

Abstract
Background.
MOG antibody disease is an autoimmune disease of the central nervous system (CNS) characterized by the presence of a serological antibody against myelin oligodendrocyte glycoprotein (MOG). MRI is instrumental in distinguishing neuromyelitis optica spectrum disorder (NMOSD) from multiple sclerosis (MS), but MRI features of MOG disease appear to overlap with NMOSD and MS.
Objectives:
In this study we aim to characterize the radiological features of MOG antibody disease and compare the findings with those previously described.
Methods.
This is a retrospective study of 26 MOG positive patients. We aim to describe their brain, spinal and orbital MRI features and compare our findings with those previously reported in the literature.
Results.
The majority of the abnormal findings was located on orbital MRIs, with more involvement of the anterior structures and bilateral involvement of the optic nerves. Brain abnormalities were distinct from both NMOSD and MS lesions. Spinal cord was the least affected.
Conclusions.
This is a dedicated radiological study aiming to characterize the features of MOG antibody disease which might aid in the proper investigation of cases presenting with acquired demyelinating disorders.
Keywords: MOG antibody, NMOSD, magnetic resonance imaging
Introduction.
MOG antibody disease is an autoimmune disease of the central nervous system (CNS) characterized by the presence of a serological antibody against myelin oligodendrocyte glycoprotein (MOG), in the context of relapsing optic neuritis, neuromyelitis optica spectrum disorder (NMOSD), or acute disseminated encephalomyelitis (ADEM). The MOG antibody is detectable in up to 42% of NMOSD patients who test negative for the AQP4 antibody (1, 2). Although initially reported as a monophasic condition (3, 4), MOG antibody portends a relapsing course in 50–80% of cases (5). In addition to distinct immunological target, MOG antibody disease is different from related autoimmune CNS diseases in its clinical course, radiological presentation and treatment responsiveness (6).
MRI is instrumental in distinguishing NMOSD from multiple sclerosis (MS), but MRI features of MOG disease appear to overlap with NMOSD and MS (2, 7). Longitudinally extensive optic nerve involvement is common in both MOG antibody disease and aquaporin-4 (AQP4) NMOSD (8, 9), but posterior and chiasmal involvement appears to be more unique to AQP4 NMOSD (9). Despite the fact that 50% of MOG patients relapse with transverse myelitis (4, 10, 11), they are less likely to experience cord necrosis or atrophy as a sequelae, relative to AQP4 patients (12–14). Moreover, conus medullaris is a frequently involved location in MOG patients compared to cervical and thoracic involvement in NMOSD (12). Similar to MS, focal myelitis is a more common presentation in MOG antibody disease (10). Resolution of brain and spinal cord lesions in MOG positive patients as opposed to their AQP4 positive peers, was also a feature noted in many studies (4, 15). Recently, seizures with or without encephalopathy and cortical MRI changes has become a feature more frequently recognized among MOG seropositive patients (16, 17).
Age at disease onset could impact the radiological picture among MOG seropositive pediatric patients presenting with different demyelinating diseases (18). Younger MOG patients tend to present with an ADEM-like picture, while older patients are more likely to present with optic neuritis (19–21).
In this study we aimed to characterize the radiological features of MOG antibody disease and compare the findings with those previously described.
Patients and Methods.
This is a retrospective analysis of patients recruited from the Johns Hopkins Hospital between 2015 and 2018, or recruited remotely through review of records by the principle investigator (ML). Inclusion criteria were: 1. MOG antibody seropositivity by cell-based assay with IgG1 secondary antibody from the Mayo Medical Lab, Quest Diagnostics or the Oxford University Neuroimmunology Laboratory (UK); 2. Disease phenotype of relapsing CNS disease that prompted consideration of MOG antibody testing by the treating neurologist. We did not necessarily exclude patients who also met criteria for multiple sclerosis (MS) as there is no consensus-based distinction between MS and MOG antibody disease. All subjects provided consent to participate in this study, which was approved by the Johns Hopkins University institutional review board.
The MR exams were performed with different scanners at either 1.5T or 3T: Philips Healthcare (Best, the Netherlands), GE Healthcare (Milwaukee, Wisconsin), and Siemens (Erlangen, Germany). For brain MRI, sagittal T1WI, axial fast spin-echo T2WI, axial/sagittal fast spin-echo FLAIR, axial diffusion and ADC map and axial/coronal post-gadolinium T1WI were analyzed. Small field of view axial and coronal T2W and post contrasted images were obtained with fat saturation for orbital evaluation. Sagittal T1, T2, STIR and axial T1, T2 weighted images were obtained through the spine without contrast followed by sagittal and axial T1 weighted images obtained post gadolinium administration. All patients were given intravenous gadolinium-based contrast media.
