

Abstract
Lyme disease is the most prevalent vector-borne disease in North America and continues to spread. The disease was first clinically described in the 1970s in Lyme, Connecticut, but the origins and history of spread of the Lyme disease bacteria, Borrelia burgdorferi sensu stricto, are unknown. To explore the evolutionary history of B. burgdorferi in North America, we collected ticks from across the United States and southern Canada from 1984 to 2013 and sequenced the largest ever collection of 146 B. burgdorferi s.s. genomes. Here, we show that B. burgdorferi s.s. has a complex evolutionary history with previously undocumented levels of migration. Diversity is ancient and geographically widespread, well predating the Lyme disease epidemic of the last ~40 years, as well as the Last Glacial Maximum ~20,000 years ago. This means the recent emergence of human Lyme disease likely reflects ecological change—climate change and land use changes over the last century—rather than evolutionary change of the bacterium.
Introduction
Lyme disease is the most prevalent vector-borne disease in North America and continues to expand across the northern United States and into southern Canada1. Lyme disease was first identified in a bizarre cluster of juvenile arthritis cases in Lyme, Connecticut in 19762. Quickly, it became clear that cases were not isolated to a single town nor to children and, by 1979, several hundred cases were reported in a cluster circling the Long Island Sound3. Since the 1970s, the disease has rapidly spread from disease foci in the Northeast and Midwest4, and to a lesser extent, in northern California. Now, over 30,000 cases of Lyme disease are reported each year and incidence is estimated to be ten times as high5.
The Lyme disease spirochete, Borrelia burgdorferi sensu stricto (s.s.), is maintained in an enzootic transmission cycle, in which the bacteria is transmitted between Ixodes tick vectors and a community of vertebrate reservoir hosts6. Humans are incidental hosts; we are susceptible to infection but do not contribute significantly to transmission nor spread. Despite its epidemiological importance, little is known about the evolutionary history of B. burgdorferi in North America. This limits our ability to track and predict the direction of ongoing spread and implement public health interventions.
Pathogen genomes can reveal epidemic histories. Phylogeography provides a powerful framework for inferring pathogen evolutionary dynamics including probable epidemic origins, rates and patterns of spread, and the distribution of highly virulent clades7–10. However, the evolutionary history of B. burgdorferi has not been explored using high resolution molecular markers such as whole genomes sampled from a broad spatial area. Sampling and sequencing B. burgdorferi genomes directly from ticks is challenging; bacterial DNA is swamped by tick and environmental DNA11 and culturing the bacteria introduces biases12. Only 15 genomes of B. burgdorferi s.s. existed previously (Table S1) and previous phylogeographic study has relied on single locus13,14 or MLST (multi-locus sequence typing) markers15–17.
Here, we use a population genomics approach to investigate outstanding questions about the processes shaping B. burgdorferi variation, patterns of gene flow across North America, and the timescale of bacterial evolution. (1) How is B. burgdorferi variation generated and maintained? Previous study of 13 B. burgdorferi s. s. genomes found frequent small-scale recombination18,19, but the relative importance of mutation to recombination in shaping population-level diversity is unknown. (2) Where did North American B. burgdorferi originate and how has the bacterium spread? A previous MLST-based study suggests that B. burgdorferi was historically introduced to the Midwest from the Northeast and the two regions are now isolated15. However, rates of gene flow of the bacteria between these two Lyme endemic regions and the relative role of different vertebrate hosts in bacterial dispersal is unknown. (3) Finally, how old is B. burgdorferi diversity? Museum specimens reveal the bacteria was present on Cape Cod, Massachusetts in the 1890s20 and Long Island, New York in the 1940s21, long before clinical recognition of the disease. Phylogeography suggests a deeper evolutionary history—ticks and B. burgdorferi may have survived the Last Glacial Maximum in the southern United States and spread across North America after glacial retreat, ~20,000 years ago13,15. Reconstructing the timescale of diversification will indicate if B. burgdorferi variation holds a signature of recent emergence of human Lyme disease in the last 40 years or if diversity reflects more ancient processes.
To address these three epidemiologically relevant aspects of the evolutionary history of B. burgdorferi in North America, we sampled and sequenced 146 B. burgdorferi genomes directly from tick vectors from across the northeastern and midwestern United States and from southern Canada, including samples from 1984. With this genomic collection and all previously published B. burgdorferi genomes, including samples from Europe and the western US (California), we reconstructed the evolutionary history of the Lyme disease bacteria in North America.
Results
Field sampling and variation.
We collected 146 B. burgdorferi-infected nymphal Ixodes scapularis ticks from across Lyme disease endemic areas in the Northeast and Midwest United States and southern Canada, spanning thirty years (1984–2013) and representing the widest spatial and temporal genomic collection of B. burgdorferi yet published (Fig. 1, Supplementary File 1). We used a hybrid capture method we previously developed11 to enrich for and efficiently sequence B. burgdorferi-derived reads from a mixed DNA template (~73 % capture efficiency), allowing us, for the first time, to screen B. burgdorferi directly from ticks. Our genomic collection allowed us to simultaneously study DNA from B. burgdorferi, the tick vector, and a co-vectored parasite, Babesia microti. To extend the geographic range of samples, we added all 15 published B. burgdorferi s. s. genomes(Table S1), including two samples from Europe and three from the western US (California), and used B. finlandensis22 as an outgroup.
Figure 1. Map of B. burgdorferi samples.
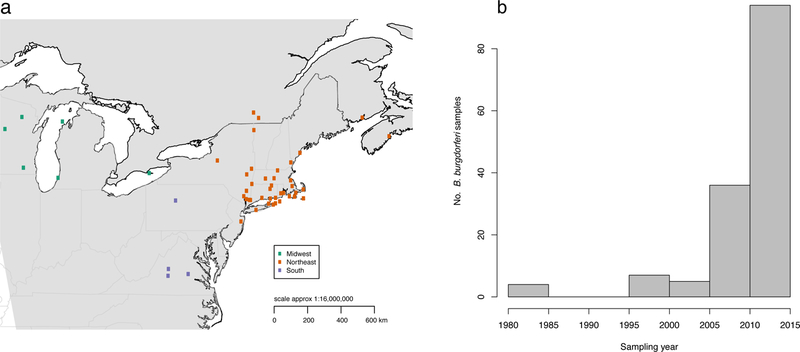
(a) Distribution of 146 Ixodes scapularis field-collected ticks. Samples are colored by major sampling region. (b) Distribution of samples collected each year.
