

Abstract
Podocyte injury is the major cause of proteinuria in primary glomerular diseases. Oxidative stress has long been thought to play a role in triggering podocyte damage; however, the underlying mechanism remains poorly understood. Here we show that the Wnt/ β- catenin pathway is involved in mediating oxidative stress-induced podocyte dysfunction. Advanced oxidation protein products, a marker and trigger of oxidative stress, were increased in the serum of patients with chronic kidney disease and correlated with impaired glomerular filtration, proteinuria, and circulating level of Wnt1. Both serum from patients with chronic kidney disease and exogenous advanced oxidation protein products induced Wnt1 and Wnt7a expression, activated β-catenin, and reduced expression of podocyte-specific markers in vitro and in vivo. Blockade of Wnt signaling by Klotho or knockdown of β-catenin by shRNA in podocytes abolished β-catenin activation and the upregulation of fibronectin, desmin, matrix metalloproteinase-9, and Snail1 triggered by advanced oxidation protein products. Furthermore, conditional knockout mice with podocyte-specific ablation of β-catenin were protected against podocyte injury and albuminuria after treatment with advanced oxidation protein products. The action of Wnt/ β- catenin was dependent on the receptor of advanced glycation end products (RAGE)-mediated NADPH oxidase induction, reactive oxygen species generation, and nuclear factor-κB activation. These studies uncover a novel mechanistic linkage of oxidative stress, Wnt/ β-catenin activation, and podocyte dysfunction.
Keywords: Oxidative stress, podocyte, Wnt, β-catenin, AOPPs, proteinuria
Graphical Abstract
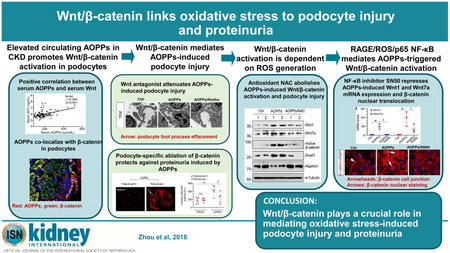
Proteinuria occurs in the early stage of many forms of chronic kidney disease (CKD).1-3 A large body of evidence indicates that proteinuria is a sensitive marker for kidney dysfunction, a strong predictor of morbidity, a potent trigger of renal inflammation.4, 5 Although proteinuria could be caused by a variety of distinct mechanisms such as a defective glomerular filtration barrier or an impaired tubular reabsorption, mounting evidence suggests that podocyte dysfunction/injury is the most important early event linked to the development of proteinuria.6-8 Depending on the nature, severity and duration of the injury, podocytes often undergo a range of different responses, including hypertrophy, autophagy, dedifferentiation, mesenchymal transition, detachment and apoptosis.4, 9-12 While cell hypertrophy and increased autophagy could be adaptive and beneficial changes, dedifferentiation and expression of mesenchymal markers represent early maladaptive responses that are destined to impairment of glomerular filtration.13
Podocytes are especially vulnerable to oxidative stress, which often leads to their dysfunction and causes proteinuria.14, 15 For instances, knockout mice lacking extracellular superoxide dismutase (EC-SOD), an antioxidant expressed at high levels in normal adult kidneys, are sensitized to various glomerular injury induced by adriamycin, chronic angiotensin II infusion or protein overload, and develop more severe proteinuria, compared with wild-type controls.16 However, EC-SOD null mice exhibit no difference with wild-type controls in kidney injury after unilateral ureteral obstruction (UUO).16 These tissue compartment-specific differences indicate that podocytes are particularly susceptible to oxidative stress due to a decreased antioxidant generation. It is also of interest to point out that injurious stimuli known to damage podocytes, such as adriamycin, puromycin, angiotensin II, TGF-β, high glucose and advanced oxidation protein products (AOPPs), are all able to increase intracellular production of reactive oxygen species (ROS).11, 12, 17, 18 In this context, it is conceivable to speculate that oxidative stress due to excessive ROS generation could be a common culprit that causes podocyte dysfunction in proteinuric CKD. However, exactly how oxidative stress triggers podocyte injury is poorly understood.
Wnt/β-catenin is evolutionarily conserved signal pathway and plays a critical role in organ development, tissue homeostasis, and injury repair.19-21 Wnt/β-catenin signaling is silent in normal adults, but reactivated after kidney injury in a wide range of CKD models, such as adriamycin nephropathy, ischemia-reperfusion injury, 5/6 nephrectomy and UUO.22-24 In human kidney biopsies, multiple Wnt ligands are upregulated and β-catenin is activated in diabetic nephropathy,25 IgA nephropathy,26 and lupus nephritis,27 in which podocyte injury and oxidative stress are common features. However, whether there is a link between oxidative stress and Wnt/β-catenin activation in the setting of CKD remains unknown.
In this study, we report that AOPPs, a marker and trigger of oxidative stress, activated Wnt/β-catenin through ROS generation and NF-κB activation. Blockade of Wnt signaling by Klotho or genetic ablation of β-catenin protected against AOPPs-induced podocyte dysfunction and proteinuria. Our results illustrate that Wnt/β-catenin plays a crucial role in mediating oxidative stress-induced podocyte damage and proteinuria.
RESULTS
Increased circulating AOPPs in CKD promotes Wnt/β-catenin activation in podocytes
To investigate the role of oxidative stress in podocyte injury, we first carried out the analysis of serum AOPPs, a marker of oxidative stress, in CKD patients. The demographic and clinical data of the patients are presented in Supplemental Table 1 and Table 2. As shown in Figure 1, a and b, the level of serum AOPPs was closely correlated with the severity of CKD and proteinuria. Furthermore, there was a correlation between AOPPs and Wnt1 protein in the circulation of CKD patients (Figure 1c), suggesting a possible connection between oxidative stress and Wnt signaling.
Figure 1. Circulating AOPPs are increased in CKD patients and induce Wnt/β-catenin activation in podocytes.

(a) Serum AOPPs levels inversely correlate with kidney function. Significant difference in serum AOPPs levels was observed in patients with different stages of CKD. Patients were grouped according to their eGFR (ml/min per 1.73m2) as indicated. Healthy controls: n=22, CKD patients: n=94. (b) Linear regression shows a correlation between serum AOPPs and urine albumin-to-creatinine ratio (UACR). (c) Linear regression shows a correlation between serum AOPPs and serum Wnt1 protein. The Spearman correlation coefficient (rs) and P value are shown. (d) Western blot analyses show that CKD serum induced active β-catenin and repressed ZO-1 and podocalyxin. Ctrl, control serum from normal subjects. (e, f) Graphic presentation of the relative abundances of active β-catenin (e), ZO-1 and podocalyxin (f) in different groups as indicated. *P< 0.05 versus controls (n = 3). (g) Western blot analyses show that CKD serum induced Wnt/β-catenin activation in podocytes, which was blocked by αRAGE. Podocyte lysates were immunoblotted with antibodies against Wnt1, Wnt7a, active β-catenin, podocalyxin, ZO-1 and α-tubulin, respectively. (h-k) Graphic presentations show the relative abundances of Wnt1 and Wnt7 (h), active β-catenin (i), podocalyxin (j) and ZO-1 (k) in different groups as indicated. *P< 0.05 versus controls; †P< 0.05 versus CKD serum alone (n = 3). (I) Representative micrographs show AOPPs and β-catenin staining in human focal and segmental glomerulosclerosis (FSGS). Human kidney biopsies from the patient with FSGS were stained with specific antibodies against AOPPs (red) and β-catenin (green), respectively. Arrows indicate positive staining in podocytes. Scale bar, 10 μm.
