

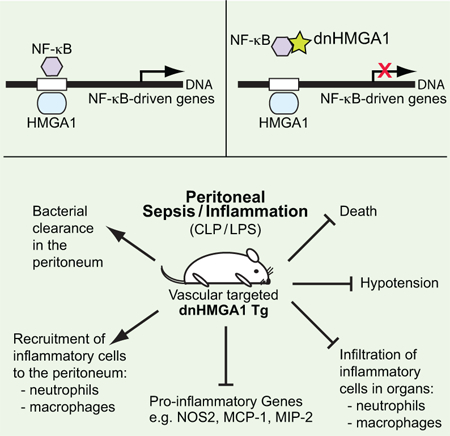
Abstract
High mobility group (HMG) proteins are a family of architectural transcription factors, with HMGA1 playing a role in the regulation of genes involved in promoting systemic inflammatory responses. We speculated that blocking HMGA1-mediated pathways might improve outcomes from sepsis. To investigate HMGA1 further, we developed genetically modified mice expressing a dominant negative (dn) form of HMGA1 targeted to the vasculature. In dnHMGA1 transgenic (Tg) mice, endogenous HMGA1 is present, but its function is decreased due to the mutant transgene. These mice allowed us to specifically study the importance of HMGA1 not only during a purely pro-inflammatory insult of endotoxemia, but also during microbial sepsis induced by implantation of a bacterial-laden fibrin clot into the peritoneum. We found that the dnHMGA1 transgene was only present in Tg and not wild-type (WT) littermate mice, and the mutant transgene was able to interact with transcription factors (such as NF-κB), but was not able to bind DNA. Tg mice exhibited a blunted hypotensive response to endotoxemia, and less mortality in microbial sepsis. Moreover, Tg mice had a reduced inflammatory response during sepsis, with decreased macrophage and neutrophil infiltration into tissues, which was associated with reduced expression of monocyte chemotactic protein-1 and macrophage inflammatory protein-2. Collectively, these data suggest that targeted expression of a dnHMGA1 transgene is able to improve outcomes in models of endotoxin exposure and microbial sepsis, in part by modulating the immune response and suggest a novel modifiable pathway to target therapeutics in sepsis.
Keywords: transgenic mice, architectural transcription factor, immune response, chemokines
Graphical Abstract
Summary statement:
High mobility group A1 is a modifiable pathway for therapeutics in sepsis, using a dominant negative approach to regulate the immune response.
INTRODUCTION
High mobility group (HMG) proteins are a family of architectural transcription factors that have been recognized to play a role in gene regulation [1, 2], and an important member of this family is HMGA1 (previously known as HMG-I/Y). HMGA1 binds to AT-rich regions in the minor groove of DNA via motifs known as AT-hooks [3–5], and facilitates the assembly of functional nucleoprotein complexes by inducing changes in DNA structure [6, 7]. HMGA1 also recruits nuclear proteins to enhancers [8–10], and augments the binding of transcription factors to their binding sites [2, 11]. Our laboratory and others have shown that HMGA1 is vital for the regulation of mouse [12–14], rat [15], and human [16] nitric oxide synthase (NOS)2 in the setting of inflammatory stimuli. HMGA1 has also been reported to regulate genes involved in the pathobiology of a number of critical illnesses, including mediators that promote proliferation, differentiation, antigen presentation, inflammation, and cancer [17–19].
Minor groove binding (MGB) drugs, such as Distamycin A and Netropsin, interfere with binding at AT-rich sites in the minor groove of DNA, and are capable of altering gene transcription, in part, by altering the binding of HMGA1 to DNA [2]. By administering MGB drugs systemically, we showed previously that inhibition of HMGA1 binding decreased hypotension and mortality association with the systemic administration of Escherichia (E.) coli lipopolysaccharide (LPS, or endotoxin), by reducing expression of NOS2 [20, 21]. More recently, we revealed that the effects of MGB drugs on HMGA1 can attenuate tissue inflammation during endotoxemia in vivo [22] by decreasing expression of P-selectin. Since MGB drugs (Distamycin A and Netropsin) have antibiotic properties, elucidating the specific role of HMGA1 in microbial sepsis has been more of a challenge.
HMGA1 has been disrupted in mice to explore its function in vivo, however both homozygous and heterozygous mice for the HMGA1 null allele showed cardiac hypertrophy, and the mice developed hematologic malignancies [23]. To overcome these underlying abnormalities, we generated transgenic (Tg) mice overexpressing a dominant-negative (dn) form of HMGA1 (dnHMGA1), which allowed us to further elucidate the importance of HMGA1 not only during a purely pro-inflammatory and more acute insult of Escherichia (E.) coli endotoxemia, but also during a more prolonged microbial sepsis model induced by E. coli fibrin clot. In the Tg mice, endogenous HMGA1 is not absent, however its function is decreased due to overexpression of a mutant transgene [24–26]. In the present study, Tg mice had a blunted hypotensive response to endotoxemia, and in a model of microbial sepsis, exhibited less mortality. Moreover, Tg mice had a reduced inflammatory response during sepsis, with a prominent decrease in macrophage and neutrophil infiltration into organs. These data suggest that targeted expression of a dnHMGA1 transgene is able to improve outcomes in models of endotoxin exposure and microbial sepsis, with an important component of modulating the immune response.
MATERIALS and METHODS
Reagents.
Murine recombinant interleukin (IL)-1β and tumor necrosis factor (TNF)-α was purchased from PeproTech Inc. E. coli lipopolysaccharide (LPS, serotype O26:B6) was purchased from Sigma-Aldrich.
Generation of the transgenic construct and mice.
To generate the transgenic construct (Figure 1A), we used a dnHMGA1 construct containing four proline to alanine substitutions introduced at amino acids 57, 61, 83, and 87 located in the second and third DNA-binding domains (AT-hooks), as described previously [25]. A hemagglutinin (HA) tag was placed on dnHMGA1 construct, in addition to a 300-bp DNA fragment containing bovine growth hormone polyadenylation sequences (bGHpA) that was ligated 3’ to the mutated HMGA1 cDNA. The fragment containing dnHMGA1/HA/bGHpA was then ligated downstream of the 2.5 kb mouse aortic carboxypeptidase-like protein (ACLP) promoter [27, 28]. The construct was subsequently injected into the pronuclei of fertilized C57BL/6 mouse eggs (Brigham and Women’s Hospital, Core Transgenic Mouse Facility). Two separate injections were performed, generating different lines of mice, and two independent lines of mice (Tg1 and Tg2) were selected for studies after expression of the transgene was confirmed. Transgenic mice harboring the ACLP promoter/dnHMGA1 cDNA were identified by Southern blot analysis [29] using a 32P-labeled probe from the ACLP promoter, after Sac1 digestion of genomic DNA prepared from tail biopsies (Figure 1B). Southern blotting recognized an endogenous (En) and a transgenic (Tg) fragment (Figure 1B). To determine the transgene copy number, radioactivity was measured on a PhosphorImager running ImageQuant software (Molecular Dynamics). Transgene copy number was determined by first calculating the ratio of the Tg/En bands, and then multiplying by a factor of 2 (for the two endogenous copies). Tg and littermate WT control mice were used at 8–12 weeks of age.
Figure 1. Characterization of dominant negative (dn) HMGA1 transgenic mice.
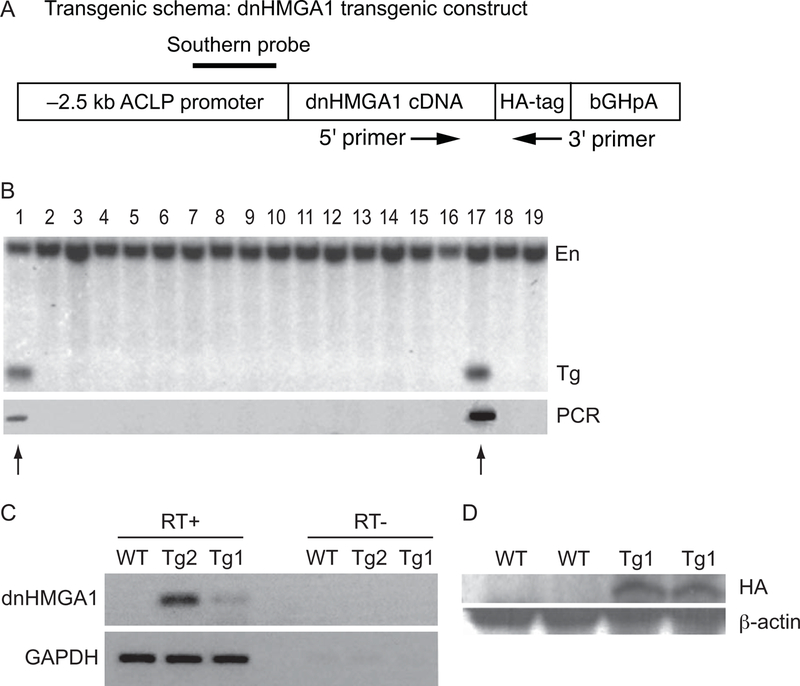
A) Schema of dnHMGA1 transgenic construct. Location of Southern probe (solid line) and PCR primers (5’ and 3’, forward and reverse arrows) is depicted. B) Upper panel demonstrates Southern blot genotyping after Sac1 digestion of the genomic DNA and using the proximal promoter region of ACLP as a probe to confirm transgene integration and copy number. A 781 base pair band was demonstrated in founder mice. Lower panel demonstrates PCR genotyping using 5’ and 3’ primers designed according to the sequences of the dnHMGA1 cDNA and the HA-tag, respectively (see A). Arrows depict positive founders. C) Total RNA was extracted from aortas of WT, Tg1, and Tg2 mice, and reverse transcriptase (RT)-PCR was performed. As a control to exclude amplification from genomic DNA, PCR was also performed in the absence of reverse transcriptase (RT). D) Protein was also extracted from aortic SMCs of WT and Tg1 mice, and Western blotting was performed using an antibody directed against the HA-tag (see A). Blotting with an antibody against β-actin was used to assess protein loading.