We identified single nucleotide polymorphisms (SNPs) along the B. burgdorferi linear chromosome (910,724-bp) and the two best-characterized and most conserved plasmids23, cp26 (26,498-bp) and lp54 (53,657-bp) by mapping short reads to the B31 reference genome24 (variant filters in Methods). We identified 18,740; 959; and 1034 SNPs distributed along the chromosome, plasmid cp36, and plasmid lp54 respectively (Methods).
Recombination detection.
Both de novo mutation and recombination contribute to B. burgdorferi variation18. Early studies described B. burgdorferi as “clonal,”14 while recent studies identified small scale recombination25,18. However, the contribution of recombination to B. burgdorferi genomic variation has not been examined in a large population sample. We evaluated the contribution of both processes to simultaneously infer recombinant tracts and the underlying maximum likelihood phylogeny of the B. burgdorferi chromosome and plasmids cp26 and lp54 (Supplementary Figure 1).
Recombination is frequent along the chromosome (763 recombination events) and plasmids cp26 (68 recombination events) and lp54 (53 recombination events) (Supplementary Figure 1) and it has shaped the B. burgdorferi genome over its evolutionary history. For each branch of the chromosomal and plasmid trees, we determined the ratio of SNPs introduced via recombination to the number introduced via point mutations, ur/m, as well as the total number of recombination events per branch. The chromosome had a mean ur/m of 1.55 (SD=3.83), and had a mean of 2.63 recombination events per branch. Despite the recombination detected, 93.18% (846,801 of 910,724 bp) of the chromosome lies within a clonal frame, outside of predicted recombinant regions (Supplementary Figure 1a). The relative frequency of recombination to point mutation is lower on the plasmids than on the chromosome: ur/m of 0.70 (plasmid cp26) and 0.259 (plasmid lp54) (Figs. S1b, c).
Recombinant hotspots on the plasmids contain several known antigens. A recombination peak exists at the ospC (outer surface protein C) gene on plasmid cp26 (Supplementary Figure 1b). ospC is a major B. burgdorferi antigen required for transmission from tick to vertebrate host and dissemination within vertebrates26. ospC defines B. burgdorferi serotype: many ospC serotypes (alleles) circulate and vertebrates develop type-specific immunity protecting them from re-infection with the same ospC serotype27. ospC serotypes vary in virulence in humans: “disseminating” serotypes are associated with spread from the skin (the initial site of infection) through the bloodstream, causing more severe disease, while non-disseminating serotypes may remain as localized skin infections28. Recombinant tracts vary in length: some are tightly centered on ospC (632-bp), while others are up to 5000-bp long, including the neighboring guaA (GMP synthase) and guaB (dehydrogenase) genes essential for infectivity in vertebrate hosts29. A recombination hotspot on plasmid cp26 also includes resT, a gene involved in recombination (Supplementary Figure 1b).
Two sharp recombination peaks on plasmid lp54 correspond to two known B. burgdorferi antigens (Supplementary Figure 1c): dbpA, an adhesin that enables B. burgdorferi dissemination in vertebrate hosts30, and BB_A05, an antigen involved in transmission from ticks to mammals12. Recombinant tracts in dbpA also vary in size from 70-bp to ~8000-bp and frequently include the neighboring dbpB, another adhesin. A second recombination hotspot includes two other antigens that elicit human antibody responses: S1 (BB_A05), highly expressed in feeding nymphal ticks during transmission to mammals31; and S2 (BbuZS7_A03).
Phylogenetic structure.
To investigate B. burgdorferi population structure, we built a maximum likelihood phylogeny of the 16,370-SNP recombination-free alignment. The chromosomal phylogeny reveals a high degree of B. burgdorferi population structure divided into four major clades, which do not always reflect geography (Fig. 2a). Samples from the Northeast and Midwest are not monophyletic, in contrast to previous MLST-based analysis suggesting the two regions had a shared past and are now isolated15. Similarly, southern samples are not monophyletic and are distributed across the tree.
Figure 2. Reconstruction of B. burgdorferi dispersal across North America.
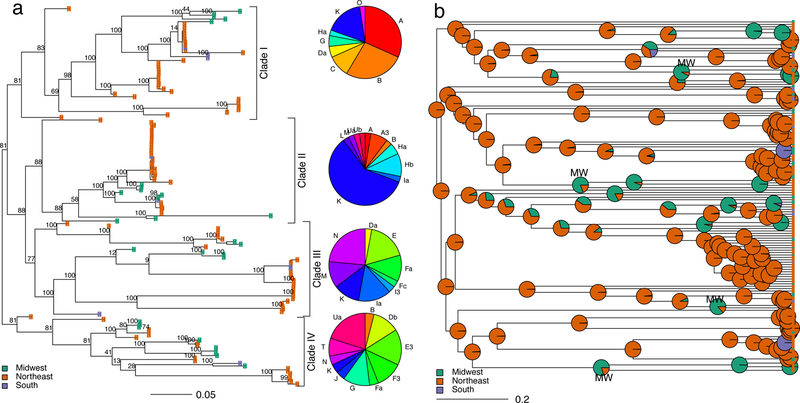
(a) Maximum likelihood, midpoint-rooted tree of B. burgdorferi inferred from a recombination-free alignment of 16,370 SNPs. Tips are colored by sampling region. Numbers at the branches represent bootstrap support. Branch lengths indicate the estimated number of substitutions per variable site. Pie charts depict the distribution of ospC serotypes within each clade. (b) Ancestral state reconstruction of B. burgdorferi geographic spread visualized on the BEAST maximum clade credibility tree. Pie charts at each internal node represent the likelihood the node was found in each geographic region. Evidence of ancient Midwestern ancestry (the oldest internal node in each clade with support for ancestral location in the Midwest) is labeled “MW.” The tree was forced to be ultrametric for ancestral state reconstruction; branch lengths represent the fraction of total root-to-tip distance. Posterior probability values shown in Figure 3.
