

 Structural/electronic influence of metalloenzyme-inspired synthetic heme–peroxo–copper models containing intramolecular hydrogen bonding interactions.
Structural/electronic influence of metalloenzyme-inspired synthetic heme–peroxo–copper models containing intramolecular hydrogen bonding interactions.
Abstract
Dioxygen reduction by heme–copper oxidases is a critical biochemical process, wherein hydrogen bonding is hypothesized to participate in the critical step involving the active-site reductive cleavage of the O–O bond. Sixteen novel synthetic heme–(μ-O22–)–Cu(XTMPA) complexes, whose design is inspired by the cytochrome c oxidase active site structure, were generated in an attempt to form the first intramolecular H-bonded complexes. Derivatives of the “parent” ligand (XTMPA, TMPA = (tris((2-pyridyl)methyl)amine)) possessing one or two amine pendants preferentially form an H-bond with the copper-bound O-atom of the peroxide bridge. This is evidenced by a characteristic blue shift in the ligand-to-metal charge transfer (LMCT) bands observed in UV-vis spectroscopy (consistent with lowering of the peroxo π* relative to the iron orbitals) and a weakening of the O–O bond determined by resonance Raman spectroscopy (rR), with support from Density Functional Theory (DFT) calculations. Remarkably, with the TMPA-based infrastructure (versus similar heme–peroxo–copper complexes with different copper ligands), the typically undetected Cu–O stretch for these complexes was observed via rR, affording critical insights into the nature of the O–O peroxo core for the complexes studied. While amido functionalities have been shown to have greater H-bonding capabilities than their amino counterparts, in these heme–peroxo–copper complexes amido substituents distort the local geometry such that H-bonding with the peroxo core only imparts a weak electronic effect; optimal H-bonding interactions are observed by employing two amino groups on the copper ligand. The amino-substituted systems presented in this work reveal a key orientational anisotropy in H-bonding to the peroxo core for activating the O–O bond, offering critical insights into effective O–O cleavage chemistry. These findings indirectly support computational and protein structural studies suggesting the presence of an interstitial H-bonding water molecule in the CcO active site, which is critical for the desired reactivity. The results are evaluated with appropriate controls and discussed with respect to potential O2-reduction capabilities.
Introduction
In protein-mediated reactions, hydrogen bonding is well known to be critical for active site chemistry, including directing substrate binding and facilitating key catalytic steps.1–5 This is especially important for metalloenzymes involved in dioxygen processing (such as in O2-carriers, oxidases, or oxygenases). For instance, protonation and/or hydrogen bonding is critical in directing O–O bond scission in heme-containing enzymes such as cytochrome P450s, catalases, and peroxidases, by lowering the barrier to O–O reductive cleavage and/or selectively enhancing heterolytic (over homolytic) cleavage.4,6,7
Catalytic reduction of O2 to water by heme–copper oxidases (HCOs) involves 8H+ and 4e–, wherein 4H+ are translocated across the inner mitochondrial membrane, driving ATP synthesis.5,8–12 The pertinent mammalian terminal respiratory enzyme is cytochrome c oxidase (CcO), which facilitates O2 reduction following dioxygen binding to the heterobinuclear active site comprised of a reduced heme (FeII) and copper (CuI) moiety, initially forming the ferric superoxide species (A, Scheme 1).5 In the established catalytic mechanism of CcO, the four electrons derive from iron (2e–; FeII → FeIV), copper (1e–; CuI → CuII), and a tyrosine residue cross-linked to one His ligand bound to Cu (1e–/1H+; TyrOH → TyrO˙).5,8,9,11 A unique covalent cross-link between the tyrosine and one of the Cu-bound histidine residues is believed to modulate both the redox potential and the pKa of the TyrO-H.13–15 It is also postulated that an H-bonding network of water molecules exists in the active site, linking the TyrOH residue to a putative metal-(hydro)peroxide species (IP), facilitating the necessary proton coupled electron transfer (PCET) from the tyrosine to achieve homolytic O–O cleavage resulting in the formation of the high-valent intermediate PM (Scheme 1). Following a subsequent series of proton (PT) and electron (ET) transfer reactions, also accompanied by membrane proton translocation steps, the initial state of reduced heme and copper (R, Scheme 1) reforms providing for the overall release of two mole-equivalents of water from one mole-equivalent O2.16–24 Interestingly, H-bonding interactions involving the active site Tyr have also been shown to be important for controlling proton gating and delivery from the K-channel to the active site for complete protonation-reduction of O2 to H2O.16
Scheme 1. A portion of the catalytic cycle of cytochrome c oxidase, emphasizing the key steps in which H-bonding (highlighted in blue, IP) mitigates homolytic O2 reductive cleavage, forming the tyrosyl radical and high valent iron(iv) species (PM). See text for further discussion.

Synthetic heme–peroxo–copper model systems can be strategically designed to probe fundamental chemical questions, including the factors that influence reductive O–O cleavage, such as H-bonding and/or proton relay processes. These are often accompanied by electron transfer reactions that result in, and can be applied to, practical catalytic systems; synthetic systems may enhance the understanding of these PT and ET steps to better grasp aspects that may be detrimental to efficiency or longevity. Results from biochemical and computational studies on HCOs, as well as various synthetic heme–peroxo–copper systems, have provided significant insights into Nature's rationale for taking advantage of hydrogen bonding to easily impart significant electronic or geometric alterations to tune the enzymatic active site to facilitate O2 reduction chemistry (heterolytic vs. homolytic cleavage) at metal sites, a biochemical process critical to all aerobic organisms.17,18,25–27 However, a deeper understanding of how H-bonding impacts O–O cleavage in HCOs is needed, as parts of this mechanism have remained elusive due to the fast kinetics of the homolytic cleavage step (from A to PM) in the enzyme, which for example has prevented thorough characterization of a widely proposed 2e–-reduced peroxo level intermediate20,28,29 (IP in Scheme 1, which has recently been supported by experimental and computational studies by Brzezinski, Blomberg, and coworkers)30 and the subtleties relevant to the further ET, PT or PCET step(s).
While previously reported heme–peroxo–copper model studies have presented a valuable new route for investigating O–O cleavage by observing H-bonding interactions with exogenous substrates,26,27,31 they are complicated by entropic factors (i.e., bimolecular interactions) not present in the enzyme. Thus, herein we develop intramolecular H-bonding heme–peroxo–copper systems in the hope that mechanistic insights can be gained toward understanding how H-bonding could: (1) stabilize the formation of the proposed, but not spectroscopically observed, IP intermediate,5,20,30 (2) help gain structural and electronic perspectives on the influence of H-bonding in O2-reduction chemistries, and (3) further the development of a more biomimetic, CcO-inspired, synthetic model in order to do corresponding reactivity studies. The results presented herein provide insights into how the incorporation of intramolecular H-bonding moieties affect the electronic structure and core vibrations of the observed low-spin heme–peroxo–copper complexes. In most cases, density functional theory calculations of the energy-minimized structures accompany the spectroscopic observations and lend support for the conclusions drawn.
Results and discussion
Development of synthetic hydrogen bonding complexes
Mechanistic insights into O2 activation and reduction from promising reactivity studies of heme–peroxo–copper synthetic systems utilizing the copper ligand TMPA (and also a porphyrinate with a tethered TMPA moiety; TMPA = tris(2-pyridylmethyl)amine) have been reported.32–43 These include synthesis of reduced heme(FeII)–CuI complexes, O2 reactivity leading to μ-oxo [heme–FeIII–(μ-O2–)–CuII]+ and conjugate acid [heme–FeIII–(μ-OH–)–CuII]2+ compounds, spectroscopic-structural characterization of discrete peroxo-bridged [heme–FeIII–(μ-O22–)–CuII]+ complexes and (electro)catalytic O2-reduction studies. In this current investigation, a series of Cu(XTMPA) derivatives are paired with tetrakis(2,6-difluorophenyl)porphyrinate (F8) heme for generation of low-spin heme–peroxo–copper synthetic model complexes [(DCHIm)(F8)FeIII–(O22–)–CuII(TMPA)]+, (DCHIm = 1,5-dicyclohexylimidazole) in 2-methyltetrahydrofuran (MeTHF) solvent under cryogenic conditions (–90 °C, Fig. 1). Most heme-containing metalloenzymes that perform dioxygen chemistry (e.g., catalases, peroxidases, P450's, oxidases, and globins) require a proximal axial base; hence, to generate low-spin species and understand the H-bonding interactions in these heme-based metalloenzyme mimics, the strong electron donating axial base, DCHIm, was employed, mirroring the histidine proximal ligand in CcO.