To explore the potential interplay between AOPPs and Wnt/β-catenin signaling, we used cultured podocytes in vitro and treated them with serum from CKD patients at different stages. As shown in Figure 1, d and e, serum from CKD patients clearly induced β-catenin activation in podocytes. Meanwhile, incubation with serum from CKD patients reduced the expression of podocyte epithelial markers of ZO-1 and podocalyxin (Figure 1, d and f). To elucidate whether AOPPs in the serum are responsible for β-catenin activation, we pretreated podocytes with a neutralizing antibody against the receptor of advanced glycation end products (αRAGE), a major receptor of AOPPs in podocyte.12 As shown in Figure 1g, CKD serum was able to induce Wnt1 and Wnt7a expression in podocytes, and activated β-catenin and repressed ZO-1 and podocalyxin. However, blockade of RAGE signaling with aRAGE abolished the CKD serum-induced Wnt1, Wnt7a and active β-catenin, and restored podocalyxin and ZO-1 (Figure 1, g-k), suggesting an association between AOPPs in serum and Wnt/β-catenin activation in podocytes. Consistent with this finding, double immunofluorescence staining on human kidney biopsies from patient with focal and segmental glomerulosclerosis (FSGS) confirmed the co-localization of AOPPs and β-catenin in podocytes (Figure 1l, arrows).
AOPPs promotes Wnt/β-catenin activation in podocytes in vivo
To provide direct evidence for AOPPs mediating Wnt/β-catenin activation in vivo, we administered exogenous AOPPs to mice by intravenous injections at 50 mg/kg/day for 4 weeks. As shown in Supplementary Figure S1, AOPPs induced RAGE expression in glomerular podocytes. Similarly, AOPPs also induced Wnt1, Wnt7a and activated β-catenin in mouse glomeruli (Figure 2a). Judged from localization and morphology, it appeared that these proteins were specifically induced in podocytes, suggesting a podocyte-specific activation of Wnt/β- catenin. Kidney homogenates were then immunoblotted for Wnt1, Wnt7a, active β-catenin and Snail1, respectively. As shown in Figure 2, b-f, injections of AOPPs induced renal expression of these proteins in vivo.
Figure 2. AOPPs induce Wnt/β-catenin activation and cause podocyte injury in vivo.

(a) Representative micrographs show renal Wnt1, Wnt7a and β-catenin expressions in mice after injections of AOPPs. Paraffin sections were stained with different antibodies. Arrows indicate Wnt1, Wnt7a and β-catenin expression specifically in glomerular podocytes. Scale bar, 25 μm.(b-f) Western blot analyses show that AOPPs induced Wnt1 and Wnt7a expression, and activated β-catenin and its downstream Snaill. Numbers (1 to 3) in (b) represent different animals in a given group. Graphic presentations show the relative abundances of Wnt1 (c), Wnt7a (d), active β-catenin (e) and Snail1 (f) proteins in different groups as indicated. *P < 0.05 versus controls. (g) Representative micrographs of desmin and nephrin staining in normal control and AOPPs groups. Scale bar, 20 μm. Arrows indicate positive staining. (h-l) Western blot analyses show that AOPPs induced α-SMA and fibronectin and repressed podocalyxin and nephrin. Numbers (1 to 3) in (h) represent different animals in a given group. Graphic presentations show the relative abundances of α-SMA (i), fibronectin (j), podocalyxin (k) and nephrin (l) proteins in different groups as indicated. *P < 0.05 versus controls.
We then examined podocyte lesions in mice injected with AOPPs. As shown in Figure 2g, AOPPs induced desmin, an injury marker for podocytes. Consistently, AOPPs impaired the linear distribution of nephrin in podocytes (Figure 2g). Western blotting demonstrated that AOPPs reduced the expression of podocalyxin and nephrin, and induced renal expression of α-smooth muscle actin (α-SMA) and fibronectin (Figure 2, h-l).
Blockade of Wnt signaling by Klotho protects against podocyte injury by AOPPs
To validate the role of Wnt signaling in AOPPs-induced podocyte injury, we sought to block this pathway by Klotho, an antiaging protein which is an endogenous Wnt antagonist through binding and sequestration of Wnt ligands.28 To this end, AOPPs-injected mice were subjected to weekly injection of the expression vector encoding the secreted form of Klotho (pV5-sKlotho) through hydrodynamic-based gene delivery approach (Figure 3a). As shown in Figure 3b, injection of pV5-sKlotho plasmid resulted in robust sKlotho expression in the glomeruli. In addition, sKlotho expression also induced endogenous, full-length Klotho expression (Figure 3b), consistent with previous report.28
Figure 3. Wnt ligands blocade attenuates AOPPs-induced podocyte injury and proteinuria.

(a) Experimental design. Red arrows indicate intravenous injection of either emty vector pcDNA43 or pV5-sKlotho at 1, 2 and 3 weeks after vehicle or AOPPs administration. (b) Western blot analyses show that both full-length and secreted Klotho were induced in isolated glomeruli from mice injected with pV5-sKlotho plasmid, compared to pcDNA3. (c) Delivery of sKlotho reduced AOPPs-induced albuminuria. Urinary albumin was expressed as μg/mg creatinine. *P < 0.05 versus control group; †P< 0.05 versus AOPPs group (n = 6). (d-h) Western blot analyses show that sKlotho restored AOPPs-induced loss of full-length Klotho and inhibited active β-catenin and its downstream fibronectin and α-SMA. Numbers (1 to 3) in (d) represent different animals in a given group. Graphic presentations show the relative abundances of full-length Klotho (e), active β-catenin (f), fibronectin (g) and α-SMA (h) proteins in different groups as indicated. (i) Antagonism of Wnt signaling by Klotho preserves podocyte integrity after AOPPs treatment. PAS staining and immunofluorescence staining for podocalyxin and nephrin revealed that sKlotho attenuated glomerular matrix accumulation and restored podocalyxin and nephrin. Arrows indicate positive staining. Scale bar, 25 μm. Representative transmission electron microscopy (TEM) microgarphs show podocyte ultrastructure after various treatments. Arrow indicates foot process effacement. Scale bar, 1 μm. (j-l) Western blot analyses show that sKlotho largely prevented AOPPs-induced loss of WT-1 and podocalyxin. Numbers (1 to 3) in (j) represent different animals in a given group. Graphic presentations show the relative abundances of WT-1 (k) and podocalyxin (l) proteins in different groups as indicated. *P < 0.05 versus controls; †P< 0.05 versus AOPPs (n = 6).
We found that expression of sKlotho in vivo reduced proteinuria in mice after AOPPs injections (Figure 3c). Furthermore, expression of sKlotho largely restored endogenous full-length Klotho expression and inhibited β-catenin activation, and abolished fibronectin and α-SMA induction by AOPPs (Figure 3, d-h). We further assessed podocyte injury. As shown in Figure 3i, AOPPs induced matrix accumulation and inhibited podocalyxin and nephrin expression in the glomeruli, and sKlotho expression prevented these lesions and restored the integrity of podocytes. Consistently, sKlotho preserved the fine ultrastructure of podocytes by ameliorating foot process effacement induced by AOPPs (Figure 3i). Similar results were obtained by western blot analysis of WT-1 and podocalyxin (Figure 3, j-l).