To explore the population structure of B. burgdorferi at a broader spatial scale, we built a maximum likelihood phylogeny including all 15 previously published B. burgdorferi s.s. genomes in addition to the 146 samples collected for this study (Supplementary Figure 2). The three samples from the western US (California) fall in two distinct clades, indicating that western B. burgdorferi are not genetically isolated from midwestern and northeastern samples (Supplementary Figure 2). Although B. burgdorferi is maintained in a different ecological cycle in the western US, where it primarily cycles between Ixodes pacificus ticks and wood rats32, the bacteria do not appear to be genetically differentiated from other North American samples. The two European B. burgdorferi s.s. genomes are closely related to each other and monophyletic (Supplementary Figure 2, pink tips), nesting in a clade that contains midwestern samples. Previous MLST-based analysis suggested a European origin of all B. burgdorferi16. Although we cannot formally test this hypothesis because of the limited European sampling for this study, the placement of the two European samples within a clade including only midwestern samples suggests a more complex pattern of genetic differentiation than the one hypothesized with MLST-based analysis.
B. burgdorferi chromosomal and plasmid phylogenies are broadly similar, though there is evidence of several historic plasmid exchanges between lineages (Supplementary Figure 3).
To assess whether clades vary in virulence, we investigated their ospC serotype composition by serotyping samples in silico. ospC serotypes26 cluster imperfectly within the B. burgdorferi tree, reflecting recombination discussed above (Fig. 2a, Supplementary Figure 2). Virulent, disseminating ospC serotypes A, B, H, I and K occur within each phylogenetic cluster. However, the clades have different virulence profiles. Disseminating strains make up the majority of samples in Clade I (72.1 %, 95 % confidence interval (CI): 56.1 – 84.2 %) and Clade II (72.5 %, 95 % CI: 55.9 – 84.9 %) but only a minority of Clades III (27.8 %, 95 % CI: 14.8 – 45.4 %) and Clade IV (7.7 %, 95 % CI: 1.34 – 26.6 %) (Fig. 2a). Virulent serotypes are more common in the Northeast, where 35.8 % (95 % CI: 28.6 – 43.6 %) serotyped samples are disseminating compared to 15. 8 % (7.91 – 28.4 %) disseminating in the Midwest.
Discordant tick and pathogen phylogenies.
Although the efficiency of our hybrid capture protocol was high (~ 70 % of reads mapped to B. burgdorferi)11, the majority of remaining sequence data corresponded to the tick, I. scapularis, genome33 (Supplementary Figure 4).
Coverage of 3 tick mitochondrial genes (16S, cytochrome oxidase II, and the control region, Table S2) enabled us to examine potential co-evolution of the bacteria and its vector34 (Supplementary Figure 5), demonstrating the biological relevance of sequence by-catch, unintentionally captured DNA sequence. We constructed a maximum likelihood tree based on 118 concatenated SNPs identified in 3 tick mitochondrial genes, including 46 samples with no more than 50% missing data (Supplementary Figure 6). As observed for B. burgdorferi (Fig. 2a), ticks from the Northeast and Midwest are not monophyletic (Supplementary Figure 6), suggesting a recent shared history or frequent migration between these regions, consistent with an earlier, single locus analysis13. However, in contrast to this earlier work13, we find that southern ticks are not monophyletic. Tick and bacterial phylogenies are incongruent (Supplementary Figure 6).
In addition to capturing B. burgdorferi, we captured sequence derived from Babesia microti35, a co-emerging tick-borne parasite4. To test for evidence of shared evolutionary history of the two pathogens, we constructed a maximum likelihood tree based on the complete genome of the B. microti apicoplast (a small organelle found in most Apicoplast parasites), 28.7 kbp, and compared it to the B. burgdorferi phylogeny (Supplementary Figure 7). Despite our limited sample size of 9 co-infected ticks, due to the relatively low prevalence of B. microti, we found incongruence between the two parasite phylogenies.
Migration patterns.
We used ancestral state reconstruction to explore the geographic origins and gene flow patterns of North American B. burgdorferi (Fig. 2b).
The most recent common ancestor of all sampled B. burgdorferi most likely existed in the Northeast (posterior probability, 77.2 %) as did the most recent common ancestor of each B. burgdorferi clade (Clade I: 78.4 %, Clade II: 89.3 %, Clade III: 76.7 %, Clade IV: 77.6 %) (Fig. 2b). Our phylogeny reveals a pattern of gene flow more complex than the previously described unidirectional pattern from Northeast to Midwest 13,15. We uncover a signal of bidirectional gene flow between the three regions surveyed (Supplementary Figure 9). Though migration rates between regions are not statistically different, there is a clear hierarchy in estimated rates of gene flow between regions. Migration between the Northeast and Midwest is most frequent, followed by migration between the Northeast and South, and finally migration between the South and Midwest. The most likely ancestral location for interior nodes within Clades I, III, and IV (Fig. 2b, 3, nodes labeled “MW”) existed in the Midwest, revealing historic gene flow from the Northeast to the Midwest. This means that midwestern B. burgdorferi populations are not recently introduced from the Northeast and that genetically distinct B. burgdorferi populations from Clades I, II, and IV were historically endemic both in the Northeast and the Midwest (Fig. 2b). Though the northeastern US is currently the region with the highest density of tick vectors36, highest prevalence of infected ticks36, and the greatest epidemiological burden of Lyme disease37, complex patterns of historic gene flow across North America have shaped current patterns of diversity (Fig. 2b).
Figure 3. Dated phylogeny of B. burgdorferi in North America.
