

Abstract
Background
Osteosarcoma (OSA) is a common bone tumor of mesenchymal origin in dogs. Chemotherapy delays metastasis, yet most dogs die of this disease within 1 year of diagnosis. The high metabolic demand of cancer cells promotes proton pump upregulation, leading to acidification of the tumor microenvironment and chemoresistance. The potassium‐sparing diuretic amiloride is among a class of proton pump inhibitors prescribed for refractory heart failure treatment in dogs.
Objective
We hypothesized that amiloride treatment improves chemotherapy response by reducing acidification in canine OSA cells. Our objective was to assess the in vitro effects of amiloride on cell viability, apoptosis, and metabolism.
Methods
In vitro study. Assessments of cell viability and apoptosis were performed after single agent or combination treatment, along with calculations of pharmacological synergism using the combination index. Protein signaling during apoptosis was evaluated by Western blotting. Metabolic profiling was performed using a Seahorse bioanalyzer.
Results
Amiloride strongly synergized with doxorubicin in combination treatment and exhibited additive or antagonistic effects with carboplatin in canine OSA cells. Combination treatment with doxorubicin significantly upregulated p53‐mitochondrial signaling to activate apoptosis and downregulate Akt phosphorylation. Amiloride‐treated cells further exhibited metabolic switching with reductions in glycolytic capacity and maximal respiration.
Conclusion and Clinical Importance
Amiloride synergized with doxorubicin to potentiate apoptosis in canine OSA cells. These results justify further investigation into repurposing of amiloride as an oncology drug for the treatment of OSA in dogs.
Keywords: chemoresistance, dog, doxorubicin, acidification, proton pumps, sensitization
Abbreviations
- 2‐DG
2‐deoxy‐D‐glucose
- A590
relative absorbance at 590 nm
- ATP
adenosine triphosphate
- Bax
Bcl‐2‐associated X protein
- Bcl‐xL
B‐cell lymphoma‐extra large
- BSA
bovine serum albumin
- Cmax
peak serum concentration
- CI
combination index
- DMEM
Dulbecco's modified eagle medium
- DMSO
dimethyl sulfoxide
- ECAR
extracellular acidification rate
- ENaC
epithelial sodium channel
- FBS
fetal bovine serum
- FCCP
carbonyl cyanide‐4‐(trifluoromethoxy)phenylhydrazone
- FITC
fluorescein isothiocyanate
- HIF
hypoxia‐inducible factor
- IC50
half‐maximal inhibitory concentration
- MFI
mean fluorescence intensity
- NHE
sodium‐hydrogen exchanger
- OCR
oxygen consumption rate
- OSA
osteosarcoma
- PBS
phosphate‐buffered saline
- PI
propidium iodide
- PK
pharmacokinetics
- RT
room temperature
- TBST
tris‐buffered saline/tween
- uPA
urokinase plasminogen activator
1. INTRODUCTION
Osteosarcoma (OSA) is the most common primary bone malignancy in dogs. Most tumors affect the appendicular skeleton, and the standard of care involves amputation of the affected limb with adjuvant chemotherapy.1 Current research efforts are aimed at delaying pulmonary metastasis and the onset of chemoresistance.
The high metabolic demand of cancer cells promotes aerobic glycolysis irrespective of oxygen tension, a phenomenon well known as the Warburg effect.2 This metabolic stress has been linked to tumor acidosis and hypoxia, hallmarks of solid tumors that contribute to increased immune evasion and chemoresistance. The development and maintenance of an intracellular pH gradient in cancer cells can be attributed, at least in part, to the action of sodium‐hydrogen exchangers (NHEs).3 Sodium‐hydrogen exchangers are ubiquitous, adenosine triphosphate (ATP)‐independent antiporters that exchange Na+ for H+ at 1 : 1 stoichiometry. Physiologically, NHEs regulate pH homeostasis and cellular response to mitogens and are critical for acid‐base balance and sodium reabsorption.4 NHE1 is the most widely expressed isoform, which, under low oxygen tension, is upregulated by hypoxia‐inducible factors (HIFs), in particular HIF‐1α.5 The amiloride family of drugs are inhibitors of NHEs, and chemical substitutions increase the affinity for inhibition of the exchanger. Amiloride hydrochloride (amiloride) is the only FDA‐approved NHE inhibitor.
Tumor acidosis in solid tumors contributes to acquire chemoresistance under selective pressures.6 Comparable to the reported evolution of multidrug resistance transporters, studies in human cancer cells now independently show that common chemotherapeutics, such as doxorubicin,7 mitoxantrone,8 and vincristine,9 are actively sequestered within acidic lysosomes. This sequestration prevents these drugs from exerting their DNA damaging or crosslinking effects and leads to their eventual degradation within cancer cells. Likewise, platinum agents, such as cisplatin and carboplatin, are actively transported into lysosomes through the endosomal expression of copper transporters.10 As such, targeting proton pumps responsible for endosomal acidification has been suggested to improve chemotherapy response in cancer cells.11
Identification of the role for NHEs in ion balance has led to the development of NHE inhibitors for the treatment of hypertension in humans and dogs.12, 13 Among this family of drugs is amiloride. Long used as a kaliuretic, amiloride is well tolerated in humans and dogs and is currently listed on the WHO's List of Essential Medicines in the universal health care system.13, 14, 15 Additionally, this drug is a classical inhibitor of NHE1, where the amiloride‐binding region resides within the transmembrane domain of the transporter, and it has been used/studied widely in vertebrates.16
Although inhibition of vacuolar‐type H+ (V)‐ATPases has recently been suggested to exhibit anticancer activity in a pilot study involving dogs,11 the use of amiloride in treatment of cancer in dogs has not been extensively studied. Amiloride sensitizes human pancreatic cancer cells to erlotinib by downregulation of PI3K/Akt signaling17 and possesses antimyeloma activity in cells with wild‐type or mutated TP53.18 However, the impact of amiloride on cell signaling during apoptosis and metabolism is poorly understood. Our current study aimed to evaluate the effects of amiloride on chemotherapy response in vitro by elucidating its influence on primary and metastatic canine OSA cells during apoptosis and its functional consequences on cancer cell metabolism.
2. MATERIALS AND METHODS
2.1. Cell lines and cell culture conditions
Canine OSA cell lines D17, Abrams, and Dharma were used. D17 was obtained from Sigma‐Aldrich/European Collection of Cell Cultures (ECACC; Salisbury, United Kingdom). Abrams was kindly provided by Dr Michael K. Huelsmeyer at the University of Wisconsin. Both D17 and Abrams cell lines were characterized and published as canine OSA cells based on morphology and xenograft analysis.19 Dharma was isolated from a primary tumor by Dr Anthony J. Mutsaers and verified to be OSA by histopathology of tumors produced from xenograft implantation in nude mice.20 All cells were cultured in high glucose Dulbecco's Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS) and 100 U/mL penicillin/streptomycin (Thermo Fisher Scientific, Waltham, Massachusetts) and maintained in a humidified incubator at 37°C with 5% CO2.
2.2. Chemical reagents
Amiloride hydrochloride was purchased from Tocris Bioscience (Bristol, United Kingdom) and resuspended at a stock concentration of 100 mM with dimethyl sulfoxide (DMSO; Sigma‐Aldrich, St. Louis, Missouri). Doxorubicin and carboplatin (both from Accord Healthcare, Kirkland, Quebec, Canada) were obtained from the Ontario Veterinary College pharmacy and maintained at their stock concentrations of 2 and 10 mg/mL, respectively, in isotonic solution. Pifithrin‐μ was purchased from Sigma‐Aldrich and maintained in UltraPure DNase/RNase‐free distilled water at 1 mM.