MRIs were performed for clinical purposes either during an acute neurological presentation or for follow up. The images were reviewed blindly by two independent raters (II, MK). Brain lesions were described in regards to location and enhancement pattern. Spinal lesions were described according to location, length, cord expansion and enhancement pattern. Longitudinally extensive transverse myelitis was defined by myelitis extending 3 or more spinal segments. Optic nerve lesions were characterized by their location, length of involved segment, enhancement, bilaterality of involvement, and T2 signal abnormality. Long segment optic neuritis was defined by enhanced segment length of 17.6 mm or more (22). When there was a mismatch between MRI findings, the images were reviewed by both readers and a consensus was achieved.
Results.
A total of 209 MRIs belonging to 26 MOG patients were retrospectively reviewed; 87 brain, 76 spinal and 46 orbit MRIs. The time lapse between clinical presentation and imaging was no more than 30 days. Demographic and clinical information about these patients is detailed in Table 1. Twenty-three patients met the study definition of MOG antibody disease. The majority were females: 18 as opposed to only 7 males, with a ratio of 2.6:1. The mean age of the cohort was 40.6 years, while the mean age at onset was 37.5 years. Fifteen patients (57.7 %) were Caucasians. The most frequent initial presentation was optic neuritis, reported in 11 patients (42.3 % of all cases). ADEM-like encephalopathic presentation followed by transverse myelitis were next, experienced by 6 patients (23%), and 5 patients (19%) respectively. Simultaneous optic neuritis and transverse myelitis were present initially in one patient (3.8%). The average number of relapses was 4 over an average disease duration of 5.7 years, with an annualized relapse rate of 0.7. Optic neuritis was the most frequent relapse, followed by ADEM-like encephalopathy and transverse myelitis. Brainstem relapses were the least common. Nine patients (34.6%) experienced immediate relapses on withdrawal of steroids. Five patients (19%) had a monophasic disease course over a disease duration of 24 months (range: 5–36 months).
Table 1.
Summary of the epidemiologic and clinical characteristics of the cohort.
| Characteristic | Number of patients (%) |
|---|---|
| Sex | |
| Female | 18 (69.2) |
| Race | |
| Caucasian | 15 |
| African/American | 5 |
| Latino | 1 |
| Other | 5 |
| Age | |
| Mean | 40.6 years |
| Median | 41.5 years |
| Range | 7–72 years |
| Age at onset | |
| Mean | 35 years |
| Median | 37.5 years |
| Range | 3.5 – 66 years |
| EDSS | |
| Mean | 2 |
| Median | 2 |
| Range | 0 – 4 |
| Number of relapses | |
| Mean | 4 |
| Median | 3.5 |
| Range | 1 – 18 |
| Autoimmune disease | |
| Thyroid disease | 5 (21.7%) |
| Ulcerative colitis | 1 (4.3%) |
| Presentation at disease onset | |
| Optic neuritis | 11(42.3%) |
| Encephalopathy | 6 (23%) |
| Transverse myelitis | 5(19%) |
| ON + TM | 1 (3.8%) |
BRAIN MRI.
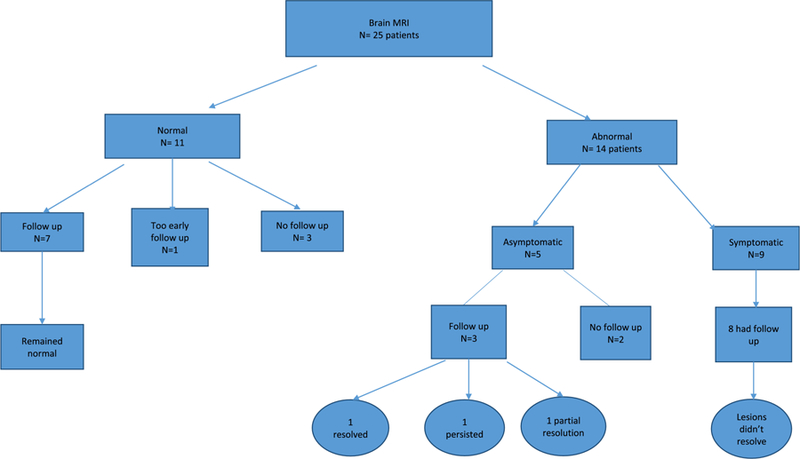
Eighty-seven brain MRIs were reviewed, of which 23 were performed at disease onset, 40 during follow up and 24 at time of a subsequent relapse. In this study, we divided patients into those who had normal brain MRIs all along their disease course and those who had any abnormal MRI at any point during their disease. Then those with abnormal MRIs were subdivided into symptomatic or asymptomatic according to the contribution of the brain lesions to their clinical symptomatology. In MOG patients, out of 25 patients with brain MRIs, eleven had completely normal MRIs all along their available disease course of 17 months (3–46 months) even though three of those eleven subjects had complaints that localized to the brain at some time in their disease course. Interestingly none of those patients developed any asymptomatic brain lesions during the follow up with serial brain scans.