Mice with podocyte-specific ablation of β-catenin are protected against podocyte injury by AOPPs
To further confirm a role for Wnt/β-catenin in mediating oxidative stress-induced podocyte injury, we utilized conditional knockout mice in which β-catenin gene was selectively disrupted in podocytes via the Cre-LoxP system. As previously reported,25 mice with podocyte-specific deletion of β-catenin (Podo-β-cat−/−) displayed no overt abnormality under basal conditions. These mice and their control littermates (Podo-β-cat+/+) were challenged with AOPPs at 50 mg/kg/day by tail vein injection for 4 weeks (Figure 4a). As shown in Figure 4b, compared with Podo-β-cat+/+ littermates, urinary albumin in Podo-β-cat−/− mice was reduced after AOPPs injections.
Figure 4. Podocyte-specific ablation of β-catenin protects against proteinuria induced by AOPPs in vivo.

(a) Scheme depicts experimental design. Conditional knockout mice with podocyte-specific deletion of β-catenin (podo-βcat−/−) and control littermates (podo-β-cat+/+) were described previously.25 (b) Podocyte-specific deletion of β-catenin protects mice from developing proteinuria after AOPPs administration in vivo. Urinary albumin was expressed as μg/mg creatinine. *P < 0.05 versus vehicle group; †P< 0.05 versus AOPPs group (n = 5 to 6). (c-f) Western blot analyses show that podocyte-specific deletion of β-catenin prevented β-catenin activation and inhibited fibronectin and α-SMA induction by AOPPs. Numbers (1 to 2) in (c) represent different animals in a given group. Graphic presentations show the relative abundances of active β-catenin (d), fibronectin (e) and α-SMA (f) expressions in different groups as indicated. *P< 0.05 versus vehicle group; †P < 0.05 versus AOPPs alone (n = 6). (g) Immunohistochemical and immunofluorescence staining shows β-catenin and fibronectin expression in different groups as indicated. AOPPs promoted active β-catenin and fibronectin expression in podocytes of podo-β-cat+/+ mice, but not in podo-β-cat−/− mice. Arrows indicate positive staining. Scale bar, 25 μm.
We also examined renal β-catenin activation and its downstream fibronectin and α-SMA expression by western blot analyses. As show in Figure 4, c-f, AOPPs induced active β-catenin, fibronectin and α-SMA expression in Podo-β-cat+/+ mice. However, their induction after AOPPs was abolished in Podo-β-cat−/− mice under the same conditions (Figure 4, c-f). Immunostaining for β-catenin and fibronectin gave rise to similar results (Figure 4g), suggesting that selective ablation of β-catenin in podocytes protects glomerular integrity after AOPPs challenge.
Renal expression of podocyte-specific proteins in these mice was assessed as well. As shown in Figure 5a, glomerular expression of nephrin and podocalyxin was reduced in Podo-β-cat+/+ mice after AOPPs challenge. However, they were largely preserved in Podo-β-cat−/− mice under the same conditions (Figure 5a). We then analyzed podocyte injury markers of vimentin, desmin and MMP-9. As shown in Figure 5, a through d, AOPPs induced vimentin, desmin and MMP-9 expressions in glomerular podocytes in Podo-β-cat+/+ mice, but not in Podo-β-cat−/− mice. Therefore, these data suggest that β-catenin activation is indispensable for AOPPs-induced podocyte dysfunction and injury in vivo.
Figure 5. Mice with podocyte-specific ablation of β-catenin are protected against podocyte injury by AOPPs.

(a) Representative micrographs show that administration of AOPPs repressed nephrin and podocalyxin, and upregulated vimentin and MMP-9. However, podocyte-specific deletion of β-catenin largely restored nephrin and podocalyxin and inhibited vimentin and MMP-9. Arrows indicate positive staining. Scale bar, 25 μm. (b-d) Western blot analyses show that podocyte-specific deletion of β-catenin abolished desmin and MMP-9 induction by AOPPs. Numbers (1 to 2) in (b) represent different animals in a given group. Graphic presentations show the relative abundances of desmin (c) and MMP-9 (d) expressions in different groups as indicated. *P< 0.05 versus vehicle group; †P < 0.05 versus AOPPs (n = 5).
AOPPs activate Wnt/β-catenin signaling in cultured podocytes
We next performed a comprehensive survey of the mRNA expression of all 19 Wnt genes in cultured podocytes after incubation with AOPPs. As shown in Figure 6a, several Wnt ligands including Wn1, Wnt2, Wnt7a and Wnt9a were upregulated in a time-dependent manner by AOPPs, but not by native MSA. Among them, the induction of Wnt1 mRNA by AOPPs was most predominant. Western blot analyses also confirmed that Wnt1 and Wnt7a proteins were induced after AOPPs treatment (Figure 6b-d). Accordingly, AOPPs induced β-catenin nuclear translocation in podocytes (Figure 6e). Consistently, AOPPs induced active β-catenin and its downstream Snail1 in a time-dependent manner (Figure 6, f-h).
Figure 6. Wnt/β-catenin signaling is activated by AOPPs in cultured podocytes.

(a) RT-PCR analyses show that AOPPs induced multiple Wnt ligands expression in cultured podocytes in a time-dependent manner. Unmodified mouse serum albumin (MSA) incubated for 24 hours was used as a negative control. (b-d) Western blot analyses show the induction of Wnt1 and Wnt7a proteins in podocytes after AOPPs treatment. Graphic presentation of Wnt1 (c) and Wnt7a (d) proteins in different groups as indicated. *P < 0.05 versus controls (n = 3). (e) Immunofluorescence staining shows that AOPPs induced β-catenin activation and its nuclear translocation. Podocytes were treated with AOPPs for 12 hours. Cells were then immunostained for β-catenin and DAPI. Arrows indicate nuclear staining of β-catenin. Scale bar, 5 μm. (f-h) Western blot analyses of cell lysates show that AOPPs time-dependently induced active β-catenin and its downstream Snail1 protein expression. Graphic presentations of active β-catenin (g) and Snail1 (h) protein expressions in different groups as indicated. *P < 0.05 versus controls (n = 3).
We next assessed whether Wnt/β-catenin activation is required for AOPPs-mediated podocyte injury in vitro. As shown in Figure 7, a through f, Klotho blocked β-catenin activation and its downstream Snail1, and restored podocalyxin and ZO-1. Moreover, transfection of constitutively activated β-catenin (pDel-β-cat) decreased ZO-1, podocalyxin and nephrin (Figure 7g), and triggered the reorganization of the F-actin cytoskeleton (Figure 7h), suggesting that β-catenin activation is sufficient for causing podocyte dysfunction. Knockdown of endogenous β-catenin by shRNA abolished AOPPs-induced fibronectin, desmin, MMP-9 and Snail1 expression (Figure 7, i-m). Together, these results establish a critical role for Wnt/β-catenin in mediating oxidative stress-induced podocyte injury.