2.3. Crystal violet cytotoxicity assay
The half‐maximal inhibitory concentration (IC50) for all drugs tested was determined systematically by the crystal violet cytotoxicity assay. D17, Abrams, and Dharma were seeded at 2 x 103, 2 x 103, and 4 x 103 cells per well, respectively, and incubated for 16 hours in 96‐well plates. For dose‐response curves, each cell line was treated with increasing doses of amiloride (10‐300 μM), doxorubicin (10‐100 nM), or carboplatin (10‐100 μM) for 72 hours. The cells were then fixed with 0.5% crystal violet in 20% methanol (Thermo Fisher Scientific), and the dye was eluted with 10% acetic acid after drying overnight. The absorbance at 590 nm (A590) was read in triplicate and normalized to a vehicle control containing 0.3% DMSO at the highest tested dose. Half‐maximal inhibitory concentration curves were fit by nonlinear regression and averaged by experiments conducted in triplicate.
2.4. Combination index computations
Combination drug treatments were performed for amiloride with doxorubicin or carboplatin compared to single agent treatment. Five drug concentration pairings near the determined IC50 were chosen for each drug. Synergism and efficacy of the drug combinations were assessed by computing combination index (CI) values using the Chou‐Talalay method.21
2.5. Annexin V/Propidium iodide flow cytometry
Apoptotic cells and dead cells were quantified by flow cytometry using fluorescein isothiocyanate (FITC)‐conjugated annexin V and propidium iodide (PI), respectively (eBioscience, San Diego, California). Each cell line was treated with single agent or combination drugs for 72 hours before staining with Annexin V‐FITC (1:400) and 2 μg/mL PI for 15 minutes at room temperature (RT). Cell fractions and conditioned media were run on the BD Accuri C6 flow cytometer (BD Biosciences, Franklin Lakes, New Jersey), with laser intensities set based on control readings and threshold at 5 x 104 events per group. Compensation was set for each data set to account for spillover between FITC and PI emission overlap (FL1 and FL3 channels).
2.6. Immunoblotting
The following antibodies were purchased from Cell Signaling Technology (Danvers, Massachusetts): Akt (pan) (11E7), p‐Akt S473 (D9E), Bcl‐xL (54H6), Bcl‐2‐associated X protein (Bax; D2E11), and Beta‐actin (polyclonal). Wt‐p53 (polyclonal) antibody was purchased from Bioss (Woburn, Massachusetts). All antibodies were validated by Western blotting for cross‐reactivity with the canine species, and BLASTp alignments were performed to support their cross‐reactivity. Canine OSA cells were treated with amiloride and doxorubicin for 48 hours, then blocked with 2 mM sodium orthovanadate (Alfa Aesar, Haverhill, Massachusetts) for 15 minutes before protein extraction. Protein was harvested in complete lysis buffer (Cell Signaling Technology) containing 20 mM Tris‐HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton‐X 100, 2.5 mM sodium pyrophosphate, 1 mM β‐glycerophosphate, 1 mM sodium orthovanadate, 1 μg/mL leupeptin, 1 mM PMSF, 2 μg/mL aprotinin, and 1% phosphatase inhibitor cocktail II. Protein concentrations were determined using the bicinchoninic acid assay and a bovine serum albumin (BSA) standard curve. Lysates were resolved by 10% or 15% reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis using 25‐30 μg of protein, followed by a wet or dry transfer to a polyvinylidene difluoride membrane. The membranes were blocked in 5% skim milk in Tris‐buffered saline/Tween (TBST) or 5% BSA (VWR International, Radnor, Pennsylvania) in TBST (for p‐Akt). Secondary antibodies were conjugated to horseradish peroxidase for chemiluminescence imaging.
2.7. Immunofluorescence
D17, Abrams, and Dharma cells were seeded on 22 mm borosilicate glass coverslips (Thermo Fisher Scientific), fixed with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, Pennsylvania), permeabilized with 0.33% Triton‐X (Thermo Fisher Scientific) in phosphate‐buffered saline (PBS), and blocked with 5% donkey serum (Sigma‐Aldrich) in PBS. Cells were incubated with wt‐p53 (Bioss) diluted 1:200 in PBS, blotted on parafilm in a humidified chamber at 4°C. Alexa‐488‐conjugated donkey anti‐rabbit IgG (H + L) (Thermo Fisher Scientific) was used as the secondary antibody (1:800, 1 hour at RT). Cells were subsequently counterstained with 300 nM 4′,6‐diamidino‐2‐phenylindole dilactate (Thermo Fisher Scientific) for 10 minutes. All cover slips were mounted with DAKO fluorescent mounting medium (Agilent Technologies, Santa Clara, California). Slides were imaged with the Leica (Wetzlar, Germany) DMLB fluorescent microscope using the 100x/1.3 aperture oil immersion lens or 10x/0.7 aperture lens for quantification with ImageJ/Fiji. An omission control was used to control for binding of the secondary antibody.
2.8. Metabolic analysis
Cell metabolic analysis was performed in real‐time as per the manufacturer's protocol using a Seahorse XFe24 analyzer (Agilent Technologies). D17, Abrams, and Dharma cells were seeded at 4 x 104 cells/well for 24 hours. To measure the effects of amiloride on glycolytic capacity in real‐time, extracellular acidification rate (ECAR) was measured and a glycolytic stress test was performed. Cells were resuspended in Seahorse XF media (cat# 102353‐100), a DMEM formulation without FBS and sodium bicarbonate. Various chemical compounds were admixed with the media in real time, in this chronological order: Amiloride or vehicle (port A), 10 mM glucose (port B), 1 μM oligomycin (port C), and 100 mM 2‐Deoxy‐D‐glucose (2‐DG) (port D). A mitochondrial stress test was then used to measure oxygen consumption rate (OCR) after amiloride pretreatment, and cells were exposed to amiloride or vehicle (port A), 1 μM oligomycin (port B), 0.5 μM carbonyl cyanide‐4‐(trifluoromethoxy)phenylhydrazone (FCCP) (port C), and 0.5 μM rotenone/antimycin A (port D). Background wells were set in wells A1, B3, C2, and D4 as per manufacturer settings. All cells were assayed in Seahorse XF media supplemented with 2 mM L‐glutamine, 10 mM glucose, or 1 mM pyruvate (Thermo Fisher Scientific), as required. Seahorse data were analyzed with Wave 2.0 to report glycolytic capacity, coupling efficiency and maximal respiration. Coupling efficiency was defined as the ratio of the ATP production rate to the basal respiration rate. Maximal respiration was defined as the maximum OCR after FCCP injection, minus rates of nonmitochondrial respiration.
2.9. Statistical and software analysis
Statistical testing was performed using GraphPad Prism 6 (GraphPad Software Inc, La Jolla, California), including linear or nonlinear regression, one‐way or two‐way anova, unpaired t tests, and multiple comparisons tests. Means were reported with SD for descriptive statistics. Flow cytometry analyses were performed with FlowJo V10 (FlowJo LLC, Ashland, Oregon). ImageJ/Fiji was used for quantifying mean fluorescence intensities in immunofluorescence studies. CompuSyn (ComboSyn Inc, Paramus, New Jersey) software based on the Chou‐Talalay method was used for the computation of CI.
