

To the Editor,
Dowling-Degos disease (DDD; MIM 179850, 615327, and 615696) is a rare autosomal-dominant hereditary pigmentation disorder. Affected individuals present with varying degrees of progressive reticulate hyperpigmentation, primarily affecting the flexures, large skin folds, trunk, face, and extremities. Genetic research has identified associations between DDD and mutations in KRT5 (Betz et al., 2006), POFUT1 (Li et al., 2013), POGLUT1 (Basmanav et al., 2014), and PSENEN (Ralser et al., 2017). Mutations in these genes, which are implicated in Notch signaling, underlie clinically and histologically distinct DDD subtypes (Basmanav et al., 2015, Betz, 2017, Ralser et al., 2017).
Following our identification of POGLUT1 mutations in DDD (Basmanav et al., 2014), we studied additional DDD patients. Ethical approval was obtained from the ethics committee of the Medical Faculty of the University of Bonn. All participants provided written informed consent prior to blood sampling. The study was conducted in accordance with Declaration of Helsinki principles. All participants were examined and sub-phenotyped by a dermatologist, as suggested previously (Basmanav et al., 2015, Betz, 2017). The predominant presentation was reticulate hyperpigmentation confined to the non-flexural areas of the body, in particular trunk and extremities (Figure 1a-h, Supplemental Table 2). All patients agreed to the publication of their photographs.
Figure 1. Clinical presentation and location of mutations in POGLUT1.

(a-c) German female with reticulate hyperpigmentation in the (a) infra- and intermammary regions, (b) on the dorsal aspects of the feet, and (c) upper extremities. (d-f) British male with hyperpigmented lesions on the (d) trunk and (e-f) lower extremities. Turkish male with hyperpigmentation and dark brown papules on the (g) trunk and (h) upper extremities. All patients agreed to the publication of their photographs. (i) Cartoon depicting the exon/intron-structure of POGLUT1. Mutations identified in this study are marked by arrow heads and are defined on the nucleotide and protein level.
Sanger sequencing revealed 12 to our knowledge previously unreported heterozygous mutations in POGLUT1: (i) five nonsense mutations, c.20C>A (p.Ser7*), c.138C>G (p.Tyr46*), c.1051C>T (p.Gln351*), c.628C>T (p.Arg210*), and c.130G>T (p.Glu44*); (ii) three frameshift mutations, c.121_121delA (p.Arg41Glyfs*21), c.292_292delC (p.R98Gfs*21) and c.1080_1081insG (p.Asn361Glufs*5); (iii) one splice site mutation, c.320+1G>C; and (iv) three missense mutations, c.983T>G (p.Val328Gly), c.836G>A (p.Arg279Gln), and c.653G>A (p.Arg218Gln) (Figure 1i, Supplemental Figure 1). Interestingly, five of these mutations are found in gnomAD with an allele frequency range of 1,321e−5 to 8,153e−6 (Supplemental Table 1). Still, it is conceivable that these mutations are pathogenic, due to i) the relatively late age of onset; and ii) the comparatively mild clinical phenotype. Therefore, DDD might be more common than reported to date.
The nonsense and frameshift mutations are likely to result either in nonsense-mediated mRNA decay or the formation of a truncated protein, as exemplarily demonstrated in Supplemental Figure 3a and b. To determine the structural consequences of the splice site mutation c.320+1G>C, exon trapping was used (Ralser et al., 2017). This revealed complete skipping of exon 3, resulting in disruption of the functionally important POGLUT1 N-terminal domain (Supplemental Figure 2a and b). To assess the consequences of two of the here described missense mutations and the previously reported mutation p.Arg279Trp (Basmanav et al., 2014), wild-type (WT) POGLUT1 and the mutants p.Arg279Gln, p.Val328Gly, and p.Arg279Trp were each fused to a C-terminal MycHis epitope and overexpressed in HEK293T cells. The enzymatic activities resulting from the WT and mutant POGLUT1 constructs were evaluated using an enzymatic assay, as described elsewhere (Takeuchi et al., 2012). Western Blot showed that both p.Arg279Gln and p.Arg279Trp resulted in a protein product whose secretion level is similar to that of WT POGLUT1, indicating that it is folded. However, this mutant protein was enzymatically dysfunctional (Figure 2a, Supplemental Figures 4-6). In contrast, the construct encoding p.Val328Gly was detected in cell lysates but not in media, suggesting that the mutant protein is unstable and eventually degraded (Supplemental Figures 4-6). To demonstrate that loss of function of the mutant p.Val328Gly protein did not result from the introduction of an additional mutation during PCR-mediated site-directed mutagenesis, a revertant was constructed. Secretion levels of the revertant and the WT were comparable (Supplemental Figures 4 and 6).
