

Abstract
Background
The effects of synthetic brain natriuretic peptide (BNP1‐32) on cardiorenal and renin angiotensin aldosterone system in dogs with naturally occurring congestive heart failure (CHF) are unknown.
Objectives
To evaluate the cardiorenal and endocrine effects of SC administered synthetic canine BNP1‐32, with or without furosemide, in dogs with CHF caused by myxomatous mitral valve disease (MMVD).
Animals
Seven client‐owned male dogs with compensated American College of Veterinary Internal Medicine stage C CHF caused by MMVD on chronic treatment with furosemide, benazepril, and pimobendan.
Methods
A single‐dose, crossover, pilot study. Each dog received a dose of BNP1‐32 (5 μg/kg), furosemide (2 mg/kg), and both BNP1‐32/furosemide (5 μg/kg and 2 mg/kg, respectively) SC with a 2‐week washout period among each treatment. Between‐ and within‐treatment effects were evaluated using linear mixed modeling with restricted maximum likelihood estimation and evaluation of least square differences.
Results
Rapid absorption of BNP1‐32 and a corresponding rise in urinary cyclic guanosine monophosphate excretion was observed at 1‐2 hours after any treatment containing BNP1‐32 (P < .05). However, BNP1‐32 did not influence measured cardiorenal variables. Plasma aldosterone concentrations were below quantifiable levels in majority of the samples.
Conclusions and Clinical Importance
No beneficial cardiorenal effects were detected. It is possible that dogs with chronic CHF have a reduction in natriuretic peptide responsiveness.
Keywords: aldosterone, brain natriuretic peptide, canine, cGMP, RAAS
Abbreviations
- ACE‐I
angiotensin‐converting enzyme inhibitor
- AEBSF
4‐(2‐aminoethyl)benzenesulfonyl fluoride hydrochloride
- BNP
5 μg/kg synthetic canine BNP 1‐32 given SC
- BNP/FRUS
5 μg/kg synthetic canine BNP1‐32 and 2 mg/kg furosemide given SC
- BNP1‐32
brain natriuretic peptide 1–32
- BP
blood pressure
- cGMP
cyclic guanosine monophosphate
- CHF
congestive heart failure
- DBP
diastolic blood pressure
- EDTA
ethylenediaminetetraacetic acid
- FRUS
2 mg/kg furosemide given SC
- HR
heart rate
- LLOQ
lowest limit of quantification
- MBP
mean blood pressure
- MMVD
myxomatous mitral valve disease
- NPR‐A
natriuretic peptide receptor A
- RAAS
renin‐angiotensin‐aldosterone system
- SBP
systolic blood pressure
- TP
total protein
- UHPLC
ultra‐high‐performance liquid chromatography
- UOP
urine output
- UVTHS
University Veterinary Teaching Hospital, Sydney
1. INTRODUCTION
Congestive heart failure (CHF) caused by myxomatous mitral valve disease (MMVD) is the leading cause of cardiac death in older, small‐breed dogs.1 Medical treatment of CHF remains the predominant form of treatment in veterinary medicine, with current guidelines recommending a combination treatment consisting of a diuretic, an angiotensin‐converting enzyme inhibitor (ACE‐I) and an inodilator.2 Though survival times using this treatment regimen have not yet been published, an estimation of around 1 year is considered standard among experts, based on extrapolations from clinical trials and retrospective studies evaluating survival outcomes on components of this treatment regimen.3, 4, 5, 6 The reasonably poor prognosis associated with this condition has prompted ongoing efforts in the discovery of additional or alternative therapeutics, including various inhibitors of the renin‐angiotensin‐aldosterone system (RAAS),7, 8 newer diuretics,9, 10 and surgical repair.11, 12, 13
In these research efforts, a somewhat neglected therapeutic target is the natriuretic peptide system. Brain natriuretic peptide (BNP1‐32) is an active component of this system. Binding to natriuretic peptide receptor A (NPR‐A) causes activation of guanylyl cyclase that converts the cytosolic nucleotide guanosine triphosphate (GTP) into cyclic guanosine monophosphate (cGMP). From there, cGMP acts as an intracellular secondary messenger and causes diuresis, natriuresis, vasodilation, and RAAS antagonism. These are desirable properties to combat the progression of CHF and, indeed, experimental animal studies,14, 15, 16, 17, 18, 19, 20 and early clinical trials in humans21, 22, 23, 24 have shown the benefits of BNP1‐32 in the treatment of CHF. This led to the registration of Nesiritide in the United States (Natrecor; Scios, Inc, Mountain View, California), a recombinant human BNP1‐32 product, for IV use in people with decompensated CHF displaying clinical signs of dyspnea at rest.
Despite its initial success, an ultimate reduction in the rate of case fatalities or rehospitalization was not observed in humans with CHF.25 This caused Nesiritide to fall out of favor in human medicine and likely contributed toward reduced interest for use in companion animals. Another limiting factor is its formulation—an injectable preparation—which has traditionally resulted in use as an IV drug. Yet, there is growing evidence of its safety and efficacy when administered SC.16, 18, 23, 24, 26, 27 SC administration facilitates its use in chronic treatment, and its potential role in the treatment of subclinical heart disease28 and in humans with diastolic dysfunction and right‐sided CHF15, 24, 29 (Kwon et al., 2003; Tong et al., 2004; Zakir, 2005 [abstracts]) has also been explored. Although no solid conclusions to support its benefits in these contexts have been reported to date, there is enough theoretical and experimental data to support further investigation into its use in dogs with naturally occurring CHF caused by MMVD.
In this study, the cardiorenal and endocrine effects of synthetic canine BNP1‐32, given SC with or without furosemide, were evaluated in dogs with compensated CHF caused by MMVD.
2. MATERIALS AND METHODS
2.1. Animals
Seven client‐owned geriatric (10.5‐17 years old) male dogs with compensated American College of Veterinary Internal Medicine (ACVIM) stage C (chronic) CHF caused by MMVD were recruited in this study. Ethics approval was obtained from the University of Sydney Animal Ethics Committee before commencement of the study (approval number 2016/1113).