3. RESULTS
Amiloride and carboplatin treatment yielded IC50 values in the micromolar range for D17, Abrams, and Dharma cells (Figure 1A, B). In contrast, doxorubicin treatment yielded nanomolar IC50 values in all 3 cell lines (Figure 1C). In combination treatment, amiloride plus doxorubicin significantly reduced cell viability compared to doxorubicin or amiloride treatment alone in all 3 cell lines (P < .0001; Figure 2A). Statistically insignificant differences in cell viability were observed for amiloride plus carboplatin when compared to amiloride treatment alone (P = .24), and significant decreases were observed when compared to carboplatin treatment alone (P = .01; Figure 2B).
Figure 1.
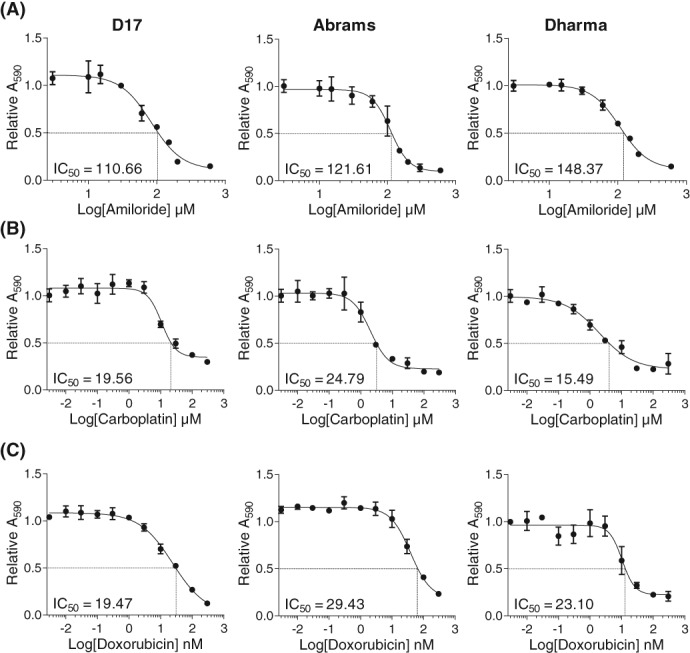
Crystal violet assays show that the potassium sparing diuretic amiloride reduces viability of canine OSA cells. Representative IC50 curves of cells treated with increasing concentrations of (A) amiloride (B) carboplatin, or (C) doxorubicin for 72 hours and the corresponding cell viability measured by crystal violet staining. Graphs represent mean ± SD of 3 technical replicates and 3 biological replicates from each cell line. IC50, half‐maximal inhibitory concentration; OSA, osteosarcoma
Figure 2.
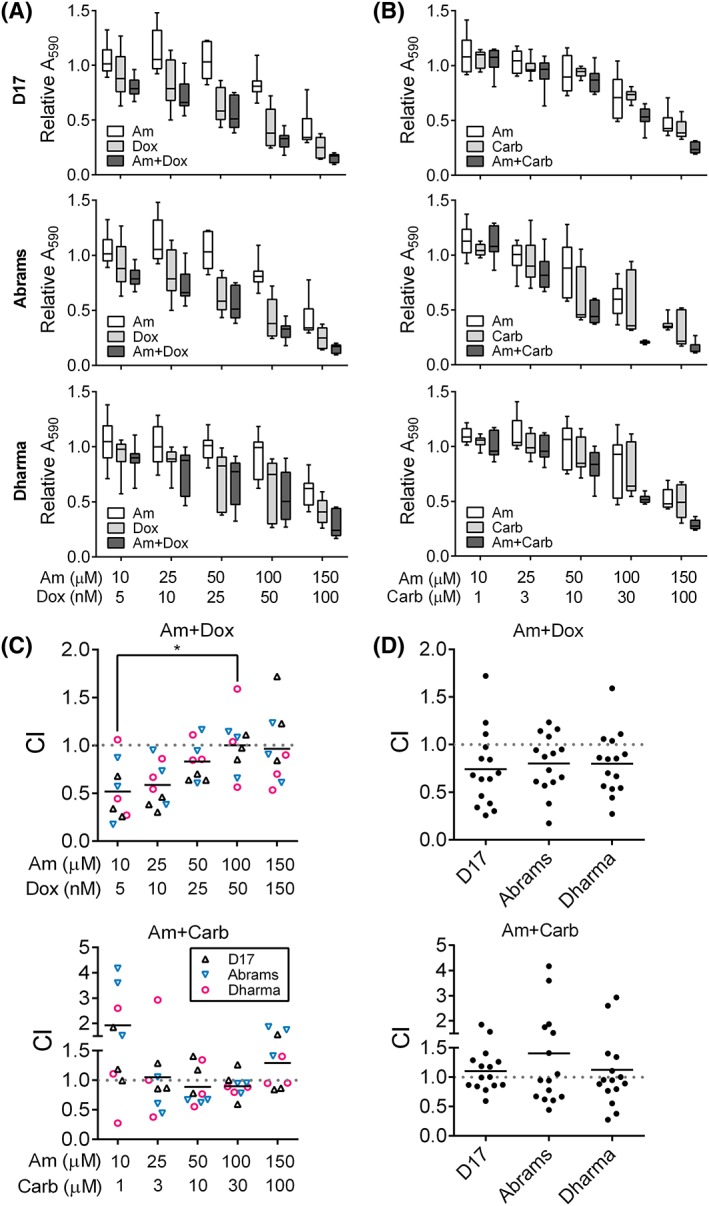
Combination treatment with calculations of the combination index (CI) in D17, Abrams, and Dharma treated with amiloride and doxorubicin, or amiloride and carboplatin. Decreased cell viability in each cell line after treatment with increasing doses of (A) amiloride and doxorubicin or (B) amiloride and carboplatin for 72 hours. X‐axis shows dose pairings for amiloride (Am, μM), doxorubicin (Dox, nM) and carboplatin (Carb, μM), which were chosen from IC50 curves. Relative absorbance values at 590 nm (A590) were presented with median ± range (N = 9). (C) Mean CI values from 3 independent experiments, stratified by dose pairing. CI < 1 was synergistic, C = 1 additive, and C > 1 antagonistic. Significance was recorded at *P < .05. (D) Mean CI values stratified by cell line independent of drug pairings. N = 15 with N = 3/drug pairing. IC50, half‐maximal inhibitory concentration
Pharmacological synergism (CI < 1) was observed with amiloride plus doxorubicin combination treatment. In amiloride plus doxorubicin‐treated groups, the lowest CI observed was 0.43 ± 0.13 with the lowest combination dose, significantly lower than the CI of 0.98 ± 0.13 at the highest dose (P < .05; Figure 2C). Regardless of drug ratios, mean CI values of 0.74 ± 0.10, 0.80 ± 0.08, and 0.79 ± 0.08 were observed for amiloride plus doxorubicin in D17, Abrams, and Dharma cells, respectively (Figure 2D). In contrast, amiloride plus carboplatin yielded mean CI values of 1.10 ± 0.09, 1.40 ± 0.28, and 1.12 ± 0.19 in the same respective cell lines. We conclude from CI calculations that amiloride synergizes with doxorubicin treatment in primary and metastatic canine OSA cells.