Fig. 1. The established parent TMPA heme–peroxo–copper complex and eight new low-spin heme–peroxo–copper complexes bearing derivatized TMPA-based copper chelates, including those with potential H-bonding modules, whose steric and H-bonding influences are elucidated in this work.

Novel low-spin heme–peroxo–copper synthetic complexes using eight TMPA-derivatized copper ligands (Fig. 1) have been prepared in order to develop a series of compounds that have the potential or do in fact demonstrate significant H-bonding capabilities; H-bonding has previously been shown in copper(ii) chloride complexes bearing some of these ligands.44 A systematic study presented here enables us to examine the effects of secondary coordination sphere substitutions on the structural and electronic properties such as (1) intramolecular H-bonding preference for the Cu–O or Fe–O oxygen-atom, (2) structural variations caused and influencing the dihedral angle of the Fe–O–O–Cu core, (3) changes in Fe–O, Cu–O, and O–O bond lengths, and (4) the impact of one vs. two H-bonding functionalities. Taken together, these results are crucial to thinking about how H-bonding dictates reactivity in the enzyme. The use of intramolecular H-bonding amino moieties incorporated on the TMPA ligand template, positioned to enable an interaction with the peroxo core, aims to further understand how H-bonding interactions may facilitate reductive O–O cleavage.
As such, TMPA-based copper ligands, with various substituents in the 6-pyridyl position, were thoughtfully synthesized and utilized: CH3TMPA, (CH3)2TMPA, OCH3TMPA, PVTMPA, A2TMPA, NH2TMPA, (NH2)2TMPA, and NH2,CH3TMPA (Fig. 1; see ESI† for synthetic and characterization details). In particular, comparison of heme–peroxo–copper complexes bearing the ligands NH2TMPA, CH3TMPA, and OCH3TMPA can help differentiate steric vs. H-bonding influences; we can rule out electron-donating effects on the basis that the CuII/I reduction potential of complexes bearing these ligands are very similar (E1/2 = –0.34 or –0.35 V) (Fig. S3 & Table S2†), indicating the three ligands donate electron density to the copper ion in a nearly identical manner.§ Another important property is revealed by the present study and data obtained; the metal–oxygen stretching frequencies (especially Cu–O) are typically not observed via resonance Raman spectroscopy (rR) in previously studied synthetic heme–peroxo–copper complexes.36,39,45–48 However, as demonstrated herein (vide infra), we have found that systems with the TMPA infrastructure yield strong resonance enhancement of both the Fe–O and Cu–O stretches, offering unique and valuable insights into the bonding in the heme–peroxo–copper core.
Azido-Cu(ii) complexes to assess ligand H-bonding capabilities
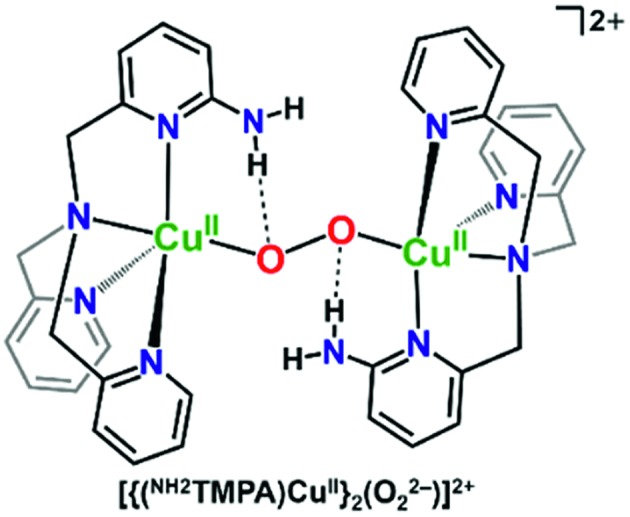
To assess the H-bonding capabilities of these ligands, the azido complexes, [(XTMPA)CuII(N3–)]+, many of which were previously reported and characterized by X-ray crystallography by Masuda and coworkers (Fig. 2),49,50 were prepared and, for the first time, analyzed by IR spectroscopy to observe steric, electronic, or H-bonding influences on the vibrational stretching frequency characteristic of the N3– moiety (Fig. 3 and S2 and Table S1†). Substitution of an amino moiety on the TMPA framework, [(NH2TMPA)CuII(N3–)]+, vs. the parent TMPA azido complex showed that the former possesses a higher ν(N–N) stretching frequency by 10 cm–1 (Fig. 3A and Table S1†). Addition of a second amino group, in [((NH2)2TMPA)CuII(N3–)]+, further increased the ν(N–N) by 8 cm–1 (18 cm–1 greater than the parent TMPA azido complex) (Fig. 3A and Table S1†), consistent with previously reported X-ray structures50 that showed H-bonding to the proximal (bound to Cu) N-atom of the azido ligand (with N(H)···Nazido distances of 2.93 and 2.95 Å and ∠NHNazido = 146.1 and 158.1° observed for [((NH2)2TMPA)CuII(N3–)]+).
Fig. 2. H-bonding interactions of cupric azido complexes observed via X-ray crystallography by Masuda and coworkers.50.

Fig. 3. Infrared spectra of [(XTMPA)CuII(N3–)]+ complexes in CH3CN, showing the increase of the ν(N–N) stretch upon incorporation of amino substituents (A) and incorporation of steric functionalities (B).

As evidenced by comparing the ν(N–N) values for the cupric azido complexes bearing the ligands NH2TMPA and TMPA (Fig. 3A) vs.NH2,CH3TMPA and CH3TMPA (Fig. 3B), the NH2TMPA and NH2,CH3TMPA ligands have the same H-bonding capabilities (each shows a 10 cm–1 blue-shift when compared to their respective “base” ligand). The azido derivative with CH3TMPA has a ν(N–N) value slightly higher than with TMPA, implicating a negligible steric influence (Fig. S2A & Table S1†). The PVTMPA cupric azido complex has a ν(N–N) stretching frequency similar to [((NH2)2TMPA)CuII(N3–)]+ (within 1 cm–1, Fig. S2B & Table S1†), suggesting that one pivalamido group can exert an H-bonding strength roughly equivalent to two –NH2 groups.
Masuda and coworkers also previously showed that some of the same H-bonding copper ligands utilized herein (PVTMPA, NH2TMPA, and (NH2)2TMPA) provide for enhanced stability of dicopper(ii) trans-peroxide complexes complexes (Fig. 4) due to H-bonding to the peroxo core.49,50 Further, PVTMPA has also been shown to stabilize a cupric superoxide complex (vide infra).51
Fig. 4. H-bonding interactions enhance the stability of dicopper(ii) trans-peroxide complexes as reported by Masuda and coworkers.50.