2.2. Study design
This study was a single‐dose, cross‐over, pilot study evaluating the efficacy of synthetic canine BNP1‐32, given with or without furosemide, in the target population. It was conducted at the University Veterinary Teaching Hospital, Sydney (UVTHS) between May and September, 2017.
2.3. Enrolment criteria
Before enrolment, each dog had a screening examination which consisted of a review of the history, physical examination, and oscillometric blood pressure (BP) measurements (VetHDO; Memo Diagnostic, Darmstadt, Germany). Thoracic radiography, echocardiography, ECG, and select blood tests (alanine aminotransferase, alkaline phosphatase, blood urea nitrogen, creatinine, glucose, albumin, globulin, total protein [TP], PCV, electrolytes) were also performed.
The inclusion criteria consisted of a diagnosis of CHF caused by MMVD and good control of clinical signs with pimobendan (Vetmedin; Boehringer Ingelheim Pty Ltd, North Ryde, NSW, Australia), benazepril (Fortekor; Elanco Australasia Pty Ltd, West Ryde, NSW, Australia), and furosemide (APO Furosemide; Apotex Pty Ltd, Macquarie Park, NSW, Australia). The exclusion criteria included previously diagnosed or suspected renal disease, adrenal disease, diabetes mellitus, systemic hyper/hypotension, primary or concurrent right‐sided CHF, and arrhythmias resulting in hemodynamic compromise. In addition, dogs were not enrolled if concurrent treatments with sildenafil, spironolactone, amlodipine, thiazide diuretics, or other medications that might interfere with cGMP metabolism, BP regulation, or renal function were required.
The diagnosis of CHF caused by MMVD was made on characteristic history and physical examination findings (dyspnea, tachypnea, or both responsive to treatment with furosemide; presence of abnormal lung sounds; at least a grade 4/6 systolic heart murmur loudest at the left apical region; and tachycardia on examination); echocardiographic evidence of severe MMVD and cardiac remodeling due to volume overload (ie, characteristic lesions of the mitral valves, severe mitral valve regurgitation, left atrial and ventricular enlargements) on an echocardiogram performed by a board‐certified veterinary cardiologist (Niek Beijerink); and radiographic evidence of cardiomegaly with left atrial enlargement, pulmonary edema, and pulmonary vasodilation. Two dogs did not have radiography performed before commencement of treatment for CHF. However, serial radiographies depicting pulmonary edema which resolved with commencement of treatment were available for the remainder of dogs enrolled.
2.4. Study outline
After enrolment, dogs presented to the UVTHS on 3 days, each 2 weeks apart, for this study. Benazepril was dosed once daily at night, including the night before each visit. The morning doses of pimobendan and furosemide were withheld on each study day. On the morning of each visit, history and physical examinations were performed to ensure well‐being of the dogs, and peripheral IV and indwelling urinary catheters were placed. At the end of a 1‐hour control period, heart rate (HR), BP, and urine output (UOP) were measured; blood and urine samples were collected for endocrine, electrolyte, biochemistry, and cGMP testing. The amount of blood collected was replaced with 7 mL saline. One of the following treatments was administered:
BNP: 5 μg/kg synthetic canine BNP1‐32 (Canine BNP1‐32 [011‐22]; Phoenix Pharmaceuticals Inc, Burlingame, California) SC (visit 1)
FRUS: 2 mg/kg furosemide (Ileum Furosemide; Troy Laboratory Ltd., Glendenning, NSW, Australia) SC (visit 2)
BNP/FRUS: 5 μg/kg synthetic canine BNP1‐32 and 2 mg/kg furosemide SC (visit 3)
Treatment order was not randomized, and all dogs received all 3 treatments in the order listed above.
Measurement of HR and BP, collection of 7 mL blood, and IV administration of 7 mL saline were repeated at 1 and 3 hours after treatment. Measurement of UOP and collection of 5 mL urine were performed at 1, 2, and 3 hours after treatment. PCV and TP were monitored for safety purposes throughout. At the completion of this sampling period, IV and urinary catheters were removed, and dogs were treated with their usual morning medications (pimobendan and furosemide, if furosemide had not been given as a part of the treatment) and then fed. Dogs were then discharged home to continue routine medical treatment with pimobendan, benazepril, and furosemide, and owners were advised to monitor for any abnormalities to the injection site, changes in behavior, or development of signs of decompensated CHF until the next scheduled visit.
HR and systolic, diastolic, and mean BP (systolic blood pressure [SBP], diastolic blood pressure [DBP], mean blood pressure [MBP]) were measured in triplicate and averaged before statistical analysis.
2.5. Sample processing and storage
Blood samples were obtained via jugular venipuncture and immediately placed into plastic blood tubes containing EDTA (ethylenediaminetetraacetic acid) or lithium heparin. Urine samples were collected into sterile plastic urine tubes. EDTA‐anticoagulated blood samples and urine samples were immediately centrifuged at or below 4°C at 5500 relative centrifugal force (RCF) for 10 minutes for collection of plasma and sediment‐free urine. After obtaining a sample for PCV and TP, the lithium heparin‐anticoagulated blood samples were centrifuged at 4000 RCF for 4 minutes at room temperature for collection of plasma. For BNP1‐32 testing, additional protease inhibition was achieved by adding 1 mL EDTA plasma to a preprepared mixture of 4‐(2‐aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF) and benzamidine (Sigma Aldrich, Castle Hill, NSW, Australia) to give a final concentration of 5 mM AEBSF and 10 mM benzamidine.30 The remaining plasma and urine samples were also transferred into sterile, plain tubes and frozen for later analysis. Before analysis, samples were stored at −80°C for up to 136 days for BNP‐32; at or below −30°C for up to 144 days for cGMP; at −80°C for up to 125 days for sodium, potassium, and creatinine; and at or below −40°C for up to 270 days for aldosterone assays. Stability testing was not performed in this study; however, samples treated and held under similar conditions were reportedly stable in human plasma samples for 6 months for BNP‐3230 and plasma and urinary cGMP.31, 32, 33 Furthermore, aldosterone concentrations in human serum control samples stored at −30°C at the particular laboratory consulted for aldosterone assays were noted to be stable for at least 9 months, with no drift observed at 0.12 and 0.68 nM/L (n = 78).