Fourteen MOG patients had abnormal brain MRIs, of which five were asymptomatic and nine were symptomatic. In the asymptomatic group, brain lesions were scattered and punctate without enhancement in three. Surprisingly, among the other two, one showed only localized leptomeningeal enhancement (Figure 1) while the other had a pontine lesion with incomplete ring enhancement (which was asymptomatic). All asymptomatic lesions were observed during relapses of optic neuritis. On follow up in three of the asymptomatic subjects with repeat scans obtained over an average duration of 37 months (1–96 months), one patient had resolution of all lesions, one had persistent T2 lesions and the third showed partial resolution.
Figure 1.
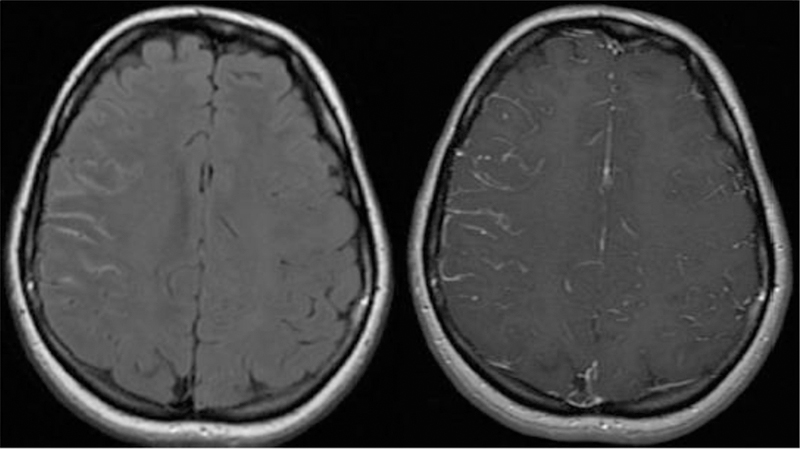
Axial FLAIR and axial post contrast brain MRI with FLAIR image showing increased leptomeningeal signal on the right with linear enhancement seen within the sulci on post contrast image.
Nine MOG patients had abnormal and symptomatic brain MRIs, of which eight were followed with repeat scans over an average duration of 15 months (1–67). All eight, showed persistent lesions on final MRI performed during remission with various degrees of improvement. Among the symptomatic patients, four presented with an encephalopathic brain picture (with or without seizures clinically). Among the cohort of 26 MOG patients, 10 presented with brain disease at their clinical onset, 5 were scanned with brain MRI at disease onset. All except one had symptomatic brain MRI (ADEM like or isolated brain stem involvement). The remaining one had a completely normal MRI inspite of the clinical presentaion. Median age at disease onset was significantly younger for patients who presented with brain disease (23.5 vs 41 years, p = 0.03). There was no significant race prediliction for brain disease presentation at clinical onset.
In the combined cohort of 14 MOG patients with either symptomatic and asymptomatic brain MRIs, there were a total of 48 abnormal MRIs over an average duration of 17.8 months. The most common lesion location was the supratentorial deep white matter (WM), observed in nine patients (64%), followed by cortical grey/juxtacortical white matter (cortical GM/juxtacortical WM) (Figure 2) and pons in eight patients (57%) each. The cerebellum, midbrain, medulla and corpus callosum were each affected in six patients (43%). The least frequently involved locations were periventricular white matter, area postrema, and basal ganglia (Table 2).
Figure 2.

A: Axial T2-weighted image showing confluent deep white matter hyperintensities extending from subcortical to periventricular white matter in a patient presenting with an encephalopathic clinical picture. B: Axial T1 post contrast image showing avid nodular enhancement pattern.
Table 2.
Location of brain lesions in MOG antibody disease.
| Location involved | Number of patients (n=14, %) |
|---|---|
| Deep WM | 9 (64%) |
| Pons | 8 (57%) |
| Cortical GM/juxtracortical WM | 8 (57%) |
| Cerebellum | 6 (43%) |
| Corpus callosum | 6 (43%) |
| Midbrain | 6 (43%) |
| Medulla | 6 (43%) |
| Periventricular WM | 5 (35.7%) |
| Basal ganglia | 2 (14.2%) |
| Area postrema | 1 (7%) |
Among the 9 patients with cortical grey/juxtacortical WM involvement, 3 presented with encephalopathy and seizures, while 2 presented with encephalopathy without seizures. One pediatric patient among our cohort, showed a leukodystrophy like pattern on brain MRI, that coincided with an ADEM-like clinical attack. It started as unilateral involvement and with disease progression started to involve the contralateral side of the brain (Figure 2).