Thus, we hypothesized that amino functionalities may stabilize a heterobinuclear peroxide complex as well, and thereby generate good systems to carry out future reactivity studies; one would expect the barrier to reduce the O–O bond would be less if H-bonding caused bond elongation. Therefore, these interactions may facilitate O2-reduction chemistry by these systems. Ligands bearing methyl (CH3TMPA & (CH3)2TMPA) or methoxy substituents (newly synthesized OCH3TMPA) (Fig. 1) were included in the series to serve as appropriate controls, wherein the methyl groups or methoxy group may influence the Cu-geometry in a similar manner as in the other derivatives, however without any hydrogen bonding interaction. The mixed derivative, NH2,CH3TMPA, was newly synthesized (Fig. S1†)§ to probe whether both amino moieties in LS-(NH2)2TMPA interact with the O–O peroxo core (and to assess if the second amino group doubles the magnitude of the H-bonding effects or interacts to a lesser degree).
Results collected in this work, further described below, (1) demonstrate the balance between sterics and H-bonding in developing appropriate synthetic systems to model behaviors featured in metalloenzymes, (2) establish the importance of corresponding controls in verifying novel synthetic models, and (3) show evidence supporting the H-bonding capabilities of the CcO-inspired low-spin heme–peroxo–copper complexes bearing the ligands NH2TMPA, (NH2)2TMPA, and NH2,CH3TMPA.
Synthesis and UV-vis spectroscopic characterization
The spectroscopic properties of the title compounds are evaluated in the context of established (heme-)copper chemistry.26,27,33,36,37,52–56 Following well-known procedures, the high-spin heme–peroxo–copper complexes ([(F8)FeIII–(O22–)–CuII(XTMPA)]+, Fig. S4†) were generated by bubbling dioxygen through an equimolar mixture of the reduced metal–ligand complexes (Fig. 5 and S4†);36 these all are stable at –90 °C in MeTHF, where the [(XTMPA)CuI]+ precursors utilized were B(C6F5)4– ( BArF) salts.§ Addition of one equivalent of DCHIm affords the corresponding low-spin analogs [(DCHIm)(F8)FeIII–(O22–)–CuII(XTMPA)]BArF (LS-XTMPA, Fig. 1 and 5) which are the focus of this work as most heme metalloenzymes can only function with (and depend on) a proximal axial base.5,6,28
Fig. 5. Established synthetic approach for heme–peroxo–copper complexes, wherein addition of dioxygen to the reduced iron and copper complexes (in a 1 : 1 stoichiometry) results in formation of the high-spin complexes, HS-XTMPA. Addition of 1 equiv. of 1,5-dicyclohexylimidazole (DCHIm, which acts as an axial base) affords low-spin heme–peroxo–copper species (LS-XTMPA, with X as depicted in Fig. 1). As shown in the bottom left inset (as one possible example of the present study), we are investigating the influence of H-bonding on the peroxo core.

The binding mode of the peroxide ligand in the parent HS-TMPA complex has been previously well established to possess a μ-η2:η1 (side-on to iron, end-on to copper) bridge (Fig. 5), based on a great deal of spectroscopic data, DFT calculations, and precedent from an early X-ray structure of an analog compound characterized by Naruta and coworkers.33,40,57,58 The high-spin state of the parent heme–peroxo–copper complex HS-TMPA (overall S = 2) was also previously confirmed by the rR spin state marker band, ν2,31 by 2H-NMR spectroscopy (observation of a paramagnetic peak using pyrrole-deuterated F8 porphyrin)59 and via EPR spectroscopy (the observed X-band silent spectrum was ascribed to anti-ferromagnetic coupling between the iron and copper ions).59 The peroxo binding mode in the low-spin heme–peroxo–copper complex LS-TMPA (Fig. 5) was previously determined to be end-on to both copper and iron (trans-μ-η1:η1) based on the combination of spectroscopic data, DFT calculations, and analogy to previously characterized low-spin species.36 Based on DFT energy-minimized structures for HS- vs.LS-TMPA,33,37,59,60 the end-on formulation in the low-spin species also results in an elongated metal–metal distance (HS-TMPA: r(Fe···Cu) = 3.982 Å (3.72 Å by EXAFS),37LS-TMPA: r(Fe···Cu) = 4.485 Å).
Eight high-spin heme–peroxo–copper complexes [(F8)FeIII–(O22–)–CuII(XTMPA)]+ were newly generated at –90 °C in MeTHF (structures depicted in Fig. S4†), and subsequent UV-vis monitoring of their formation indicated they all had comparable spectra with a Soret ∼416 nm and a split Q-band λmax ∼ 560 nm and a diagnostic ∼650 nm absorption feature (a porphyrin to iron transition often called CT2 in heme literature)61 regardless of the TMPA substituent (Table 1, Fig. S5 and S6† in blue). Due to the comparable UV-vis spectra of these HS-XTMPA complexes, [(F8)FeIII–(O22–)–CuII(XTMPA)]+ (Fig. S4–S6†), the paramagnetic 2H-NMR shifts observed (Fig. S7† in blue), and their silent EPR spectra, these new species were confirmed to be high-spin. It is of note that the similar UV-vis spectra, paramagnetic 2H-NMR signal, and silent EPR spectra (X-band) are consistent with many other high-spin heme–peroxo–copper complexes previously reported (not just with HS-TMPA).27,36,53,59,62
Table 1. Spectroscopic properties of low-spin heme–peroxo-complexes with TMPA-derived copper chelates (br = broad; f.d. = Fermi doublet) a .
| (DCHIm)F8FeIII–O2–CuII(L), Identity of L | UV-vis (nm) | Raman 16O2 (cm–1) |
|||
| ν(O–O) | ν(Fe–O) | ν(Cu–O) | v 2;v4 | ||
| TMPA | 426, 535, 850, 950, 1020 (br) | 812 | 623 | 535 | 1572; 1364 |
| NH2TMPA | 426, 536, 790, 900 (br) | 775 | 625 | 533 | 1570; 1364 |
| (NH2)2TMPA | 425, 538, 785, 870 (br) | 760 | 614 | 506 | 1571; 1367 |
| NH2,CH3TMPA | 425, 533, 789, 897 (br) | 765 | 610 | 510 | 1571; 1367 |
| CH3TMPA | 422, 534, 855, 980 (br) | 789 (f.d.) | 612 | 525 | 1570; 1363 |
| (CH3)2TMPA | 420, 538, 845, 940 (br) | 776 | 593 | 501 | 1570; 1363 |
| PVTMPA | 429, 536, 840 (br) | 806 | 573 | 489 | 1571; 1365 |
| A2TMPA | 426, 535, 823 (br) | 802 | 580 | 484 | 1571; 1366 |
| OCH3TMPA | 424, 539, 853, 946 (br) | 805 | 575 | 483 | 1569; 1365 |
aSee the ESI for the UV-vis spectra (Fig. S5 and S6) and for the (16O2–18O2) data and raw rR spectra (Fig. S8).
The UV-vis spectra show a red shift in both the Soret (∼7–12 nm) and Q-band (∼27 nm) after addition of one equivalent of DCHIm (Fig. 5) to the HS-XTMPA complexes (Fig. S5 and S6† in green), consistent with previously reported shifts of synthetic heme–peroxo–copper complexes going from HS to LS-XTMPA species.26,36,62 Thus, with eight new HS (Fig. S4†) and then eight corresponding LS complexes, a total of sixteen new heme–peroxo–copper species are described here. However, this work focuses particularly on the imidazole-ligated low-spin complexes, which possess a heme structure comparable to the CcO binuclear active site (Fig. 1). The low-spin state (S = 0) of these novel LS-XTMPA complexes was confirmed by the rR spin state marker band, ν2 (Table 1),31 by a diamagnetic 2H-NMR signal from employing the d8(pyrrole)-F8 analog (i.e., [(DCHIm)–(d8F8)FeIII–(O22–)–CuII(XTMPA)]BArF, thus the observed signal is from the pyrrole deuterons, Fig. S7† in green), and from silent EPR spectra being observed for all the complexes, indicating antiferromagnetic coupling between the low-spin iron(iii) and copper(ii) sites, mediated by the peroxo bridging ligand. These findings are consistent with the previous spectral characterization of the parent heme–peroxo–copper complex LS-TMPA.36,40,59 The UV-vis spectral changes from the high-spin to low-spin species in each case are analogous to those reported for similar model complexes (vide infra), which have been shown to be concomitant with a change in binding mode from μ-η2:η1 to trans-μ-η1:η1.26,27,33,36,37,59,60 Therefore, we propose at this time that the core peroxo binding mode in the novel LS-XTMPA complexes is also end-on coordinated to both metal ions, as depicted in Fig. 1 and 5.