2.6. Blood and urine analysis
Plasma immunoreactive BNP‐32 concentrations were measured at a commercial laboratory (Phoenix Pharmaceuticals, Inc) using a validated34 radioimmunoassay kit made by the company (BNP‐32 Canine RIA kit, RK‐011‐22; Phoenix Pharmaceuticals Inc). The samples were transported frozen to the laboratory, and each sample was assayed in duplicate and averaged for analysis.
Plasma and urine cGMP concentrations were assayed using a commercially available competitive enzyme‐linked immunosorbent assay kit using the manufacturer's guidelines (Parameter cGMP Assay, KGE003; R&D Systems Inc, Minneapolis, MN, USA). Low, medium, and high controls (QC54 cGMP controls; R&D Systems Inc) were added to each plate. Briefly, urine and plasma samples were thawed at room temperature, and 25 μL of each sample was mixed with 475 μL of diluent. Samples were further diluted 1:2 using the diluent provided if final concentrations exceeded the range of the assay. Of note, 100 μL of each standard, sample, and control were mixed with 50 μL of the cGMP conjugate and 50 μL of the primary antibody solution. After incubating for 3 hours on a microplate shaker at 450 rpm at room temperature, wells were washed 4 times, and 200 μL of substrate solution were added. This was incubated for 30 minutes in the dark, and 50 μL of stop solution were added. The plate was read using a microplate reader (Infinite M1000 PRO; Tecan, Männedorf, Switzerland) set at 450 and 540 nm. Readings made at 540 nm were subtracted from those made at 450 nm. A standard curve was generated using a 4‐parameter logistic curve fit using GraphPad Prism V7.02 (La Jolla, California). Measured concentrations were derived using this standard curve and multiplied by the dilution factor. All samples and standards were assayed in triplicate, all controls in duplicate, and results were averaged for analysis. All standard curves had R2 > 0.987. The %CV of standard, sample, and control replicates were < 10%, and the %CV of standards between plates were < 10%. Each control fell within the expected concentration range. Plasma concentrations of cGMP were reported in pM/mL. In order to account for differences in concentration due to rate of urine production, urinary excretion of cGMP (UEcGMP; pM/kg/min) was also calculated by multiplying the UOP by the urinary concentration of cGMP.
Plasma and urine sodium (Na), potassium (K), and creatinine concentrations were measured at a commercial veterinary diagnostic laboratory (Veterinary Pathology Diagnostic Service, University of Sydney, Australia). Fractional excretion of sodium (FENa) and potassium (FEK) were calculated using the formula:
where X indicates sodium or potassium.
Plasma concentrations of aldosterone were determined at the Radboud University Medical Centre laboratory (Nijmegen, the Netherlands) using a validated aldosterone assay involving ultra‐high‐performance liquid chromatography (UHPLC) coupled with tandem mass spectrometry (Rotteveel‐de Groot et al., 2018 manuscript in preparation). Briefly, 500 μL of plasma was prepared with protein precipitation, supported liquid extraction (Isolute SLE+; Biotage, Uppsala, Sweden) and solid phase extraction (Oasis HLB; Waters, Etten‐Leur, the Netherlands). Eluates were injected on an Agilent Technologies 1290 Infinity VL UHPLC system (Agilent Technologies, Santa Clara, California) equipped with a CSH Phenyl Hexyl (1.7 μm, 2.1 × 100 mm) analytical column (Waters) and coupled to an Agilent 6490 tandem mass spectrometer (Agilent Technologies). Settings included a capillary voltage of 3.0 kV, fragmentor voltage of 380 V, sheath gas temperature of 350°C, and gas temperature of 200°C. N2 was used as the collision gas. Aldosterone was quantified using both aldosterone‐d4 and aldosterone‐d7 internal standards (IsoSciences, Ambler, Pennsylvania). Two transitions (qualitative and quantitative) were measured for each analyte. The transitions (Q1 > Q3) were m/z 366.1 > 338.2 (15 kEV) and m/z 366.1 > 194.1 (15 kEV) for aldosterone‐d7; m/z 363.1 > 335.2 (15 kEV) and m/z 363.1 > 190.1 (15 kEV) for aldosterone‐d4; m/z 359.1 > 331.2 (15 kEV) and m/z 359.1 > 189.1 (15 kEV) for aldosterone. The dwell time was 60 milliseconds. Sample concentrations were derived using a 7‐point calibration curve generated from aldosterone reference standards (Batch H158; Steraloids, Newport, Rhode Island) using Microsoft Excel and MassHunter (Agilent Technologies). All samples and standards were assayed in duplicate and averaged. The lowest limit of quantification (LLOQ) was estimated at 0.070 nM/L, with total %CV of 9.0 and 6.9 at 0.12 and 0.68 nM/L, respectively (n = 78).
2.7. Statistical analysis
Descriptive analysis consisted of visual inspection of trends over time and summary statistics. The median and range were reported. Genstat 16th edition (VSN International Ltd, Hemel) was used for statistical analysis. Data were evaluated for normality via inspection of Q‐Q plots. Where the data were not normally distributed, transformations (loge for UOP, plasma immunoreactive BNP‐32 concentrations, FEK, urinary cGMP concentrations, urinary excretion of cGMP; Logit transformation for FENa) were performed to approximate normality before statistical analysis.
Between‐ and within‐treatment effects were evaluated via linear mixed modeling with restricted maximum likelihood estimation using the following equation:
Significance was set at α of 0.05. If the interaction term was significant, significant interactions were identified using the least square difference method post hoc. If this term was not significant, it was dropped from the model. Model assumptions were assessed through visual inspection of residual plots.