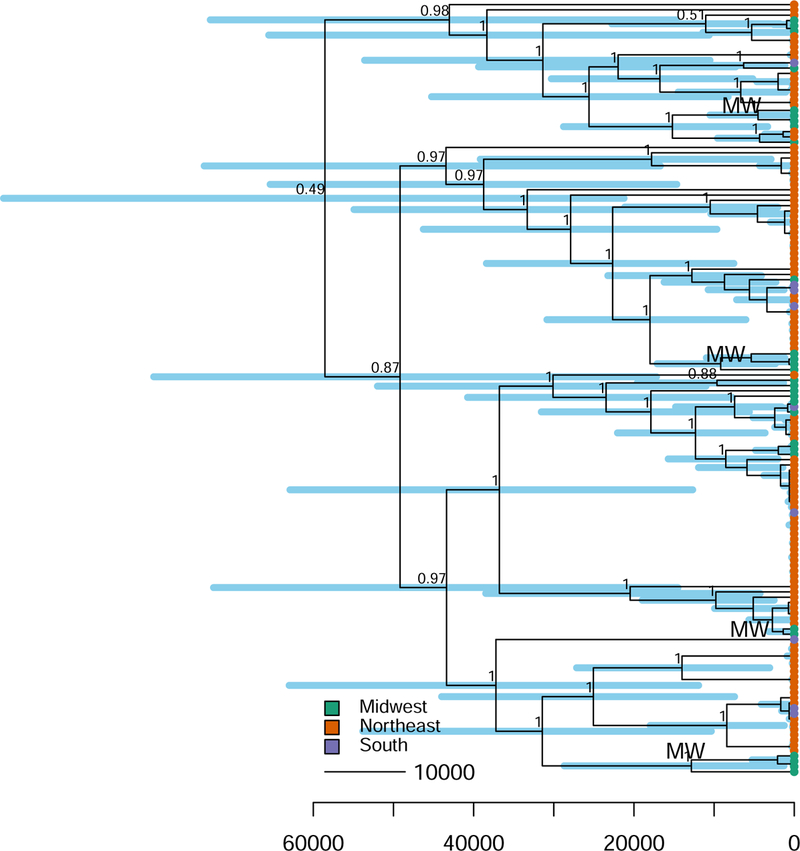
Maximum clade credibility tree of the best-fitting model inferred with BEAST (relaxed lognormal molecular clock model; Bayesian skyline population size). Tips are colored by sampling region. Evidence of ancient Midwestern ancestry (the oldest internal node in each clade with support for ancestral location in the Midwest) is labeled “MW.” Branch lengths are in years before the present. Blue bars at each interior node represent 95 % credible intervals of estimated node age. Branches are labeled with posterior probability values (not shown for shallow nodes for clarity).
Emergence timing.
Estimating the timing of the North American B. burgdorferi diversification requires knowledge of how fast the bacteria evolves. However, the substitution rate of this bacteria has not been measured directly, and available estimates vary widely from 10−9 to 10−6 substitutions per site per year15,38. In bacteria, where fossils are not available to calibrate node ages, serially sampled tips (i.e. samples collected at several time points) may enable estimation of substitution and diversification rates along the rest of the tree. This is possible if sufficient substitutions accumulate over the sampling period, within a measurably evolving population39,40. In order to maximize our sampling period, we included 4 ticks from 1984, the oldest available B. burgdorferi DNA in our dataset (Fig. 1b) and the oldest existing B. burgdorferi whole genomes besides the B31 reference strain cultured from a tick in 198224. We simultaneously estimated phylogenies, substitution rates, and demography, with a Bayesian approach implemented in BEAST41.
The observed substitution rate of 7.49 × 10−6 substitutions per site per year (95 % highest posterior density, HPD, 3.32 × 10−6 to 1.34 × 10−5; Table S3) is faster than previously estimated for B. burgdorferi15. This likely reflects the bacterium’s ecology. When B. burgdorferi is transmitted from tick to vertebrate host to tick, its population experiences severe bottlenecks, so that only a fraction of the variants transmit to the next host. Genetic drift in this severely bottlenecked bacterial population may increase the observed substitution rate42, though the within-host B. burgdorferi substitution rate is likely much slower.
We estimate the most recent common ancestor (MRCA) of the B. burgdorferi analyzed in this study existed ~60,000 years ago (95 % HPD 20,000 to 98,000) (Fig. 3, Supplementary Table 3). B. burgdorferi diversity is ancient and long predates not only the Lyme epidemic of the last ~40 years but also the last glacial maximum ~20,000 years ago13. Current Lyme disease foci are in regions once covered by the Pleistocene ice sheets13. Earlier studies suggest that the South was a refugia for many North American animals during the Last Glacial Maximum (~20,000 years ago) 13,43, and after glacial retreat, southern migrants colonized the Northeast and Midwest13,15. The ancient timescale of B. burgdorferi diversification in North America suggests that B. burgdorferi, in addition to ticks, was endemic in North America before the Pleistocene glaciation and its evolutionary history was shaped by ancient geological events.
Our estimates of substitution rate and tree age have substantial uncertainty (Fig. 3, Supplementary Table 3), as we do not have independent data to calibrate divergence time estimates, and the 30-year sampling interval which separate our samples is orders of magnitude shorter than the estimated tree age. However, even if B. burgdorferi substitution rates are slower than estimated in this study (i.e. ~ 10−7 - 10−8 substitutions per site per year), the age of the most recent common ancestor would still be quite old, dating to several hundred thousand or a million years old. Thus, our finding that B. burgdorferi diversity is ancient and predates the ongoing Lyme epidemic seems to be robust to uncertainty in the substitution rate estimate.
Demographic change.
B. burgdorferi population size expanded dramatically ~20,000 years ago and remained relatively stable thereafter (Supplementary Figure 10a), according to the skyline plot, a piecewise population model that allows effective population size to vary over time44, estimated in BEAST45. This signal may reflect population expansion from refugial populations in the South, after Pleistocene ice sheets retreated, allowing ticks and vertebrate hosts of B. burgdorferi to repopulate wide regions of northern North America13. This is consistent with previous studies reporting historic population size expansions in B. burgdorferi 13–15. However, this result must be interpreted with caution because the bacterial population is geographically structured both across space and between hosts, violating the coalescent model assumption of a single, panmitic population, which can potentially bias effective population size estimates.
To complement the skyline plot analyses, we used the mismatch distribution—the distribution of pairwise SNP differences between samples—to test for historic changes in B. burgdorferi population size (Methods). The mismatch distribution was unimodal (Supplementary Figure 10b) and consistent with past demographic or spatial expansion. The peak of the mismatch distribution (τ= 389) corresponds to the timing of historic population expansion, in units of mutational distance. Separate analyses of the mismatch distribution for samples from the Northeast, Midwest, and South reveal a similar pattern of historic population expansion in each region.
Discussion
Here, we reconstruct the evolutionary history of the Lyme disease bacteria in North America from the widest yet collection of B. burgdorferi genomes sequenced directly from field-collected ticks. By mining sequence by-catch and co-captured DNA in our genomic collection, we explore patterns of potential co-evolution with the tick vector and another co-infecting human parasite, B. microti.
We find that although B. burgdorferi is largely clonal, both recombination of short genomic tracts along the chromosome and plasmids (Supplementary Figure 1) and shuffling of entire plasmids (Supplementary Figure 3) shapes genomic diversity. We also found that recombination hotspots include the major B. burgdorferi antigens ospC, dbpA, and dbpB (Supplementary Figures1b, c). These surface-expressed antigens are likely experiencing balancing selection imposed by vertebrate host immune responses.