All of the LS-XTMPA complexes have similar UV-vis spectral features with a Soret absorption around 425 nm and a Q-band λmax around 533 nm regardless of the Cu ligand identity (Fig. S5 & S6,† Table 1); however, the absorption bands observed at lower energy show variation in both wavelength and intensity (Fig. 6 and S6† & Table 1). These unique low energy bands have been attributed to peroxo-to-Fe charge transfer (CT) transitions and serve as a diagnostic fingerprint for all synthetic low-spin heme–O22––Cu species of this nature characterized to date,36,63 providing valuable evidence confirming the low-spin state and showing how variations in Cu ligand substitution influence the peroxo core. Relative to the parent LS-TMPA and the steric control compounds, LS-CH3TMPA, LS-(CH3)2TMPA, and LS-OCH3TMPA, the CT bands for LS-NH2TMPA, LS-(NH2)2TMPA, and LS-NH2,CH3TMPA are observed at higher energy (i.e., amine substitution on the pyridyl donor(s) results in a blue shift; this ligand-to-metal charge transfer (LMCT) blue shift has also been previously reported by Masuda and coworkers for the copper(ii)-azido and dicopper(ii) trans-peroxo complexes bearing the ligands NH2TMPA and (NH2)2TMPA).49,50 This is attributed to the H-bond interaction lowering the peroxo π* donor orbital energies more than the Fe acceptor orbitals (see Fig. 6, S6† & Table 1). Importantly, this diagnostic marker, the blue shift of the low energy absorption λmax, increases systematically with the number of –NH2 substituents (Fig. 6, S6† & Table 1), providing compelling evidence that all –NH2 groups employed interact with the peroxo core. In line with these trends, LS-NH2,CH3TMPA shows low energy features similar to LS-NH2TMPA, while those of LS-(NH2)2TMPA are shifted to even higher energy, suggesting that both amino groups of LS-(NH2)2TMPA are H-bonding to the O–O peroxo core and that it is not due to the additional sterics from incorporating a second functionality on the TMPA framework (Fig. 6, S6† & Table 1). These conclusions about H-bonding to the peroxo core by the ligand-amino pendants are supported by resonance Raman spectroscopic interrogation (vide infra).
Fig. 6. Zoomed in UV-vis spectra of the low-energy region (∼700–1000 nm) for the low-spin heme–peroxo–copper complexes with various TMPA-based copper ligands, demonstrating the characteristic blue shift observed with substituents that H-bond to the O–O peroxo core. Bottom left: LS-TMPA (black), LS-CH3TMPA (blue), and LS-(CH3)2TMPA (orange). Bottom right: LS-TMPA (black), LS-NH2TMPA (red), and LS-(NH2)2TMPA (green).

Masuda and co-workers49 previously demonstrated increased H-bonding capabilities from PVTMPA (with pivalamido potential H-bonding moiety (Fig. 1)) relative to NH2TMPA by comparing their respective dicopper(ii) trans-peroxide complexes; this is expected since –pivalamido groups are more acidic than –amino substituents. Thus, to optimize the amount of H-bonding interactions between the intramolecular functionality and the O–O peroxo core, the low-spin heme–peroxo–copper complex with PVTMPA (i.e., [(DCHIm)(F8)FeIII–(O22–)–CuII(PVTMPA)]BArF, LS-PVTMPA) was synthesized (Fig. 1). The UV-vis spectrum exhibits similar Soret (429 nm) and Q-band λmax (533 nm) absorptions as all the other LS-XTMPA derived complexes (Table 1), however, the low-energy region exhibits uniquely one broad intense peak around 840 nm (Fig. S5G, S6G† & Table 1). While this effect could indicate H-bonding to the peroxo core, it is notable that this low-energy band for LS-PVTMPA is markedly more intense than observed for the other LS-XTMPA complexes (ε ∼ 6000 mM–1 cm–1vs. 2000–4000 mM–1 cm–1). This deviation may indicate a structural change (or distortion of the dihedral Fe–O–O–Cu angle), that is likely imparted by the presence of the very bulky tert-butyl group. To probe this possibility, a new ligand with a less bulky carbonyl substituent (a methyl instead of a tert-butyl group) was synthesized (A2TMPA, with a 6-pyridyl –acetylamido moiety) and the corresponding DCHIm-bound low-spin heme–peroxo–copper complex was prepared (Fig. 1). The UV-vis spectrum of this complex ([(DCHIm)(F8)FeIII–(O22–)–CuII(A2TMPA)]BArF, LS-A2TMPA) was similar to LS-PVTMPA, exhibiting one broad, intense low-energy feature around 823 nm (Fig. S5F, S5G, S6F and S6G,† Table 1). This indicates that the amido functionality impacts the peroxo core in a manner that is distinct from amino or methyl groups alone, potentially arising from variation in the geometric and electronic structure around the Cu (vide infra). This finding demonstrates the careful balance between sterics and electronics (i.e., H-bonding) in our synthetically derived heme–peroxo–Cu complexes (highlighting the intricacies in developing synthetic models), which nature has also mastered in its design of metalloenzymes in order to dictate biologically required catalytic transformations.
These diagnostic low-energy CT bands (Fig. 6 & S6† and Table 1) provide valuable evidence for how variations in Cu ligand substitution influence the peroxo core and offer indirect evidence for intramolecular H-bonding interactions for the three novel low-spin heme–peroxo–copper complexes containing amino groups (LS-NH2TMPA, LS-(NH2)2TMPA, and LS-NH2,CH3TMPA, Fig. 1), and potentially two amido-containing complexes (LS-PVTMPA and LS-A2TMPA, Fig. 1). As other research labs, such as those of Masuda, Nocera, and Dey,50,64–67 have demonstrated inter- and intramolecular H-bonding in dicopper–peroxide and heme (hydro)peroxide complexes through the use of resonance Raman spectroscopy, we have therefore also evaluated our new low-spin heme–peroxo–copper complexes (Fig. 1) using rR spectroscopy and corresponding computational analysis (vide infra).
Impact of H-bonding and sterics on M–O and O–O bonds in the peroxo core as determined from rR spectroscopy
Insight into the effect of these H-bonding interactions is provided by rR spectroscopy, which can serve as a valuable probe of O–O and M–O bond strengths, and distinguish between H-bonding to the proximal or distal O-atom (with respect to Fe).26,27,68,69 Importantly, we find that significant differences in the rR data for these low-spin complexes are observed (vide infra) as a consequence of the derivatization of the TMPA ligands as outlined in Fig. 1.
Among both high-spin and low-spin synthetic model complexes compiled from 15 reports in the literature, a Cu–O stretch has been observed in only three LS species out of 24 HS and LS complexes.36,39,45,46,48,53,55,56,59,70–75 Further, of those three, only one employed a copper ligand that was not TMPA (the other two being the HS and LS parent TMPA complexes).36,59,72 Thus, this report more than doubles the description and characterization of known synthetic low-spin heme–peroxo–copper complexes in the literature, with 8 newly prepared species. The remarkably strong resonance enhancement of both the Fe–O and Cu–O stretches with the TMPA ligand may be due to vibrational coupling and/or the presence of an LMCT band masked by the Soret absorption used for excitation. Thus, the rR data on these complexes with derivatized TMPA ligands afford critical insights into the nature of the O–O peroxo core.