3. RESULTS
Breeds represented in the study cohort included the Cavalier King Charles Spaniel (n = 2), Kelpie, Maltese cross, Jack Russell Terrier cross, and Miniature Poodle cross (n = 2). Baseline characteristics and doses of medications have been summarized in Table 1. At the time of the first study day (visit 1), dogs had been on treatment with furosemide for a median of 59 days (range 18‐133 days), pimobendan for 63 days (range 26‐252 days), and benazepril for 33 days (range 16‐62 days). The median number of days between the diagnosis of CHF and the first study day was 59 days (range 18‐133 days).
Table 1.
Baseline characteristics at the time of screening in 7 dogs enrolled in the study
| Median | Range | |
|---|---|---|
| Weight at screening (kg) | 8.9 | 5.2‐14.4 |
| Age (y) | 12 | 10.5‐17 |
| Daily pimobendan dose (mg/kg/d) | 0.45 | 0.32‐0.56 |
| Daily furosemide dose (mg/kg/d) | 3.85 | 1.94‐5.06 |
| Daily benazepril dose (mg/kg/d) | 0.32 | 0.17‐0.56 |
| LA : Ao | 2.3 | 1.8‐2.5 |
| wnLVIDd | 2.16 | 1.84‐2.44 |
| wnLVIDs | 0.95 | 0.83‐1.45 |
| FS (%) | 53 | 35.3‐56.9 |
| HR (bpm) | 120 | 114‐150 |
| SBP (mm Hg) | 138 | 120‐148 |
| DBP (mm Hg) | 80 | 66‐85 |
| MBP (mm Hg) | 100 | 92‐105 |
| BG (mg/dL) | 104.5 | 82.88‐120.72 |
| BUN (mg/dL) | 33.33 | 24.65‐48.74 |
| Crea (mg/dL) | 1.14 | 0.74‐1.67 |
| TP (g/dL) | 7.3 | 5.1‐7.8 |
| Albumin (g/dL) | 3.2 | 2.5‐4.2 |
| Globulin (g/dL) | 3.6 | 2.7‐4.5 |
| ALT (U/L) | 95 | 24‐293 |
| ALKP (U/L) | 101 | 12‐341 |
| Na (mEq/L) | 149 | 145‐153 |
| K (mEq/L) | 3.6 | 2.7‐4.2 |
| Cl (mEq/L) | 112 | 106‐120 |
| PCV (%) | 47 | 37‐54 |
Abbreviations: ALKP, alkaline phosphatase; ALT, alanine aminotransferase; BG, blood glucose; BUN, blood urea nitrogen; Crea, creatinine; FS, fractional shortening; HR, heart rate; LA : Ao, left atrium to aorta ratio; SBP/DBP/MBP, systolic/diastolic/mean blood pressure; TP, total protein; wnLVIDd/s, weight‐normalized left ventricular internal diameter in diastole/systole.
A deviation from the protocol occurred during visit 1 (BNP treatment) in 1 dog when urine samples were erroneously collected from the end of an extension line holding 10 mL of volume which was attached to the urinary catheter instead of emptying the contents of the line each time. As the line was filled with urine before collection of data, the measurement of UOP was not affected. However, the volume held in this line exceeded the amount of urine production in each hour; thus, urinary analyte concentrations did not accurately reflect expected concentrations for the period in which the sample was taken. As such, urinary endocrine, cGMP, and fractional excretions at visit 1 for this dog were not calculated, and these data were excluded from the overall analysis.
Administration of synthetic canine BNP1‐32 was well tolerated by all dogs in the study. There were no systemic or local adverse effects encountered during the study.
3.1. Effect of treatments on endocrine variables, cGMP, and fractional excretions
The effect of each treatment on endocrine, cGMP, and FENa/K are depicted in Figures 1, 2, 3.
Figure 1.

Plasma concentrations of immunoreactive BNP‐32 (pg/mL; graph A) and cGMP (pM/mL; graph B) after administration of BNP1‐32 (5 μg/kg, solid black line), furosemide (2 mg/kg, solid gray line), or BNP1‐32 + furosemide (5 μg/kg + 2 mg/kg, dashed black line) SC in 7 dogs with chronic congestive heart failure caused by myxomatous mitral valve disease. The median and range were plotted. Asterisks denote significant (P < .05) differences between BNP1‐32 and furosemide (*) and BNP1‐32 + furosemide and furosemide (***). Hats denote significant (P < .05) differences from baseline for BNP1‐32 (∧) and BNP1‐32 + furosemide (∧ ∧ ∧)
Figure 2.

Urinary excretion of cGMP (pM/kg/min; graph A) and fractional excretions of sodium (FENa, %; graph B) and potassium (FEK, %; graph C) after administration of BNP1‐32 (5 μg/kg, solid black line), furosemide (2 mg/kg, solid gray line), or BNP1‐32 + furosemide (5 μg/kg + 2 mg/kg, dashed black line) SC in 7 dogs with chronic congestive heart failure caused by myxomatous mitral valve disease. The median and range were plotted. Asterisks denote significant (P < .05) differences between BNP1‐32 and furosemide (*); BNP1‐32 and BNP1‐32 + furosemide (**); and BNP1‐32 + furosemide and furosemide (***). Hats denote significant (P < .05) differences from baseline for BNP1‐32 (∧); furosemide (∧ ∧); and BNP1‐32 + furosemide (∧ ∧ ∧)
Figure 3.

Plasma concentrations of aldosterone (nM/L) after administration of BNP1‐32 (5 μg/kg, solid black line), furosemide (2 mg/kg, solid gray line), or BNP1‐32 + furosemide (5 μg/kg + 2 mg/kg, dashed black line) SC in 7 dogs with chronic congestive heart failure caused by myxomatous mitral valve disease. The lowest limit of quantification (LLOQ) of the assay was 0.070 nM/L. All concentrations < LLOQ were estimated to be half the LLOQ
Plasma concentrations of immunoreactive BNP‐32 rose significantly within an hour of BNP and FRUS/BNP treatments but not with FRUS (Figure 1). Immunoreactive BNP‐32 levels returned to baseline by 3 hours after treatment.