Cell culture of vascular smooth muscle cells (SMCs).
Primary vascular SMCs were harvested from WT and Tg mouse aortas, using collagenase and elastase digestion of aortas as described previously [30]. The cells were expanded using Dulbecco’s Modified Eagle Medium with 20% fetal bovine serum and Penicillin-Streptomycin-Glutamine.
RNA isolation and RT-PCR analysis.
RNeasy Mini RNA isolation kit (Qiagen) was used to extract total RNA, according to the manufacturer’s instructions, from mouse aortas harvested from WT, Tg2, or Tg1 mice. After either reverse transcriptase (RT+) to generate complementary DNA from the RNA, or no reverse transcriptase (RT–), PCR was performed using a forward primer from the sequence of HMGA1 (5’ GCCTCCAAGCAGGAAAAGG 3’) and a reverse primer from the sequence of the HA tag (5’ GGGACGTCGTATGGGTACTG 3’). The RT– samples exclude PCR amplification from genomic DNA contamination. For quantitative real time-PCR (qRT-PCR), RNA was extracted using TRIzol® Reagent from SMCs (after decreasing FBS to 0.5 % FBS) stimulated with vehicle (Veh, PBS), IL-1β (10 ng/ml), or TNF-α (10 ng/ml) for various time points. The samples were treated with DNase I to degrade any genomic DNA contamination, and cDNA synthesized by iScript™ Reverse Transcription Supermix. qRT-PCR as described [31] was performed for NOS2, monocyte chemotactic protein (MCP-1), and macrophage inflammatory protein 2 (MIP-2) using the primers mouse NOS2 forward 5’-GCCACCAACAATGGCAACA-3’ and reverse 5’-CGTACCGGATGAGCTGTGAATT-3’; mouse MCP-1 forward 5’-ACTGAAGCCAGCTCTCTCTTCCTC-3’ and reverse 5’-TTCCTTCTTGGGGTCAGCACAGAC-3’; and mouse MIP-2 forward 5’-CCACCAACCACCAGGCTACAGGGGC-3’ and reverse 5’-AGGCTCCTCCTTTCCAGGTCAGTTAGC-3’. qRT-PCR of β-actin was employed for normalization of NOS2, MCP-1, and MIP-2 expression by the comparative Ct method, using primers mouse β-actin forward 5’-ACCAACTGGGACGATATGGAGAAGA-3’ and reverse 5’-TACGACCAGAGGCATACAGGGACAA-3’.
Western blot analysis.
Western immunoblotting was performed as previously described [32]. Briefly, the cells were harvested using RIPA buffer (Tris/Cl (pH 7.6)), 100 mmole/L; EDTA, 5 mmole/L; NaCl, 50 mmole/L; β-glycerophosphate, 50 mmole/L; NaF, 50 mmole/L; Na3VO4, 0.1 mmole/L; NP-40, 0.5%; sodium deoxycholate, 0.5%) with protease inhibitors (Roche Applied Science). Protein concentrations of cell lysates were determined using a BCA protein assay kit (Thermo Scientific). The samples were resolved by NuPAGE® Novex 4~12% Bis-Tris gels (Invitrogen, Carlsbad, CA), and transferred to PVDF membranes (Bio-Rad, Hercules, CA). The transferred membranes were hybridized with an antibody against HA (Roche Applied Science), followed by HRP-conjugated IgG, and visualized with SuperSignal West Pico Chemiluminescent Substrate (Pierce).
Transfection assays.
Mouse NOS2 luciferase reporter plasmid (base pairs −1485/+31) [33], was transiently transfected into SMCs (FuGENE 6 transfection reagent, Roche Applied Science), as described previously [34]. The transfection was performed in the presence of the dnHMGA1 expression plasmid (at a ratio of 4:1 to the promoter), or a control plasmid. A β-galactosidase expression vector was also co-transfected to normalize for luciferase activity. The cells were treated with IL-1β or vehicle (PBS). Twenty-four hours following treatment, the cells were harvested and assessed for luciferase activity and β-galactosidase, as described previously [21, 34].
Co-immunoprecipitation (co-IP) Assays.
Co-IP assays were performed as previously described [35]. In brief, Protein G plus/Protein A-Agarose (Calbiochem) beads were washed with PBS and resuspended with dilution buffer (PBS+1 mg/ml BSA). An antibody against the p50 subunit of NF-κB (Santa Cruz Biotechnology), 5 µg in dilution buffer, was added at a 1:1 ratio to the beads. Dimethyl pimelimidate 1 mg/ml was added to prevent IgG binding to the beads. The beads were agitated for 30 minutes at room temperature, and then washed three times with buffer (0.2 M triethanolamine in PBS) prior to quenching the crosslinking with 50 mM ethanolamine in PBS. Finally, unlinked antibody was washed out with 1 M glycine (pH 3) and the crosslinked beads were reconstituted in PBS. SMCs from WT or dnHMGA1 Tg mice were then harvested and the nuclear protein extract was isolated with lysis buffer (20 mM Tris HCl pH 7.5, 25% sucrose, 420 mM NaCl, 5 mM dithiothreitol, 2 mM MgCl2 0.2 mM EDTA, 1x protease inhibitor mixture (Roche Applied Science)). Nuclear protein lysates (400 µg) were incubated with the antibody-crosslinked beads at 4°C overnight. The beads were then washed four times with a cold buffer (20 mM Tris HCl pH 7.2, 1 mM EDTA, 0.1% Triton X-100, 150 mM NaCl, 1 mg/ml BSA, and 1x protease inhibitor mixture), and Western blot analysis was subsequently performed on the bound proteins using an HA antibody (Roche Applied Science).
Blood pressure analysis.
To evaluate blood pressure (BP) of mice, intra-arterial catheters were placed in the carotid arteries, as we have performed previously [20, 36, 37]. For studies in mice exposed to endotoxemia (E. coli lipopolysaccharide, LPS), the mice were allowed to recover overnight after catheter placement, and mean arterial pressure was assessed following intraperitoneal (i.p.) endotoxin administration (10 mg/kg). The mice then had their blood pressure assessed at serial time points.
Fibrin clot model of peritoneal sepsis in mice.
For the fibrin clot model [28], E. coli bacteria (strain MMB1287) were grown in LB media for 24 hours at 37°C, and 1 ml bacteria were regrown in 9 ml LB media for 3 hours at 37°C. The bacteria were pelleted by centrifugation at 3500 rpm for 10 minutes, washed in 10 ml PBS twice, and resuspended in 1 ml PBS. The bacteria were diluted in 1% bovine fibrinogen (Sigma, St. Louis, MO), and the final bacterial concentrations of the experiments were 2.2 ± 0.2 × 109 cfu/clot. Human thrombin (Sigma, St. Louis, MO) was added at a final concentration of 2 U/ml, and the mixture was allowed to clot for 10 minutes [38]. The bacterial concentration in the suspension and the clot was confirmed in each experiment by plating and overnight incubation at 37°C and counting CFUs. Under ketamine/xylazine anesthesia and sterile conditions, the fibrin clot was placed within the peritoneal cavity of WT and Tg mice via a 1.5 cm abdominal incision that was closed in layers with 6–0 surgical sutures (Harvard Apparatus, Hilliston, MA).
Assessment of bacterial colony forming units and inflammatory cells from peritoneal lavage.
WT and Tg mice were sacrificed 24 hours after fibrin clot placement, and peritoneal lavage was performed. The peritoneal fluid was assessed for colony forming units (CFUs) of bacteria, and also for total numbers and percentages of macrophages and neutrophils. Serial dilutions were made from an aliquot of peritoneal fluid, and then incubated overnight at 37°C on LB agar plates and CFUs of bacteria were counted and calculated [39]. Cells from the remaining fluid were stained with antibodies targeting F4/80-APC (BioLegend) and Ly6G-PE (BD Biosciences), to identify macrophages and neutrophils respectively, and were then assessed by flow cytometry using a BD FACS Canto II, and analyzed by FlowJo software.
Phagocytosis assay.
In vivo phagocytosis was performed as described [39]. Mice were injected with 5×106 live GFP-labeled E. coli as described. After one hour of incubation, the mice were anesthetized and a peritoneal lavage performed. The cells from the lavage (predominantly neutrophils) were treated with 0.2% of trypan blue to quench extracellular fluorescence, and then stained for Ly6G-Alexa 647 (BioLegend). Following staining, the cells were fixed (BD Cytofix/Cytoperm, BD Biosciences), and phagocytosis was measured by flow cytometry. The percentage of double positive (GFP and Alexa 647) populations is an index of neutrophil phagocytosis. To assess macrophage phagocytosis, macrophages in the peritoneal lavage were identified by forward and side scatter characteristics after live cell gating. This gating strategy was verified by staining peritoneal lavage with the F4/80 macrophage marker. Macrophage phagocytosis was determined as the percentage of macrophages positive for GFP. Flow cytometry was performed using a BD FACS Canto II, and analysis using FlowJo software.
Histology.
Mice were sacrificed 24 hours following fibrin clot placement, and the spleens and lungs were harvested for histological evaluation. The tissues were fixed in either 10% formalin or methyl Carnoy’s solution, processed, embedded in paraffin, and sectioned (5 μm). Tissues were immunostained with F4/80 (AbD Serotec) or Ly6G (BioLegend) antibodies, for assessment of macrophage or neutrophil infiltration respectively, and scored by an investigator who was blinded to the group. The area of positively stained cells was calculated per 40X objective using FRIDA Software (FRamework for Image Dataset Analysis, [40]), and numerous random fields were assessed per tissue section. Immunostaining using an isotype control antibody was also performed to demonstrate a lack of non-specific staining (Supplemental Figure 1).