The complex phylogenetic structure of B. burgdorferi (Fig. 2a) together with the migration rate analyses (Fig. 2b) suggest previously undocumented levels of gene flow across North America. Since Ixodes ticks move little, B. burgdorferi spreads through the movement of its vertebrate hosts, small mammals and birds. In contrast to previous findings of strong barriers to gene flow between the Northeast and Midwest13,15, we find evidence of long-distance migration events between the three major geographic regions sampled (Fig. 2b), likely due to long-distance, bird-mediated dispersal. Local dispersal by small mammals likely also contributes to gene flow on much smaller spatial scales. One limitation to this study is that phylogenies are inferred from consensus bacterial sequences from each individual tick sample. The consensus B. burgdorferi sequence from a tick with a mixed infection46 often represents the majority strain infecting a tick (i.e. the strain comprising the largest proportion of infection). However, the consensus sequence may alternatively represent a chimeric sequence, a combination of segments of the multiple co-infecting haplotypes. With short-read sequence data used here, we are unable to reconstruct multiple bacterial haplotypes within a single sample. Longer-read sequencing will enable sequencing multiple bacterial genomes from individual vectors in the future.
We find no support for co-evolution of B. burgdorferi and its tick vector (Supplementary Figure 6). While captured sequence data enabled us to examine phylogenetic structure at only a few tick genes, we do not expect further analysis to reveal a strong pattern of co-emergence of the bacteria and its vector. This likely reflects the bacterium’s transmission cycle. In contrast to obligate parasites that co-diverge with their hosts, B. burgdorferi cycles between tick vectors and vertebrate hosts, decoupling tick and bacterial evolutionary histories. Our finding of a lack of association between tick and bacterial evolutionary histories suggests that invasion of ticks and bacteria is not coupled and may reflect distinct ecological processes. This is consistent with observations that B. burgdorferi invasion often lags behind tick invasion. This means that epidemiological surveillance should focus on areas with established I. scapularis tick populations that are potential sites for B. burgdorferi introduction.
Finally, we find B. burgdorferi diversity is ancient (~ 60,000 years old) (Fig. 3). Although there is substantial uncertainty in estimated divergence times, our phylogeny shows a clear signature of ancient diversification of B. burgdorferi that long predates the Lyme epidemic of the last ~40 years. Our reconstruction of the likely geographic origins of this diversity shows that it was geographically widespread across the Northeast and Midwest (Fig. 2b). The recent geographic and population size expansion (Supplementary Figure 10) of B. burgdorferi reflects the spread of diverse B. burgdorferi already present in each region (Figs. 2a, b). This means that the recent emergence of Lyme disease does not reflect the spread of a single epidemic B. burgdorferi lineage across North America nor recent diversification of the bacteria, but rather the spread of pre-existing, geographically widespread bacterial diversity.
Our finding of ancient B. burgdorferi diversification suggests that the recent Lyme disease epidemic does not reflect evolutionary processes but rather was driven by the ecological change in North America beginning in the colonial period ~700 years ago. Deforestation and intensive hunting47 during the colonial period followed by population explosion of white-tailed deer 47,48 and climate change49,50 in the last century likely enabled dramatic range expansion of Ixodes spp. ticks49. The spread of ticks into environments with high densities of competent vertebrate hosts substantially widened the potential geographic range of B. burgdorferi and of Lyme disease. Ticks and B. burgdorferi continue to spread into southern Canada and across the United States, putting a greater population at risk of Lyme disease.
Although the evolutionary history of B. burgdorferi occurs at a deeper timescale than the recent Lyme disease epidemic, our phylogeographic analysis provides insights into the epidemiology of Lyme disease. The high levels of B. burgdorferi genomic diversity found across North America and continued long-distance gene flow suggests that humans may be exposed to diverse bacterial lineages regardless of where they are infected. B. burgdorferi strains vary substantially in virulence and we find that disseminating strains associated with more severe disease are found in all regions surveyed and can occur on diverse genomic backgrounds (Fig. 2a). The patterns of migration uncovered here suggest continued spread of diverse B. burgdorferi not only from the Northeast across the rest of North America but also in several other directions (Fig. 2b), with gene flow occurring not only locally but also at a continental scale (Supplementary Figure 2). This finding has important epidemiological consequences as it suggests that wide regions with established tick and vertebrate host populations are potential sites of B. burgdorferi invasion and future Lyme disease.
Methods
Bacterial sampling and sequencing.
We collected 146 B. burgdorferi-infected nymphal Ixodes scapularis ticks from the widest available spatial and temporal range (i.e. including ticks sampled from 1984–2013) (Fig. 1, Supplementary File 1). Tick sampling, DNA extractions, and qPCR testing for B. burgdorferi infection followed described protocols11,51. Genomic libraries were prepared from infected tick samples. B. burgdorferi DNA was captured using a custom hybridization capture array method1. Sequencing of 75-bp paired end reads was conducted on an Illumina HiSeq 2500 at the Yale Center for Genomic Analysis. Short-read sequence data were submitted to the NCBI Short Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/), SRA accession: SRP058536 (Supplementary File 1).
Additional genomes.
We conducted analyses with samples collected for this study and added to them all 15 previously published B. burgdorferi genomes, representing cultured isolates from ticks, humans, and a song sparrow (Supplementary Table 1)24,22,52. We included B. finlandensis as an outgroup22.
Read alignment and SNP detection.
We focused analysis on single nucleotide polymorphisms (SNPs) on the B. burgdorferi linear chromosome (910724-bp) and the two best-characterized and most conserved plasmids, lp54 (53657-bp) and cp26 (26498-bp) 53,54. This represents 65% of the total B3124,52 reference genome.
Raw sequence reads for each sample were aligned to the B. burgdorferi reference genome strain B3124,52, using BWA mem (v. 0.7.7) 55. Duplicate sequence reads were excluded from downstream analysis, using the Picard Suite (v. 1.117) MarkDuplicates (http://picard.sourceforge.net). Only samples with mean coverage > 10X were retained for analysis. Variants with respect to strain B3124,52 were identified with GATK HaplotypeCaller56 (ploidy set to 1), generating sample-specific gVCF files. We conducted joint genotyping of samples with GATK GenotypeGVCFs. We excluded indels (insertions and deletions) and SNPs with signals of low mapping or genotyping quality with GATK VariantFiltration, using the following filters recommended by GATK57: quality by depth (QD < 2.0), Fisher strand bias (FS>60.0), mapping quality (MQ < 40.0), mapping quality rank sum test (MQRankSum < −12.5), the Mann-Whitney Rank Sum Test (ReadPosRankSum < −8.0), low genotype call (GQ < 20), and strand odds ratio test (SOR > 4.0), and set filtered genotypes to no call (GATK option --setFilteredGtToNocall).