Combination treatment with amiloride and doxorubicin induced early and late stages of apoptosis, as verified by the etoposide‐treated positive control (Figure 3A). Although treatment with the 2 lower doses of amiloride alone (A10 and A30) did not result in a detectable increase in apoptosis (P = .37), there was induction of early apoptosis at the higher amiloride doses (A100 and A150), compared to the vehicle control (P < .05). Furthermore, combination treatment with the 2 doses of amiloride (A100 and A150) significantly increased early apoptosis in all 3 cell lines compared to treatment with vehicle control (P < .0001) or doxorubicin alone (P < .0001). Significant increases in late apoptosis were observed in D17 and Dharma cells (P < .05) but not Abrams (P = .39; Figure 3B).
Figure 3.
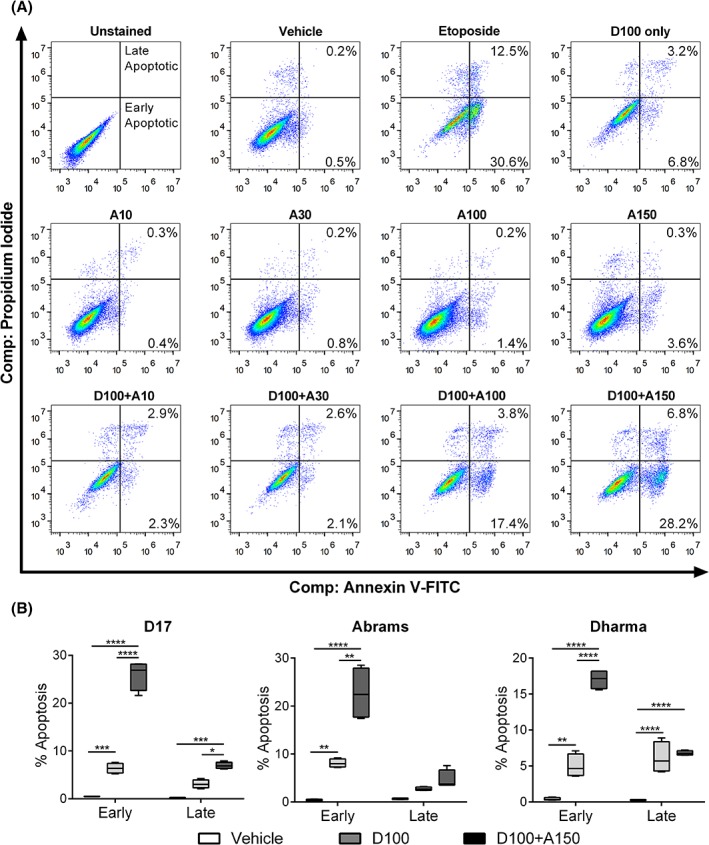
Apoptosis analysis of canine OSA cells treated with amiloride and doxorubicin alone or in combination. (A) Density plots obtained after compensation for spillover by the BD Accuri C6 flow cytometer. Combination treatments were compared to the 100 nM doxorubicin‐only control, D100, and high‐dose etoposide‐positive control (50 μM). Cells were treated with increasing doses of amiloride, from A10 to A150 (μM), or treated with increasing doses of amiloride and doxorubicin in combination, D + A. (B) Combined analysis of early and late apoptotic fractions, compared to the vehicle control with median ± range shown from 3 independent experiments. Significance was reported at *P < .05, **P < .01, ***P < .001, ****P < .0001. OSA, osteosarcoma; PI, propidium iodide
Visual upregulation of the p53 signaling pathway was observed after a 48‐hour treatment with amiloride and doxorubicin when compared to the beta‐actin housekeeping control (Figure 4A‐C). Signaling downstream of p53 activation further led to the modulation of apoptosis‐related protein expression. Upregulation of pro‐apoptotic protein Bax was observed in D17 (Figure 4A) and Abrams (Figure 4B) cells. This result coincided with downregulation of the anti‐apoptotic regulator, B‐cell lymphoma‐extra large (Bcl‐xL) at increasing combination doses tested (Figure 4A‐C). Amiloride treatment further downregulated Akt phosphorylation at serine 473 in a dose‐dependent manner in all 3 cell lines (Figure 4A‐C), leading to inactivation of native Akt at the highest doses for D17 cells (Figure 4A), but not Abrams and Dharma cells (Figure 4B, C).
Figure 4.
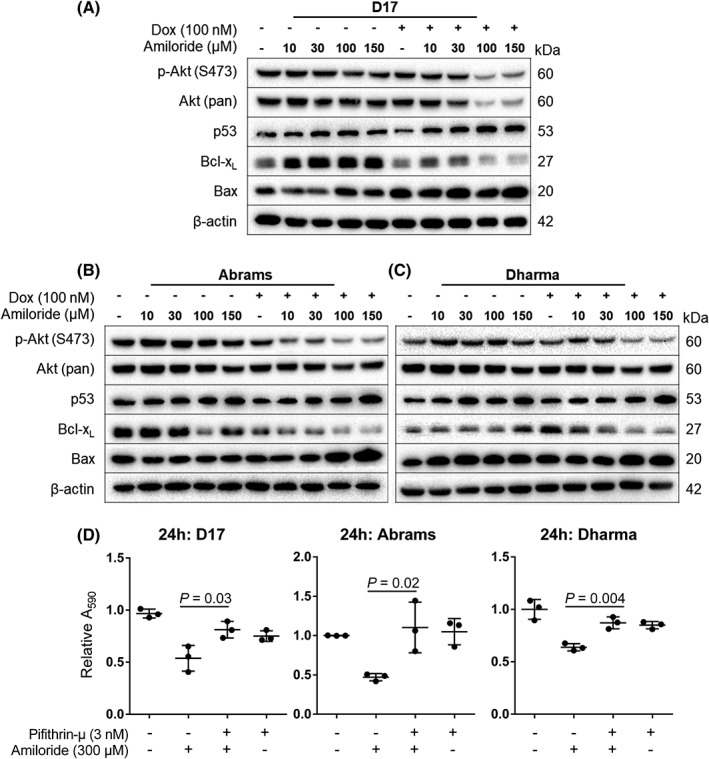
Western blot of proteins involved in p53 and Akt signaling in (A) D17, (B) Abrams, and (C) Dharma cells. (D) Viability after pharmacological inhibition of p53 translocation to the mitochondria. Cells were treated with pifithrin‐μ (3 nM) alone or in combination with 300 μM amiloride for 24 hours. Cell viability was measured with the crystal violet assay. Means were presented with SD, and significance reported at *P < .05
Combination treatment with high‐dose amiloride (300 μM) and pifithrin‐μ (3 nM) rescued D17, Abrams, and Dharma cells from amiloride‐induced reductions in cell viability. Significantly increased cell viability was observed with the drug combination compared to treatment with high dose amiloride alone (P < .05 for D17 and Abrams, P < .01 for Dharma; Figure 4D). Furthermore, treatment with pifithrin‐μ alone did not result in significant decreases in cell viability compared to the vehicle control (P = .70) (Figure 4D).
Immunofluorescence images show p53‐positive expression in both vehicle‐treated and amiloride‐treated cells, particularly in the nuclear regions where it is most active (Figure 5A). Image analysis with ImageJ verified differences by measuring mean fluorescence intensities (MFI) of each image taken, compared to the vehicle control in D17, Abrams, and Dharma cells. Significant increases in MFI in amiloride‐treated cells were observed in all 3 cell lines compared to the vehicle control (P < .05 and P < .01; Figure 5B).
Figure 5.