Chemokine / cytokine assay.
For MCP-1 and IL-10 analyses of tissue and plasma, samples were assessed using the multiplex assay technology by Luminex as described previously [41, 42]. The levels of MCP-1 and IL-10 were determined by standard curve analysis. The plate was read on a Luminex 200TM instrument (Luminex, Austin, TX). Data acquisition and analysis were conducted using StarStation software v2.3 (Applied Cytometry Systems, Dinnington, U.K.).
Statistical analyses.
Data are expressed as mean ± SEM. Comparisons of mortality were made by analyzing Kaplan-Meier survival curves, and then log-rank test to assess for differences in survival. For comparisons between two groups, we used Student’s unpaired t test. One-way analysis of variance was used for analysis of more than two groups. When data were not normally distributed, non-parametric analyses were performed using Mann-Whitney U or Kruskal-Wallis testing, respectively. The numbers of samples per group (n), or the numbers of experiments, are specified in the figure legends. Statistical significance is accepted at P<0.05.
RESULTS
Generation of the dnHMGA1 transgenic mouse.
Due to the beneficial effects of blocking HMGA1 binding to DNA by MGB drugs (such as Distamycin A and Netropsin) during endotoxemia [20–22], we hypothesized that overexpression of a mutant HMGA1 transgene incapable of binding to DNA would be beneficial not only in endotoxemia, but also in a mouse model of microbial sepsis. Sepsis is a systemic infectious process disseminated via the blood stream, thus we generated Tg mice using the promoter of aortic carboxypeptidase-like protein (ACLP) to target dnHMGA1 expression in the vasculature [27]. The Tg construct is shown in Figure 1A, and a full description of the construct is provided in Materials and Methods. Mutations in the second and third DNA-binding domains of HMGA1 prevent the dominant-negative construct from binding to DNA [25], thus altering HMGA1 function. The hemagglutinin (HA) tag placed on the construct allows identification of the transgene from endogenous HMGA1. The founder mice were identified by Southern blot analysis of genomic DNA and by PCR (Figure 1B, arrows). Analysis of the copy number demonstrated that one line of mice had twice the copy number of the other, from a separate injection of pronuclei, and we designated the dnHMGA1 mice as Tg2 and Tg1 respectively. To confirm mRNA expression of the transgene in Tg mice, total RNA was isolated from aortas of WT, Tg2 and Tg1 mice, and RT-PCR was performed using primers to HMGA1 and the HA tag. The dnHMGA1 transgene was only present in the Tg mice (Figure 1C). Also, dnHMGA1 protein was detected in vascular SMCs of Tg mice, but not WT mice, by blotting with an antibody against the HA tag (Figure 1D). Moreover, in contrast to the cardiac hypertrophy reported in HMGA1 null mice [23], dnHMGA1 Tg mice did not exhibit an increase in ratio of heart to body weight (mg/gram) compared with WT hearts (4.95 ± 0.17 versus 4.73 ± 0.06 respectively, P=0.28).
To determine the biological response of dnHMGA1, SMCs harvested from WT and Tg mice were stimulated with the inflammatory cytokines IL-1β or TNF-α (Figure 2A, left panel and right panel respectively), and assessed for levels of NOS2 mRNA by qRT-PCR. Levels of NOS2 were significantly decreased in SMCs from Tg mice compared with WT mice after exposure to the inflammatory stimuli. A NOS2 promoter/enhancer construct was also overexpressed in vascular SMCs, in the presence or absence of IL-1β. Co-expression of dnHMGA1 resulted in an attenuated transactivation of the NOS2 promoter exposed to IL-1β (Figure 2B). While the mutant (dn) HMGA1 construct has been shown previously to not bind to DNA [25], we wanted to determine whether the dnHMGA1 protein would still be able to interact with transcription factors that contribute to gene regulation. We have previously shown that HMGA1 facilitates NF-κB subunit p50 binding to DNA and transactivation of the NOS2 promoter/enhancer [14]. Thus, we performed a co-immunoprecipitation (co-IP) assay using antibodies against p50 and the HA tag of the dnHMGA1 transgene. The antibody to p50 was cross-linked to protein A/G agarose beads and incubated with nuclear protein extracts from mouse vascular SMCs. The precipitated proteins were separated by SDS-PAGE for Western blot analysis using an antibody targeted against the HA tag of dnHMGA1. As seen in the pre-IP nuclear lysates, the dnHMGA1 protein was present in cells from Tg but not from WT mice (Figure 2C). Also, the dnHMGA1 protein was present in the p50 antibody-immunoprecipitated proteins from Tg cells, but not from WT cells (Figure 2C). There was no evidence of dnHMGA1 transgene in samples from IgG immunoprecipitates of TG or WT cells. Taken together, these data show that the dnHMGA1 protein is capable of interacting with p50, independent of DNA binding, and expression of dnHMGA1 attenuates transactivation of the NOS2 promoter/enhancer and levels of NOS2 mRNA.
Figure 2. NOS2 promoter transactivation and mRNA is suppressed by expression of dnHMGA1.
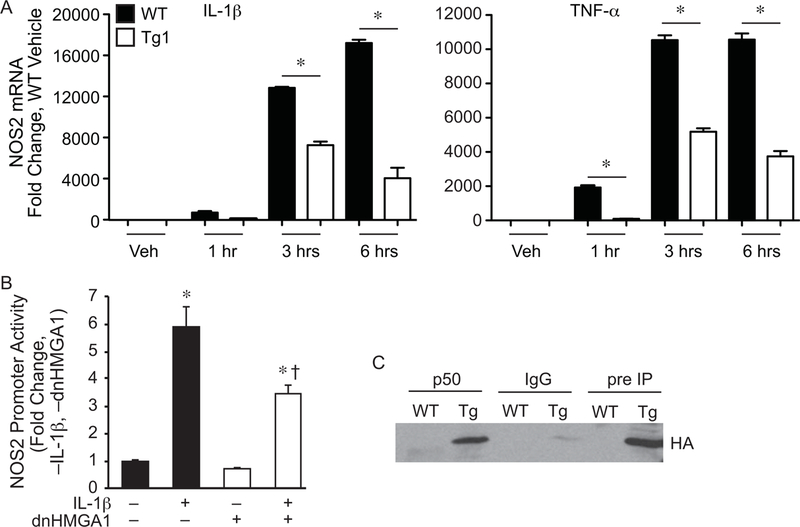
A) SMCs harvested from WT or Tg1 mice were exposed to vehicle (Veh), IL-1β (10 ng/ml, left panel), or TNF-α (10 ng/ml, right panel) for various time points (as depicted). Total RNA was extracted from the cells, and levels of NOS2 were measured by qRT-PCR. Data are presented as mean ± SEM, n=3 per group, with testing by one-way ANOVA (P<0.0001). Significant comparisons; * WT versus Tg1. B) SMCs were transiently transfected with a NOS2 luciferase reporter plasmid (base pairs −1485/+31), and either a vector control plasmid or an expression plasmid for dnHMGA1. An expression plasmid for β-galactosidase was used to correct for transfection efficiency. Cells were allowed to recover overnight and then the cells were exposed to IL-1β (10 ng/ml) or vehicle for 24 hours prior to harvest. Luciferase activity of each group is presented as mean ± SEM, n=6 per group, with testing by one-way ANOVA (P<0.0001). Significant comparisons; * versus no (–) IL-1β and no (–) dnHMGA1, and † versus IL-1β (+) and no (–) dnHMGA1. C) Co-immunoprecipitation assay (co-IP) was performed as described in the Material and Methods. SMCs from WT or dnHMGA1 Tg mice were then harvested and the nuclear protein extracts were incubated with p50 antibody-crosslinked protein A/G beads, rabbit IgG-crosslinked protein A/G beads, or protein A/G beads alone. The bound proteins were separated with SDS-PAGE. Western blot analysis of IP bound proteins (lanes 1 to 4) and pre-IP cellular proteins (lanes 5 and 6) were performed using HA antibody.
dnHMGA1 Tg mice have less severe hypotension during exposure to E. coli LPS.
To determine whether overexpression of the dnHMGA1 transgene in the vasculature of mice has a phenotype in the setting of systemic inflammation, we implanted intra-arterial catheters [20, 36, 37] into the carotid arteries of WT, Tg2, and Tg1 mice. The mice were allowed to recover overnight after catheter placement, and the next morning E. coli LPS (10 mg/kg i.p.) was administered. The mice subsequently had their blood pressure assessed at serial time points. Figure 3 demonstrates that during the first 6 hours after LPS, mean arterial pressure was not different between the groups. However, by 8 hours, arterial pressure was significantly higher in Tg2 and Tg1 mice compared with WT mice. Moreover, by 24 hours the blood pressure stabilized in Tg2 and Tg1 mice, with no further decrease, while the arterial pressure in WT mice continued to fall and was significantly lower than Tg2 and Tg1 mice. Thus, Tg2 and Tg1 mice have a blunted hypotensive response compared with WT mice exposed to systemic LPS. While we previously showed that a MGB drug (Distamycin A) protected against severe acute hypotension during endotoxemia in mice [20], the present study in Tg mice demonstrated that this beneficial response occurred not only early (8 hours), but was also maintained throughout 24 hours after administration of E. coli LPS. Next, we transitioned to a model of sepsis in mice using live E. coli bacteria.
Figure 3. dnHMGA1 Tg mice have less severe hypotension when exposed to E. coli LPS.