Figure 7. Wnt/β-catenin activation is required for AOPPs-mediated podocyte injury.

(a-f) Western blot analyses show that recombinant Klotho (100 ng/ml) blocked AOPPs-mediated β-catenin activation and Snail1 induction, and restored the expressions of podocalyxin and ZO-1. Representative western blots (a, d) and graphic presentation of active β-catenin (b), Snail1 (c), podocalyxin (e) and ZO-1 (f) proteins in different groups are shown. *P < 0.05 versus controls; †P < 0.05 versus AOPPs (n = 3). (g) Western blot analyses show that overexpression of Flag-tagged, constitutively activated β-catenin repressed ZO-1, podocalyxin and nephrin expression. (h) Immunofluorescence staining shows that AOPPs induced reorganization of the F-actin cytoskeleton in podocytes. Arrow indicates stress-fiber in control podocytes, whereas arrowhead denotes peripheral localization of actin cytoskeleton in cells with over-expression of active β-catenin. Scale bar, 10 μm. (i-m) Western blot analyses show that knockdown of β-catenin by lentivirus-mediated β-catenin shRNA significantly inhibited AOPPs-mediated fibronectin, desmin, MMP-9 and Snail1 induction in podocytes. Graphic presentations show the relative abundances of fibronectin (j), desmin (k), MMP-9 (l) and Snail1 (m) proteins in different groups as indicated. *P < 0.05 versus controls; † P< 0.05 versus AOPPs plus Ctrl-shRNA (n = 3).
Wnt/β-catenin activation is dependent on ROS production
To delineate the molecular details that link AOPPs to Wnt/β-catenin activation and podocyte injury, we assessed reactive oxidative species (ROS) production after AOPPs challenge. As shown in Figure 8, a and b, detection of 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA) fluorescence indicated that AOPPs promoted ROS generation in cultured podocytes in a time-dependent fashion. Because nicotinamide adenine dinucleotide phosphate (NADPH) oxidase plays a critical role in generating ROS, we next examined its expression after AOPPs challenge in podocytes. As shown in Figure 8, c-e, Nox2 and p47phox, the major subunits of NADPH oxidase complex,11 were upregulated in podocytes after AOPPs treatment.
Figure 8. Wnt/β-catenin activation is dependent on ROS generation.

(a) ROS production is increased following AOPPs treatment in cultured podocytes. ROS was assessed by detection of DCFH fluorescence. Representative micrographs show that AOPPs induced ROS production in a time-dependent manner. Scale bar, 50 μm. (b) Quantitative data show the relative DCFH fluorescence intensity in different groups. The intensity of DCFH fluorescence in cells was assessed using a flow cytometer, and expressed as relative fluorescence intensity/100μg cell protein. (c-e) Western blot analyses show that AOPPs induced Nox2 and p47phox expression in cultured podocytes. Graphic presentations show the relative abundances of Nox2 (d) and p47phox (e) proteins in different groups as indicated. *P < 0.05 versus controls (n = 3). (f-k) Western blotting shows that NAC abolished AOPPs-induced Wnt1, Wnt7a, active β-catenin, Snail1 and restored nephrin expression in cultured podocytes. Podocytes were pre-incubated with NAC (2 mM), followed by incubation with AOPPs (10 μg/ml) for 24 hours. Graphic presentations of the relative abundances of Wnt1 (g), Wnt7a (h), active β-catenin (i), Snail1 (j) and nephrin (k) in different groups as indicated. *P< 0.05 versus controls; †P< 0.05 versus AOPPs alone (n = 3). (l) Representative micrographs show that NAC restored ZO-1 expression in cultured podocytes after AOPPs treatment. Arrows indicate positive staining. Scale bar, 20 μm.
We further tested whether AOPPs-triggered ROS is required for Wnt/β-catenin activation in podocytes. To this end, podocytes were pre-treated with N-acetyl cysteine (NAC), an antioxidant compound, and followed by incubation with AOPPs. As shown in Figure 8, f-j, NAC abolished AOPPs-induced Wnt1, Wnt7a, active β-catenin and Snail1. Furthermore, NAC restored nephrin and ZO-1 expression in AOPPs-treated podocytes (Figure 8, f, k and l). These results indicate that AOPPs-induced Wnt/β-catenin activation is dependent on ROS generation.
RAGE/ROS/p65 NF-κB axis mediates AOPPs-triggered Wnt/β-catenin activation
To investigate how AOPPs transduce their signal across plasma membrane to induce Wnt expression, we studied the potential involvement of RAGE and p65 NF-κB. To this end, a neutralizing antibody to RAGE was used to block the activity of receptor-ligand interaction. As shown in Figure 9a, RAGE overexpression induced Wnt1 and activated β-catenin. Furthermore, αRAGE abolished AOPPs-induced Wnt1 expression in cultured podocytes. Accordingly, αRAGE also blocked β-catenin activation (Figure 9b). We further studied the downstream signaling of RAGE in AOPPs-induced Wnt/β-catenin activation. As shown in Figure 9c, AOPPs induced phosphorylation and activation of NF-κB p65 subunit in a time-dependent manner. Of interest, Wnt1 and Wnt7a mRNA expression, as well as Wnt1, Wnt7a and active β-catenin proteins were all inhibited by administration of SN50 (Figure 9, d-h), a highly selective peptide inhibitor that blocks NF-κB activation. Moreover, SN50 also inhibited AOPPs-induced β-catenin nuclear translocation (Figure 9i). Taken together, as summarized in Figure 9j, it is concluded that AOPPs induced Wnt/β-catenin activation through RAGE mediated ROS/p65 NF-κB signaling.
Figure 9. RAGE/ROS/p65 NF-κB mediates AOPPs-triggered Wnt/β-catenin activation in podocytes.

(a) Western blotting shows that overexpression of RAGE induced Wnt1 and active β-catenin. (b) Blockade of RAGE by neutralizing antibody abolished AOPPs-induced Wnt1 and active β-catenin expression. (c) AOPPs induced phosphorylation of NF-κB p65 subunit in a time-dependent manner. (d) RT-PCR analyses show that NF-κB inhibitor SN50 repressed Wnt1 and Wnt7a mRNA expression induced by AOPPs. (e-h) Western blotting shows that SN50 inhibited AOPPs-mediated Wnt1, Wnt7a and active β-catenin induction in cultured podocytes. Graphic presentations of the relative abundances of Wnt1 (f), Wnt7a (g) and active β-catenin (h) in different groups are shown. *P< 0.05 versus controls; †P< 0.05 versus AOPPs alone (n = 3). (i) Representative micrographs show that SN50 blocked AOPPs-induced β-catenin nuclear translocation. Arrowheads indicate β-catenin in cell-cell junction, whereas arrows denote its nuclear staining. Scale bar, 20 μm. (j) Schematic presentation depicts the potential mechanism by which oxidative stress induces podocyte injury. AOPPs bind to plasma membrane receptor RAGE and trigger the activation of NADPH oxidase, thereby leading to an increased generation of ROS. This causes activation of p65 NF-κB, which in turn induces Wnt ligands such as Wnt1 and Wnt7a, and subsequently activates β-catenin. The activation of β-catenin triggers podocyte injury through inducing their dedifferentiation and mesenchymal transition. Multiple approaches such as blockade RAGE by neutralizing antibody, reduction of ROS by NAC, and genetic or pharmacologic ablation of β-catenin protect podocytes from injury.