Significant between‐treatment or within‐treatment effects were not observed with regard to plasma concentrations of cGMP, as indicated by the lack of a significant treatment‐time interaction (Figure 1). In contrast to plasma concentrations of cGMP, a significant rise in UEcGMP occurred after administration of BNP and FRUS/BNP treatments, with observable differences arising between these treatments and FRUS (Figure 2). Maximal effects were reached 1 hour after treatment and decreased to baseline levels by 3 hours after treatment. In contrast, UEcGMP reduced after FRUS treatment.
Both FENa and FEK increased when dogs were given any treatment containing furosemide (Figure 2). However, administration of BNP did not cause a significant change in these variables, and the addition of BNP did not result in significant changes in the excretion of these electrolytes when compared to treatment with furosemide alone.
Around 60% of plasma samples had aldosterone concentrations < LLOQ. Because of this large number, only a descriptive analysis was performed. Traditional approaches for handling such data in bioanalytics include reporting values as missing, “rounding down” values to zero, or estimating these values to be half the LLOQ.35 As the data was not missing, and as aldosterone concentrations would not be expected to be zero from a biological perspective, the value of half the LLOQ was assigned where concentrations < LLOQ were obtained, and plasma aldosterone concentrations were graphically depicted (Figure 3).
3.2. Effect of treatments on cardiorenal variables
The UOP increased significantly from baseline after administration of FRUS and FRUS/BNP and remained increased at the end of the study; in contrast, BNP alone did not result in changes to UOP (Figure 4). Maximal effects were reached at 1 hour after treatment in the FRUS and FRUS/BNP groups, with a 1246% and 887% increase in median UOP compared to baseline, respectively. Though these treatments resulted in significantly higher UOP compared to BNP, there were no differences between the 2 furosemide containing treatments. None of the treatments resulted in significant changes in HR or BP (Figure 5).
Figure 4.
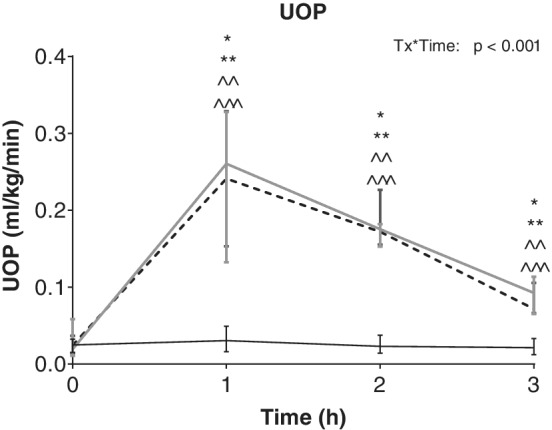
Urine output (UOP) after administration of BNP1‐32 (5 μg/kg, solid black line), furosemide (2 mg/kg, solid gray line), or BNP1‐32 + furosemide (5 μg/kg + 2 mg/kg, dashed black line) SC in 7 dogs with chronic congestive heart failure caused by myxomatous mitral valve disease. The median and range were plotted. Asterisks denote significant (P < .05) differences between BNP1‐32 and furosemide (*); and BNP1‐32 and BNP1‐32 + furosemide (**). Hats denote significant (P < .05) differences from baseline for furosemide (∧ ∧) and BNP1‐32 + furosemide (∧ ∧ ∧)
Figure 5.

Systolic blood pressure (SBP; graph A), diastolic blood pressure (DBP; graph B), mean blood pressure (MBP; graph C), and heart rate (HR; graph D) after administration of BNP1‐32 (5 μg/kg, solid black line), furosemide (2 mg/kg, solid gray line), or BNP1‐32 + furosemide (5 μg/kg + 2 mg/kg, dashed black line) SC in 7 dogs with chronic congestive heart failure caused by myxomatous mitral valve disease. The median and range were plotted
4. DISCUSSION
This is a report of a prospective study evaluating the cardiorenal and endocrine effects and tolerability of single doses of synthetic canine BNP1‐32 given SC to dogs with compensated ACVIM stage C CHF caused by MMVD. BNP1‐32 was well tolerated, and after subcutaneous dosing, it was well absorbed and resulted in an increase in UEcGMP; however, there were no measurable effects on biological outcomes (UOP, BP, HR, FENa/K, and plasma aldosterone concentration) in this study. These findings are in contrast to results from dogs with experimentally induced CHF, and thus, raise questions regarding the potential for natriuretic peptide resistance in dogs with naturally occurring CHF.
In particular, derangements of intracellular pathways via which cGMP mediate natriuresis and diuresis might exist in dogs with CHF caused by MMVD. The rise in plasma concentrations of immunoreactive BNP1‐32 observed upon its administration confirms successful absorption from the site of administration. Appropriate binding to NPR‐A is demonstrated through the rise in UEcGMP accompanying any treatment containing BNP1‐32 but not furosemide alone. The validity of urinary and plasma concentrations of cGMP in demonstrating this interaction is evidenced by observations from numerous previous studies relating to this matter.14, 16, 17, 18, 19, 23, 24, 26, 28, 36, 37, 38, 39, 40 These changes in cGMP also make NPR‐A receptor downregulation less likely. The process of elimination thus renders downstream intracellular mechanisms a potential source of natriuretic peptide resistance in these dogs. Although the entirety of the pathways by which cGMP mediates cellular effects has yet to be discovered, there is evidence to support involvement of cGMP‐dependent protein kinases (PKG)41, 42, 43 and amiloride‐sensitive cation channels43 in the generation of natriuretic peptide‐induced vasodilation, natriuresis, and diuresis. In addition, phosphodiesterase 5 inhibition to reduce cGMP hydrolysis appears to be of benefit in the face of CHF and augments effects of exogenous BNP1‐32.19, 26, 44, 45, 46
Alternatively, the biological effects of BNP1‐32 might have been obscured in the present study due to aspects relating to its study design. The labeled use of Nesiritide in people is for acutely decompensated CHF, to be given as an IV bolus succeeded by a continuous rate infusion. In contrast, this study utilized an SC mode of delivery in dogs with compensated chronic CHF in order to minimize variability in clinical presentation and investigated the potential for its use in chronic treatment. Despite experimental studies showing effects of BNP1‐32 in chronic CHF,18, 23, 24 it is possible that the lack of an overt state of volume overload might have resulted in lack of diuresis and natriuresis in this study, or that longer exposure to BNP1‐32 (either as a constant rate infusion or via multiple SC delivered doses of BNP1‐32) might have been required to produce detectable changes to cardiorenal variables. The noninvasive techniques of measuring BP and HR might have lacked the sensitivity to detect BNP1‐32 effects in such a small group of dogs, and only a limited number of cardiovascular variables were evaluated. Renal blood flow and measures of renal function other than UOP and FENa/K were not measured. The small number of sampling points might have also resulted in a failure to detect short‐lived effects. A crossover treatment design was used so that each dog acted as its own “control,” thereby reducing variability and increasing chances of detecting treatment effects in this small cohort of dogs; however, the inclusion of a placebo treatment stream in which no furosemide or BNP1‐32 was given could have helped to highlight smaller treatment effects. Finally, a group of healthy dogs undergoing the same study procedures would have strengthened the conclusions drawn from this study and enabled direct comparisons to be made between healthy and diseased dogs.