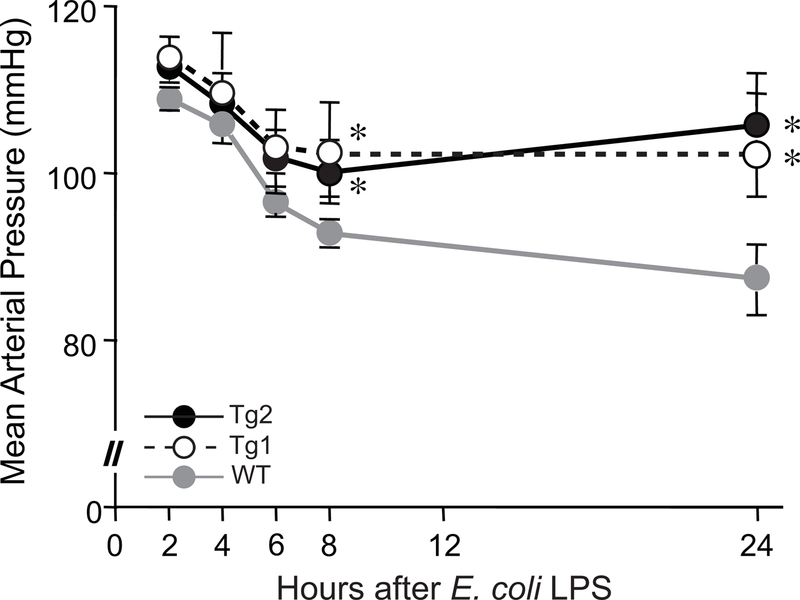
Carotid artery catheters were implanted in WT (gray line), Tg1 (black dashed line), and Tg2 (black solid line) mice. The following day, the mice received E. coli LPS (10 mg/kg) IP. Mean arterial pressure (MAP) was measured at 2, 4, 6, 8, and 24 hours after injection of LPS. Data points represent mean MAPs ± SEM, for WT (n=10), Tg1 (n=8), and Tg2 (n=11) mice. Data was assessed by two-way ANOVA (P<0.0001). Significant comparison, * versus WT mice.
dnHMGA1 Tg mice are protected against E. coli sepsis-induced mortality.
A fibrin clot model of E. coli sepsis, as described previously [28], was performed in Tg2 and Tg1 mice, compared with WT mice. This model allows a slow, progressive release of microorganisms. As shown in Figure 4, WT mice exposed to E. coli sepsis had a 37% survival at 48 hours, which continued throughout 5 days. The Tg1 line of dnHMGA1 mice had an improved survival compared with WT mice, particularly prominent between 24 and 48 hours. Survival of the Tg1 line was 56% by the end of the study. The Tg2 line of dnHMGA1 mice had an even more impressive increase in survival compared with WT mice, with a survival of 72% at 48 hours, which continued through 5 days. Due to a similar response to endotoxin exposure and microbial sepsis in Tg1 and Tg2 mice, the remaining studies were performed in Tg1 mice and cells.
Figure 4. dnHMGA1 Tg mice have increased survival to E. coli-induced sepsis compared with WT mice.
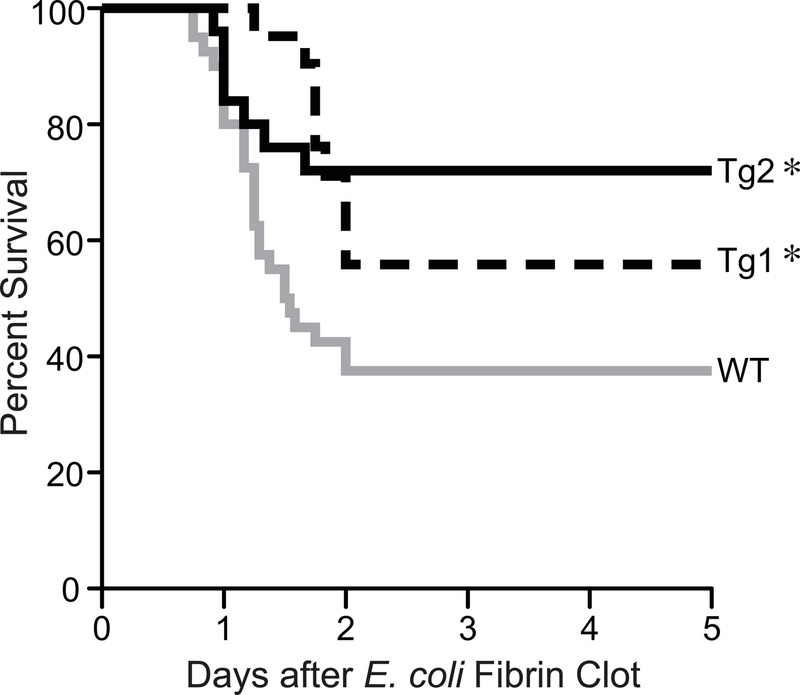
WT (gray line, n=40), Tg1 (black dashed line, n=21) and Tg2 (black solid line, n=25) mice were subjected to fibrin clot-induced sepsis using E. coli (2.2 ± 0.2 × 109 cfu/clot). Survival of mice was monitored for 5 days and data are presented as a Kaplan-Meier survival curve, and analyzed by Log-rank Test (P=0.0154). Significant comparison, ∗ versus WT group.
Peritoneal response to E. coli-induced sepsis in dnHMGA1 mice.
We subsequently investigated the peritoneal response to fibrin clot-induced sepsis by E. coli. Figure 5A demonstrates that at 24 hours after the onset of sepsis, the recruitment of innate immune cells into the peritoneal cavity was predominantly neutrophils in both WT and dnHMGA1 Tg mice. Moreover, the total numbers and percentages of neutrophils, and macrophages, were not different between the groups. In conjunction with a similar immune response to peritoneal sepsis, there was also no difference in peritoneal bacteria (Figure 5B). Likewise, in vivo phagocytosis assays in WT and Tg mice revealed no differences in the percentages of neutrophils (21±3% versus 21±7% respectively, P=0.99, Figure C) or macrophages (21± 9% versus 11±1% respectively, P=0.44) phagocytizing bacteria. Thus, to further understand the differences in the physiologic effects of WT and Tg mice to sepsis, we next assessed systemic inflammation.
Figure 5. dhHMGA1 Tg mice have no difference in inflammatory cell recruitment or bacterial clearance in the peritoneum during E. coli-induced sepsis compared with WT mice.
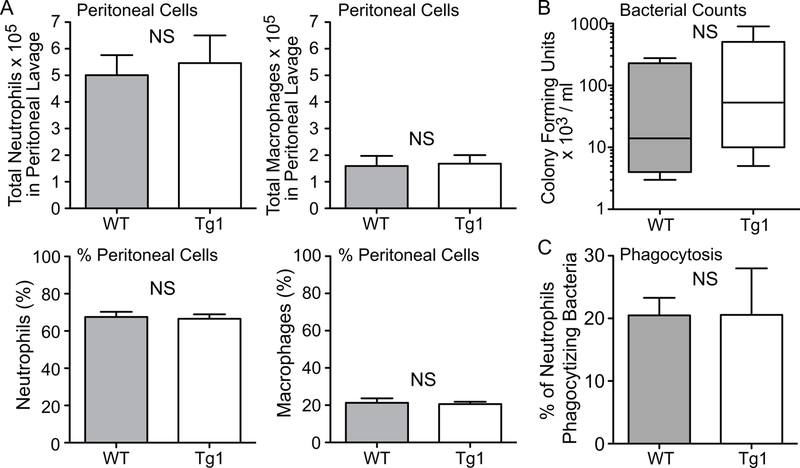
A) WT (gray bar, n=3) and Tg1 (white bar, n=4) mice were subjected to fibrin clot-induced sepsis using E. coli as above. At 24 hours, peritoneal fluid was collected and cells were analyzed for total numbers (upper panels) and percentages (%, lower panels) of neutrophils and macrophages. Data are presented at mean ± SEM. B) WT (gray bar, n=5) and Tg1 (white bar, n=7) mice were subjected to fibrin clot-induced sepsis using E. coli, and at 24 hours the peritoneal fluid was collected and analyzed for CFUs/ml. Data are presented as box plots, which show median values and interquartile ranges. C) GFP-labeled E. coli were injected i.p. into WT (n=3) and Tg1 (n=3) mice, and after 1 hour a peritoneal lavage was performed to assess neutrophil phagocytosis in vivo. Percentage of neutrophils phagocytizing bacteria are presented as mean ± SEM. NS = not significant for panels A, B, and C.
Decreased tissue infiltration of inflammatory cells in dnHMGA1 Tg mice.
The spleen is a very sensitive organ to the inflammatory effects of sepsis, resulting in injury and an alteration in cellular composition [43, 44]. Thus, we evaluated the inflammatory response and the infiltration of innate immune cells (macrophages and neutrophils) into the spleen after E. coli-induced sepsis. Compared with sham mice, there was a marked increase in macrophage infiltration into the spleens (mainly in the red pulp) of WT mice 24 hours after placement of the E. coli fibrin clot (Figure 6). In dnHMGA1 Tg mice, there was also evidence of increased splenic macrophages after E. coli-induced sepsis compared with sham mice, however the level of macrophage infiltrates was dramatically less compared with septic WT mice (Figure 6). We next examined neutrophil infiltrates into spleens of WT and Tg mice 24 hours after E. coli fibrin clot, compared with sham mice. The dramatic increase in splenic neutrophils in WT mice after the onset of E. coli sepsis was significantly less in dnHMGA1 Tg mice (Figure 7). This reduction of splenic neutrophils in dnHMGA1 mice was analogous to the reduction of splenic macrophages in dnHMGA1 mice compared with WT mice exposed to E. coli sepsis. We also assessed neutrophils in the lungs. Similar to the spleen, an increase in lung neutrophils in WT mice was blunted in Tg mice (Figure 8). Collectively, these data revealed a decrease in organ inflammation in dnHGMA1 mice (involving organs both inside and outside of the peritoneum), compared with WT mice during peritoneal sepsis.