Figure 2. Analysis of the missense mutations p.Arg279Gln, p.Arg279Trp, p.Arg218Gln, and p.Val328Gly and knockdown experiments of POGLUT1 in MZ7-MEL cells.
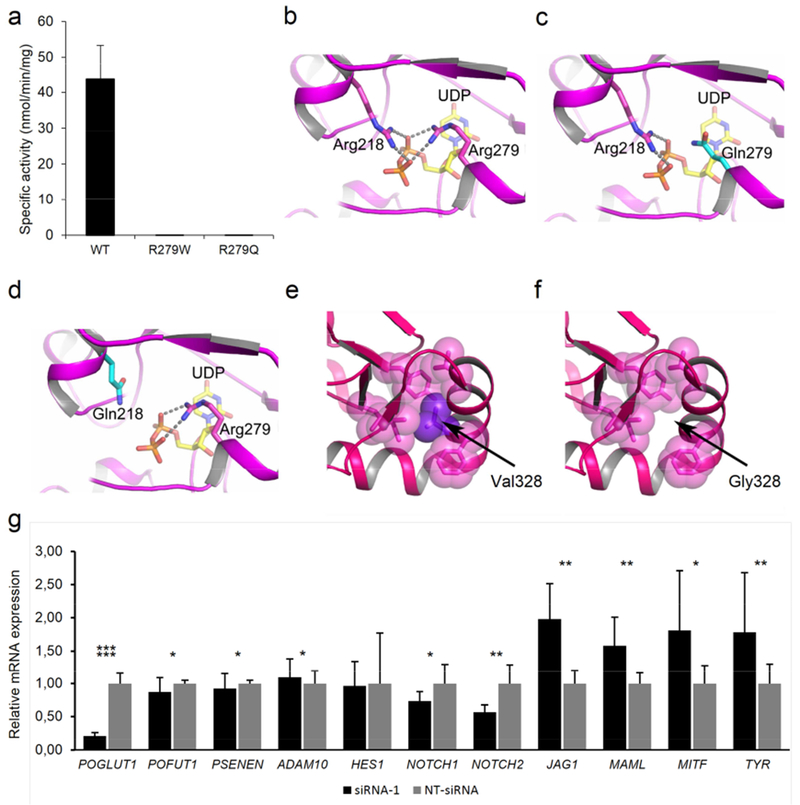
(a) Graph displaying the enzymatic activity of wild-type (WT), p.Arg279Gln, and p.Arg279Trp mutant POGLUT1 against human factor IX EGF repeat. (b-f) Protein modeling of WT and mutant POGLUT1. (b-d) The active site of POGLUT1 is shown. The residues Arg218 and Arg279 are depicted in stick representation with carbon atoms in magenta and nitrogen atoms in blue. A part of donor substrate, UDP-glucose is shown as a stick model as well, with carbon atoms in yellow, oxygen atoms in red and phosphate in orange. Both arginine residues are critical to the binding of UDP-glucose since they form salt bridges to the oxygen atoms of the UDP-glucose diphosphate moiety. The salt bridges are indicated by broken lines. Both the p.Arg218Gln and p.Arg279Gln mutation lead to substitution of a glutamine for an arginine residue (c and d, carbon atoms of Gln are depicted in cyan). Loss of the interaction between arginine and the phosphate group of UDP-glucose by substitution with glutamine will likely decrease the affinity for the substrate, and thereby the catalytic function of POGLUT1. In panel (e) the effect of mutation p.Val328Gly is modeled. The bulky sidechain of Val328 (purple) is located in a cluster of hydrophobic residues (shown in magenta as transparent spheres). (f) Mutation p.Val328Gly causes loss of this hydrophobic interaction and probably leads to the destabilization of the helix and, thus, the entire protein. (g) Analysis of siRNA-mediated POGLUT1 knockdown in MZ7-MEL cells. Relative mRNA expression levels of HES1, NOTCH1, NOTCH2, POGLUT1, POFUT1, PSENEN, ADAM10, JAG1, MAML, MITF, and TYR were evaluated by quantitative polymerase chain reaction. All experiments were carried out in triplicates. Expression levels were normalized against NT-siRNA.