The effect of BNP1‐32 on the spontaneous and furosemide‐activated RAAS in this population remains ill‐defined due to the majority of samples containing aldosterone concentrations below the LLOQ. With the reported range of plasma aldosterone concentrations being 20‐800 pM/L in dogs with naturally occurring CHF,47, 48 an LLOQ of 70 pM/L (equivalent to 0.07 nM/L) would appear to be adequate. Although in‐house stability testing showed prolonged stability of serum aldosterone, sample degradation in plasma remains a possible cause of these low concentrations. Alternative explanations include the chronicity of benazepril treatment (16‐62 days, with median 33 days) and the possibility for reduced RAAS activation in the chronic, stable phase of CHF.49 Despite this, both treatments containing BNP1‐32 appeared to demonstrate an initial suppression and then a rise in aldosterone concentrations on visual inspection of Figure 3. This observation is weak but is in contrast to previous reports of aldosterone suppression after BNP1‐32 administration in people with CHF23 and in dogs with furosemide‐induced RAAS activation.17 As a final note, the cGMP and aldosterone assays employed in this study should ideally have been fully validated for use on canine plasma and urine samples. Instead, formal validation was performed by the manufacturers of the assays utilizing samples from other species (humans and rodents), and partial validation and quality control monitoring of canine samples were employed during the course of this study. Yet, with the molecular structure of both cGMP and aldosterone being conserved between species, it is expected that the assays will perform similarly in samples from different species. These tests were thus deemed fit for use for the purposes of this study.
In summary, SC administered synthetic canine BNP1‐32 was well‐absorbed and increased UEcGMP in dogs with compensated CHF caused by MMVD. However, there were no biological effects on HR, BP, UOP, or FENa/K, and BNP1‐32 did not augment effects of furosemide on UOP or FENa/K. These findings suggest peripheral natriuretic peptide resistance and raise further questions regarding the role of BNP1‐32 in both the pathophysiology and treatment of CHF in dogs with MMVD.
CONFLICT OF INTEREST DECLARATION
Authors declare no conflict of interest.
OFF‐LABEL ANTIMICROBIAL DECLARATION
Authors declare no off‐label use of antimicrobials.
INSTITUTIONAL ANIMAL CARE AND USE COMMITTEE (IACUC) OR OTHER APPROVAL DECLARATION
Ethics approval was obtained from the University of Sydney Animal Ethics Committee (approval number 2016/1113).
HUMAN ETHICS APPROVAL DECLARATION
Authors declare human ethics approval was not needed for this study.
ACKNOWLEDGMENTS
The authors acknowledge Teun van Herwaarden for assistance with the aldosterone assays and Elisabeth Newfield and Kelly Amaro for their assistance in conducting this study. An abstract of this work was presented at the 2018 ACVIM Forum, Seattle, WA.
Yata M, Kooistra HS, Beijerink NJ. Cardiorenal and endocrine effects of synthetic canine BNP1‐32 in dogs with compensated congestive heart failure caused by myxomatous mitral valve disease. J Vet Intern Med. 2019;33:462–470. 10.1111/jvim.15416
Funding information Catherine Heather Patron Bequest; University of Sydney Compacts Grant
REFERENCES
- 1. Borgarelli M, Buchanan JW. Historical review, epidemiology and natural history of degenerative mitral valve disease. J Vet Cardiol. 2012;14:93‐101. [DOI] [PubMed] [Google Scholar]
- 2. Atkins CE, Bonagura JD, Ettinger SJ, et al. Guidelines for the diagnosis and treatment of canine chronic valvular heart disease. J Vet Intern Med. 2009;23:1142‐1150. [DOI] [PubMed] [Google Scholar]
- 3. Häggström J, Boswood A, O'Grady MR, et al. Effect of pimobendan or benazepril hydrochloride on survival times in dogs with congestive heart failure caused by naturally occurring myxomatous mitral valve disease: the QUEST study. J Vet Intern Med. 2008;22:1124‐1135. [DOI] [PubMed] [Google Scholar]
- 4. De Madron E, King JN, Strehlau G, et al. Survival and echocardiographic data in dogs with congestive heart failure caused by mitral valve disease and treated by multiple drugs: a retrospective study of 21 cases. Can Vet J. 2011;52:1219‐1225. [PMC free article] [PubMed] [Google Scholar]
- 5. Lombard CW, Jöns O, Bussadori CM. Clinical efficacy of pimobendan versus benazepril for the treatment of acquired atrioventricular valvular disease in dogs. J Am Anim Hosp Assoc. 2006;42:249‐261. [DOI] [PubMed] [Google Scholar]
- 6. Borgarelli M, Savarino P, Crosara S, et al. Survival characteristics and prognostic variables of dogs with mitral regurgitation attributable to myxomatous valve disease. J Vet Intern Med. 2008;22:120‐128. [DOI] [PubMed] [Google Scholar]
- 7. Bernay F, Bland J, Häggström J, et al. Efficacy of spironolactone on survival in dogs with naturally occurring mitral regurgitation caused by myxomatous mitral valve disease. J Vet Intern Med. 2010;24:331‐341. [DOI] [PubMed] [Google Scholar]