DISCUSSION
Increased oxidative stress, resulting from an imbalance between oxidant production and antioxidant reserves, is highly prevalent in a wide variety of CKD.29-31 Mounting evidence indicates that oxidative stress, resulting in excessive generation of ROS, have a significant role in the initiation and progression of kidney disorders.32, 33 Although podocytes are well known to be particularly vulnerable to oxidation damage,16 the mechanisms by which oxidative stress induces podocyte dysfunction was poorly defined. In this study, we demonstrate that AOPPs, a family of oxidized, dityrosine-containing protein products formed during oxidative stress,34 induces NAPDH oxidase activation via plasma membrane receptor RAGE, promotes ROS generation and activation NF-κB transcription factor, which leads to induction of Wnt ligands such as Wnt1 and Wnt7a and activation of β-catenin. This cascade of events results in podocyte injury and dysfunction, leading to impairment of glomerular filtration and onset of proteinuria. As depicted in Figure 9j, blockade of this signal cascade at several different points preserves podocyte integrity and ameliorates proteinuria. These studies uncover a previously unrecognized signal pathway in which Wnt/β-catenin plays a crucial role in mediating oxidative stress-triggered podocyte dysfunction and proteinuria. Our findings also underscore that targeting Wnt/β-catenin could be a novel therapeutic strategy for alleviating podocyte damage and proteinuria.
Podocyte dysfunction is a principal feature of many proteinuric CKD.35, 36 Although podocyte depletion could contribute to a defect in glomerular filtration, loss of podocyte-specific markers has been proposed as an early and initial response causing podocyte dysfunction, leading to an impaired glomerular filtration and proteinuria. 10,13,37 Indeed, numerous studies have demonstrated that podocytes could undergo phenotypic change under pathological conditions, such as diabetic nephropathy,10, 37, 38 focal segmental glomerulosclerosis,39 and HIV-associated nephropathy.40 In the present study, we show that the expression of nephrin, podocin and ZO-1 is decreased after AOPPs treatment, indicating the loss of epithelial characteristics. Meanwhile, this loss of podocyte features is accompanied by the expression of desmin, fibronectin, Snail 1 and MMP-9, suggesting that oxidative stress triggered by AOPPs is able to trigger phenotypic alteration. These findings are in harmony with earlier reports showing the phenotypic changes of podocytes after challenge with various ROS-generating agents such as TGF-β1, adriamycin, and high glucose.10, 25, 38 It should be pointed out that the decrease in podocyte-specific proteins is unlikely due to a reduced podocyte number, because there was little evidence for podocyte apoptosis after AOPPs at the dose used in this study (Supplementary Figure S2).
One of the novel findings in the present study is that AOPPs via RAGE/NAPDH oxidase-dependent ROS/p65 NF-κB pathway induces Wnt ligand expression (Figure 9). This observation, for the first time, unequivocally links oxidative stress to Wnt/β-catenin activation, which may have broad implications beyond podocyte pathology. The finding that oxidative stress is an upstream regulator of Wnt/β-catenin, a developmental signal usually silenced in normal adult kidney, is quite interesting and significant, because excessive ROS generation is a common feature in a variety of pathologic conditions. There are several lines of evidence supporting that AOPPs stimulate Wnt/β-catenin activation via a ROS/ p65 NF-κB-dependent pathway. First, serum AOPPs in CKD patients is correlated with the circulating level of Wnt1 protein, and blockade of RAGE, the plasma membrane receptor mediating AOPPs-induced ROS generation,12 mitigates the damaged effects of CKD serum (Figure 1). Second, AOPPs induce expression of multiple Wnt ligands including Wnt1, Wnt2, Wnt7a and Wnt9a, activate b-catenin and stimulate its downstream targets in vitro and in vivo (Figure 2 and 6). Third, incubation of podocytes with a broad spectrum anti-oxidant, NAC, abolishes AOPPs-mediated Wnt/β-catenin activation (Figure 8). Finally, RAGE neutralizing antibody and SN50 blunt Wnt induction and β-catenin activation (Figure 9). All together, these studies clearly establish a role for ROS/p65 NF-κB in activation of Wnt/β-catenin signaling.
The present study also illustrates an important role for Wnt/β-catenin in mediating AOPPs-induced podocyte injury and proteinuria, which is supported by both in vitro and in vivo evidence. Ectopic expression of Klotho in vivo or knockdown of β-catenin by shRNA-mediated inhibition prevents WT-1 and nephrin loss induced by AOPPs (Figure 3), and negates desmin, fibronectin, Snail 1 and MMP-9 expression (Figure 7), underscoring that β-catenin activation is required for mediating phenotypic change of podocytes after AOPPs challenge. More importantly, conditional knockout mice with podocyte-specific ablation of β-catenin are protected against development of proteinuria after chronic infusion of AOPPs (Figure 4 and 5). These findings are consistent with earlier studies that Wnt/β-catenin is implicated in mediating podocyte injury and proteinuria induced by adriamycin,25, 41 a podocyte toxin that also produce oxidative stress via excessive ROS generation. Given that oxidative stress is present in various pathologic condition, our studies suggest that ROS/p65 NF-κB-mediated Wnt/β-catenin activation could be a common patho-mechanism leading to podocyte injury and proteinuria in a variety of proteinuric CKD.
In summary, we have shown that Wnt/β-catenin is activated in glomerular podocytes through a cascade of signal events involving AOPPs/RAGE/NADPH oxidase-dependent ROS/p65 NF-κB and Wnt induction. Furthermore, knockdown of β-catenin in vitro by shRNA-mediated inhibition or genetic ablation of β-catenin in podocyte-specific fashion in vivo is able to preserve podocyte integrity and ameliorates proteinuria. This study provides a novel mechanistic linkage of Wnt/β-catenin activation to oxidative stress in the setting of proteinuric CKD. Given the fact that ROS generation is prevalent in a variety of kidney disorders, targeting Wnt/β-catenin could be a novel strategy for developing therapeutic modalities of proteinuric CKD.
MATERIALS AND METHODS
Human serum and kidney biopsies samples
All human samples (serum, urine and kidney biopsies) were collected from CKD patients in the National Clinical Research Center of Kidney disease at Nanfang Hospital and Huadu District People’s Hospital, with written consent of the donors. Serum and urine samples were also obtained from normal healthy volunteers. Kidney paraffin sections from diagnostic renal biopsies were used for immunohistochemical staining. Human studies were approved by the Ethics Committee at the Nanfang Hospital, Southern Medical University. The demographic and clinical data of the CKD patients involved were presented as Supplemental Table 1 and Table 2.
Serum AOPPs assay
The content of AOPPs in human serum was measured using the spectrophotometric method described by Witko-Sarsat et al,42 as previously reported.43 Briefly, serum samples were centrifuged at 10 000x g for 30 min to avoid the confounding effect of endogenous compounds. Samples below the lipid layer were sucked out and diluted 10 times with phosphate buffered saline (PBS). Then, 200 μL diluted sample, 200 μL chloramine T (0–100 μmol/L, for calibration), and 200 μL PBS (as a blank) were applied to each well of a 96-well microplate. Subsequently, 10 μL of potassium iodide (1.16 mol/L) and 20 μL of acidic acid were added to the samples and the reaction mixture was measured immediately at 340 nm absorbance. Concentrations of AOPPs were expressed in μmol/L chloramine T equivalents.