Figure 6. dnHMGA1 Tg mice have decreased macrophage infiltrates into the spleen during E. coli-induced sepsis compared with WT mice.
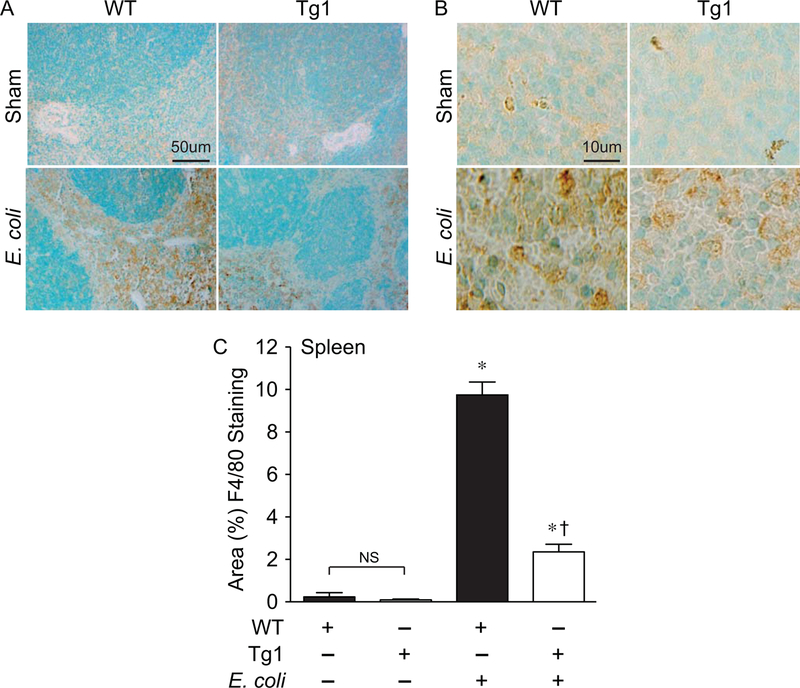
A-B) WT and dnHMGA1 Tg mice underwent sham or fibrin clot (E. coli) surgery. At 24 hours after surgery, tissues were harvested and immunostaining was performed with F4/80 antibody (brown) to assess macrophages in the spleens. Panels A and B show representative lower and higher power light microscopy images, respectively. The scale bar represents 50 µm (A) and 10 µm (B). C) The area of F4/80 positively stained cells from images of WT and Tg mice, under sham and E. coli conditions, was calculated per 40X objective, and random fields were assessed per tissue section. Data are represented as mean±SEM. sham WT, n=20 images; sham Tg, n=20 images; E. coli WT, n=40 images; E. coli Tg, n=40 images. Analyses by one-way ANOVA (P<0.0001). Significant comparison, * versus sham groups, † versus WT E. coli group.
Figure 7. dnHMGA1 Tg mice have decreased neutrophil infiltrates into the spleen during E. coli-induced sepsis compared with WT mice.
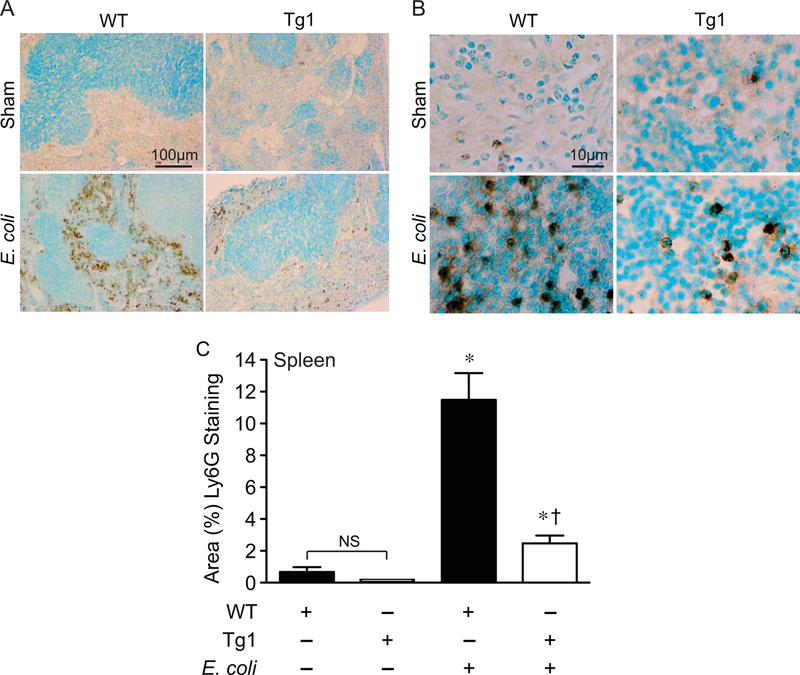
A-B) WT and dnHMGA1 Tg mice underwent sham or fibrin clot (E. coli) surgery. At 24 hours after surgery, tissues were harvested and immunostaining was performed with Ly6G antibody (brown) to assess neutrophils in the spleens. Panels A and B show representative lower and higher power light microscopy images, respectively. The scale bar represents 100 µm (A) and 10 µm (B). C) The area of Ly6G positively stained cells from images of WT and Tg mice, under sham and E. coli conditions, was calculated per 40X objective, and random fields were assessed per tissue section. Data are represented as mean±SEM. sham WT, n=20 images; sham Tg, n=20 images; E. coli WT, n=40 images; E. coli Tg, n=40 images. Analyses by one-way ANOVA (P<0.0001). Significant comparison, * versus sham groups, † versus WT E. coli group.
Figure 8. dnHMGA1 Tg mice have decreased neutrophil infiltrates into the lung during E. coli-induced sepsis compared with WT mice.
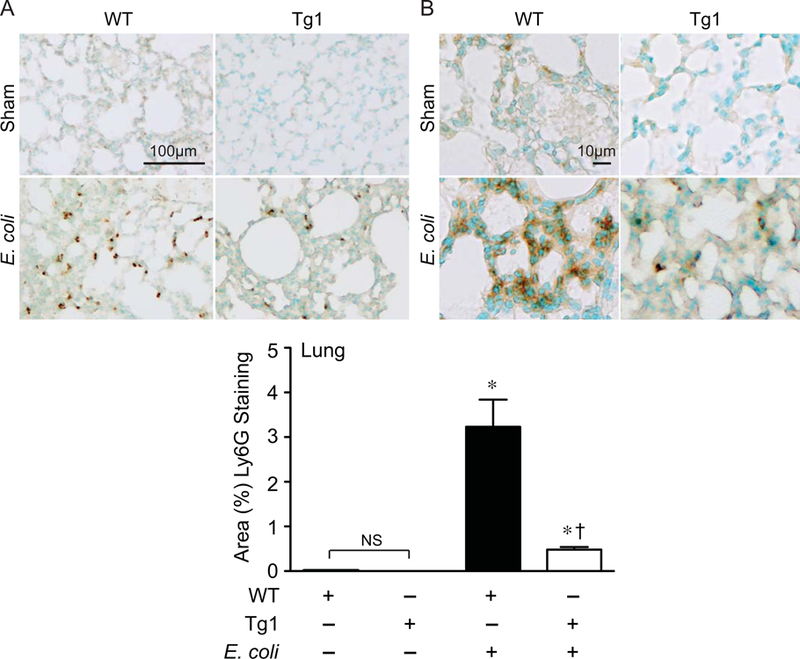
A-B) WT and dnHMGA1 Tg mice underwent sham or fibrin clot (E. coli) surgery. At 24 hours after surgery, tissues were harvested and immunostaining was performed with Ly6G antibody (brown) to assess neutrophils in the lung. Panels A and B show representative lower and higher power light microscopy images, respectively. The scale bar represents 100 µm (A) and 10 µm (B). C) The area of Ly6G positively stained cells from images of WT and Tg mice, under sham and E. coli conditions, was calculated per 40X objective, and random fields were assessed per tissue section. Data are represented as mean±SEM. sham WT, n=20 images; sham Tg, n=20 images; E. coli WT, n=40 images; E. coli Tg, n=40 images. Analyses by one-way ANOVA (P<0.0001). Significant comparison, * versus sham groups, † versus WT E. coli group.
Given the reduced infiltration of macrophages and neutrophils into tissues of Tg mice compared with WT mice during E. coli sepsis, we next assessed the levels of chemokine ligand 2 (CCL2) / monocyte chemotactic protein (MCP)-1, and C-X-C motif chemokine ligand 2 (CXCL2) / macrophage inflammatory protein 2 (MIP-2), to assess macrophage and neutrophil chemokines respectively [45–47]. We evaluated the mRNA levels of MCP-1 and MIP-2 in SMCs harvested from WT and Tg mice that were stimulated with IL-1β or TNF-α. MCP-1 message was significantly decreased in SMCs from Tg mice compared with cells from WT mice, after exposure to IL-1β or TNF-α for 3 and 6 hours (Figure 9A). In a very similar response, MIP-2 expression was also decreased in SMCs from Tg mice after exposure to inflammatory stimuli (IL-1β or TNF-α, Figure 9B). Moreover, the marked increase in MCP-1 protein levels seen in splenic tissue and plasma (Figures 9C, left panel and right panel respectively) of WT mice after E. coli-induced sepsis was considerably decreased in septic dnHMGA1 Tg mice. However, when specifically compared with each sham control, the levels of MCP-1 (from both the spleen and plasma) in the E. coli infected dnHMGA1 Tg mice remained above the levels of sham WT and sham Tg mice (Figure 9).
Figure 9. dnHMGA1 cells and mice have decreased expression of chemokines during exposure to imflammatory stimuli.