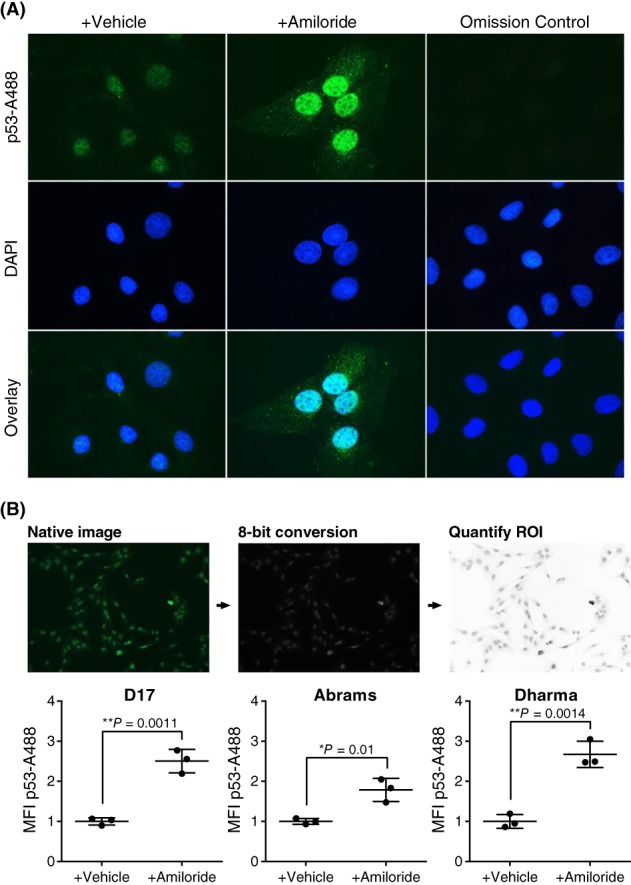
Immunofluorescence of p53 localization in canine OSA cells after amiloride treatment. (A) Representative images of p53 localization in D17 cells after treatment with high‐dose amiloride (300 μM) or vehicle for 24 hours before p53 staining. Cells were imaged with the Leica DMLB fluorescent microscope. Scale bar 10 μm. (B) Mean fluorescence intensity (MFI) of p53 staining in D17, Abrams, and Dharma cells as calculated by ImageJ/Fiji image analysis. Native images were taken at 10x/0.7 aperture, the regions of interest (ROI) were defined and quantified using ImageJ. N = 3 per group, with mean ± SD. Significance was reported at *P < .05 and **P < .01. OSA, osteosarcoma
Basal readings of ECAR and OCR were measured and established before glycolytic and mitochondrial stress tests in canine OSA cells (Figure 6). Glycolytic capacity, as measured by the ECAR was reduced compared to vehicle after amiloride exposure in all 3 cell lines (P < .05; Figure 6A). Exposure to saturating levels of glucose and oligomycin amplified the glycolytic output, and 2‐DG exposure inhibited glycolytic output. In the mitochondrial stress test, amiloride exposure had no significant effect on OCR after basal readings (P = .28). However, upon FCCP uncoupling of the mitochondria during oxidative phosphorylation, spare capacity and maximal respiration were decreased, when compared to the vehicle control (P < .0001) (Figure 6B).
Figure 6.
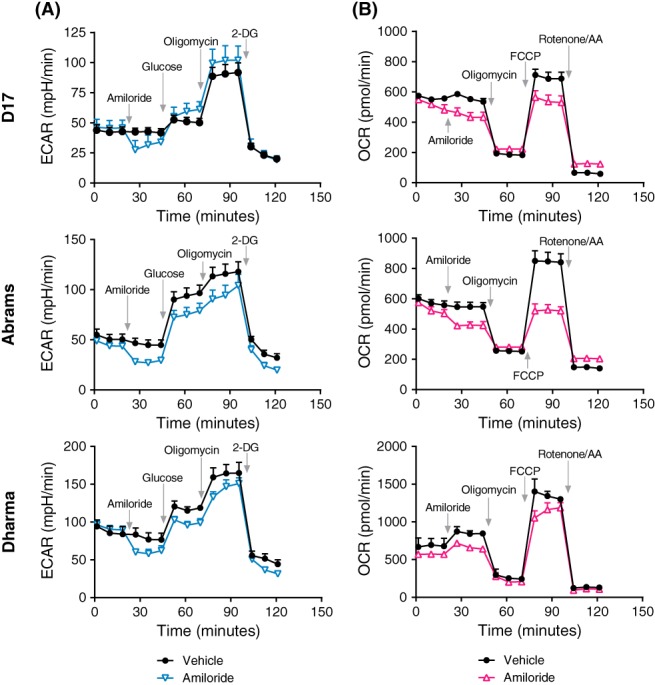
Seahorse metabolic tests of glycolytic and mitochondrial function in D17, Abrams, and Dharma cells. Cells were equilibrated in low‐buffered XF medium for basal readings of oxygen consumption rates (OCR) and extracellular acidification rates (ECAR). (A) Glycolytic test measuring ECAR in each cell line after amiloride (300 μM) or vehicle injection in real time, before the injection of glucose (10 mM), oligomycin (1 μM) and 2‐Deoxy‐D‐glucose (2‐DG) (100 mM). (B) Mitochondrial test measuring OCR in each cell line after amiloride or vehicle injection in real time, before the injection of oligomycin (1 μM), FCCP (0.5 μM), and rotenone/AA (0.5 μM). N = 3 per time point with mean ± SD. AA, antimycin A; FCCP, carbonyl cyanide‐4‐(trifluoromethoxy)phenylhydrazone
Measurements of ECAR readings during the first 45 minutes of the glycolytic test revealed a linear relationship between ECAR and dose by regression analysis (Figure 7A). Consistent with this model, a significant reduction in ECAR (P < .05) was observed after high‐dose amiloride treatment compared to vehicle (Figure 7B). Coupling efficiency (the ratio of ATP production rate to the basal respiration rate; Figure 7C) and maximal respiration rates (Figure 7D) were both significantly reduced in a dose‐dependent manner with amiloride treatment compared to the vehicle control (P < .0001). Basal readings of amiloride‐treated cells revealed a metabolic switch from an aerobic to quiescent phenotype (Figure 7E). Oligomycin exposure also resulted in a metabolic shift towards quiescence (Figure 7F).
Figure 7.