In those cases with periventricular lesions, MOG lesions appeared to extend from nearby cortical lesions, they were large in size and they were not perpendicular to the ventricular wall. None of our patients fulfilled the Macdonald’s MS imaging criteria. Gadolinium enhancement was observed in nine patients (64.3%) with nodular enhancement as the most frequent pattern (in 8 patients), followed by incomplete ring (in 3 patients). Three patients showed a leptomeningeal enhancement pattern.
SPINAL CORD MRI.
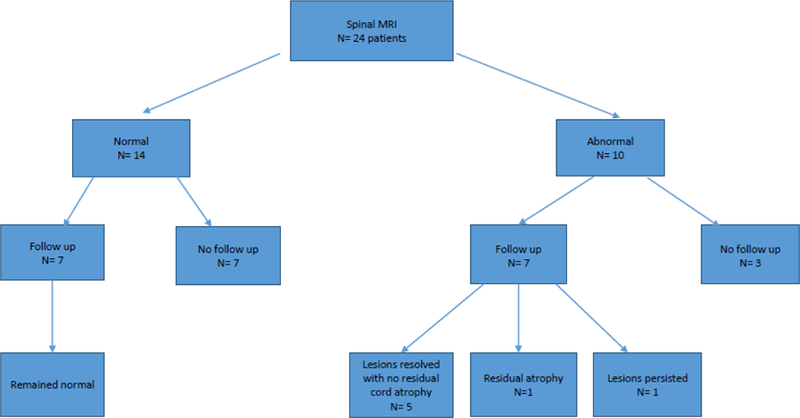
Out of the 26 patients in this cohort, 24 underwent imaging of the spinal cord for either routine purposes or for relapse investigation. A total of 75 spinal MRIs were reviewed. 22 at disease onset, 34 at time of follow up and 19 at time of subsequent relapse. Fourteen patients (58%) had normal spinal cord MRIs along their 1.5-year disease course despite a clinical suspicion of myelitis in two subjects (was ruled out by negative MRI) (23). Ten patients (38%) had abnormal spinal cord MRIs, of which seven had serial spinal cord MRIs performed either during remission or optic neuritis relapse over a mean follow up duration of 21.3 months (3 weeks - 55 months). Six of those seven had normalization of the T2 signal MRI signal on follow up, with one of them showing residual cord atrophy, while only one patient had persistent lesions. Among the six patients with normalization of their cord images, 2 were African American while four were Caucasians.
The location of spinal cord locations were as follows: two patients had isolated cervical (20%), four had isolated thoracic (40%), four had cervicothoracic (40%) and four had conus involvement (40%). The myelitis was longitudinally extensive in seven patients (70%) (Figure 3) and was short segment in four patients (40%).
Figure 3.

Sagittal and 3 axial T2 cervical spine MRIs; showing varying degrees of hyperintense signal changes in a longitudinally extensive pattern and sagittal post contrast T1 cervical spine MR image showing focal contrast enhancement at C6–7 level during acute MOG positive myelitis.
In the axial plane, eight patients (80%) had involvement of the central cord with more than 50% maximal area involved in 6 of them, while peripheral cord was involved in 5 patients (50%). Cord expansion was observed in six patients (60%) while bright spoty lesions (BSL) were observed in five (50%). T1 hypointensity was detected in 4 patients (40%). None of the previous features correlated significantly with EDSS at final follow up (Table 4).
Table 4:
Correlation between radiological spinal cord features and EDSS.
| Pearson Test | LETM vs EDSS | BSL vs EDSS | T1 hypointensity vs EDSS | Central lesions vs EDSS | Peripheral Lesion vs EDSS |
|---|---|---|---|---|---|
| 95% confidence interval | −0.4601 to 0.3094 | −0.5871 to 0.1432 | −0.4905 to 0.2735 | −0.3539 to 0.4198 | −0.2026 to 0.5455 |
| R squared | 0.007835 | 0.06683 | 0.01622 | 0.001503 | 0.04020 |
| P (two-tailed) | 0.6672 | 0.2023 | 0.5352 | 0.8509 | 0.3260 |
| P value summary | Ns | ns | ns | ns | ns |
Enhancement was notable in six patients, in five of which, it coincided with acute clinical relapse localizing to the spinal cord (60%). The sixth patient had clinical remission despite the enhancement on spinal imaging. Cord atrophy was observed in one patient after resolution of myelitis (Table 3).
Table 3.