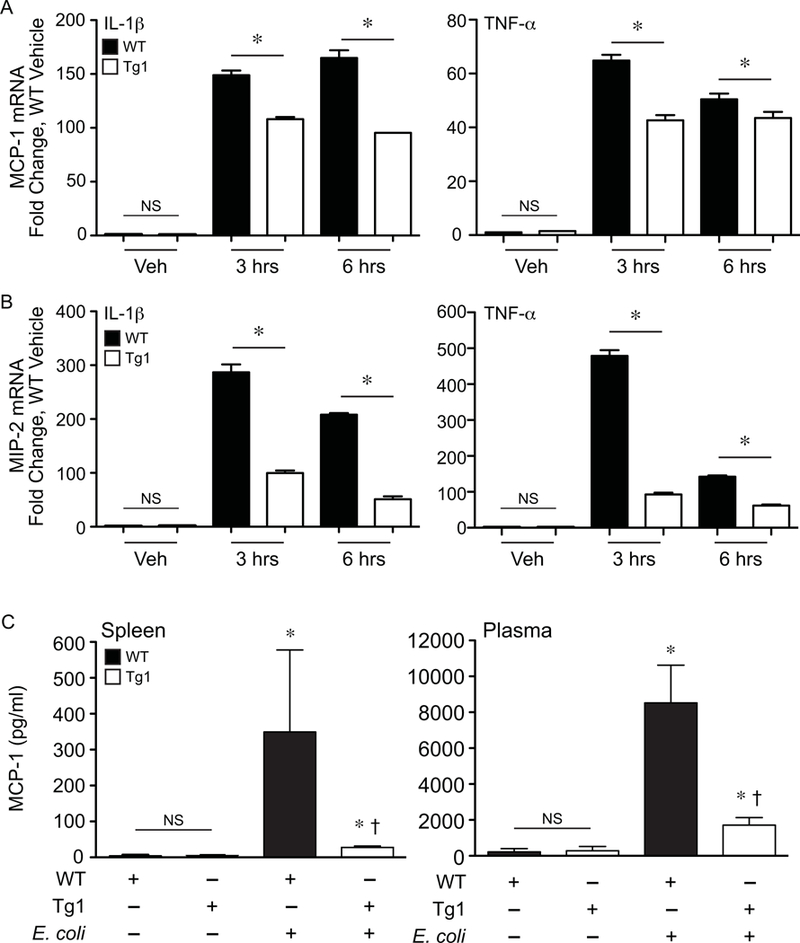
SMCs harvested from WT or dnHMGA1 (Tg1) mice were exposed to vehicle (Veh), IL-1β (10 ng/ml), or TNF-α (10 ng/ml) for various time points (as depicted). Total RNA was extracted from the cells, and levels of (A) MCP-1 and (B) MIP-2 were measured by qRT-PCR. Data are presented as mean ± SEM, n=3 per group, with testing by one-way ANOVA (P<0.0001). Significant comparisons; * WT versus Tg1. C) WT and Tg1 mice underwent sham (– E. coli) or fibrin clot (+ E. coli) surgery. At 24 hours after surgery, tissue (spleen) and plasma were collected, and the concentration of MCP-1 was assessed using a Luminex bead assay as described in the Material and Methods. Data are represented as mean±SEM, from 3 mice per group. Analyses by one-way ANOVA (P=0.0399, spleen; P=0.0253, plasma). Significant comparisons for A and B, * versus sham groups, † versus WT E. coli group. NS = not significant.
DISCUSSION
Sepsis is defined as a dysregulated host response to infection, with evidence of organ dysfunction that carries a high rate of mortality [48]. The immune reaction to infection involves both pro-inflammatory and anti-inflammatory responses, with a balance of these processes needed to prevent collateral injury and to restore homeostasis [49]. The innate immune system is the primary line of defense against invading microbes [49, 50], and an aberrant response can result in exaggerated inflammation and associated tissue damage and organ injury. Thus, further insights into the immune response to infection, and associated sepsis, will provide targets for novel therapeutic interventions. The aim of the present study was to further understand the role of HMGA1 in the pathophysiology of the immune response to E. coli-induced sepsis.
The balance between pro-inflammatory and anti-inflammatory mediators is often regulated at the level of gene expression [17], with a major component determined by transcription. While the production of RNA is controlled by the interaction of classical transcription factors with promoter/enhancer regions of DNA, architectural transcription factors play an important role in the efficiency of gene regulation by modifying DNA conformation [51]. Interestingly, a high level of expression of HMGA1 has been shown to be present during carcinogenesis and leukemic transformation [19, 52], and associated with dysregulated genes involved in the inflammatory response (including NF-κB as a major node of this gene set). The increased expression of HMGA1 is not limited to malignancies, as we have shown previously that HMGA1 expression can be induced by pro-inflammatory mediators and endotoxin, in vitro and in vivo respectively, in vascular SMCs [12]. Thus we proposed that altering a dysregulated HMGA1 response by overexpressing a dnHMGA1 transgene in the vasculature would have a beneficial response during sepsis. This concept was previously tested using minor groove binding drugs, Distamycin A and Netropsin, in models of endotoxemia [12, 20–22]. However, due to the antibiotic properties of these drugs, we developed genetically modified mice to further understand HMGA1 in microbial sepsis.
In the present study, our data demonstrate that dnHMGA1 Tg mice have a blunted hypotensive response during endotoxemia (Figure 3), and in a microbial model of sepsis, the Tg mice have improved survival (Figure 4). Interestingly, innate immune cell recruitment to the peritoneal cavity (housing the E. coli fibrin clot), along with bacterial counts and phagocytosis, was not different between WT and Tg mice (Figure 5A-C). Thus, we next assessed the inflammatory response in organs, both inside (spleen) and outside (lung) the peritoneum. In comparison with WT mice, 24 hours after the onset of E. coli-induced sepsis the infiltration of macrophages and neutrophils into the spleen was markedly less in Tg mice (Figures 6 and 7, respectively), as well as neutrophil infiltration into the lungs (Figure 8). Thus, during microbial sepsis, inflammation in critical organs was more effectively resolved in dnHMGA1 Tg mice compared with WT mice, associated with improved survival.
To further characterize the infiltration of macrophages and neutrophils into tissue during sepsis, we further assessed the levels of MCP-1 and MIP-2 in cells from Tg and WT mice. MCP-1 is a member of the C-C chemokine family, and it is known to regulate the migration and infiltration of leukocytes into tissue, with a predominant effect on monocytes [53], which subsequently differentiate into macrophages within the inflamed tissue. MCP-1 is produced in a number of cell types, including SMCs, and during an infectious insult expression is increased, resulting in high levels of MCP-1 in plasma and inflamed tissues [45, 46]. During exposure to the inflammatory cytokines IL-1β or TNF-α in vitro, SMCs harvested from dnHMGA1 Tg mice showed decreased expression of MCP-1 mRNA compared with SMCs from WT mice (Figure 9A). In addition, expression of the neutrophil chemokine MIP-2 was similarly decreased in Tg SMCs compared with WT SMCs exposed to inflammatory cytokines (Figure 9B). Furthermore, during E. coli-induced sepsis in vivo, the levels of splenic and plasma MCP-1 protein were significantly increased in WT mice, however the elevated MCP-1 levels were diminished in the dnHMGA1 Tg mice (Figure 9C).
Previous studies have demonstrated that an absence of MCP-1 in genetically modified mice leads to increased susceptibility to systemic inflammation in models of endotoxemia and polymicrobial sepsis, compared with WT mice [54]. Moreover, in the absence of MCP-1, bacterial clearance was reduced in models of peritoneal sepsis (either polymicrobial or E. coli) and E. coli-induced pneumonia [55]. In peritoneal sepsis, MCP-1 deficient mice had decreased recruitment of monocytes to the sight of injury, and lethality correlated with impaired production of the anti-inflammatory mediator IL-10, suggesting MCP-1 played an important immunomodulatory role in controlling the balance of pro-inflammatory mediators and anti-inflammatory mediators during sepsis [54]. In contrast, in the dnHMGA1 Tg mice, the expression of IL-10 was not different from WT mice during sepsis (Supplemental Figure 2). Interestingly, in the dnHMGA1 Tg mice, the induction of MCP-1 and MIP-2 was blunted, although the levels were still above that of sham mice (Figure 9). Thus, expression of dnHMGA1 was able to modulate the chemokine response with less tissue inflammation.
Since MCP-1 and MIP-2 are NK-κB regulated genes [56–58], and dnHMGA1 is able to bind to NF-κB subunits but not to DNA, this may explain the attenuated expression of MCP-1 and MIP-2 in cells of dnHMGA1 Tg mice exposed to inflammatory cytokines. The decreased expression of MCP-1 may account for more than just the reduction in macrophage infiltration into the spleen (Figure 6), as it has been shown that MCP-1 regulates neutrophil recruitment during E. coli infection [59]. Moreover, decreased expression of MIP-2, a chemokine critical for neutrophil recruitment [60, 61], contributes to less infiltration of neutrophils into organs of Tg mice during peritoneal sepsis (Figures 7-8).
HMGA1 is known to regulate a number of genes that have been implicated in the pathophysiology of sepsis [17]. HMGA1 plays an important role in the regulation of mouse [12–14] and human [16] NOS2 during exposure to pro-inflammatory mediators. In addition, using the minor groove binding drug Distamycin A, we were able to reduce the induction of NOS2, rather than eliminating its expression, resulting in an attenuation of hypotension and improved survival during endotoxemia [20]. A similar attenuation of NOS2 mRNA levels (Figure 2A) and NOS2 transactivation (Figure 2B) was seen when dnHMGA1 was expressed in SMCs in the presence of inflammatory cytokines. Distamycin A also suppressed the induction of P-selectin, resulting in decreased inflammation in key organs during endotoxemia [22]. This is very comparable to the chemokine response in the dnHMGA1 Tg mice during microbial sepsis, with diminished MCP-1 and MIP-2 induction, rather than elimination of expression, and Tg mice experiencing improved outcome.
