doi: 10.1056/NEJMoa1508054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu, C. et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nat. Commun.6, 10086 (2015). [DOI] [PMC free article] [PubMed]
- 6.Rubio-Perez, C. et al. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals novel targeting opportunities. Cancer Res.75, 2983 (2015). [DOI] [PubMed]
- 7.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 8.Stronen E, et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. 2016;352:1337–1341. doi: 10.1126/science.aaf2288. [DOI] [PubMed] [Google Scholar]
- 9.Latchman Y, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 10.Zou, W. P., Wolchok, J. D. & Chen, L. P. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 8, 328rv4 (2016). [DOI] [PMC free article] [PubMed]
- 11.Kataoka K, et al. Aberrant PD-L1 expression through 3’-UTR disruption in multiple cancers. Nature. 2016;534:402–406. doi: 10.1038/nature18294. [DOI] [PubMed] [Google Scholar]
- 12.Parsa AT, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007;13:84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 13.Akbay EA, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3:1355–1363. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roussel H, et al. Composite biomarkers defined by multiparametric immunofluorescence analysis identify ALK-positive adenocarcinoma as a potential target for immunotherapy. Oncoimmunology. 2017;6:e1286437. doi: 10.1080/2162402X.2017.1286437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soo RA, et al. Immune checkpoint inhibitors in epidermal growth factor receptor mutant non-small cell lung cancer: Current controversies and future directions. Lung Cancer. 2018;115:12–20. doi: 10.1016/j.lungcan.2017.11.009. [DOI] [PubMed] [Google Scholar]
- 16.Freeman GJ, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eroglu Z, et al. High response rate to PD-1 blockade in desmoplastic melanomas. Nature. 2018;553:347–350. doi: 10.1038/nature25187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gopalakrishnan V, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103. doi: 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matson V, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359:104–108. doi: 10.1126/science.aao3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le DT, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 22.Wei SC, et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell. 2017;170:1120–1133 e1117. doi: 10.1016/j.cell.2017.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iglesia, M. D. et al. Genomic analysis of immune cell infiltrates across 11 tumor types. J. Natl Cancer Inst.10.1093/jnci/djw144 (2016). [DOI] [PMC free article] [PubMed]
- 24.Li B, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 2016;17:174. doi: 10.1186/s13059-016-1028-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldszmid RS, Dzutsev A, Trinchieri G. Host immune response to infection and cancer: unexpected commonalities. Cell Host Microbe. 2014;15:295–305. doi: 10.1016/j.chom.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fakhry C, et al. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J. Natl Cancer Inst. 2008;100:261–269. doi: 10.1093/jnci/djn011. [DOI] [PubMed] [Google Scholar]
- 27.Lyford-Pike S, et al. Evidence for a role of the PD-1:PD-L1 pathway in immune resistance of HPV-associated head and neck squamous cell carcinoma. Cancer Res. 2013;73:1733–1741. doi: 10.1158/0008-5472.CAN-12-2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Badoual C, et al. PD-1-expressing tumor-infiltrating T cells are a favorable prognostic biomarker in HPV-associated head and neck cancer. Cancer Res. 2013;73:128–138. doi: 10.1158/0008-5472.CAN-12-2606. [DOI] [PubMed] [Google Scholar]
- 29.Chakravarthy A, et al. Human papillomavirus drives tumor development throughout the head and neck: improved prognosis is associated with an immune response largely restricted to the oropharynx. J. Clin. Oncol. 2016;34:4132–4141. doi: 10.1200/JCO.2016.68.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao S, et al. Divergent viral presentation among human tumors and adjacent normal tissues. Sci. Rep. 2016;6:28294. doi: 10.1038/srep28294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hutloff A, et al. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature. 1999;397:263–266. doi: 10.1038/16717. [DOI] [PubMed] [Google Scholar]
- 32.Fan X, Quezada SA, Sepulveda MA, Sharma P, Allison JP. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J. Exp. Med. 2014;211:715–725. doi: 10.1084/jem.20130590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parfenov M, et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc. Natl Acad. Sci. USA. 2014;111:15544–15549. doi: 10.1073/pnas.1416074111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen BJ, et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin. Cancer Res. 2013;19:3462–3473. doi: 10.1158/1078-0432.CCR-13-0855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iglesia MD, et al. Prognostic B-cell signatures using mRNA-seq in patients with subtype-specific breast and ovarian cancer. Clin. Cancer Res. 2014;20:3818–3829. doi: 10.1158/1078-0432.CCR-13-3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Palmer, C., Diehn, M., Alizadeh, A. A. & Brown, P. O. Cell-type specific gene expression profiles of leukocytes in human peripheral blood. BMC Genom.7, 115 (2006). [DOI] [PMC free article] [PubMed]