It is highly relevant to note that in order to mimic the unique tyrosine–histidine crosslink coordinated to the copper ion in CcO, the Karlin and Naruta labs, separately, have reported on synthetic heme–peroxo–copper complexes with a phenol-imidazole crosslink moiety (Fig. 7).70,74 Interestingly, no H-bonding interactions were observed between the phenol and peroxo core, based on rR spectroscopic data (i.e., no effect on the O–O vibrational frequency was observed). This would likely also be the case in the CcO active site, where the nearby tyrosine residue is too far away (∼6 Å) to directly H-bond to the O–O peroxo core of a putative IP intermediate (Scheme 1). However, an H-bonded network of interstitial water molecules has been suggested, in many computational studies,28,76,77 to provide the means for proton transfer (from tyrosine) and to facilitate O2 reductive cleavage. Addressing the growing interest in understanding possible interactions with such active site water molecules, this study presents the first demonstration of intramolecular hydrogen bonding in a synthetic heme–peroxo–copper model system incorporating H-bonding groups.
Fig. 7. Previously reported high-spin heme–peroxo–copper complexes and their O–O vibrational frequencies as detected by rR spectroscopy. Incorporation of the phenol-imidazole moiety (vs. the parent TMPA complex) showed a similar ν(O–O) stretch, suggesting the phenol is too far away to directly interact with the peroxo core (similar to what is observed in CcO), providing indirect evidence for an interstitial water.

As mentioned, significant differences were observed in the rR data (Fig. 8 and S8† and Table 1)¶ for these low-spin complexes with derivatized TMPA ligands (i.e., LS-XTMPA, Fig. 1). The data indicate that increasing steric factors (i.e., the presence of ligand methyl-substituents) consistently results in weaker O–O, Fe–O, and Cu–O bonds, based on the progressively lower bond stretches observed for LS-TMPAvs.LS-CH3TMPAvs.LS-(CH3)2TMPA, as well as LS-NH2TMPAvs.LS-NH2,CH3TMPA (Fig. 8, S8A–C, & S8E,† and Table 1). In contrast, substitution of an H-bonding amino group into the complex (i.e., LS-TMPAvs.LS-NH2TMPA, LS-NH2,CH3TMPA) appears to minimally impact the Fe–O and Cu–O bond strengths, while greatly weakening the O–O bond. This is a particularly important finding, as a similar decrease in the ν(O–O) vibrational frequency was observed for an H-bonded adduct between a heme–peroxo–Cu model complex and an exogenous phenol (a shift from 870 to 827 cm–1 upon addition of p-nitrophenol, Scheme 2).26
Fig. 8. Comparison of rR data (16O2–18O2) for the LS-XTMPA complexes, which are normalized to equal ν(Fe–O) and ν(O–O) intensities to aid in visual comparison. Isotope-sensitive features are labelled by their frequency in 16O2. Note that some modes (e.g. 789 cm–1 in red spectrum) are split by Fermi coupling. Asterisks (*) denote a ferric superoxo impurity. Laser excitation was 413 nm or 780–890 nm, see ESI.† .

Scheme 2. Previously reported hydrogen-bonding adduct that formed upon addition of 10 equivalents of 4-nitrophenol to a low-spin heme–peroxo–copper complex. Resonance Raman results reported were consistent with the rR in this work (Table 1), where a decrease is observed in the ν(O–O) when H-bonding groups interact with the peroxo core.

The differences observed between LS-CH3TMPA and LS-NH2TMPA are attributable to H-bonding in the latter, since there is no electronic difference between the two ligands (as determined from CuII/I reduction potentials, as mentioned above§). Interestingly, incorporating a second H-bonding group (as in LS-(NH2)2TMPA) acts to weaken all three core modes (Fig. 8 and S8D,† & Table 1) and in fact yields the lowest ν(O–O) among the complexes studied herein.
Resonance Raman data for LS-PVTMPA showed significantly lower ν(Fe–O) and ν(Cu–O) modes (573 cm–1 and 489 cm–1, respectively), compared to values for LS-TMPA, but a relatively high ν(O–O) (806 cm–1), close to that for LS-TMPA (812 cm–1, Fig. S8G† and Table 1). The less sterically hindered amido-substituted complex LS-A2TMPA nevertheless yielded similar rR data to LS-PVTMPA (Fig. S8F† and Table 1), suggesting that these two complexes impact the peroxo core in a comparable manner. However, the rR data obtained for LS-OCH3TMPA (steric control bulkier than CH3TMPA) are remarkably similar to LS-PVTMPA and LS-A2TMPA, including isotope-sensitive modes as well as heme bands (Fig. S8H† and Table 1), suggesting that the effects of the PVTMPA ligand on the peroxo core involve little or no H-bonding, but are predominantly steric in nature.
Further, valuable information is provided by resonance Raman data profiled over the low energy (700–900 nm) region (with excitation at various wavelengths, Fig. S9†). In particular, the complexes LS-(NH2)2TMPA and LS-NH2,CH3TMPA show preferential enhancement of the ν(Fe–O) and ν(O–O) modes at lower energy excitation, with enhancement of the ν(Cu–O) and ν(Fe–O) (and some ν(O–O)) observed at higher energy excitation. This indicates the presence of two distinguishable LMCT absorption bands (illustrated for LS-(NH2)2TMPA in Fig. S10†), wherein the lower energy transition involves excitation from a peroxo orbital that is donating primarily into Fe, while the higher energy transition involves a peroxo orbital donating into both metals. While the remaining complexes studied herein did not exhibit rR profiles indicating two distinct bands, it is worth noting that the complexes LS-PVTMPA and LS-A2TMPA yielded exceptionally strong ν(Fe–O) and ν(O–O) enhancement, but very weak Cu–O enhancement, suggesting primarily O22– to Fe CT character.
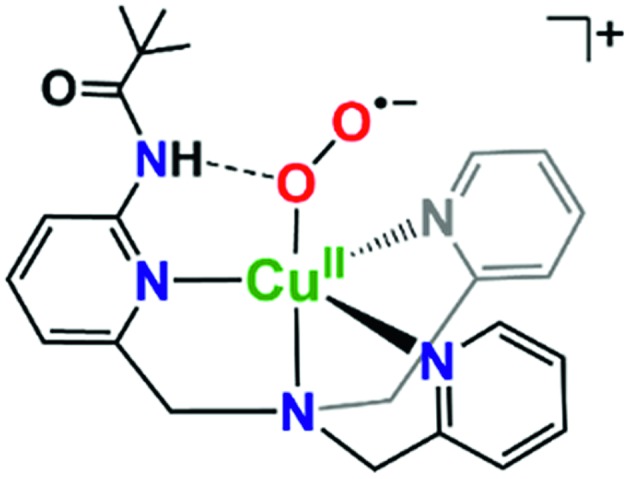
It is valuable to note that our analysis leading to the conclusion that the pivalamido group in LS-PVTMPA does not or only weakly H-bonds to the peroxo bridging ligand (and see the DFT calculations, below) stands in strong contrast to what we know for the cupric superoxo complex (without any heme present) with the PVTMPA ligand, Fig. 9.51,78 The reduced copper(i) complex, [(PVTMPA)CuI]+ reacts with O2 at or below –125 °C in MeTHF giving [(PVTMPA)CuII(O2˙–)]+ with rR stretching values of ν(Cu–O) = 482 {Δ18O2 = –20} cm–1 and ν(O–O) = 1130 {Δ18O2 = –63} cm–1, clearly shifted significantly from the values for the parent superoxide complex [(TMPA)CuII(O2˙–)]+ possessing no H-bonding modalities, ν(Cu–O) = 463 {Δ18O2 = –20} and ν(O–O) = 1119 {Δ18O2 = –61} cm–1.51Fig. 9 depicts the structure assigned to [(PVTMPA)CuII(O2˙–)]+ based on DFT calculations and other spectroscopic or physical characterization. Clearly, the presence of the heme moiety laying in close proximity to the pivalamido tert-butyl group in LS-PVTMPA, inhibits H-bonding, vide infra.