Wnt1 ELISA
Serum Wnt1 was measured by using a human Wnt1 enzyme-linked immunosorbent assay (ELISA) kit (CSB-EL026128HU; Cusabio Technology, China), according to the manufacturer’s protocol. The concentration of Wnt1 was calculated according to standard curve and expressed as nanograms per illiliter.
Cell culture and treatment
The conditionally immortalized mouse podocyte cell line was described previously.44 Experiments were performed under differentiated conditions. In some experiments, podocytes were transfected with N-terminally truncated, constitutively activated β-catenin expression vector (pDel-β-cat) by using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) or incubated with recombinant protein of Klotho (5334-KL-025, R&D system, Minneapolis, MN) or a neutralizing antibody to mouse RAGE (AF1179, R&D system, Minneapolis, MN).
AOPPs-MSA preparation
AOPPs-MSA was prepared in vitro by incubation of mouse serum albumin (MSA) (Sigma, St Louis, MO) with hypochlorous acid (Fluke, Buchs, Switzerland) in the absence of free amino acid/carbohydrates/lipids to exclude the formation of AGEs-like structures as previously described.11 Prepared samples were dialyzed against PBS to remove free HOCl and passed through a DetoxiGel column (Pierce, Rockford, IL) to remove contaminated endotoxin. The components of AGEs, including Nε (carboxylmethyl) lysine, pentosidine, pyridine, or glyoxal-, glycolaldehyde-, and glyceraldehyde- modified proteins were determined as described previously,45 and were found to be undetectable in the prepared samples.
Animal models
Mouse model of podocyte injury and proteinuria was established by intravenous injection of AOPPs, as described previously.11, 12 Male C57BL/6 mice weighing 22-24g were obtained from Southern Medical University Animal Center and housed in the standard environment with regular light/dark cycles and free access to water and chow diet. Three groups of mice were used: 1) normal controls injected with vehicle (endotoxin-free PBS, pH 7.4) (n=6), 2) AOPPs mice injected intravenously with 50 mg/kg/day of AOPPs-MSA and 1 mg/kg/week of pcDNA3 plasmid (n=6), 3) AOPPs/Klotho mice injected intravenously with 50 mg/kg/day of AOPPs-MSA and 1 mg/kg/week of pV5-sKlotho plasmid (n=6). The mice were sacrificed at the end of week 4. Urine and kidney tissue were collected for various analyses. All animal studies were approved by the Animal Experimentation Ethic Committee at the Nanfang Hospital, and were performed in compliance with the NIH’s Guidelines for the Care and Use of Laboratory Animals.
Conditional knockout mice with podocyte-specific deletion of β-catenin (Podo-β-cat−/−) were previously reported.25 Briefly, Podo-β-cat−/− mice were obtained by mating β-catenin floxed mice with podocin-Cre transgenic mice to inactivate both β-catenin alleles by Cre-mediated excision in glomerular podocytes (genotype: β-catfl/fl, Cre). Littermates with β-catenin floxed but lack of Cre (genotype: β-catfl/fl) were used as controls (Podo-β-cat+/+). Both Podo-β-cat+/+ and Podo-β-cat−/− mice were received daily intravenous injection of either vehicle (endotoxin-free PBS, pH 7.4) or AOPPs-MSA (50 mg/kg/day) for 4 weeks. Animal studies involving conditional knockout mice were performed by use of the procedures approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh.
Isolation of mouse glomeruli
We isolated glomeruli from mice following the method described with some modifications.25 Briefly, male C57BL/6 mice weighing 22-24g were injected intravenously with 1 mg/kg of pcDNA3 plasmid or pV5-sKlotho plasmid. After 24 hours, mice were anesthetized and then perfused cardiovascularly with 10 ml PBS, followed by Dynabead (00388551; Invitrogen) perfusion. The kidney were excised and cut into small pieces (∼1 mm3). Kidney pieces were then digested by collagenase IV at 37°C for 15 min. The digests were gently pressed with a spatula through a 100 μm sieve and the glomeruli collected by magnetic concentrator. The glomeruli were centrifuged at 500x g for 5 min at 4°C and lysed in a buffer for Western blot analysis.
Urinary albumin and creatinine assay
Urinary albumin was measured by using a mouse Albumin ELISA Quantitation kit, according to the manufacturer's protocol (Bethyl Laboratories, Inc., Montgomery, TX). Urine creatinine was determined by a routine procedure as described previously.25 Urinary albumin was standardized to creatinine and expressed as μg/mg Ucr.
Western blot analysis
Protein expression was analyzed by Western blot analysis as described previously.12 The primary antibodies used were as follows: anti-fibronectin (F3648, Sigma-Aldrich), anti-desmin (PB0095; Boster, Wuhan, China), anti-vimentin (SAB1305447, Sigma-Aldrich), anti-MMP-9 (AB805; Millipore), anti-podocin (sc-22298; Santa Cruz Biotechnology, Santa Cruz, CA), anti-podocalyxin (AF1556; R&D Systems), anti-ZO-1(QF215185; Life technologies), anti-nephrin(ab58968; Abcam), anti-Klotho (AF1819; R&D Systems), anti-p47phox (07-500; Millipore, Billerica, MA), anti-Nox2 (BA2811; Boster), anti-active β-catenin (05-665; Millipore), anti-Snail1 (ab17732; Abcam), anti-Wnt1 (ab15251; Abcam), anti-Wnt7a (sc-26361; Santa Cruz Biotechnology), anti-Flag(F3165; Sigma-Aldrich), anti-p65(4764S; Cell Signaling Technology), anti-p-p65(3031S; Cell Signaling Technology), anti-α-Tubulin (RM2007; Ray Antibody, Beijing, China) and anti-β-actin (MAB1501; Millipore, Billerica, MA).
Immunofluorescence staining and confocal microscopy
Podocytes cultured on coverslips were fixed with cold methanol: acetone (1:1) for 10 min at −20°C, followed by blocking with 20% normal donkey serum in PBS. Slides were incubated with various primary antibodies: rabbit polyclonal anti-fibronectin (F3648, Sigma-Aldrich), rabbit polyclonal anti-ZO-1 (61-7300; Invitrogen), and anti-β-catenin (ab15180; Abcam). After washing, the slides were then incubated with Alexa Fluor 488-conjugated chicken anti-rabbit IgG or Cy3-conjugated donkey anti-rabbit IgG. For F-actin staining, podocytes were fixed with 4% paraformaldehyde and incubated with 100 nM TRITC-Phalloidin (40734ES75; Yeasen, Shanghai, China) for 30 min at room temperature. For staining of kidney tissue, paraffin-embedded tissue sections or frozen sections were fixed with 4% paraformaldehyde, and permeabilized by 0.5% TritonX-100 and blocked with donkey serum. Primary antibodies used were as follows: anti-podocalyxin (AF1556; R&D Systems), anti-vimentin (5741S; Cell Signaling Technology), and anti-nephrin (20R-NP002; Fitzgerald Industries International). For staining of human kidney biopsies, sections were stained with mouse monoclonal anti-AOPPs (Clone 3F2) and rabbit monoclonal anti-β-catenin (AF1329, R&D Systems), respectively. The anti-AOPPs monoclonal antibody was described previously and demonstrated to react specifically with hypochlorous acid-modified proteins but not with the native form or other oxidant-modified proteins.43 Second antibodies from Jackson ImmunoResearch were as follows: Cy3-conjugated donkey anti-goat IgG, Cy3-conjugated donkey anti-rabbit IgG, Cy2-conjugated donkey anti-rabbit IgG, Cy3-conjugated donkey anti-mouse IgG and ALexa Fluro594-conjugated donkey anti-guinea pig IgG. Nuclei were stained with DAPI (Sigma-Aldrich) according to manufacturer’s instruction. Images were taken by confocal microscopy (Leica TCS SP2 AOBS; Leica Microsystems, Buffalo Grove, IL) or Olympus DP80 microscope with EMCCD camera.