The dominant negative construct, while not being able to bind to DNA, can still interact with transcription factors. As seen in Figure 2, the dnHMGA1 protein co-immunoprecipitates with NF-κB subunit p50 in extracts from dnHMGA1 Tg SMCs, but not WT SMC. Thus, we hypothesize that one mechanism by which the dnHMGA1 transgene may function is by sequestering transcription factors from binding to DNA (for example NF-κB subunits), and altering the expression of genes (such as NOS2, MCP-1, MIP-2 and inflammatory mediators) contributing to the pathophysiologic response to microbial sepsis [17]. Taken together, our data suggest that HMGA1 is a novel modifiable pathway that may be targeted for therapeutics in sepsis.
Supplementary Material
dnHMGA1 Tg mice underwent fibrin clot (E. coli) surgery. At 24 hours after surgery, spleen tissue was harvested and immunostaining was performed with an isotype control antibody (rat IgG2). There was no evidence of brown staining using the isotype control antibody. The scale bar represents 100 µm.
WT and dnHMGA1 Tg mice underwent sham (– E. coli) or fibrin clot (+ E. coli) surgery. At 24 hours after surgery, spleen (A) and plasma (B) were collected, and the concentration of IL-10 was assessed using a Luminex bead assay as descriibed in the Material and Methods. Data are represented as mean±SEM, from 3 mice per group. Analyses by one-way ANOVA. NS = not significant.
ACKNOWLEDGMENTS
We thank Dr. Manuela Cernadas for her help with the analysis of the flow cytometry data. This work was supported by National Institutes of Health grants P01HL108801 to M.A.P. and R.M.B.; U01AI061246 and R01GM118456 to M.A.P.; R01HL091957 to R.M.B., T32HD007466 to A.P.C., X.L., and S.G.; and K08GM126313 to S.G.
Abbreviations:
- ACLP
aortic carboxypeptidase-like protein
- bGHpA
bovine growth hormone polyadenylation sequences
- BP
blood pressure
- CCL2
chemokine ligand 2
- CFU
colony forming units
- CXCL2
chemokine (C-X-C motif) ligand 2
- Dn
dominant negative
- E. coli
Escherichia coli
- En
endogenous
- HMG
high mobility group
- HA
hemagglutinin
- IL
interleukin
- IP
immunoprecipitation
- LPS
lipopolysaccharide
- MCP-1
monocyte chemotactic protein
- MIP-2
macrophage inflammatory protein 2
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NOS
nitric oxide synthase
- PBS
phosphate buffered saline
- qRT-PCR
quantitative real time polymerase chain reaction
- RT
reverse transcriptase
- SMC
smooth muscle cells
- Tg
transgenic
- Veh
vehicle
- WT
wild-type
Footnotes
DISCLOSURE
The authors declare no conflict of interest.
REFERENCES
- 1.Bianchi ME and Agresti A (2005) HMG proteins: dynamic players in gene regulation and differentiation. Curr Opin Genet Dev 15, 496–506. [DOI] [PubMed] [Google Scholar]
- 2.Reeves R and Beckerbauer L (2001) HMGI/Y proteins: flexible regulators of transcription and chromatin structure. Biochim. Biophys. Acta 1519, 13–29. [DOI] [PubMed] [Google Scholar]
- 3.Eckner R and Birnstiel ML (1989) Cloning of cDNAs coding for human HMG I and HMG Y proteins: both are capable of binding to the octamer sequence motif. Nucleic Acids Res 17, 5947–5959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson KR, Disney JE, Whatt CR, Reeves R (1990) Expression of mRNAs encoding mammalian chromosomal proteins HMG-I and HMG-Y during cellular proliferation. Exp. Cell Res 187, 69–76. [DOI] [PubMed] [Google Scholar]
- 5.Reeves R and Wolffe AP (1996) Substrate structure influences binding of the non-histone protein HMG-I(Y) to free nucleosomal DNA. Biochemistry 35, 5063–74. [DOI] [PubMed] [Google Scholar]
- 6.Giese K, Cox J, Grosschedl R (1992) The HMG domain of lymphoid enhancer factor 1 bends DNA and facilitates assembly of functional nucleoprotein structures. Cell 69, 185–195. [DOI] [PubMed] [Google Scholar]
- 7.Falvo JV, Thanos D, Maniatis T (1995) Reversal of intrinsic DNA bends in the IFN beta gene enhancer by transcription factors and the architectural protein HMG I(Y). Cell 83, 1101–1111. [DOI] [PubMed] [Google Scholar]
- 8.Thanos D and Maniatis T (1992) The high mobility group protein HMG I(Y) is required for NF-kappa B-dependent virus induction of the human IFN-beta gene. Cell 71, 777–789. [DOI] [PubMed] [Google Scholar]
- 9.Du W, Thanos D, Maniatis T (1993) Mechanisms of transcriptional synergism between distinct virus-inducible enhancer elements. Cell 74, 887–898. [DOI] [PubMed] [Google Scholar]
- 10.Thanos D and Maniatis T (1995) Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 83, 1091–1100. [DOI] [PubMed] [Google Scholar]
- 11.Bustin M and Reeves R (1996) High-mobility-group chromosomal proteins: architectural components that facilitate chromatin function. Prog. Nucleic Acid Res. Mol. Biol 54, 35–100. [DOI] [PubMed] [Google Scholar]
- 12.Pellacani A, Chin MT, Wiesel P, Ibanez M, Patel A, Yet S-F, Hsieh C-M, Paulauskis JD, Reeves R, Lee M-E, Perrella MA (1999) Induction of high mobility group-I(Y) protein by endotoxin and interleukin-1b in vascular smooth muscle cells: Role in activation of inducible nitric oxide synthase. J. Biol. Chem 274, 1525–1532. [DOI] [PubMed] [Google Scholar]
- 13.Pellacani A, Wiesel P, Razavi S, Vasilj V, Feinberg MW, Chin MT, Reeves R, Perrella MA (2001) Downregulation of high mobility group-I(Y) protein contributes to the inhibition of nitric oxide synthase 2 by transforming growth factor-b1. J. Biol. Chem 276, 1653–1659. [DOI] [PubMed] [Google Scholar]
- 14.Perrella MA, Pellacani A, Wiesel P, Chin MT, Foster LC, Ibanez M, Hsieh C-M, Reeves R, Yet S-F, Lee M-E (1999) High mobility group-I(Y) protein facilitates nuclear factor-kappaB binding and transactivation of the inducible nitric-oxide synthase promoter/enhancer. J. Biol. Chem 274, 9045–9052. [DOI] [PubMed] [Google Scholar]
- 15.Darville MI, Terryn S, Eizirik DL (2004) An octamer motif is required for activation of the inducible nitric oxide synthase promoter in pancreatic beta-cells. Endocrinology 145, 1130–1136. [DOI] [PubMed] [Google Scholar]
- 16.Takamiya R, Baron RM, Yet SF, Layne MD, Perrella MA (2008) High mobility group A1 protein mediates human nitric oxide synthase 2 gene expression. FEBS Lett 582, 810–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carvajal IM, Baron RM, Perrella MA (2002) High Mobility Group-I/Y Proteins: Potential role in the pathophysiology of critical illnesses. Crit Care Med 30, S36–S42. [PubMed] [Google Scholar]
- 18.Resar LM (2010) The high mobility group A1 gene: transforming inflammatory signals into cancer? Cancer Res 70, 436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schuldenfrei A, Belton A, Kowalski J, Talbot CC Jr., Di Cello F, Poh W, Tsai HL, Shah SN, Huso TH, Huso DL, Resar LM (2011) HMGA1 drives stem cell, inflammatory pathway, and cell cycle progression genes during lymphoid tumorigenesis. BMC Genomics 12, 549. doi: 10.1186/1471-2164-12-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baron RM, Carvajal IM, Liu X, Okabe RO, Chen Y-H, Ejima K, Layne MD, Perrella MA (2004) Reduction of nitric oxide synthase 2 expression by distamycin A improves survival from endotoxemia. J. Immunol 173, 4147–4153. [DOI] [PubMed] [Google Scholar]
- 21.Grant MA, Baron RM, Macias AA, Layne MD, Perrella MA, Rigby AC (2009) Netropsin improves survival from endotoxemia by disrupting HMGA1-binding to the NOS2 promoter. Biochem J 418, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baron RM, Lopez-Guzman S, Riascos DF, Macias AA, Layne MD, Cheng G, Harris C, Chung SW, Reeves R, von Andrian UH, Perrella MA (2010) Distamycin A inhibits HMGA1-binding to the P-selectin promoter and attenuates lung and liver inflammation during murine endotoxemia. PLoS One 5, e10656. doi: 10.1371/journal.pone.0010656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fedele M, Fidanza V, Battista S, Pentimalli F, Klein-Szanto AJ, Visone R, De Martino I, Curcio A, Morisco C, Del Vecchio L, Baldassarre G, Arra C, Viglietto G, Indolfi C, Croce CM, Fusco A (2006) Haploinsufficiency of the Hmga1 gene causes cardiac hypertrophy and myelo-lymphoproliferative disorders in mice. Cancer Res 66, 2536–2543. [DOI] [PubMed] [Google Scholar]
- 24.Andreucci A, Reeves R, McCarthy KM, Nikolajczyk BS (2002) Dominant-negative HMGA1 blocks µ enhancer activation through a novel mechanism. Biochem. Biophys. Res. Commun 292, 427–433. [DOI] [PubMed] [Google Scholar]
- 25.Himes SR, Reeves R, Attema J, Nissen M, Li Y, Shannon MF (2000) The role of high-mobility group I(Y) proteins in expression of IL-2 and T cell proliferation. J. Immunol 164, 3157–3168. [DOI] [PubMed] [Google Scholar]
- 26.Reeves R, Edberg DD, Li Y (2001) Architectural transcription factor HMGI(Y) promotes tumor progression and mesenchymal transition of human epithelial cells. Mol Cell Biol 21, 575–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Layne MD, Yet S-F, Maemura K, Hsieh C-M, Liu X, Ith B, Lee M-E, Perrella MA (2002) Characterization of the mouse aortic carboxypeptidase-like protein promoter reveals activity in differentiated and dedifferentiated vascular smooth muscle cells. Circ. Res 90, 728–736. [DOI] [PubMed] [Google Scholar]