To create an alignment of consensus sequences from the variant file, we used the GATK tool FastaAlternateReferenceMaker. This resulted in multiple alignments of consensus sequences of the chromosome (910724-bp), plasmid cp26 (26498-bp), and plasmid lp54 (53657-bp).
Phylogenetic analysis.
It is critical to account for recombination when reconstructing bacterial evolutionary histories because horizontal gene transfer can overwhelm the evolutionary signal from vertically inherited mutations and skew inferred phylogenies58. Therefore, we used GUBBINS59 to infer recombinant tracts along the chromosome in addition to plasmids cp26 and lp54. We used RAxML60 to construct maximum-likelihood phylogenetic trees from the 16,370-bp recombination-free SNP alignment of all 146 isolates. We used the generalized time-reversible model and a gamma distribution to model site-specific rate variation (the GTR + Γ substitution model) and corrected for using only variant sites in the tree, using the Lewis ascertainment bias correction61. Support for each branch was assessed using 100 bootstrapped samples of the multiple alignment. Recombination hotspots and RAxML phylogenies are visualized in Supplmentary Figure 1 with Phandango (http://jameshadfield.github.io/phandango/).
To infer rates of gene flow between geographic regions, we performed discrete ancestral state reconstruction62,63, coding the region of origin of tick samples as a discrete trait. We fit a Markov model of character state evolution63 to the underlying BEAST phylogeny with the diversitree package62 in R and found that posterior probability distributions of migration rates between regions overlapped significantly (Supplementary Figure 8). Therefore, we fit an equal rates model that considered a single long-distance migration rate between regions in phytools and simulated 1000 stochastic character maps, or reconstructions of ancestral geographic location, on our phylogeny with the phytools64 function make.simmap (Fig. 2b).
We identified ospC serotypes in silico with srst265. Trees were visualized in R with the packages phytools and ape64,66 in R.
Sequence by-catch and co-capture.
Although our hybrid capture approach was efficient and the majority of sequence reads corresponded to B. burgdorferi, ~30% of reads did not map to B. burgdorferi and constitute metagenomic data from our tick samples. We tested whether by-catch—unintentionally captured sequence data—corresponded to the tick vector by mapping sequence reads to the tick, Ixodes scapularis, genome33 with BWA mem (v. 0.7.7) 55, as described above. To investigate whether by-catch sequence contained sufficient coverage of I. scapularis genes for phylogenetic inference, we mapped reads to 10 tick genes previously used for population genetic study (Table S2). Genbank Accession numbers of the I. scapularis haplotypes used as references for mapping are in Table S2.
Three tick mitochondrial genes (16S, cytochrome oxidase II, and the control region) had a mean coverage > 1.5 X (Supplementary Figure 5, Table S2), enabling us to use them in phylogenetic analyses to examine potential co-evolution of the bacteria and its vector34.We called variants on the three mitochondrial genes and used RAxML to infer a maximum likelihood tree, using the methods described above and including all tick samples with < 40% missing data.
As we simultaneously co-captured both B. burgdorferi and the co-vectored pathogen Babesia microti35, we were able to explore patterns of potential co-evolution of the two co-vectored pathogens. Read mapping, variant calling, and tree building of the B. microti apicoplast, a small organelle found in most Apicoplast parasites, (28.7 kbp) was conducted as previously described35.
We tested for evidence of co-evolution of B. burgdorferi and both I. scapularis and B. microti with a global test of co-evolution implemented in ParaFit67. The two null hypotheses that B. burgdorferi and its tick vector and that B. burgdorferi and B. microti have independent evolutionary histories were tested by permuting the observed associations of parasite-vector or parasite-parasite 999 times.
Phylogeographic and demographic analysis.
We used a coalescent approach implemented in BEAST 241,45 to simultaneously explore the phylogenetic and demographic history of sampled B. burgdorferi. Recombination-free SNP alignments were used to fit four alternate models including either a strict molecular clock or a relaxed (uncorrelated lognormal) molecular clock68 (allowing substitution rates to vary across tree branches) and a constant population size or a Bayesian skyline model of changing population size. We used dates of tick collection to calibrate tips. We used an ascertainment bias correction to account for the invariant sites not included in the SNP alignment. All phylogeographic models used the HKY substitution model, allowing for gamma-distributed rate variation across sites.
For each model, we ran chains of 100 million iterations or until convergence. Chains were thinned by sampling every 1000 iterations and 10% of each chain was discarded as burn-in. Maximum-clade credibility (MCC) trees were generated using TreeAnnotator41,45. Tracer v. 1.6 was used to assess convergence visually and confirm effective sample sizes were greater than 200 for each parameter. To test the influence of prior distributions specified for evolutionary and demographic model parameters, we reran BEAST in triplicate for each model with no input alignment, sampling only from prior distributions and compared results to the posterior parameter estimates of fitted models (Supplementary Figure 11).
To identify the best fitting model, we used path sampling69 to estimate the marginal likelihood of each model. We compared alternative models with Bayes Factors and identified the relaxed log-normal clock, skyline population size as the best-fitting model for the recombination-free chromosomal SNP alignment (Bayes factors > 80, Supplementary Table 3), used in all above analyses. All four coalescent models estimated highly congruent tree topologies and differed only in placement of deep branches, expected because of the high degree of uncertainty at deeper timescales within the B. burgdorferi tree.
To speed computational time, we performed BEAST analyses on randomly sampled 5000-bp subsets of the full 16,370-bp SNP alignment. We resampled and reran analyses 10 times for the best-fitting model and compared maximum clade credibility (MCC) trees to confirm that subsampling the alignment did not distort phylogenetic inference. The MCC tree depicted in Figures 2b and 3 was inferred from a single 5000-bp subset of the SNP alignment.
To investigate B. burgdorferi demographic history, we complemented the coalescent based approach with mismatch distribution analysis, as implemented in DnaSP v570 and tested if our observed data were consistent with a mismatch distribution expected from a sudden population expansion or a spatial expansion71,72. The mismatch distribution and expected mismatch distribution for a stable population was visualized in the pegas package73 in R.