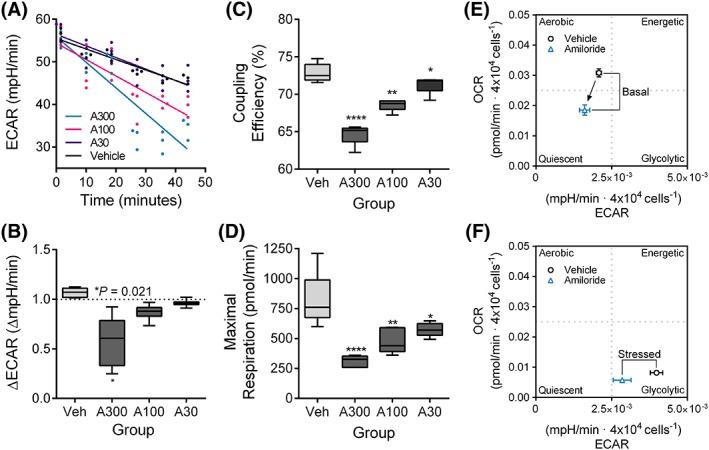
Combined energy phenotype analysis reveals a dose‐dependent effect of amiloride on ECAR and OCR in D17 cells. (A) Linear regression model of ECAR during the first 50 minutes, representing readings after amiloride injection. (B) Fold changes in ECAR expressed across all treatment groups tested after amiloride injection. Coupling efficiency (C) and maximal respiration (D) as measured using the mitochondrial stress test. (E‐F) Energy phenotype analysis after a 24‐hour pretreatment with amiloride (100 μM). ECAR and OCR were normalized to cell density (4 x 10−4 cells), and vehicle‐treated cells were compared to amiloride‐treated cells after oligomycin injection. A metabolic switch is indicated by the black arrow between the phenotype markers. Median ± range values were presented in box plots. Mean ± SD values were presented in energy phenograms. Significance was recorded at *P < .05, **P < .01, ****P < .0001. ECAR, extracellular acidification rates; OCR, oxygen consumption rates
4. DISCUSSION
To investigate acidification as a potential therapeutic target, we determined the effects of amiloride on carboplatin and doxorubicin response in canine OSA cells. After demonstrating that amiloride decreases cell viability in a dose‐dependent manner, we evaluated its pharmacological synergism with carboplatin and doxorubicin (Figure 1). Based on CI studies, amiloride exhibited synergism in combination with doxorubicin in canine OSA cells but could possess merely additive or possible antagonistic interactions with carboplatin (Figure 2). Not surprisingly, the hydrophobicity of doxorubicin suggests that it is passively partitioned into lysosomes and sequestered through protonation, whereas platinum agents like carboplatin are actively transported into lysosomes through the action of copper transporters.22 Therefore, although we hypothesized that amiloride would reduce the trapping of both doxorubicin and carboplatin, our results suggest that lysosomal trapping of carboplatin might not be a driving mechanism for its chemoresistance in cancer cells. Furthermore, mechanisms other than inhibition of NHEs could be responsible for the sensitization effect we observed with doxorubicin.
The interaction of amiloride with chemotherapy was investigated using CI calculations. A limitation of the Chou‐Talalay method21 is that the CI is inherently contingent on the inhibitory performance of both single agent drugs. Buffering antagonism is a scenario where the dominant effect of one drug effectively masks the effect of the other. Consistent with our findings, we observed higher antagonism (CI > 1) at the highest combination doses, supporting the buffering antagonism phenomenon that excessive killing from one compound can mask the contributory effects of the other compound tested. Historically, multiple chemotherapy drugs and protocols have been evaluated for efficacy in the adjuvant treatment of canine OSA, such as alternating carboplatin and doxorubicin,1 or their use in single‐agent protocols.23 Overall, no superior adjuvant chemotherapy protocol currently exists, although single‐agent carboplatin may often be chosen for dogs due its relative ease of administration and better tolerability, including lack of cardiotoxicity compared to doxorubicin and reduced nephrotoxicity compared to cisplatin.24 Despite this potential carboplatin preference, if doxorubicin plus amiloride were to be proven more effective than either doxorubicin or carboplatin alone in clinical trial evaluation, this result could lead to a superior chemotherapy protocol choice for OSA treatment in dogs.
After cell viability results suggested synergism between amiloride and doxorubicin in combination treatment, we determined the effect of this combination on apoptosis (Figure 3). As expected, combination treatment with doxorubicin significantly increased early apoptosis in canine OSA cells compared to doxorubicin or vehicle treatment alone. To elucidate potential mechanisms behind amiloride‐induced increases in apoptosis, we investigated molecular targets that could be activated during apoptosis. Previous studies that have suggested indirect, downstream effects of amiloride on Akt signaling and p53 signaling prompted investigation into this pathway in canine OSA.25 Up to 50% of OSA tumors have mutated TP53.26 Because amiloride exhibits antimyeloma activity and synergizes with dexamethasone, melphalan, lenalidomide, and pomalidomide,18 we investigated changes in p53 signaling during amiloride‐induced apoptosis in canine OSA cells and the downstream impact on pro‐ and anti‐apoptotic family members (Figure 4). Upon exposure to amiloride, the pro‐apoptotic family member Bax was upregulated, whereas the anti‐apoptotic protein Bcl‐xL was downregulated in a dose‐dependent manner, suggesting that amiloride potentiates p53‐dependent apoptosis. Pharmacological disruption of p53 translocation to the mitochondria by pifithrin‐μ inhibition of p53 binding to Bcl family proteins rescued canine OSA cells from apoptosis. This result suggests that translocation of p53 to the mitochondria is required for amiloride‐induced apoptosis. From immunofluorescence experiments, we further demonstrated that localization of p53 within the cytosol increases after amiloride exposure, further supporting that p53‐mitochondrial signaling is upregulated compared to vehicle‐treated cells (Figure 5). Finally, downregulation of Akt phosphorylation at serine 473 was also observed after amiloride treatment. It is known that this site‐specific phosphorylation is required for full activation of Akt in PI3K/mTOR signaling.27
Metabolic phenotypes for cancer cells are glycolytic because of Warburg metabolism.28 We have determined with the Seahorse analyzer that under basal conditions, canine OSA cells can be considered to be quite glycolytic, with ECAR rates above 50 mpH/min (Figure 6). By performing energetic stress tests, we further showed that amiloride decreased ECAR in a dose‐dependent manner, suggesting an inhibition of proton extrusion (Figure 7). Glycolytic capacity is inferred from the extracellular production of protons, and reduced NHE activity can contribute to reduced proton extrusion through the endolysosomal pathway,29 a gradient that leads to extracellular acidification. NHEs are transported to the cell's basal surface, where inhibition could be further increased on the plasma membrane. Coupling is critical for powering the electrochemical gradient that drives ATP synthesis. The proportion of oxygen consumed, compared to the leakage of protons, is defined as the coupling efficiency. From mitochondrial function tests, we further observed impaired coupling efficiency in canine OSA cells after amiloride treatment. Because uncoupling of oxidative phosphorylation requires the transport of protons across the inner mitochondrial membrane by an ionophore,30 disrupting the normal flow of proton output during oxidative phosphorylation may be a mechanism to explain why canine OSA cells exhibit reduced maximal respiration, even under conditions of persistent oxygen availability and uncoupling. Lastly, energy phenograms after amiloride exposure show an increased shift towards quiescence, suggesting that reduced glycolytic capacity coincides with reductions in metabolic uptake of glucose in canine OSA cells (Figure 7).
4.1. Limitations
Amiloride was originally discovered as MK‐870 for correcting the profound hypokalemia and alkalosis induced by thiazide and other diuretics.31 Since its discovery, it was tested in a canine cardiac study, where a blood concentration of 0.2 μmoL/mL, corresponding to 200 μmol/L was used to perfuse isolated AV node, SA node, and papillary muscle preparations.12 At this dose, amiloride prolonged the functional refractory period at atrial pacing rates of 100, 150, and 200 stimuli/min. The study also noted that a blood concentration of 0.2 μmoL/mL corresponded to 46 μg/mL. Earlier studies of MK‐870 in guinea pigs in 1977 have dose escalated up to 200 μg/mL but found no significant effects in the upstroke velocity of ventricular muscle.32 To the best of our knowledge, although amiloride has been extensively tested in the dog, no specific pharmacokinetic (PK) study of amiloride in the dog is published. Rather, variables such as renal clearance and sodium excretion have been widely reported in dogs given high doses of amiloride.33 In human PK studies, peak reported serum concentrations (Cmax) of amiloride varies depending on the route of administration and dose. A single oral 10 mg dose leads to a Cmax of 20.6 ± 10.0 ng/mL in adults,34 and an oral dose of 20 mg alone leads to a Cmax of 47.5 ± 13.0 ng/mL35 both achieved shortly after 4 hours. In contrast, PK data in rats demonstrates that an amiloride of 10 mg/kg results in a Cmax of 390.0 ng/mL after 4 hours.36 From human PK data, we propose that there could be limitations as to how much amiloride could be given to dogs, especially on a consistent basis in oncology practice.