- 39.Schmidt M, et al. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 2008;68:5405–5413. doi: 10.1158/0008-5472.CAN-07-5206. [DOI] [PubMed] [Google Scholar]
- 40.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liberzon A, et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aran D, Sirota M, Butte AJ. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015;6:8971. doi: 10.1038/ncomms9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rimsza LM, et al. Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: a follow-up study from the Leukemia and Lymphoma Molecular Profiling Project. Blood. 2004;103:4251–4258. doi: 10.1182/blood-2003-07-2365. [DOI] [PubMed] [Google Scholar]
- 46.Kinsey C, et al. Plac8 links oncogenic mutations to regulation of autophagy and is critical to pancreatic cancer progression. Cell Rep. 2014;7:1143–1155. doi: 10.1016/j.celrep.2014.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 49.Appay V, et al. Memory CD8+T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 2002;8:379–385. doi: 10.1038/nm0402-379. [DOI] [PubMed] [Google Scholar]
- 50.Badve S, et al. FOXA1 expression in breast cancer–correlation with luminal subtype A and survival. Clin. Cancer Res. 2007;13:4415–4421. doi: 10.1158/1078-0432.CCR-07-0122. [DOI] [PubMed] [Google Scholar]
- 51.Kimple RJ, et al. Enhanced radiation sensitivity in HPV-positive head and neck cancer. Cancer Res. 2013;73:4791–4800. doi: 10.1158/0008-5472.CAN-13-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ichim G, Tait SW. A fate worse than death: apoptosis as an oncogenic process. Nat. Rev. Cancer. 2016;16:539–548. doi: 10.1038/nrc.2016.58. [DOI] [PubMed] [Google Scholar]
- 53.Lin, R. J. et al. Cyclin D1 overexpression is associated with poor prognosis in oropharyngeal cancer. J. Otolaryngol. Head Neck Surg.42, 23 (2013). [DOI] [PMC free article] [PubMed]
- 54.Sekiya T, et al. The nuclear orphan receptor Nr4a2 induces Foxp3 and regulates differentiation of CD4+T cells. Nat. Commun. 2011;2:269. doi: 10.1038/ncomms1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamazaki T, et al. A butyrophilin family member critically inhibits T cell activation. J. Immunol. 2010;185:5907–5914. doi: 10.4049/jimmunol.1000835. [DOI] [PubMed] [Google Scholar]
- 56.Hsiao HW, et al. Deltex1 is a target of the transcription factor NFAT that promotes T cell anergy. Immunity. 2009;31:72–83. doi: 10.1016/j.immuni.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 57.Rahrmann EP, et al. Identification of PDE4D as a proliferation promoting factor in prostate cancer using a sleeping beauty transposon-based somatic mutagenesis screen. Cancer Res. 2009;69:4388–4397. doi: 10.1158/0008-5472.CAN-08-3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Herbst RS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ferris RL, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2016;375:1856–1867. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harkins L, et al. Specific localisation of human cytomegalovirus nucleic acids and proteins in human colorectal cancer. Lancet. 2002;360:1557–1563. doi: 10.1016/S0140-6736(02)11524-8. [DOI] [PubMed] [Google Scholar]
- 61.Bender C, et al. Analysis of colorectal cancers for human cytomegalovirus presence. Infect. Agent. Cancer. 2009;4:6. doi: 10.1186/1750-9378-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Collins D, Hogan AM, Winter DC. Microbial and viral pathogens in colorectal cancer. Lancet Oncol. 2011;12:504–512. doi: 10.1016/S1470-2045(10)70186-8. [DOI] [PubMed] [Google Scholar]
- 63.Mertins P, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55–5. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cao, S. & Ding, L. VirusScan Pipeline, https://github.com/ding-lab/VirusScan (2016).
- 65.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ye K, Schulz MH, Long Q, Apweiler R, Ning ZM. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wyczalkowski MA, et al. BreakPoint Surveyor: a pipeline for structural variant visualization. Bioinformatics. 2017;33:3121–3122. doi: 10.1093/bioinformatics/btx362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nielsen M, Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016;8:33. doi: 10.1186/s13073-016-0288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thorsson V, et al. The immune landscape of cancer. Immunity. 2018;48:812–830. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Broad firehose, http://gdac.broadinstitute.org/runs/stddata__2016_01_28/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
We collected gene expression (RSEM), and clinical data from Broad firehose70 across six cancer types including cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), colon adenocarcinoma and rectal adenocarcinoma (COADREAD), esophageal cancer (ESCA), head/neck squamous cell carcinoma (HNSC), stomach adenocarcinomas (STAD), and liver hepatocellular carcinoma (LIHC) from The Cancer Genome Atlas (TCGA). The aligned TCGA RNA-Seq bams included in this study can be downloaded from the NCI’s Genomic Data Commons (GDC). The source gene expression data are available in the Supplementary Data 1.