Supplementary Material
Acknowledgements.
This study was supported by the National Institutes of Health Ecology and Evolution of Infectious Disease Program grant R01 GM105246 and by NIH grant R21AI112938. KSW was supported by the NIH Ruth L. Kirschstein National Research Service Award (F31 AI118233–01A1) and the NSF Doctoral Dissertation Improvement Grant (DEB-1401143). GC was supported by the Gaylord Donnelley Postdoctoral Environmental Fellowship (the Yale Institute for Biospheric Studies). The authors would like to thank Nicholas Ogden, Jory Brinkerhoff, Susan Paskewitz, Durland Fish, and Steven Bent for providing tick samples; Peter Flynn and James Underwood for laboratory work; Choukri Ben Mamoun and Peter Krause for discussions; and Robert Bjornson and the Yale High Performance Computing Center for computational support.
Footnotes
Data availability. Short-read sequence data were submitted to the NCBI Short Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/), SRA accession: SRP058536 (Supplementary File 1).
Competing interests
The authors declare no competing financial interests.
References
- 1.Steere A, Coburn J & Glickstein L The emergence of Lyme disease. J. Clin. Invest 113, 1093–1101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steere ACC, Malawista SE, Snydman DR & Andiman WA A cluster of arthritis in children and adults in Lyme, Connecticut. Arthritis Rheum 19, 824 (1976). [DOI] [PubMed] [Google Scholar]
- 3.Steere AC & Malawista SE Cases of Lyme disease in the United States: locations correlated with distribution of Ixodes dammini. Ann. Intern. Med 91, 730–3 (1979). [DOI] [PubMed] [Google Scholar]
- 4.Walter KS et al. Invasion of two tick-borne diseases across New England: harnessing human surveillance data to capture underlying ecological invasion processes. Proc. Biol. Sci 283, S301–S327 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention. How many people get Lyme disease? (2015). [Google Scholar]
- 6.Spielman A & Wilson M Ecology of Ixodes Dammini-Borne Human Babesiosis and Lyme disease. Annu. Rev. Entomol 30, 439–460 (1985). [DOI] [PubMed] [Google Scholar]
- 7.Worobey M et al. Direct evidence of extensive diversity of HIV-1 in Kinshasa by 1960. Nature 455, 661–664 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris SR et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science (80-. ) 327, 469–74 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gray RR et al. Testing spatiotemporal hypothesis of bacterial evolution using methicillin-resistant Staphylococcus aureus ST239 genome-wide data within a bayesian framework. Mol. Biol. Evol 28, 1593–603 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Biek R et al. Whole Genome Sequencing Reveals Local Transmission Patterns of Mycobacterium bovis in Sympatric Cattle and Badger Populations. PLoS Pathog 8, e1003008 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carpi G et al. Whole genome capture of vector-borne pathogens from mixed DNA samples: a case study of Borrelia burgdorferi. BMC Genomics 16, 434 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu G et al. Detection of heterogeneity of Borrelia burgdorferi in Ixodes ticks by culture-dependent and culture-independent methods. J. Clin. Microbiol 51, 615–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Humphrey PT, Caporale DA & Brisson D Uncoordinated phylogeography of Borrelia burgdorferi and its tick vector, Ixodes scapularis. Evolution (N. Y) 64, 2653–63 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qiu W-G, Dykhuizen DE, Acosta MS & Luft BJ Geographic uniformity of the Lyme disease spirochete (Borrelia burgdorferi) and its shared history with tick vector (Ixodes scapularis) in the Northeastern United States. Genetics 160, 833–49 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoen AGA et al. Phylogeography of Borrelia burgdorferi in the eastern United States reflects multiple independent Lyme disease emergence events. Proc. Natl. Acad. Sci. U. S. A 106, 15013–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Margos G et al. MLST of housekeeping genes captures geographic population structure and suggests a European origin of Borrelia burgdorferi. Proc. Natl. Acad. Sci. U. S. A 105, 8730–8735 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mechai S, Margos G, Feil EJ, Lindsay LR & Ogden NH Complex Population Structure of Borrelia burgdorferi in Southeastern and South Central Canada as Revealed by Phylogeographic Analysis. Appl. Environ. Microbiol 81, 1309–1318 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haven J et al. Pervasive recombination and sympatric genome diversification driven by frequency-dependent selection in Borrelia burgdorferi, the Lyme disease bacterium. Genetics 189, 951–66 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mongodin EEF et al. Inter- and intra-specific pan-genomes of Borrelia burgdorferi sensu lato: genome stability and adaptive radiation. BMC Genomics 14, 693 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marshall WF et al. Detection of Borrelia burgdorferi DNA in museum specimens of Peromyscus leucopus. J. Infect. Dis 170, 1027–32 (1994). [DOI] [PubMed] [Google Scholar]
- 21.Persing DH et al. Detection of Borrelia burgdorferi DNA in museum specimens of Ixodes dammini ticks. Science (80-. ) 249, 1420–3 (1990). [DOI] [PubMed] [Google Scholar]
- 22.Casjens SR et al. Whole genome sequence of an unusual Borrelia burgdorferi sensu lato isolate. J. Bacteriol 193, 1489–90 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casjens SR et al. Plasmid diversity and phylogenetic consistency in the Lyme disease agent Borrelia burgdorferi. BMC Genomics 18, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fraser CM et al. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature 390, 580–6 (1997). [DOI] [PubMed] [Google Scholar]
- 25.Qiu W-G et al. Genetic exchange and plasmid transfers in Borrelia burgdorferi sensu stricto revealed by three-way genome comparisons and multilocus sequence typing. Proc. Natl. Acad. Sci. U. S. A 101, 14150–5 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwan TG, Piesman J, Golde WT, Dolan MC & Rosa PA Induction of an outer surface protein on Borrelia burgdorferi during tick feeding. Proc. Natl. Acad. Sci. U. S. A 92, 2909–13 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barbour AAG & Travinsky B Evolution and distribution of the ospC gene, a transferable serotype determinant of Borrelia burgdorferi. MBio 1, e00153–10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wormser GP et al. Borrelia burgdorferi genotype predicts the capacity for hematogenous dissemination during early Lyme disease. J. Infect. Dis 198, 1358–64 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jewett MW et al. GuaA and GuaB are essential for Borrelia burgdorferi survival in the tick-mouse infection cycle. J. Bacteriol 191, 6231–6241 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi Y, Xu Q, McShan K & Fang TL Both decorin-binding proteins A and B are critical for the overall virulence of Borrelia burgdorferi. Infect. Immun 76, 1239–1246 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu H et al. Characterization of the highly regulated antigen BBA05 in the enzootic cycle of Borrelia burgdorferi. Infect. Immun 78, 100–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown RN & Lane RS Lyme disease in California: a novel enzootic transmission cycle of Borrelia burgdorferi. Science 256, 1439–42 (1992). [DOI] [PubMed] [Google Scholar]