Histology and immunohistochemical staining
Paraffin-embedded mouse kidney sections (3 μm thickness) were prepared by a routine procedure. Immunohistochemical staining was performed using routine protocol. Antibodies used were as follows: rabbit polyclonal anti-β-catenin (ab15180; Abcam), rabbit polyclonal anti-Wnt1 (ab15251; Abcam), goat polyclonal anti-Wnt7a (sc-26361; Santa Cruz Biotechnology), rabbit polyclonal anti-MMP-9 (AB805; Millipore), rabbit polyclonal anti-FN (F3648; Sigma), goat polyclonal anti-podocalyxin (AF1556; R&D Systems).
Intracellular production of reactive oxygen species
Intracellular reactive oxygen species (ROS) was detected in podocytes by analyzing the fluorescence intensity of the intracellular fluoroprobe DCFH (10 μM) after incubation with AOPPs for varying periods of time as indicated.46 Representative images were obtained by confocal microscopy (Leica TCS SP2 AOBS). For further quantification, the intensity of cellular DCF fluorescence was detected using a flow cytometer (BD FACS Calibur System, Franklin Lakes, NJ) with a wavelength of 488 nm for excitation and 530 nm for emission.
Reverse transcriptase (RT)-PCR
Total RNA was prepared using TRIzol RNA isolation system (Life Technologies, Grand Island, NY) according to the manufacturer’s instruction. The first strand of complementary DNA was synthesized using 1 μg of RNA in 20 μl of reaction buffer using AMV-RT and random primers at 42 °C for 60 min. PCR amplification was performed using a HotStar Taq Master Mix kit (Qiagen, Valencia, CA). The sequences of the primers for 19 different Wnt ligands and β-actin were described previously.30
Lentivirus transduction
Lentiviral shRNA particles targeting β-catenin (SC-29209-V) and a scrambled non-targeting control (SC-108080) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and infected into cultured podocytes according to the manufacturer’s instruction. Briefly, cells were plated approximately 50% confluent in a 6-well plate 24 hours before viral infection, then 2 ml of mixture of polybrene (5 μg/ml) (sc-134220) and lentiviral particles were added to the culture and incubated for 48 hours. Cells were then harvested for protein analyses.
Statistical analyses
All data examined were expressed as mean ± SEM. Statistical analysis of the data was carried out using SPSS 13.0 (SPSS Inc, Chicago, IL). Comparison between groups was made using one-way ANOVA followed by Student-Newman-Kuels test or Dunnett’s T3 procedure. P< 0.05 was considered significant.
Supplementary Material
Figure S1. AOPPs induce RAGE expression in the kidney of mice. (a) Immunohistochemical staining show that AOPPs induced RAGE expressions in glomerular podocytes. Arrows indicate positive staining. Scale bar, 20 μm. (b-c) Western blotting shows that AOPPs induced RAGE protein expression. Graphic presentations of the relative abundances of RAGE (c) in two groups are shown. *P< 0.05 versus controls (n = 6).
{kind=link}
Figure S2. AOPPs do not induce podocyte apoptosis. Representative miscrographs show that AOPPs did not cause podocyte apoptosis. Kidney sections were subjected to TUNEL staining. Arrows indicate TUNEL-positive cells in renal tubules. Scale bar, 50 μm.
{kind=link}
Table S1. Demographic characteristics of the participants in the study.
Table S2. Parameter values of healthy subjects and CKD patients.
ACKNOWLEDGEMENTS
This work was supported by National Natural Science Foundation of China Grant 81521003 and 81570620, and Guangdong Science Foundation grant 2014A030312014, and National Institute of Health grant DK064005 and DK091239.
Footnotes
DISCLOSURE
The authors declared no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain
Supplementary information is available at Kidney International's website.
REFERENCES
- 1.Fogo AB. Causes and pathogenesis of focal segmental glomerulosclerosis. Nat Rev Nephrol 2015;11:76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miner JH. Podocyte biology in 2015: New insights into the mechanisms of podocyte health. Nat Rev Nephrol 2016;12:63–64. [DOI] [PubMed] [Google Scholar]
- 3.Mathieson PW. The podocyte as a target for therapies--new and old. Nat Rev Nephrol 2012;8:52–56. [DOI] [PubMed] [Google Scholar]
- 4.Kuusniemi AM, Lapatto R, Holmberg C, et al. Kidneys with heavy proteinuria show fibrosis, inflammation, and oxidative stress, but no tubular phenotypic change. Kidney Int 2005;68:121–132. [DOI] [PubMed] [Google Scholar]
- 5.Vepsalainen T, Laakso M, Kantola I, et al. Proteinuria modifies the effect of systolic blood pressure on total and cardiovascular disease mortality in patients with type 2 diabetes. J Intern Med 2012;272:611–619. [DOI] [PubMed] [Google Scholar]
- 6.Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol 2006;17:3438–3446. [DOI] [PubMed] [Google Scholar]
- 7.Matsui I, Hamano T, Tomida K, et al. Active vitamin D and its analogue, 22-oxacalcitriol, ameliorate puromycin aminonucleoside-induced nephrosis in rats. Nephrol Dial Transplant 2009;24:2354–2361. [DOI] [PubMed] [Google Scholar]
- 8.Zou J, Yaoita E, Watanabe Y, et al. Upregulation of nestin, vimentin, and desmin in rat podocytes in response to injury. Virchows Arch 2006;448:485–492. [DOI] [PubMed] [Google Scholar]
- 9.Yasuda-Yamahara M, Kume S, Tagawa A, et al. Emerging role of podocyte autophagy in the progression of diabetic nephropathy. Autophagy 2015;11:2385–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, Kang YS, Dai C, et al. Epithelial-to-mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. Am J Pathol 2008;172:299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou LL, Hou FF, Wang GB, et al. Accumulation of advanced oxidation protein products induces podocyte apoptosis and deletion through NADPH-dependent mechanisms. Kidney Int 2009;76:1148–1160. [DOI] [PubMed] [Google Scholar]
- 12.Zhou LL, Cao W, Xie C, et al. The receptor of advanced glycation end products plays a central role in advanced oxidation protein products-induced podocyte apoptosis. Kidney Int 2012;82:759–770. [DOI] [PubMed] [Google Scholar]
- 13.Zhou L, Liu Y. Wnt/beta-catenin signalling and podocyte dysfunction in proteinuric kidney disease. Nat Rev Nephrol 2015;11:535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piwkowska A Role of protein kinase G and reactive oxygen species in the regulation of podocyte function in health and disease. J Cell Physiol 2017;232:691–697. [DOI] [PubMed] [Google Scholar]