Although we tested both low and high doses of amiloride typically used in in vitro doses, we found from CI calculations that the strongest synergism with doxorubicin was achieved at the lowest doses of amiloride that were tested (Figure 2C). Therefore, to achieve a potential sensitization effect in vivo, it might not be necessary to dose‐escalate to the highest in vitro doses tested in this study. Treatment of mice with amiloride at a dose of 15 mg/kg/d resulted in significant antitumor efficacy in a model of multiple myeloma.18 In comparison to early pharmacological studies in dogs with doses of 8 mg/kg/day, similar doses could be applied in combination with chemotherapy in the clinical setting.33 Overall, the outstanding limitation of using in vitro doses is that the effects of amiloride observed might not translate to the same effects observed in vivo. Thus, the clinical evaluation of amiloride as a repurposed drug in an oncology setting would likely proceed in a dose‐escalation format, and possibly in combination with chemotherapy, to maximize any potential chemosensitization benefit.
Apart from NHE targeting, other off‐targets of amiloride have been documented, including its competitive inhibition of the urokinase plasminogen activator (uPA) and the epithelial sodium channel (ENaC).37, 38 The uPA axis has been recently causally associated with the progression of OSA from a nonmetastatic to metastatic phenotype.39 Thus, the potentiation of doxorubicin cytotoxicity by amiloride at high doses could extend beyond inhibition of the transporter itself. Although the goal of this study was to evaluate the impact of amiloride on chemotherapy response in canine OSA cells, it can be hypothesized that amiloride also inhibits uPA to decrease invasion in canine OSA cells. Furthermore, although amiloride is also a potent inhibitor of ENaC in the context of its diuretic effects, the resulting antikaliuresis counters hypokalemia. Because the main potential side effect of amiloride administration is hyperkalemia, amiloride is often prescribed in combination with a thiazide diuretic, such as hydrochlorothiazide.
Previous studies on cardiac effects and immune function in mice and dogs do not show systemic toxicosis after amiloride treatment, even at millimolar doses.15, 18 For dogs with OSA, chemotherapy is the principal treatment to decelerate metastatic progression. Because OSA in dogs is highly metastatic early in disease, novel approaches to improve chemotherapy response, prevent chemoresistance, or both are needed. Along with the historical use of amiloride in dogs and the accessibility of this approved drug, the specific pharmacological evaluation of amiloride in oncology is worthy of future investigation.
Future studies of the molecular mechanisms involved in the potentiation of doxorubicin cytotoxicity by amiloride in canine OSA cells are warranted. NHEs are critically involved in invasion and metastasis of human breast cancer cells.40 Furthermore, inhibition of other targets, such as the uPA signaling axis, could also contribute to the anticancer effects of amiloride reported in this study and are also worthy of further investigation.
5. CONCLUSION
This study determined that amiloride treatment improved the response of primary and metastatic canine OSA cells to doxorubicin in vitro. Furthermore, amiloride treatment activated p53 signaling and downregulated Akt phosphorylation, leading to increased apoptosis in combination treatment. Metabolic profiling of amiloride‐treated cells revealed that canine OSA cells exhibited reduced glycolytic capacity, coupling efficiency, and maximal respiration. Taken together, these results conclude that amiloride synergizes with doxorubicin to increase killing in canine OSA cells, which could have clinical implications for the potential translation of this drug to veterinary oncology.
CONFLICT OF INTEREST DECLARATION
Authors declare no conflict of interest.
OFF‐LABEL ANTIMICROBIAL DECLARATION
Authors declare no off‐label use of antimicrobials.
INSTITUTIONAL ANIMAL CARE AND USE COMMITTEE (IACUC) OR OTHER APPROVAL DECLARATION
Authors declare no IACUC or other approval was needed.
HUMAN ETHICS APPROVAL DECLARATION
Authors declare human ethics approval was not needed for this study.
ACKNOWLEDGMENTS
The authors acknowledge Charlotte Mitz, Dr Robert Jones, Allison MacKay, and Jodi Morrison for their equipment expertise. The authors also thank OVC Pet Trust and the University of Guelph Alumni Affairs and Development office for their financial support of this project. All work from this manuscript was performed in the Department of Biomedical Sciences, Ontario Veterinary College at the University of Guelph. A portion of this work was presented on as an oral abstract at the 2018 ACVIM Forum, Seattle, WA.
Poon AC, Inkol JM, Luu AK, Mutsaers AJ. Effects of the potassium‐sparing diuretic amiloride on chemotherapy response in canine osteosarcoma cells. J Vet Intern Med. 2019;33:800–811. 10.1111/jvim.15382
Funding information Memorial donation through University of Guelph Alumni Affairs and Development; OVC Pet Trust, Grant/Award Number: 053524
REFERENCES
- 1. Bacon NJ, Ehrhart NP, Dernell WS, Lafferty M, Withrow SJ. Use of alternating administration of carboplatin and doxorubicin in dogs with microscopic metastases after amputation for appendicular osteosarcoma: 50 cases (1999‐2006). J Am Vet Med Assoc. 2008;232(10):1504‐1510. [DOI] [PubMed] [Google Scholar]
- 2. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8(6):519‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cardone RA, Casavola V, Reshkin SJ. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat Rev Cancer. 2005;5(10):786‐795. [DOI] [PubMed] [Google Scholar]
- 4. Slepkov ER, Rainey JK, Sykes BD, Fliegel L. Structural and functional analysis of the Na+/H+ exchanger. Biochem J. 2007;401(3):623‐633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shimoda LA, Fallon M, Pisarcik S, Wang J, Semenza GL. HIF‐1 regulates hypoxic induction of NHE1 expression and alkalinization of intracellular pH in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol. 2006;291(5):L941‐L949. [DOI] [PubMed] [Google Scholar]
- 6. Corbet C, Feron O. Tumour acidosis: from the passenger to the driver's seat. Nat Rev Cancer. 2017;17(10):577‐593. [DOI] [PubMed] [Google Scholar]
- 7. Herlevsen M, Oxford G, Owens CR, Conaway M, Theodorescu D. Depletion of major vault protein increases doxorubicin sensitivity and nuclear accumulation and disrupts its sequestration in lysosomes. Mol Cancer Ther. 2007;6(6):1804‐1813. [DOI] [PubMed] [Google Scholar]
- 8. Smith PJ, Sykes HR, Fox ME, Furlong IJ. Subcellular distribution of the anticancer drug mitoxantrone in human and drug‐resistant murine cells analyzed by flow cytometry and confocal microscopy and its relationship to the induction of DNA damage. Cancer Res. 1992;52(14):4000‐4008. [PubMed] [Google Scholar]