- 33.Gulia-Nuss M et al. Genomic insights into the Ixodes scapularis tick vector of Lyme disease. Nat. Commun 7, 10507 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khatchikian CE et al. Recent and rapid population growth and range expansion of the Lyme disease tick vector, Ixodes scapularis, in North America. Evolution (N. Y) 69, 1678–1689 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carpi G et al. Babesia microti from humans and ticks hold a genomic signature of strong population structure in the United States. BMC Genomics 17, 888 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diuk-Wasser MA et al. Field and climate-based model for predicting the density of host-seeking nymphal Ixodes scapularis, an important vector of tick-borne disease agents in the eastern United States. Glob. Ecol. Biogeogr 19, 504–514 (2010). [Google Scholar]
- 37.Kugeler KJ, Farley GM, Forrester JD & Mead PS Geographic Distribution and Expansion of Human Lyme Disease, United States. Emerg. Infect. Dis 21, 1455–1457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qiu W-G et al. Wide distribution of a high-virulence Borrelia burgdorferi clone in Europe and North America. Emerg. Infect. Dis 14, 1097–104 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Biek R, Pybus OG, Lloyd-Smith JO & Didelot X Measurably evolving pathogens in the genomic era. Trends Ecol. Evol 30, 306–313 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Drummond A, Pybus OG & Rambaut A Inference of viral evolutionary rates from molecular sequences. Adv. Parasitol 54, 331–58 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Bouckaert R et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol 10, e1003537 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Funk DJ, Wernegreen JJ & Moran NA Intraspecific Variation in Symbiont Genomes: Bottlenecks and the Aphid-Buchnera Association. Genetics 157, (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hewitt G The genetic legacy of the Quaternary ice ages. Nature 405, 907–913 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Drummond AJ, Rambaut A, Shapiro B & Pybus OG Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol 22, 1185–92 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Drummond AJ & Rambaut A BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol 7, 214 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walter KS et al. Vectors as Epidemiological Sentinels: Patterns of Within-Tick Borrelia burgdorferi Diversity. PLOS Pathog 12, e1005759 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cronon W Changes in the Land: Indians, Colonists, and the Ecology of New England (Hill and Wnag, 1983). [Google Scholar]
- 48.Matuschka F & Spielman A The emergence of Lyme disease in a changing environment in North America and central Europe. Exp. Appl. Acarol 2, 337–353 (1986). [DOI] [PubMed] [Google Scholar]
- 49.Eisen RJ, Eisen L & Beard CB County-Scale Distribution of Ixodes scapularis and Ixodes pacificus (Acari: Ixodidae) in the Continental United States. J. Med. Entomol tjv237 (2016). doi: 10.1093/jme/tjv237 [DOI] [PMC free article] [PubMed]
- 50.Ogden NH et al. A dynamic population model to investigate effects of climate on geographic range and seasonality of the tick Ixodes scapularis. Int. J. Parasitol 35, 375–89 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Walter KS, Carpi G, Evans BR, Caccone A & Diuk-Wasser MA Vectors as Epidemiological Sentinels: Patterns of Within-Tick Borrelia burgdorferi Diversity. PLOS Pathog 12, 868–878 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schutzer SE et al. Whole-genome sequences of thirteen isolates of Borrelia burgdorferi. J. Bacteriol 193, 1018–20 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Casjens S et al. A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol. Microbiol 35, 490–516 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Casjens SR et al. Genome stability of Lyme disease spirochetes: comparative genomics of Borrelia burgdorferi plasmids. PLoS One 7, e33280 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H & Durbin R Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–60 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McKenna A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.DePristo MA et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feil E et al. Recombination within natural populations of pathogenic bacteria: short-term empirical estimates and long-term phylogenetic consequences. Proc. R. Soc. B (2001). [DOI] [PMC free article] [PubMed]
- 59.Croucher NJ et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res gku1196- (2014). doi: 10.1093/nar/gku1196 [DOI] [PMC free article] [PubMed]
- 60.Stamatakis A RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lewis PO A Likelihood Approach to Estimating Phylogeny from Discrete Morphological Character Data. Syst. Biol 50, 913–925 (2001). [DOI] [PubMed] [Google Scholar]
- 62.FitzJohn RG Diversitree : comparative phylogenetic analyses of diversification in R. Methods Ecol. Evol 3, 1084–1092 (2012). [Google Scholar]
- 63.Pagel M Detecting Correlated Evolution on Phylogenies: A General Method for the Comparative Analysis of Discrete Characters. Proc. R. Soc. London B Biol. Sci 255, (1994). [Google Scholar]
- 64.Revell LJ phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol 3, 217–223 (2012). [Google Scholar]
- 65.Inouye M, Dashnow H, Raven L & Schultz M SRST2 : Rapid genomic surveillance for public health and hospital microbiology labs. bioRxiv 0–16 (2014). [DOI] [PMC free article] [PubMed]
- 66.Paradis E, Claude J & Strimmer K APE: Analyses of Phylogenetics and Evolution in R language. Bioinforma. Appl. NOTE 20, 289–290 (2004). [DOI] [PubMed] [Google Scholar]
- 67.Legendre P, Desdevises Y & Bazin E A Statistical Test for Host–Parasite Coevolution. Syst. Biol 51, 217–234 (2002). [DOI] [PubMed] [Google Scholar]
- 68.Lemey P, Rambaut A, Welch JJ & Suchard MA Phylogeography takes a relaxed random walk in continuous space and time. Mol. Biol. Evol 27, 1877–85 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baele G et al. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol 29, 2157–67 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Librado P & Rozas J DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009). [DOI] [PubMed] [Google Scholar]
- 71.Rogers AR & Harpending H Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol 9, 552–69 (1992). [DOI] [PubMed] [Google Scholar]
- 72.Slatkin M & Hudson RR Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129, 555–62 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Paradis E pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics 26, 419–420 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.