- 15.Gorin Y, Wauquier F. Upstream regulators and downstream effectors of NADPH oxidases as novel therapeutic targets for diabetic kidney disease. Mol Cells 2015;38:285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan RJ, Zhou D, Xiao L, et al. Extracellular superoxide dismutase protects against proteinuric kidney disease. J Am Soc Nephrol 2015;26:2447–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma T, Zhu J, Chen X, et al. High glucose induces autophagy in podocytes. Exp Cell Res 2013;319:779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marshall CB, Pippin JW, Krofft RD, et al. Puromycin aminonucleoside induces oxidant-dependent DNA damage in podocytes in vitro and in vivo. Kidney Int 2006;70:1962–1973. [DOI] [PubMed] [Google Scholar]
- 19.Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol 2009;10:468–477. [DOI] [PubMed] [Google Scholar]
- 20.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell 2012;149:1192–1205. [DOI] [PubMed] [Google Scholar]
- 21.Zhou D, Tan RJ, Fu H, et al. Wnt/beta-catenin signaling in kidney injury and repair: a double-edged sword. Lab Invest 2016;96:156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou L, Li Y, Hao S, et al. Multiple genes of the renin-angiotensin system are novel targets of Wnt/beta-catenin signaling. J Am Soc Nephrol 2015;26:107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao L, Zhou D, Tan RJ, et al. Sustained Activation of Wnt/beta-Catenin Signaling Drives AKI to CKD Progression. J Am Soc Nephrol 2016;27:1727–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou L, Mo H, Miao J, et al. Klotho ameliorates kidney injury and fibrosis and normalizes blood pressure by targeting the renin-angiotensin system. Am J Pathol 2015;185:3211–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dai C, Stolz DB, Kiss LP, et al. Wnt/beta-catenin signaling promotes podocyte dysfunction and albuminuria. J Am Soc Nephrol 2009;20:1997–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cox SN, Sallustio F, Serino G, et al. Altered modulation of WNT-beta-catenin and PI3K/Akt pathways in IgA nephropathy. Kidney Int 2010;78:396–407. [DOI] [PubMed] [Google Scholar]
- 27.Wang XD, Huang XF, Yan QR, et al. Aberrant activation of the WNT/beta-catenin signaling pathway in lupus nephritis. PLoS One 2014;9:e84852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou L, Li Y, Zhou D, et al. Loss of Klotho contributes to kidney injury by derepression of Wnt/beta-catenin signaling. J Am Soc Nephrol 2013;24:771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Small DM, Coombes JS, Bennett N, et al. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology (Carlton) 17:311–321. [DOI] [PubMed] [Google Scholar]
- 30.Ramos LF, Shintani A, Ikizler TA, et al. Oxidative stress and inflammation are associated with adiposity in moderate to severe CKD. J Am Soc Nephrol 2008;19:593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agarwal R Proinflammatory effects of oxidative stress in chronic kidney disease: role of additional angiotensin II blockade. Am J Physiol Renal Physiol 2003;284:F863–869. [DOI] [PubMed] [Google Scholar]
- 32.Sachse A, Wolf G. Angiotensin II-induced reactive oxygen species and the kidney. J Am Soc Nephrol 2007;18:2439–2446. [DOI] [PubMed] [Google Scholar]
- 33.Gorin Y, Block K, Hernandez J, et al. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J Biol Chem 2005;280:39616–39626. [DOI] [PubMed] [Google Scholar]
- 34.Cao W, Hou FF, Nie J. AOPPs and the progression of kidney disease. Kidney Int Suppl 2014;4:102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grahammer F, Schell C, Huber TB. The podocyte slit diaphragm--from a thin grey line to a complex signalling hub. Nat Rev Nephrol 2013;9:587–598. [DOI] [PubMed] [Google Scholar]
- 36.Mathieson PW. The podocyte as a target for therapies--new and old. Nat Rev Nephrol 2011;8:52–56. [DOI] [PubMed] [Google Scholar]
- 37.Yamaguchi Y, Iwano M, Suzuki D, et al. Epithelial-mesenchymal transition as a potential explanation for podocyte depletion in diabetic nephropathy. Am J Kidney Dis 2009;54:653–664. [DOI] [PubMed] [Google Scholar]
- 38.Kang YS, Li Y, Dai C, et al. Inhibition of integrin-linked kinase blocks podocyte epithelial- mesenchymal transition and ameliorates proteinuria. Kidney Int 78:363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohtaka A, Ootaka T, Sato H, et al. Phenotypic change of glomerular podocytes in primary focal segmental glomerulosclerosis: developmental paradigm? Nephrol Dial Transplant 2002;17 Suppl 9:11–15. [DOI] [PubMed] [Google Scholar]
- 40.Lu TC, He JC, Klotman PE. Podocytes in HIV-associated nephropathy. Nephron Clin Pract 2007;106:c67–71. [DOI] [PubMed] [Google Scholar]
- 41.Heikkila E, Juhila J, Lassila M, et al. beta-Catenin mediates adriamycin-induced albuminuria and podocyte injury in adult mouse kidneys. Nephrol Dial Transplant 2010;25:2437–2446. [DOI] [PubMed] [Google Scholar]
- 42.Witko-Sarsat V, Friedlander M, Capeillere-Blandin C, et al. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int 1996;49:1304–1313. [DOI] [PubMed] [Google Scholar]
- 43.Wang J, Liang M, Xu J, et al. Renal expression of advanced oxidative protein products predicts progression of renal fibrosis in patients with IgA nephropathy. Lab Invest 2014;94:966–977. [DOI] [PubMed] [Google Scholar]
- 44.Zhou L, Li Y, He W, et al. Mutual antagonism of Wilms’ tumor 1 and beta-catenin dictates podocyte health and disease. J Am Soc Nephrol 2015;26:677–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hou FF, Miyata T, Boyce J, et al. beta(2)-Microglobulin modified with advanced glycation end products delays monocyte apoptosis. Kidney Int 2001;59:990–1002. [DOI] [PubMed] [Google Scholar]
- 46.Mo H, Wu Q, Miao J, et al. C-X-C chemokine receptor type 4 plays a crucial role in mediating oxidative stress-induced podocyte injury. Antioxid Redox Signal 2017;27:345–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. AOPPs induce RAGE expression in the kidney of mice. (a) Immunohistochemical staining show that AOPPs induced RAGE expressions in glomerular podocytes. Arrows indicate positive staining. Scale bar, 20 μm. (b-c) Western blotting shows that AOPPs induced RAGE protein expression. Graphic presentations of the relative abundances of RAGE (c) in two groups are shown. *P< 0.05 versus controls (n = 6).
Figure S2. AOPPs do not induce podocyte apoptosis. Representative miscrographs show that AOPPs did not cause podocyte apoptosis. Kidney sections were subjected to TUNEL staining. Arrows indicate TUNEL-positive cells in renal tubules. Scale bar, 50 μm.
Table S1. Demographic characteristics of the participants in the study.
Table S2. Parameter values of healthy subjects and CKD patients.