Fig. 9. Intramolecular H-bonding interactions in cupric superoxo complexes, such as in [(PVTMPA)CuII(O2˙–)]+ depicted here, result in enhanced reactivity towards phenolic (O–H) substrates.51,78 .
Electronic insights from DFT calculations: orbital considerations for H-bonding effects
In order to better understand how hydrogen bonding impacts the heme–peroxo–copper core, DFT calculations were performed to correlate to the spectroscopic data. Geometry optimization for LS-TMPA‖ and LS-NH2TMPA reveals distinct changes in the O–O, Fe–O, and Cu–O bond lengths that provide good agreement with the trends observed in the rR data (Table 1). Analytical frequency calculations support the observed trends, although vibrational modes are often highly mixed in the calculations. Therefore, predicted changes in vibrational frequency were also computed based on calculated structural changes using Badger's Rule (Table S3†), providing a direct correlation between bond length and stretching frequency for two-atom-centered vibrational modes.
To evaluate how the –NH2 group in LS-NH2TMPA interacts with the peroxo core, four principle orientations were considered, wherein the H-bond is directed toward either OCu (Fig. 10A and 10B) or OFe (Fig. S10A and B†), and interacting via the O22– π*v (perpendicular to Fe–O–O plane, Fig. 10A, 11B & S11A†) or the O22– π*σ (in the Fe–O–O plane, forming the metal-O σ-bonds, Fig. 10B, 11A & S11B†) orbital. Based on the calculations, binding to the OCu atom is favored by ∼2 kcal mol–1 (vs. OFe), and results in a substantially weaker O–O bond, slightly weaker Cu–O bond, and slightly stronger Fe–O bond, relative to LS-TMPA (consistent with the rR data). In contrast, H-bonding to the OFe substantially weakens the Fe–O bond and strengthens the Cu–O bond, which does not correlate with the actual rR data obtained. Calculations show that H-bonding via the O2 π*v orbital is favored by ∼5 kcal mol–1 (vs. O2 π*σ), although it is of note that both interactions yield similar effects (but greater in magnitude for O2 π*ν binding). Taken together, these results suggest that the –NH2 moiety in LS-NH2TMPA H-bonds to OCuvia the π*v orbital (Fig. 11B).
Fig. 10. Optimized DFT structures of LS-NH2TMPA (top: (A) and (B)) and LS-(NH2)2TMPA (bottom: (C) and (D)) indicating which orientation of the –NH2 moieties is preferred in H-bonding to the peroxo core.

Fig. 11. Orbital diagram for the interaction of the H-bonding amino moiety with the π* orbitals (π*σ, (A), π*ν, (B)) of the peroxo core. The latter (B) interaction is found to be the most important, see the text.

To fully understand how the H-bonding interaction impacts the core vibrations, we evaluated both structural and electronic consequences of the H-bond to the O2 π*v orbital. Regarding the structural effects of H-bonding in LS-NH2TMPA, the peroxo core rearranges to a more obtuse dihedral angle (from Fe–O–O–Cu = 141° in LS-TMPA to 152° in LS-NH2TMPA, Fig. S12†), orienting the H-bond vector to be nearly orthogonal to both M–O bonds (minimizing its impact on peroxo-to-metal σ donation). While the wider Fe–O–O–Cu dihedral angle would decrease O–O π*σ donation into Fe due to competition with donation (α-spin) into Cu, it orients the O22– π* orbitals to more efficiently overlap both Fe dz2 and dπ orbitals, which would increase donation into Fe.
With respect to the electronic effects, the H-bond lowers the energy of the peroxo π* orbitals and decreases donation into both metals, resulting in a net transfer of electron density from Fe and Cu to O22–, which weakens the O–O bond. However, the π*σ orbital (which donates into both the Fe dz2 (α/β) and Cu dz2 (α), where α-spin Fe is antiferromagnetically coupled to β-spin Cu), exhibits a slight increase in donation into Fe. Since the σ donation from O22– contributes more to the Fe–O bond strength than π donation (which decreases with H-bonding), the overall effect of the H-bond is to strengthen the Fe–O bond. Taken together, these findings demonstrate that the impact of H-bonding to the OCu atom of the peroxo core observed in LS-NH2TMPA (weaker O–O and Cu–O bonds, slightly stronger Fe–O bond) is attributable to a combination of electronic and structural effects.
The DFT calculations provide particularly valuable insights into the impact of H-bonding to the O2 π*vversus the O2 π*σ orbital with respect to O–O cleavage chemistry. As has been previously shown,27 O–O cleavage involves electron transfer from the Fe dyz orbital into the O2 σ* (which have π-symmetry overlap) and formation of a FeIV O product. The optimized structures (shown in Fig. 11A and B) reveal that binding via the O2 π*v results in a longer O–O bond and shorter Fe–O bond than O2 π*σ binding, suggesting weaker and stronger bonds, respectively. Additionally, H-bonding to O2 π*v lowers the energy of the O2 σ* orbital to a greater extent than H-bonding to O2 π*σ, resulting in a smaller energy gap between the O2 σ* and Fe dyz orbital. Applying these trends to O–O cleavage chemistry, which involves formation of a FeIV O product, these results suggest that H-bonding to the OCuatom via the O2π*vorbital (see Fig. 11B ) gives the most effective orientation for activating O–O cleavage.
Geometry optimizations of LS-(NH2)2TMPA yielded two possible structures (energetic minima) differing in H-bond orientation, one with 2 H-bonds to OCu (with N(H)···OCu distances of 2.80 and 2.83 Å and ∠OCuHN of 156.6 and 161.0°), and the other comprising of 1 H-bond to OCu and 1 H-bond to OFe (with an N(H)···OCu distance of 2.84 Å, a ∠OCuHN of 162.4°, an N(H)···OFe distance of 2.76 Å and a ∠OFeHN of 168.4°; Fig. 10C, D and Table S2†). These were found to be very similar in energy (with the former being ∼1 kcal mol–1 lower); a structure with two H-bonds to OFe was sufficiently higher in energy, such that one H-bond consistently reoriented to bind OCu upon unconstrained optimization. Comparing the OCu,OCu and OCu,OFe H-bonded structures, the former exhibits a weaker Cu–O bond and stronger Fe–O bond (as expected), as well as a weaker O–O bond. Relative to LS-NH2TMPA, both structures agree with the observed trends in the rR data (O–O, Fe–O, and Cu–O bonds all weaken, but by different magnitudes). However, comparing the rR data on LS-(NH2)2TMPA to LS-NH2,CH3TMPA suggests that replacing the methyl group in the latter with an amine results in slightly weaker O–O, stronger Fe–O, weaker Cu–O bonds, based on the differences in stretching frequencies (Table 1). Considering the findings for LS-NH2TMPA, these vibrational effects implicate H-bonding to the OCu atom (since binding OFe would be expected to weaken the Fe–O bond). Nevertheless, it is interesting that while incorporating one –NH2 group (as in LS-NH2TMPA) weakens the O–O bond and minimally affects the M–O bonds (even strengthening the Fe–O), addition of the second –NH2 group acts to weaken all three core modes (based on the rR data, Table 1).
From the rR data above, minor increases in sterics (–CH3 substitution) cause a decrease in the O–O, Fe–O, and Cu–O vibrations (Table 1), indicating that the bonds weaken and elongate. This behavior can be replicated computationally by constraining and systematically elongating the Fe···Cu distance in LS-TMPA, which results in weakening of all three core modes, therefore suggesting that the steric consequence of the methyl group in CH3TMPA is to push the Fe and Cu moieties apart.