- 28.Chung SW, Liu X, Macias AA, Baron RM, Perrella MA (2008) Heme oxygenase-1-derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J. Clin. Invest 118, 239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yet S-F, Perrella MA, Layne MD, Hsieh C-M, Maemura K, Kobzik L, Wiesel P, Christou H, Kourembanas S, Lee M-E (1999) Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J. Clin. Invest 103, R23–R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kwon MY, Liu X, Lee SJ, Kang YH, Choi AM, Lee KU, Perrella MA, Chung SW (2011) Nucleotide-binding oligomerization domain protein 2 deficiency enhances neointimal formation in response to vascular injury. Arterioscler Thromb Vasc Biol 31, 2441–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsoyi K, Geldart AM, Christou H, Liu X, Chung SW, Perrella MA (2015) Elk-3 is a KLF4-regulated gene that modulates the phagocytosis of bacteria by macrophages. Journal of leukocyte biology 97, 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chung SW, Chen Y-H, Perrella MA (2005) Role of Ets-2 in the regulation of heme oxygenase-1 by endotoxin. J. Biol. Chem 280, 4578–4584. [DOI] [PubMed] [Google Scholar]
- 33.Perrella MA, Patterson C, Tan L, Yet SF, Hsieh CM, Yoshizumi M, Lee ME (1996) Suppression of interleukin-1β-induced nitric-oxide synthase promoter/enhancer activity by transforming growth factor-β1 in vascular smooth muscle cells. Evidence for mechanisms other than NF-κB. J. Biol. Chem 271, 13776–13780. [DOI] [PubMed] [Google Scholar]
- 34.Chen Y-H, Layne MD, Chung SW, Ejima K, Baron RM, Yet S-F, Perrella MA (2003) Elk-3 is a transcriptional repressor of nitric-oxide synthase 2. J. Biol. Chem 278, 39572–39577. [DOI] [PubMed] [Google Scholar]
- 35.Hung CC, Liu X, Kwon MY, Kang YH, Chung SW, Perrella MA (2010) Regulation of heme oxygenase-1 gene by peptidoglycan involves the interaction of Elk-1 and C/EBPalpha to increase expression. Am J Physiol Lung Cell Mol Physiol 298, L870–L879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiesel P, Mazzolai L, Nussberger J, Pedrazzini T (1997) Two-kidney, one clip and one-kidney, one clip hypertension in mice. Hypertension 29, 1025–1030. [DOI] [PubMed] [Google Scholar]
- 37.Perrella MA, Hsieh CM, Lee WS, Shieh S, Tsai JC, Patterson C, Lowenstein CJ, Long NC, Haber E, Shore S, Lee ME (1996) Arrest of endotoxin-induced hypotension by transforming growth factor ß1. Proc. Natl. Acad. Sci. U.S.A 93, 2054–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matute-Bello G, Frevert CW, Kajikawa O, Skerrett SJ, Goodman RB, Park DR, Martin TR (2001) Septic shock and acute lung injury in rabbits with peritonitis. Am J Respir Crit Care Med 163, 234–243. [DOI] [PubMed] [Google Scholar]
- 39.Hall SR, Tsoyi K, Ith B, Padera RFJ, Lederer JA, Wang Z, Liu X, Perrella MA (2012) Mesenchymal Stromal Cells Improve Survival During Sepsis in the Absence of Heme Oxygenase-1: The Importance of Neutrophils. Stem Cells 31, 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fredenburgh LE, Velandia MM, Ma J, Olszak T, Cernadas M, Englert JA, Chung SW, Liu X, Begay C, Padera RF, Blumberg RS, Walsh SR, Baron RM, Perrella MA (2011) Cyclooxygenase-2 deficiency leads to intestinal barrier dysfunction and increased mortality during polymicrobial sepsis. J. Immunol 187, 5255–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tajima G, Delisle AJ, Hoang K, O’Leary FM, Ikeda K, Hanschen M, Stoecklein VM, Lederer JA (2013) Immune system phenotyping of radiation and radiation combined injury in outbred mice. Radiat Res 179, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wanke-Jellinek L, Keegan JW, Dolan JW, Guo F, Chen J, Lederer JA (2016) Beneficial Effects of CpG-Oligodeoxynucleotide Treatment on Trauma and Secondary Lung Infection. J Immunol 196, 767–777. [DOI] [PubMed] [Google Scholar]
- 43.Muenzer JT, Davis CG, Chang K, Schmidt RE, Dunne WM, Coopersmith CM, Hotchkiss RS (2010) Characterization and modulation of the immunosuppressive phase of sepsis. Infect Immun 78, 1582–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tinsley KW, Grayson MH, Swanson PE, Drewry AM, Chang KC, Karl IE, Hotchkiss RS (2003) Sepsis induces apoptosis and profound depletion of splenic interdigitating and follicular dendritic cells. Journal of immunology 171, 909–914. [DOI] [PubMed] [Google Scholar]
- 45.Kobayashi M, Tsuda Y, Yoshida T, Takeuchi D, Utsunomiya T, Takahashi H, Suzuki F (2006) Bacterial sepsis and chemokines. Current drug targets 7, 119–134. [DOI] [PubMed] [Google Scholar]
- 46.Shi C and Pamer EG (2011) Monocyte recruitment during infection and inflammation. Nature reviews. Immunology 11, 762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Driscoll KE (1994) Macrophage inflammatory proteins: biology and role in pulmonary inflammation. Exp Lung Res 20, 473–490. [DOI] [PubMed] [Google Scholar]
- 48.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC (2016) The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wiersinga WJ, Leopold SJ, Cranendonk DR, van der Poll T (2014) Host innate immune responses to sepsis. Virulence 5, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fang H, Jiang W, Cheng J, Lu Y, Liu A, Kan L, Dahmen U (2015) Balancing Innate Immunity and Inflammatory State via Modulation of Neutrophil Function: A Novel Strategy to Fight Sepsis. J Immunol Res 2015, 187048. doi: 10.1155/2015/187048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wolffe AP (1994) Architectural Transcription Factors. Science 264, 1100–1101. [DOI] [PubMed] [Google Scholar]
- 52.Fusco A and Fedele M (2007) Roles of HMGA proteins in cancer. Nat Rev Cancer 7, 899–910. [DOI] [PubMed] [Google Scholar]
- 53.Deshmane SL, Kremlev S, Amini S, Sawaya BE (2009) Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 29, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gomes RN, Figueiredo RT, Bozza FA, Pacheco P, Amancio RT, Laranjeira AP, Castro-Faria-Neto HC, Bozza PT, Bozza MT (2006) Increased susceptibility to septic and endotoxic shock in monocyte chemoattractant protein 1/cc chemokine ligand 2-deficient mice correlates with reduced interleukin 10 and enhanced macrophage migration inhibitory factor production. Shock 26, 457–463. [DOI] [PubMed] [Google Scholar]
- 55.Gomes RN, Teixeira-Cunha MG, Figueiredo RT, Almeida PE, Alves SC, Bozza PT, Bozza FA, Bozza MT, Zimmerman GA, Castro-Faria-Neto HC (2013) Bacterial clearance in septic mice is modulated by MCP-1/CCL2 and nitric oxide. Shock 39, 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ueda A, Ishigatsubo Y, Okubo T, Yoshimura T (1997) Transcriptional regulation of the human monocyte chemoattractant protein-1 gene. Cooperation of two NF-kappaB sites and NF-kappaB/Rel subunit specificity. The Journal of biological chemistry 272, 31092–31099. [DOI] [PubMed] [Google Scholar]
- 57.Wang Y, Rangan GK, Goodwin B, Tay YC, Harris DC (2000) Lipopolysaccharide-induced MCP-1 gene expression in rat tubular epithelial cells is nuclear factor-kappaB dependent. Kidney Int 57, 2011–2022. [DOI] [PubMed] [Google Scholar]
- 58.Widmer U, Manogue KR, Cerami A, Sherry B (1993) Genomic cloning and promoter analysis of macrophage inflammatory protein (MIP)-2, MIP-1 alpha, and MIP-1 beta, members of the chemokine superfamily of proinflammatory cytokines. Journal of immunology 150, 4996–5012. [PubMed] [Google Scholar]
- 59.Balamayooran G, Batra S, Balamayooran T, Cai S, Jeyaseelan S (2011) Monocyte chemoattractant protein 1 regulates pulmonary host defense via neutrophil recruitment during Escherichia coli infection. Infection and immunity 79, 2567–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kobayashi Y (2006) Neutrophil infiltration and chemokines. Crit Rev Immunol 26, 307–316. [DOI] [PubMed] [Google Scholar]
- 61.Kobayashi Y (2008) The role of chemokines in neutrophil biology. Frontiers in bioscience : a journal and virtual library 13, 2400–2407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
dnHMGA1 Tg mice underwent fibrin clot (E. coli) surgery. At 24 hours after surgery, spleen tissue was harvested and immunostaining was performed with an isotype control antibody (rat IgG2). There was no evidence of brown staining using the isotype control antibody. The scale bar represents 100 µm.
WT and dnHMGA1 Tg mice underwent sham (– E. coli) or fibrin clot (+ E. coli) surgery. At 24 hours after surgery, spleen (A) and plasma (B) were collected, and the concentration of IL-10 was assessed using a Luminex bead assay as descriibed in the Material and Methods. Data are represented as mean±SEM, from 3 mice per group. Analyses by one-way ANOVA. NS = not significant.