MRI characteristics of MOG antibody disease spinal cord lesions.
| Feature | Number of patients (n=10, %) |
|---|---|
| Central cord lesion | 8 (80%) |
| Peripheral cord lesion | 5 (50%) |
| Cord expansion | 6 (60%) |
| Bright spots | 5 (50%) |
| T1 hypointensity | 4 (40%) |
| Enhancement | 6 (60%) |
| Cord atrophy | 1 (10%) |
ORBITAL MRI.
Forty-six orbital MRIs among 21 patients were available for analysis. A total of 24 MRIs belonging to 18 patients, were obtained during a relapse of acute optic neuritis. Within the optic nerves, lesions were divided among the five segments in regards to T2 hyperintensity: orbital (75% of 24 images), intracanalicular (54%), pre-chiasmal (37.5%), chiasmal (8.3%) and optic tracts (4%). Gadolinium enhancement was evident in all 24 images (100 %), with the orbital segment being the most commonly involved, in 20 MRIs (83.3%). Canalicular and pre-chiasmal enhancement was observed in 14 (58.3%) and 10 (41.7%) MRIs, respectively. Chiasmal enhancement was evident in only 2 MRIs while optic tract enhanced in one.
Based on the extent of gadolinium enhancement, 35 optic nerve measurements of the involved optic segment(s) were obtained, 17 (48.6%) showed longitudinally extensive lesions (Figure 4), while 18 (51.4%) were short segment optic neuritis. The average lesion length was of 20.5 mm (4 – 41.7 mm). Eight patients had serial MRI imaging of the optic nerves, of whom two showed complete resolution of optic nerve lesions within an average of 4.25 months, two showed residual optic atrophy by an average of 8.5 months and five patients had persistent T2 hyperintense optic nerve lesions on follow up over 11.9 months average duration. The follow up MRI was performed during remission in two patients, during optic neuritis relapse in 2 and during ADEM relapse in one patient.
Figure 4.

Coronal fat saturated T2, axial and coronal fat saturated contrast-enhanced T1 weighted images of the orbits showing bilateral longitudinally extensive acute optic neuritis with T2 hyperintense signal, enlargement and contrast enhancement of the optic nerves in orbital and canalicular segments in a MOG positive patient.
Twelve patients (63%) had involvement of both optic nerves at least once along their disease course (either simultaneously or sequentially). All of them experienced bilateral simultaneous optic neuritis at least once while 2 patients showed alternating optic neuritis.
Discussion.
In this study we characterized MRI features of optic nerves, brain and spinal cord of patients with MOG antibody disease. Overall, the location of lesions in our population of MOG antibody disease patients was in line with previously published studies. We observed that most MRI findings were localized to the optic nerves associated with attacks of optic neuritis, which is the most frequent clinical presentation of MOG antibody disease (10, 14, 24, 25). The most abnormal findings in our study were observed on the orbital MRIs, where the orbital segment was the most frequently involved on T2 (75%) and post-contrast sequences (83.3%) while the chiasm and optic tracts were the least frequently involved parts. This observation could be of value in differentiating AQP4 optic neuritis from that of MOG, where in the former, posterior structures are more frequently affected (22, 26, 27). Similarly, bilateral optic neuritis was a feature that used to distinguish NMO from MS, but now it appears that bilateral optic neuritis is even more common in MOG antibody disease than AQP4 NMO. Our results are concordant with a similar finding by Kitley et al, where bilateral optic neuritis occured in up to 63% of MOG positive patients (4).
The average lesion length within the optic nerve among our cohort was 20.5 mm; however, MOG lesions were segregated into long lesions with an average of 30.8 mm and short lesions with an average of 10.8 mm. According to our previous study in NMOSD (majority AQP4 antibody seropositive), we calculated 17.6 mm to be a cutoff point differentiating NMO from MS optic nerve lesions with a sensitivity of 80.8 % and a specificity of 76.9%. Thus, in this cohort of MOG patients, the average lesion length in the optic nerve is even longer than our combined NMOSD cohort (22).
In the French MOGADOR study (24), similar to our study, the most and the least frequently involved locations of lesions on brain MRI were in the deep white matter and the area postrema, respectively. In a German study, supratentorial deep white matter lesions was the most frequently involved location, observed in 47% of patients in final MRI (10). Leptomeningeal enhancement was observed in 3 patients in one study (7) and one patient in another (10) as compared to three patients in our study. Among those 3 patients, two presented with seizures and encephalopathy, while the third was completely asymptomatic and never had relapses localizing to the brain. In a previous study, this sign was thought to be related to leptomeningeal blood brain barried disruption in NMOSD and was associated in all patients with encephalopathy and/or myelopathy (16, 28). In our cohort it was not observed on spinal MRIs.