- 8. Ovaert P, Elliott J, Bernay F, et al. Aldosterone receptor antagonists – how cardiovascular actions may explain their beneficial effects in heart failure. J Vet Pharmacol Ther. 2010;33:109‐117. [DOI] [PubMed] [Google Scholar]
- 9. Peddle GD, Singletary GE, Reynolds CA, Trafny DJ, Machen MC, Oyama MA. Effect of torsemide and furosemide on clinical, laboratory, radiographic and quality of life variables in dogs with heart failure secondary to mitral valve disease. J Vet Cardiol. 2012;14:253‐259. [DOI] [PubMed] [Google Scholar]
- 10. Chetboul V, Pouchelon JL, Menard J, et al. Short‐term efficacy and safety of torasemide and furosemide in 366 dogs with degenerative mitral valve disease: the TEST study. J Vet Intern Med. 2017;31:1629‐1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Uechi M. Mitral valve repair in dogs. J Vet Cardiol. 2012;14:185‐192. [DOI] [PubMed] [Google Scholar]
- 12. Uechi M, Mizukoshi T, Mizuno T, et al. Mitral valve repair under cardiopulmonary bypass in small‐breed dogs: 48 cases (2006–2009). J Am Vet Med Assoc. 2012;240:1194‐1201. [DOI] [PubMed] [Google Scholar]
- 13. Mizuno T, Mizukoshi T, Uechi M. Long‐term outcome in dogs undergoing mitral valve repair with suture annuloplasty and chordae tendinae replacement. J Small Anim Pract. 2013;54:104‐107. [DOI] [PubMed] [Google Scholar]
- 14. Grantham JA, Borgeson DD, Burnett JC Jr. BNP: pathophysiological and potential therapeutic roles in acute congestive heart failure. Am J Physiol Regul Integr Comp Physiol. 1997;272:R1077‐R1083. [DOI] [PubMed] [Google Scholar]
- 15. Yamamoto K, Burnett JC Jr, Redfield MM. Effect of endogenous natriuretic peptide system on ventricular and coronary function in failing heart. Am J Physiol. 1997;273:H2406‐H2414. [DOI] [PubMed] [Google Scholar]
- 16. Chen HH, Grantham JA, Schirger JA, Jougasaki M, Redfield MM, Burnett JC Jr. Subcutaneous administration of brain natriuretic peptide in experimental heart failure. J Am Coll Cardiol. 2000;36:1706‐1712. [DOI] [PubMed] [Google Scholar]
- 17. Cataliotti A, Boerrigter G, Costello‐Boerrigter LC, et al. Brain natriuretic peptide enhances renal actions of furosemide and suppresses furosemide‐induced aldosterone activation in experimental heart failure. Circulation. 2004;109:1680‐1685. [DOI] [PubMed] [Google Scholar]
- 18. Chen HH, Schirger JA, Cataliotti A, Burnett JC Jr. Intact acute cardiorenal and humoral responsiveness following chronic subcutaneous administration of the cardiac peptide BNP in experimental heart failure. Eur J Heart Fail. 2006;8:681‐686. [DOI] [PubMed] [Google Scholar]
- 19. Forfia PR, Lee M, Tunin RS, Mahmud M, Champion HC, Kass DA. Acute pohosphodiesterase 5 inhibition mimics hemodynamic effects of B‐type natriuretic peptide and potentiates B‐type natriuretic peptide effects in failing but not normal canine heart. J Am Coll Cardiol. 2007;49:1079‐1088. [DOI] [PubMed] [Google Scholar]
- 20. Thireau J, Karam S, Roberge S, et al. β‐Adrenergic blockade combined with subcutaneous B‐type natriuretic peptide: a promising approach to reduce ventricular arrhythmia in heart failure? Heart. 2014;100:833‐841. [DOI] [PubMed] [Google Scholar]
- 21. Colucci WS, Elkayam U, Horton DP, et al. Intravenous Nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure. N Engl J Med. 2000;343:246‐253. [DOI] [PubMed] [Google Scholar]
- 22. Publication Committee for the VMAC Investigators . Intravenous nesiritide vs nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. J Am Med Assoc. 2002;287:1531‐1540. [DOI] [PubMed] [Google Scholar]
- 23. Chen HH, Redfield MM, Nordstrom LJ, Horton DP, Burnett JC Jr. Subcutaneous administration of the cardiac hormone BNP in symptomatic human heart failure. J Card Fail. 2004;10:115‐119. [DOI] [PubMed] [Google Scholar]
- 24. Chen HH, Glockner JF, Schirger JA, Cataliotti A, Redfield MM, Burnett JC Jr. Novel protein therapeutics for systolic heart failure: chronic subcutaneous B‐type natriuretic peptide. J Am Coll Cardiol. 2012;60:2305‐2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. O'Connor CM, Starling RC, Hernandez AF, et al. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. 2011;365:32‐43. [DOI] [PubMed] [Google Scholar]
- 26. Chen HH, Huntley BK, Schirger JA, Cataliotti A, Burnett JC Jr. Maximizing the renal cyclic 3′‐5′‐guanosine monophosphate system with type V phosphodiesterase inhibition and exogenous natriuretic peptide: a novel strategy to improve renal function in experimental overt heart failure. J Am Soc Nephrol. 2006;17:2742‐2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oyama MA, Solter PF, Thorn CL, Stern JA. Feasibility, safety, and tolerance of subcutaneous synthetic canine B‐type natriuretic peptide (syncBNP) in healthy dogs and dogs with stage B1 mitral valve disease. J Vet Cardiol. 2017;19:211‐217. [DOI] [PubMed] [Google Scholar]