These findings were supported by protein modeling based on the structures of human POGLUT1 co-crystallized with substrates (PDB-code: 5L0T) (Li et al., 2017) (Figure 2b-f). Arg279 is involved in the high-affinity binding of the donor substrate (Figure 2b). Substitution of glutamine for both, Arg218 and Arg279, respectively, likely decreases substrate affinity, thereby impairing POGLUT1 catalytic function (Figure 2c and d). Val328 is part of a hydrophobic cluster of POGLUT1 (Figure 2e), which stabilizes the C-terminal end of a helix formed by residues 320-331 (Li et al., 2017). The substitution of glycine for valine likely causes loss of the helix and destabilization of the entire protein (Figure 2f). Conceivably, this unstable POGLUT1 variant is prone to rapid degradation, as observed in our immunoblotting experiments (Supplemental Figures 4-6).
POGLUT1 is involved in the posttranslational modification of Notch proteins (Acar et al., 2008, Basmanav et al., 2014, Takeuchi et al., 2011). To gain further insights into the pathogenesis of DDD, POGLUT1 was down-regulated in the melanocyte-derived cell lineage MZ7-MEL using siRNA-mediated knockdown. Adequate POGLUT1 knockdown was confirmed by quantitative polymerase chain reaction (Figure 2g). An mRNA expression analysis of selected Notch pathway genes revealed altered Notch signaling (Figure 2g). This is analogous to findings from recent POFUT1 knockdown experiments in zebrafish larvae and HaCaT cells (Liet al., 2013), and PSENEN mutation carriers (Pavlovsky et al., 2017). These findings support the hypothesis that dysfunctional Notch signaling is a common pathomechanism in DDD (Frank et al., 2017).
Interestingly, mRNA expression of MITF, which encodes microphthalmia-associated transcription factor, was elevated in POGLUT1-deficient cells (Figure 2g). MITF is important for melanocyte development by regulating the expression of TYR, which encodes the enzyme tyrosinase. Tyrosinase is directly involved in melanin production by catalyzing the conversion of tyrosine to melanin (Hsiao and Fisher, 2014, Kawakami and Fisher, 2017, Yasumoto et al., 1995). The mRNA expression of TYR was also elevated in POGLUT1-deficient MZ7-MEL cells (Figure 2g). These data indicate a connection between altered Notch signaling and elevated levels of MITF and TYR, resulting in an increased production and misdirected deposition of melanin, as observed in patients with DDD. Detailed studies on these processes are needed.
In conclusion, our analyses demonstrate a gene-phenotype correlation in DDD patients with POGLUT1 mutations, enabling clinical sub-phenotyping, as suggested previously (Basmanav et al., 2015, Betz, 2017). The data presented here reveal 12 to our knowledge previously unreported POGLUT1 mutations in DDD, and evidence for the causal involvement of missense mutations. The association between altered Notch signaling and elevated mRNA levels of MITF and TYR, which are directly involved in melanogenesis, may be implicated in DDD hyperpigmentation.