Callosal lesions in our MOG cohort were distinct from the dawson fingers pattern seen in MS and the arch of bridge sign in AQP4 NMOSD. They tended to be focal, discrete and nodular without a specific orientation around the ventricles. This finding was also reported by Jurynczyk, where dawson’s fingers were only detected in MS patients and to a much lesser extent in AQP4 NMOSD, but in none of the MOG patients (15).
The leukodystrophy-like pattern which was observed in one pediatric patient among our cohort, was perviously reported by Hacohen et al., in 7 children presenting with ADEM (29). Similar to those patients, our patient was given an initial diagnosis of ADEM, which was multiphasic. MOG antibody testing was warranted when she started experiencing isolated optic neuritis relapses.
A recent study concluded that age at disease onset could impact the radiological picture among MOG seropositive pediatric patients presenting with different demyelinating diseases (18). This could reflect what was reported by previous studies, where younger MOG patients tended to present with ADEM while older patients presented more likely with optic neuritis (19–21). Interestingly that clinical observation was also noted among our patients, where the 5 subjects in our cohort who had their disease onset before 20 years, presented with an encephalopathic clinical picture with or without seizures at their disease onset. Despite that fact, we did not note any radiological difference based on age at disease onset. Similar to recent studies that reported cortical encephalitis on brain MRIs in MOG patients presenting with encephalopathy and/or seizures (16, 17), these findings were observed among five patients in our cohort.
In a Korean study, MOG patients showed less spinal cord involvement and fewer brain abnormalities compared to NMO and MS groups, which is also consistent with our findings (14). As opposed to our study, Jarius el al reported cervical cord to be the most frequently involved part of myelitis attacks (82.1% of patients), while in our study it was the least frequently involved (20%) (10). In a study conducted in Netherlands, the thoracic cord was the most frequently involved segment (25). Although originally thought to be a seminal feature of MOG myelitis, isolated lesions of the conus medullaris were seen in four patients (40%) among our cohort (5).
Normalization of the T2 signal on MRI is not typical for NMOSD lesions associated with AQP4, but is more commonly seen in patients with MOG antibody disease (15). Similar to our findings, five out of six MOG patients in the U.K. showed resolution of their brain lesions on follow up in comparison to NMO patients (4). In our cohort, resolution of brain lesions were limited to clinically asymptomatic lesions only while the symptomatic ones improved but did not resolve completely. Within the spinal cord, complete resolution of spinal lesions was observed within a median of 2 months after onset, similar to findings reported by Kitley et al reported in which 6 out of 7 MOG positive patients showed complete resolution of lesions within a median of 9.5 months, and no evidence of cord atrophy (4).
Longitudinally extensive lesions are more common in MOG antibody attacks of the spinal cord than short lesions. Jarius et al reported longitudinally extensive lesions in the spinal cord at least once in 72.4% of German MOG patients and short segment myelitis in 41.3 % of patients (10). These findings are similar to our results where we observed LETM in 70% of our cohort and short segment myelitis in 40%. In the same German study, 20 out of 28 patients had two simultaneous spinal cord lesions on the same MRI, compared to only 1 patient out of 24 among our cohort (10). Although it has been reported that a small percentage (7%) of MOG positive patients may present with short myelitis that can be confused with MS (30, 31), more than a third (40%) of our cohort experienced short segment involvement of their spinal cords.This finding could be related to the timing of spinal MRI in relation to onset of clinical symptoms (32–34), but it might also reflect a true observation and supports the overlap theory between MOG disease and MS. Bright spotty lesions (BSLs) was reported to be a differentiating imaging finding between NMOSD and MS, especially when combined with LETM and T1 dark spots.(35, 36) Similarly, BSLs and T1 dark spots were evident in 50% and 40% of MOG patients who had abnormal spinal MRIs, respectively.
In conclusion, our findings support the previously published data on MOG antibody disease. We identified unique MRI features of MOG antibody disease that could aid in the differentiation of MOG from both MS and AQP4 NMO. First, brain lesions tend to involve the deep white matter in a pattern distinct from MS and NMOSD. Second, resolution of brain and spinal cord lesions on follow up may reflect an underlying repair process unique to MOG, Third, longitudinal extensive optic neuritis tends to be bilateral and affect the anterior structures, Fourth, focal myelitis is common among MOG patients, And lastly, chiasmal involvement is rather uncommon in MOG positive optic neuritis.
Figure 5:
summary of findings in patients who underwent brain MRIs.
Figure 6:
summary of findings in patients who underwent spinal MRIs
Figure 7:

summary of findings in patients who underwent orbital MRIs.
HIGHLIGHTS.
MOG antibody disease is an emerging entity that yet needs to be characterized.
Although previously thought to be a monophasic disease, relapsing course is a common presentation among such patients.