- 28. McKie PM, Schirger JA, Benike SL, et al. Chronic subcutaneous brain natriuretic peptide therapy in asymptomatic systolic heart failure. Eur J Heart Fail. 2016;18:433‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Clarkson PBM, Wheeldon NM, MacLeod C, Coutie W, MacDonald TM. Brain natriuretic peptide: effect on left ventricular filling patterns in healthy subjects. Clin Sci. 1995;88:159‐164. [DOI] [PubMed] [Google Scholar]
- 30. Niederkofler EE, Kiernan UA, O'Rear J, et al. Detection of endogenous B‐type natriuretic peptide at very low concentrations in patients with heart failure. Circ Heart Fail. 2008;1:258‐264. [DOI] [PubMed] [Google Scholar]
- 31. Zhang Y, Dufield D, Klover J, et al. Development and validation of an LC–MS/MS method for quantification of cyclic guanosine 3′,5′‐monophosphate (cGMP) in clinical applications: a comparison with a EIA method. J Chromatogr B. 2009;877:513‐520. [DOI] [PubMed] [Google Scholar]
- 32. Vorderwinkler KP, Artner‐Dworzak E, Jakob G, et al. Release of cyclic guanosine monophosphate evaluated as a diagnostic tool in cardiac diseases. Clin Chem. 1991;37:186‐190. [PubMed] [Google Scholar]
- 33. W‐c L, Vesterqvist O, Delaney C, et al. Pharmacokinetics and pharmacodynamics of the vasopeptidase inhibitor, omapatrilat in healthy subjects. Br J Clin Pharmacol. 2003;56:395‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schellenberg S, Grenacher B, Kaufmann K, Reusch CE, Glaus TM. Analytical validation of commercial immunoassays for the measurement of cardiovascular peptides in the dog. Vet J. 2008;178:85‐90. [DOI] [PubMed] [Google Scholar]
- 35. Keizer RJ, Jansen RS, Rosing H, et al. Incorporation of concentration data below the limit of quantification in population pharmacokinetic analyses. Pharmacol Res Perspect. 2015;3:e00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boerrigter G, Costello‐Boerrigter LC, Harty GJ, Lapp H, Burnett JC Jr. Des‐serine‐proline brain natriuretic peptide 3–32 in cardiorenal regulation. Am J Physiol Regul Integr Comp Physiol. 2007;292:R897‐R901. [DOI] [PubMed] [Google Scholar]
- 37. Jensen KT, Eiskjær H, Carstens J, et al. Renal effects of brain natriuretic peptide in patients with congestive heart failure. Clin Sci. 1999;96:5‐15. [PubMed] [Google Scholar]
- 38. Nakamura M, Arakawa N, Yoshida H, Makita S, Niinuma H, Hiramori K. Vasodilatory effects of B‐type natriuretic peptide are impaired in patients with chronic heart failure. Am Heart J. 1998;135:414‐420. [DOI] [PubMed] [Google Scholar]
- 39. Stasch J‐P, Kazda S, Neuser D. Different effects of ANP and nitroprusside on cyclic GMP extrusion of isolated aorta. Eur J Pharmacol. 1989;174:279‐282. [DOI] [PubMed] [Google Scholar]
- 40. Wong KR, Xie MH, Shi LB, et al. Urinary cGMP as biological marker of the renal activity of atrial natriuretic factor. Am J Physiol Renal Physiol. 1988;255:F1220‐F1224. [DOI] [PubMed] [Google Scholar]
- 41. Potter LR, Abbey‐Hosch S, Dickey DM. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate‐dependent signaling functions. Endocr Rev. 2006;27:47‐72. [DOI] [PubMed] [Google Scholar]
- 42. Theilig F, Wu Q. ANP‐induced signaling cascade and its implications in renal pathophysiology. Am J Physiol Renal Physiol. 2015;308:F1047‐F1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Martel G, Hamet P, Tremblay J. Central role of guanylyl cyclase in natriuretic peptide signaling in hypertension and metabolic syndrome. Mol Cell Biochem. 2010;334:53‐65. [DOI] [PubMed] [Google Scholar]
- 44. Katz SD, Balidemaj K, Homma S, Wu H, Wang J, Maybaum S. Acute type 5 phosphodiesterase inhibition with sildenafil enhances flow‐mediated vasodilation in patients with chronic heart failure. J Am Coll Cardiol. 2000;36:845‐851. [DOI] [PubMed] [Google Scholar]
- 45. Takimoto E, Champion HC, Li M, et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11:214‐222. [DOI] [PubMed] [Google Scholar]
- 46. Guazzi M, Vicenzi M, Arena R, Guazzi MD. PDE5‐inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry and clinical status in patients with stable systolic heart failure: results of a 1‐year prospective, randomized, placebo‐controlled study. Circ Heart Fail. 2011;4:8‐17. [DOI] [PubMed] [Google Scholar]
- 47. Häggström J, Hansson K, Kvart C, Karlberg BE, Vuolteenaho O, Olsson K. Effects of naturally acquired decompensated mitral valve regurgitation on the renin‐angiotensin‐aldosterone system and atrial natriuretic peptide concentration in dogs. Am J Vet Res. 1997;58:77‐82. [PubMed] [Google Scholar]
- 48. Häggström J, Lord PF, Höglund K, et al. Short‐term hemodynamic and neuroendocrine effects of pimobendan and benazapril in dogs with myxomatous mitral valve disease and congestive heart failure. J Vet Intern Med. 2013;27:1452‐1462. [DOI] [PubMed] [Google Scholar]
- 49. Hezzell MJ, Boswood A, Chang YM, Moonarmart W, Elliott J. Associations among serum N‐terminal procollagen type III concentration, urinary aldosterone‐to‐creatinine ratio, and ventricular remodeling in dogs with myxomatous mitral valve disease. Am J Vet Res. 2012;73:1765‐1774. [DOI] [PubMed] [Google Scholar]