- 9. Groth‐Pedersen L, Ostenfeld MS, Høyer‐Hansen M, Nylandsted J, Jäättelä M. Vincristine induces dramatic lysosomal changes and sensitizes cancer cells to lysosome‐destabilizing siramesine. Cancer Res. 2007;67(5):2217‐2225. [DOI] [PubMed] [Google Scholar]
- 10. Polishchuk EV, Concilli M, Iacobacci S, et al. Wilson disease protein ATP7B utilizes lysosomal exocytosis to maintain copper homeostasis. Dev Cell. 2014;29(6):686‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Walsh M, Fais S, Spugnini EP, et al. Proton pump inhibitors for the treatment of cancer in companion animals. J Exp Clin Cancer Res. 2015;34:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamashita S, Motomura S, Taira N. Cardiac effects of amiloride in the dog. J Cardiovasc Pharmacol. 1981;3(4):704‐715. [DOI] [PubMed] [Google Scholar]
- 13. Vidt DG. Mechanism of action, pharmacokinetics, adverse effects, and therapeutic uses of amiloride hydrochloride, a new potassium‐sparing diuretic. Pharmacotherapy. 1981;1(3):179‐187. [DOI] [PubMed] [Google Scholar]
- 14. Bull MB, Laragh JH. Amiloride: a potassium‐sparing natriuretic agent. Circulation. 1968;37(1):45‐53. [DOI] [PubMed] [Google Scholar]
- 15. Duff HJ, Lester WM, Rahmberg M. Amiloride. Antiarrhythmic and electrophysiological activity in the dog. Circulation. 1988;78(6):1469‐1477. [DOI] [PubMed] [Google Scholar]
- 16. Wakabayashi S, Shigekawa M, Pouyssegur J. Molecular physiology of vertebrate Na+/H+ exchangers. Physiol Rev. 1997;77(1):51‐74. [DOI] [PubMed] [Google Scholar]
- 17. Zheng Y, Yang H, Li T, et al. Amiloride sensitizes human pancreatic cancer cells to erlotinib in vitro through inhibition of the PI3K/AKT signaling pathway. Acta Pharmacol Sin. 2015;36(5):614‐626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rojas EA, Corchete LA, San‐Segundo L, et al. Amiloride, an old diuretic drug, is a potential therapeutic agent for multiple myeloma. Clin Cancer Res. 2017;23(21):6602‐6615. [DOI] [PubMed] [Google Scholar]
- 19. Legare ME, Bush J, Ashley AK, Kato T, Hanneman WH. Cellular and phenotypic characterization of canine osteosarcoma cell lines. J Cancer. 2011;2:262‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mantovani FB, Morrison JA, Mutsaers AJ. Effects of epidermal growth factor receptor kinase inhibition on radiation response in canine osteosarcoma cells. BMC Vet Res. 2016;12(1):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chou T‐C. Drug combination studies and their synergy quantification using the Chou‐Talalay method. Cancer Res. 2010;70(2):440‐446. [DOI] [PubMed] [Google Scholar]
- 22. Kilari D, Guancial E, Kim ES. Role of copper transporters in platinum resistance. World J Clin Oncol. 2016;7(1):106‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saam DE, Liptak JM, Stalker MJ, Chun R. Predictors of outcome in dogs treated with adjuvant carboplatin for appendicular osteosarcoma: 65 cases (1996‐2006). J Am Vet Med Assoc. 2011;238(2):195‐206. [DOI] [PubMed] [Google Scholar]
- 24. Yasumasu T, Ueda T, Uozumi J, Mihara Y, Kumazawa J. Comparative study of cisplatin and carboplatin on pharmacokinetics, nephrotoxicity and effect on renal nuclear DNA synthesis in rats. Pharmacol Toxicol. 1992;70(2):143‐147. [DOI] [PubMed] [Google Scholar]
- 25. Kim KM, Lee YJ. Amiloride augments TRAIL‐induced apoptotic death by inhibiting phosphorylation of kinases and phosphatases associated with the P13K‐Akt pathway. Oncogene. 2005;24(3):355‐366. [DOI] [PubMed] [Google Scholar]
- 26. Kirpensteijn J, Kik M, Teske E, Rutteman GR. TP53 gene mutations in canine osteosarcoma. Vet Surg. 2008;37(5):454‐460. [DOI] [PubMed] [Google Scholar]
- 27. Persad S, Attwell S, Gray V, et al. Regulation of protein kinase B/Akt‐serine 473 phosphorylation by integrin‐linked kinase: critical roles for kinase activity and amino acids arginine 211 and serine 343. J Biol Chem. 2001;276(29):27462‐27469. [DOI] [PubMed] [Google Scholar]
- 28. Parks SK, Chiche J, Pouysségur J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat Rev Cancer. 2013;13(9):611‐623. [DOI] [PubMed] [Google Scholar]
- 29. Steffan JJ, Snider JL, Skalli O, Welbourne T, Cardelli JA. Na+/H+ exchangers and RhoA regulate acidic extracellular pH‐induced lysosome trafficking in prostate cancer cells. Traffic. 2009;10(6):737‐753. [DOI] [PubMed] [Google Scholar]
- 30. Terada H. Uncouplers of oxidative phosphorylation. Environ Health Perspect. 1990;87:213‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Antcliff AC, Beevers DG, Hamilton M, Harpur JE. The use of amiloride hydrochloride in the correction of hypokalaemic alkalosis induced by diuretics. Postgrad Med J. 1971;47(552):644‐647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lüderitz B, Naumann d'Alnoncourt C, Steinbeck G. Effects of antikaliuretic agents on cardiac electrophysiology—measurements in papillary heart muscle and in purkinje fibers. Klin Wochenschr. 1977;55(9):423‐427. [DOI] [PubMed] [Google Scholar]
- 33. Goldman WJ, Skeggs HR, Scott W, Baer JE, Mattis PA. Effects of the chronic administration of amiloride hydrochloride (N‐amidino‐3,5‐diamino‐6‐chloropyrazinecarboxamide hydrochloride dihydrate) on electrolyte balance in the dog. J Pharmacol Exp Ther. 1971;179(2):438‐446. [PubMed] [Google Scholar]
- 34. Jones KM, Liao E, Hohneker K, et al. Pharmacokinetics of amiloride after inhalation and oral administration in adolescents and adults with cystic fibrosis. Pharmacother J Hum Pharmacol Drug Ther. 1997;17(2):263‐270. [PubMed] [Google Scholar]
- 35. Smith AJ, Smith RN. Kinetics and bioavailability of two formulations of amiloride in man. Br J Pharmacol. 1973;48(4):646‐649. [PMC free article] [PubMed] [Google Scholar]
- 36. Segre G, Cerretani D, Bruni G, Urso R, Giorgi G. Amiloride pharmacokinetics in rat. Eur J Drug Metab Pharmacokinet. 1998;23(2):218‐222. [DOI] [PubMed] [Google Scholar]
- 37. Jankun J, Skrzypczak‐Jankun E. Molecular basis of specific inhibition of urokinase plasminogen activator by amiloride. Cancer Biochem Biophys. 1999;17(1–2):109‐123. [PubMed] [Google Scholar]
- 38. Canessa CM, Schild L, Buell G, et al. Amiloride‐sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994;367(6462):463‐467. [DOI] [PubMed] [Google Scholar]
- 39. Endo‐Munoz L, Cai N, Cumming A, et al. Progression of osteosarcoma from a non‐metastatic to a metastatic phenotype is causally associated with activation of an autocrine and paracrine uPA axis. PLoS One. 2015;10(8):e0133592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Amith SR, Wilkinson JM, Baksh S, Fliegel L. The Na+/H+ exchanger (NHE1) as a novel co‐adjuvant target in paclitaxel therapy of triple‐negative breast cancer cells. Oncotarget. 2015;6(2):1262‐1275. [DOI] [PMC free article] [PubMed] [Google Scholar]