Most of imaging findings are localized within the optic nerves which correlates with optic neuritis as the most common clinical relapse.
The encephalopathic presentation with an ADEM-like MRI picture is more common among such patients and might help in its distinction from MS and AQP4 NMOSD.
Resolution of MRI findings on follow up might explain the relatively good long term recovery.
Acknowledgments
Funding: This work was supported by a scholarship from the Egyptian ministry of higher education, JS-3725 (SS), as well as a grant from the National Institute of Neurological Disease and Stroke, NS-078555 (ML).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References.
- 1.Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: Frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflammation 2016;13(1):279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Probstel AK, Rudolf G, Dornmair K, Collongues N, Chanson JB, Sanderson NS, et al. Anti-MOG antibodies are present in a subgroup of patients with a neuromyelitis optica phenotype. J Neuroinflammation 2015;12:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kitley J, Woodhall M, Waters P, Leite MI, Devenney E, Craig J, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology 2012;79(12):1273–7. [DOI] [PubMed] [Google Scholar]
- 4.Kitley J, Waters P, Woodhall M, Leite MI, Murchison A, George J, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol 2014;71(3):276–83. [DOI] [PubMed] [Google Scholar]
- 5.Sato DK, Callegaro D, Lana-Peixoto MA, Waters PJ, de Haidar Jorge FM, Takahashi T, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 2014;82(6):474–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Narayan R, Simpson A, Fritsche K, Salama S, Pardo S, Mealy M, et al. MOG antibody disease: A review of MOG antibody seropositive neuromyelitis optica spectrum disorder. Mult Scler Relat Disord 2018;25:66–72. [DOI] [PubMed] [Google Scholar]
- 7.Spadaro M, Gerdes LA, Krumbholz M, Ertl-Wagner B, Thaler FS, Schuh E, et al. Autoantibodies to MOG in a distinct subgroup of adult multiple sclerosis. Neurology(R) neuroimmunology & neuroinflammation 2016;3(5):e257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akaishi T, Sato DK, Nakashima I, Takeshita T, Takahashi T, Doi H, et al. MRI and retinal abnormalities in isolated optic neuritis with myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies: a comparative study. J Neurol Neurosurg Psychiatry 2016;87(4):446–8. [DOI] [PubMed] [Google Scholar]
- 9.Ramanathan S, Prelog K, Barnes EH, Tantsis EM, Reddel SW, Henderson AP, et al. Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. Mult Scler 2016;22(4):470–82. [DOI] [PubMed] [Google Scholar]
- 10.Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflammation 2016;13(1):280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mariotto S, Ferrari S, Monaco S, Benedetti MD, Schanda K, Alberti D, et al. Clinical spectrum and IgG subclass analysis of anti-myelin oligodendrocyte glycoprotein antibody-associated syndromes: a multicenter study. J Neurol 2017;264(12):2420–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitley J, Leite MI, Kuker W, Quaghebeur G, George J, Waters P, et al. Longitudinally extensive transverse myelitis with and without aquaporin 4 antibodies. JAMA neurology 2013;70(11):1375–81. [DOI] [PubMed] [Google Scholar]
- 13.Kim HJ, Paul F, Lana-Peixoto MA, Tenembaum S, Asgari N, Palace J, et al. MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology 2015;84(11):1165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim SM, Woodhall MR, Kim JS, Kim SJ, Park KS, Vincent A, et al. Antibodies to MOG in adults with inflammatory demyelinating disease of the CNS. Neurol Neuroimmunol Neuroinflamm 2015;2(6):e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jurynczyk M, Geraldes R, Probert F, Woodhall MR, Waters P, Tackley G, et al. Distinct brain imaging characteristics of autoantibody-mediated CNS conditions and multiple sclerosis. Brain 2017;140(3):617–27. [DOI] [PubMed] [Google Scholar]
- 16.Ogawa R, Nakashima I, Takahashi T, Kaneko K, Akaishi T, Takai Y, et al. MOG antibody-positive, benign, unilateral, cerebral cortical encephalitis with epilepsy. Neurol Neuroimmunol Neuroinflamm 2017;4(2):e322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamid SHM, Whittam D, Saviour M, Alorainy A, Mutch K, Linaker S, et al. Seizures and Encephalitis in Myelin Oligodendrocyte Glycoprotein IgG Disease vs Aquaporin 4 IgG Disease. JAMA Neurol 2018;75(1):65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baumann M, Grams A, Djurdjevic T, Wendel EM, Lechner C, Behring B, et al. MRI of the first event in pediatric acquired demyelinating syndromes with antibodies to myelin oligodendrocyte glycoprotein. J Neurol 2018;265(4):845–55. [DOI] [PubMed] [Google Scholar]