Supplementary Material
Acknowledgments
We thank all patients for their participation. The study was supported by local funding (BONFOR to R.C.B. and D.J.R.) and NIH funding (GM061126 to R.S.H.). R.C.B. is a member of the DFG-funded Excellence Cluster ImmunoSensation, and is a past recipient of a Heisenberg-Professorship from the DFG (BE 2346/4-2).
We dedicate this manuscript to the memory of Prof. Dr. Martin Leverkus, who died while this work was in progress.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
None declared.
References
- Acar M, Jafar-Nejad H, Takeuchi H, Rajan A, Ibrani D, Rana NA, et al. Rumi is a CAP10 domain glycosyltransferase that modifies Notch and is required for Notch signaling. Cell 2008;132(2):247–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basmanav FB, Fritz G, Lestringant GG, Pachat D, Hoffjan S, Fischer J, et al. Pathogenicity of POFUT1 in Dowling-Degos disease: additional mutations and clinical overlap with reticulate acropigmentation of kitamura. J Invest Dermatol 2015;135:615–8. [DOI] [PubMed] [Google Scholar]
- Basmanav FB, Oprisoreanu AM, Pasternack SM, Thiele H, Fritz G, Wenzel J, et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomal-dominant Dowling-Degos disease. Am J Hum Genet 2014;94:135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz RC. A path through the reticulate pigmentation disorder jungle. Br J Dermatol 2017;177:893–4. [DOI] [PubMed] [Google Scholar]
- Betz RC, Planko L, Eigelshoven S, Hanneken S, Pasternack SM, Bussow H, et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet 2006;78:510–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J, Ralser DJ, Betz RC. Intra- and interfamilial phenotype variability associated with mutations in gamma-secretase subunit-encoding PSENEN. J Invest Dermatol 2017; doi: 10.1016/j.jid.2017.09.050. [DOI] [PubMed] [Google Scholar]
- Hsiao JJ, Fisher DE. The roles of microphthalmia-associated transcription factor and pigmentation in melanoma. Arch Biochem Biophys 2014;563:28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami A, Fisher DE. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab Invest 2017;97:649–56. [DOI] [PubMed] [Google Scholar]
- Li M, Cheng R, Liang J, Yan H, Zhang H, Yang L, et al. Mutations in POFUT1, encoding protein O-fucosyltransferase 1, cause generalized Dowling-Degos disease. Am J Hum Genet 2013;92:895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Fischer M, Satkunarajah M, Zhou D, Withers SG, Rini JM. Structural basis of Notch O-glucosylation and O-xylosylation by mammalian protein-O-glucosyltransferase 1 (POGLUT1). Nat commun 2017;8(1):185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlovsky M, Sarig O, Eskin-Schwartz M, Malchin N, Bochner R, Mohamad J, et al. A phenotype combining hidradenitis suppurativa with Dowling-Degos disease caused by a founder mutation in PSENEN. Br J Dermatol 2017;doi: 10.1111/bjd.16000. [DOI] [PubMed] [Google Scholar]
- Ralser DJ, Basmanav FB, Tafazzoli A, Wititsuwannakul J, Delker S, Danda S, et al. Mutations in gamma-secretase subunit-encoding PSENEN underlie Dowling-Degos disease associated with acne inversa. J Clin Invest 2017;3;127:1485–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Fernandez-Valdivia RC, Caswell DS, Nita-Lazar A, Rana NA, Garner TP, et al. Rumi functions as both a protein O-glucosyltransferase and a protein O-xylosyltransferase. Proc Natl Acad Sci U S A 2011;108(40):16600–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Kantharia J, Sethi MK, Bakker H, Haltiwanger RS. Site-specific O-glucosylation of the epidermal growth factor-like (EGF) repeats of notch: efficiency of glycosylation is affected by proper folding and amino acid sequence of individual EGF repeats. J Biol Chem 2012;287:33934–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasumoto K, Yokoyama K, Shibata K, Tomita Y, Shibahara S. Microphthalmia-associated transcription factor as a regulator for melanocyte-specific transcription of the human tyrosinase gene. Mol Cell Biol 1995; 15:1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.