However, incorporating substituents with greater steric contributions, as in the amido-based groups of PVTMPA and A2TMPA, resulted in a different behavior of the core vibrational frequencies. As shown above, these bulkier ligands yielded very low frequency M–O stretches and high frequency O–O stretches, suggesting weak M–O bonds and strong O–O bonds. The DFT-optimized structure of LS-PVTMPA exhibits an exceptionally small Fe–O–O–Cu dihedral angle (100°), a wide Fe–O–O angle (119°), and a seemingly weak H-bonding interaction to the OCu atom of the peroxo (O···N(H) = 2.90 Å; the ∠OCuHN = 164.4°) via the O22– π*σ orbital (Fig. 12A). In contrast, LS-NH2TMPA showed a wider Fe–O–O–Cu dihedral angle (152°), smaller Fe–O–O angle (115°) and a stronger H-bond (O···N(H) = 2.72 Å, vide supra) via the O22– π*v orbital.
Fig. 12. Optimized structure for (A) LS-PVTMPA and (B) LS-A2TMPA. Note that the Cu geometry is now skewed toward square pyramidal (τ5 = 0.22 and 0.16 for (A) and (B), respectively), unlike the optimized structures for LS-NH2TMPA (Fig. S11†). Further, note the distorted dihedral angle of 100° and 107° (for (A) and (B), respectively) vs. 141° for LS-TMPA (Fig. S13A†).

In agreement with the observed vibrational frequencies, the optimized LS-PVTMPA structure exhibits relatively long M–O bonds and a short O–O bond (Table S5†), which would suggest low ν(M–O) and high ν(O–O) stretches (according to Badger's rule). Furthermore, a wider Fe–O–O angle (as in LS-PVTMPA) would increase vibrational coupling of the Fe–O and O–O modes, resulting in even lower ν(Fe–O) and higher ν(O–O) frequencies. Interestingly, the optimized structure of LS-A2TMPA (Fig. 12B) exhibits a shorter O···N(H) distance (2.73 Å), but very similar core structure (with slightly shorter M–O and O–O bond lengths compared to LS-PVTMPA, in excellent agreement with the trend in vibrational frequencies from the rR data), suggesting that the H-bond in these amido complexes has a minor impact on the core bond strengths. Offering insight into the intense low-energy band observed by UV-Vis for LS-PVTMPA and LS-A2TMPA (vide supra), the calculations also show that H-bonding to the O2 π*σ orbital polarizes the O2 π*v orbital toward the OFe atom, which would result in a more intense LMCT band. Taken together, these results suggest that the effect of H-bonding to the peroxo core in the amido-appended complexes differs from the amino-appended complexes by a combination of structural and electronic aspects.
Conclusions
Hydrogen bonding is prevalent in biology, especially concerning dioxygen chemistry in metalloenzymes (i.e., globins, peroxidases, catalases, cytochrome P450's, oxidases).6 The effects of intramolecular H-bonding will allow for interesting mechanistic studies for reductive O2 cleavage; this could be invaluable in the design of more efficient fuel cells in which O2 reduction is the key reaction occurring at the cathode. This study has fully characterized eight novel synthetic low-spin heme–peroxo–copper models inspired by cytochrome c oxidase active site chemistry; overall 16 complexes have been newly synthesized, which more than doubles reported analogous complexes in the literature.
The present work demonstrates the flexibility to design and synthesize various novel TMPA-based ligands and observe a range of effects (elucidated here), reinforcing the importance of a careful balance between sterics and hydrogen bonding being critical in enzyme active site reactions, those requiring specific H-bonding or protonation situations. This is especially shown using the ligands containing appended amido moieties, which have been shown to be strong H-bonding groups in previous studies (with copper-only complexes) but herein perturb the heme–peroxo–copper complex geometry, hindering significant H-bonding to the peroxo core.
The effects of intramolecular H-bonding on the heme–peroxo–Cu core were examined by UV-vis and rR spectroscopies and DFT calculations. It is shown that the H-bonding moieties (–NH2, –(NH2)2, and –NH2,CH3) reproduce experimental data for intermolecular H-bonding effects reported for a related heme–peroxo–copper systems, and preferentially interact with the OCu atom of the peroxo; these findings offer a critical correlation to studies showing that an H-bonding interaction can lower the O–O cleavage barrier.26,27 Importantly, while the nature of the H-bonding interaction could not be well understood in past studies due to complications including poorly defined Cu ligation and entropic costs, the ligand system employed herein provides a well-defined platform that is reliably modelled by computational means. This has delivered critical insight into an orientational anisotropy in H-bond activating the O–O bond for cleavage. Furthermore, while the metal–oxygen bond vibrations (especially Cu–O) are not observed for most synthetic heme–peroxo–copper species, these low-spin complexes for the first time allowed direct insight into the Cu–O bond; this is a paramount step towards future development of a more biomimetic, efficient model of CcO (with full access to modifications of new ligand systems and a deeper understanding of what is occurring during O2 reduction studies). Overall, systematic control over H-bonding ability of the XTMPA ligands provides a foundation to draw conclusions about the effects of H-bonding on the thermodynamic stability, electronic structure, and reactivity of corresponding heme–peroxo–copper complexes.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The research support of the U.S. National Institutes of Health (GM60353 to K. D. K., DK031450 to E. I. S.) is gratefully acknowledged.
Footnotes
†Electronic supplementary information (ESI) available: Synthetic and analytical details (methodologies and UV-vis, EPR, NMR, rR, and IR spectra). See DOI: 10.1039/c8sc05165h
§See ESI† for more details.
¶It should be noted that enhancement of all three core vibrational modes simultaneously (e.g., with 413 nm excitation) may indicate significant coupling between these modes, and that this coupling can contribute to the observed vibrational frequencies (in addition to the bond strength).79 However, the relative enhancement of the Fe–O/O–O/Cu–O modes at 413 nm excitation remains similar across these LS complexes (except the bulkier derivatives noted in the text), and it is therefore unlikely that the amount of coupling varies drastically.
‖Adapted from ref. 36, but re-optimized here.