- 19.Fernandez-Carbonell C, Vargas-Lowy D, Musallam A, Healy B, McLaughlin K, Wucherpfennig KW, et al. Clinical and MRI phenotype of children with MOG antibodies. Mult Scler 2016;22(2):174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hennes EM, Baumann M, Schanda K, Anlar B, Bajer-Kornek B, Blaschek A, et al. Prognostic relevance of MOG antibodies in children with an acquired demyelinating syndrome. Neurology 2017;89(9):900–8. [DOI] [PubMed] [Google Scholar]
- 21.Hacohen Y, Mankad K, Chong WK, Barkhof F, Vincent A, Lim M, et al. Diagnostic algorithm for relapsing acquired demyelinating syndromes in children. Neurology 2017;89(3):269–78. [DOI] [PubMed] [Google Scholar]
- 22.Mealy MA, Whetstone A, Orman G, Izbudak I, Calabresi PA, Levy M. Longitudinally extensive optic neuritis as an MRI biomarker distinguishes neuromyelitis optica from multiple sclerosis. J Neurol Sci 2015;355(1–2):59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kessler RA, Mealy MA, Levy M. Early indicators of relapses vs pseudorelapses in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm 2016;3(5):e269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cobo-Calvo A, Ruiz A, Maillart E, Audoin B, Zephir H, Bourre B, et al. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: The MOGADOR study. Neurology 2018;90(21):e1858–e69. [DOI] [PubMed] [Google Scholar]
- 25.van Pelt ED, Wong YY, Ketelslegers IA, Hamann D, Hintzen RQ. Neuromyelitis optica spectrum disorders: comparison of clinical and magnetic resonance imaging characteristics of AQP4-IgG versus MOG-IgG seropositive cases in the Netherlands. Eur J Neurol 2016;23(3):580–7. [DOI] [PubMed] [Google Scholar]
- 26.Khanna S, Sharma A, Huecker J, Gordon M, Naismith RT, Van Stavern GP. Magnetic resonance imaging of optic neuritis in patients with neuromyelitis optica versus multiple sclerosis. J Neuroophthalmol 2012;32(3):216–20. [DOI] [PubMed] [Google Scholar]
- 27.Levin MH, Bennett JL, Verkman AS. Optic neuritis in neuromyelitis optica. Prog Retin Eye Res 2013;36:159–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asgari N, Flanagan EP, Fujihara K, Kim HJ, Skejoe HP, Wuerfel J, et al. Disruption of the leptomeningeal blood barrier in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm 2017;4(4):e343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hacohen Y, Rossor T, Mankad K, Chong W, Lux A, Wassmer E, et al. ‘Leukodystrophy-like’ phenotype in children with myelin oligodendrocyte glycoprotein antibody-associated disease. Dev Med Child Neurol 2018;60(4):417–23. [DOI] [PubMed] [Google Scholar]
- 30.Dos Passos GR, Oliveira LM, da Costa BK, Apostolos-Pereira SL, Callegaro D, Fujihara K, et al. MOG-IgG-Associated Optic Neuritis, Encephalitis, and Myelitis: Lessons Learned From Neuromyelitis Optica Spectrum Disorder. Front Neurol 2018;9:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sepulveda M, Armangue T, Martinez-Hernandez E, Arrambide G, Sola-Valls N, Sabater L, et al. Clinical spectrum associated with MOG autoimmunity in adults: significance of sharing rodent MOG epitopes. J Neurol 2016;263(7):1349–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flanagan EP, Weinshenker BG, Krecke KN, Lennon VA, Lucchinetti CF, McKeon A, et al. Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. JAMA Neurol 2015;72(1):81–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Asgari N, Skejoe HP, Lennon VA. Evolution of longitudinally extensive transverse myelitis in an aquaporin-4 IgG-positive patient. Neurology 2013;81(1):95–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Borisow N, Mori M, Kuwabara S, Scheel M, Paul F. Diagnosis and Treatment of NMO Spectrum Disorder and MOG-Encephalomyelitis. Front Neurol 2018;9:888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yonezu T, Ito S, Mori M, Ogawa Y, Makino T, Uzawa A, et al. “Bright spotty lesions” on spinal magnetic resonance imaging differentiate neuromyelitis optica from multiple sclerosis. Mult Scler 2014;20(3):331–7. [DOI] [PubMed] [Google Scholar]
- 36.Pekcevik Y, Mitchell CH, Mealy MA, Orman G, Lee IH, Newsome SD, et al. Differentiating neuromyelitis optica from other causes of longitudinally extensive transverse myelitis on spinal magnetic resonance imaging. Mult Scler 2016;22(3):302–11. [DOI] [PMC free article] [PubMed] [Google Scholar]