References
- Shook R. L., Borovik A. S. Inorg. Chem. 2010;49:3646–3660. doi: 10.1021/ic901550k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook S. A., Borovik A. S. Acc. Chem. Res. 2015;48:2407–2414. doi: 10.1021/acs.accounts.5b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook S. A., Hill E. A., Borovik A. S. Biochemistry. 2015;54:4167–4180. doi: 10.1021/acs.biochem.5b00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borovik A. S. Acc. Chem. Res. 2005;38:54–61. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]
- Adam S. M., Wijeratne G. B., Rogler P. J., Diaz D. E., Quist D. A., Liu J. J., Karlin K. D. Chem. Rev. 2018;118:10840–11022. doi: 10.1021/acs.chemrev.8b00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X., Groves J. T. Chem. Rev. 2018;118:2491–2553. doi: 10.1021/acs.chemrev.7b00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso-Prieto M., Biarnés X., Vidossich P., Rovira C. J. Am. Chem. Soc. 2009;131:11751–11761. doi: 10.1021/ja9018572. [DOI] [PubMed] [Google Scholar]
- Wikström M., Krab K., Sharma V. Chem. Rev. 2018;118:2469–2490. doi: 10.1021/acs.chemrev.7b00664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa S., Shimada A. Chem. Rev. 2015;115:1936–1989. doi: 10.1021/cr500266a. [DOI] [PubMed] [Google Scholar]
- Quist D. A., Diaz D. E., Liu J. J., Karlin K. D. J. Biol. Inorg Chem. 2017;22:253–288. doi: 10.1007/s00775-016-1415-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon E. I., Heppner D. E., Johnston E. M., Ginsbach J. W., Cirera J., Qayyum M., Kieber-Emmons M. T., Kjaergaard C. H., Hadt R. G., Tian L. Chem. Rev. 2014;114:3659–3853. doi: 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg M. R. A. Biochemistry. 2016;55:489–500. doi: 10.1021/acs.biochem.5b01205. [DOI] [PubMed] [Google Scholar]
- Aki M., Ogura T., Naruta Y., Le T. H., Sato T., Kitagawa T. J. Phys. Chem. A. 2002;106:3436–3444. [Google Scholar]
- McCauley K. M., Vrtis J. M., Dupont J., van der Donk W. A. J. Am. Chem. Soc. 2000;122:2403–2404. [Google Scholar]
- Pratt D. A., Pesavento R. P., van der Donk W. A. Org. Lett. 2005;7:2735–2738. doi: 10.1021/ol050916g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L., Liu J., Mills D. A., Proshlyakov D. A., Hiser C., Ferguson-Miller S. Biochemistry. 2009;48:5121–5130. doi: 10.1021/bi9001387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrik I. D., Davydov R., Ross M., Zhao X., Hoffman B., Lu Y. J. Am. Chem. Soc. 2016;138:1134–1137. doi: 10.1021/jacs.5b12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg M. R. A., Siegbahn P. E. M., Wikström M. Inorg. Chem. 2003;42:5231–5243. doi: 10.1021/ic034060s. [DOI] [PubMed] [Google Scholar]
- Ravikiran B., Mahalakshmi R. RSC Adv. 2014;4:33958–33974. [Google Scholar]
- Noodleman L., Han Du W. G., Fee J. A., Götz A. W., Walker R. C. Inorg. Chem. 2014;53:6458–6472. doi: 10.1021/ic500363h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa S., Muramoto K., Shinzawa-Itoh K. Biochim. Biophys. Acta, Bioenerg. 2011;1807:1279–1286. doi: 10.1016/j.bbabio.2011.06.008. [DOI] [PubMed] [Google Scholar]
- Yoshikawa S., Muramoto K., Shinzawa-Itoh K., Mochizuki M. Biochim. Biophys. Acta, Bioenerg. 2012;1817:579–589. doi: 10.1016/j.bbabio.2011.12.012. [DOI] [PubMed] [Google Scholar]
- Dominelli-Whiteley N., Brown J. J., Muchowska K. B., Mati I. K., Adam C., Hubbard T. A., Elmi A., Brown A. J., Bell I. A. W., Cockroft S. L. Angew. Chem., Int. Ed. 2017;56:7658–7662. doi: 10.1002/anie.201703757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V., Wikström M. Biochim. Biophys. Acta, Bioenerg. 2016;1857:1111–1115. doi: 10.1016/j.bbabio.2016.02.008. [DOI] [PubMed] [Google Scholar]
- Muramoto K., Ohta K., Shinzawa-Itoh K., Kanda K., Taniguchi M., Nabekura H., Yamashita E., Tsukihara T., Yoshikawa S. Proc. Natl. Acad. Sci. 2010;107:7740–7745. doi: 10.1073/pnas.0910410107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam S. M., Garcia-Bosch I., Schaefer A. W., Sharma S. K., Siegler M. A., Solomon E. I., Karlin K. D. J. Am. Chem. Soc. 2017;139:472–481. doi: 10.1021/jacs.6b11322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer A. W., Kieber-Emmons M. T., Adam S. M., Karlin K. D., Solomon E. I. J. Am. Chem. Soc. 2017;139:7958–7973. doi: 10.1021/jacs.7b03292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg M. R. A., Siegbahn P. E. R. E. M. J. Comput. Chem. 2006;27:1373–1384. doi: 10.1002/jcc.20448. [DOI] [PubMed] [Google Scholar]
- Blomberg M. R. A., Borowski T., Himo F., Liao R.-Z., Siegbahn P. E. M. Chem. Rev. 2014;114:3601–3658. doi: 10.1021/cr400388t. [DOI] [PubMed] [Google Scholar]
- Poiana F., Von Ballmoos C., Gonska N., Blomberg M. R. A., Ädelroth P., Brzezinski P. Sci. Adv. 2017;3:e1700279. doi: 10.1126/sciadv.1700279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halime Z., Kieber-Emmons M. T., Qayyum M. F., Mondal B., Gandhi T., Puiu S. C., Chufán E. E., Sarjeant A. A. N., Hodgson K. O., Hedman B., Solomon E. I., Karlin K. D. Inorg. Chem. 2010;49:3629–3645. doi: 10.1021/ic9020993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuzumi S., Kotani H., Lucas H. R., Doi K., Suenobu T., Peterson R. L., Karlin K. D. J. Am. Chem. Soc. 2010;132:6874–6875. doi: 10.1021/ja100538x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiladi R. A., Chufán E. E., del Río D., Solomon E. I., Krebs C., Huynh B. H., Huang H.-W., Moënne-Loccoz P., Kaderli S., Honecker M., Zuberbühler A. D., Marzilli L., Cotter R. J., Karlin K. D. Inorg. Chem. 2007;46:3889–3902. doi: 10.1021/ic061726k. [DOI] [PubMed] [Google Scholar]
- Karlin K. D., Nanthakumar A., Fox S., Murthy N. N., Ravi N., Huynh B. H., Orosz R. D., Day E. P. J. Am. Chem. Soc. 1994;116:4753–4763. [Google Scholar]
- Hematian S., Garcia-Bosch I., Karlin K. D. Acc. Chem. Res. 2015;48:2462–2474. doi: 10.1021/acs.accounts.5b00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bosch I., Adam S. M., Schaefer A. W., Sharma S. K., Peterson R. L., Solomon E. I., Karlin K. D. J. Am. Chem. Soc. 2015;137:1032–1035. doi: 10.1021/ja5115198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Río D., Sarangi R., Chufán E. E., Karlin K. D., Hedman B., Hodgson K. O., Solomon E. I. J. Am. Chem. Soc. 2005;127:11969–11978. doi: 10.1021/ja043374r. [DOI] [PubMed] [Google Scholar]
- Halime Z., Kotani H., Li Y., Fukuzumi S., Karlin K. D. Proc. Natl. Acad. Sci. U. S. A. 2011;108:13990–13994. doi: 10.1073/pnas.1104698108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiladi R. A., Ju T. D., Lee D. H., Moënne-Loccoz P., Kaderli S., Neuhold Y. M., Zuberbühler A. D., Woods A. S., Cotter R. J., Karlin K. D. J. Am. Chem. Soc. 1999;121:9885–9886. [Google Scholar]
- Ghiladi R. A., Hatwell K. R., Karlin K. D., Huang H., Moënne-Loccoz P., Krebs C., Huynh B. H., Marzilli L. A., Cotter R. J., Kaderli S., Zuberbühler A. D. J. Am. Chem. Soc. 2001;123:6183–6184. doi: 10.1021/ja010602y. [DOI] [PubMed] [Google Scholar]
- Kopf M.-A., Neuhold Y.-M., Zuberbühler A. D., Karlin K. D. Inorg. Chem. 1999;38:3093–3102. [Google Scholar]
- Obias H. V., Van Strijdonck G. P. F., Lee D. H., Ralle M., Blackburn N. J., Karlin K. D. J. Am. Chem. Soc. 1998;120:9696–9697. [Google Scholar]
- Fox S., Nanthakumar A., Wikström M., Karlin K. D., Blackburn N. J. J. Am. Chem. Soc. 1996;118:24–34. [Google Scholar]
- Rivas J. C. M., Hinchley S. L., Metteau L., Parsons S. Dalton Trans. 2006:2316. doi: 10.1039/b516234c. [DOI] [PubMed] [Google Scholar]
- Sasaki T., Nakamura N., Naruta Y. Chem. Lett. 1998;27:351. [Google Scholar]
- Naruta Y., Sasaki T., Tani F., Tachi Y., Kawato N., Nakamura N. J. Inorg. Biochem. 2001;83:239–246. doi: 10.1016/s0162-0134(00)00170-7. [DOI] [PubMed] [Google Scholar]
- Liu J. G., Naruta Y., Tani F. Chem.–Eur. J. 2007;13:6365–6378. doi: 10.1002/chem.200601884. [DOI] [PubMed] [Google Scholar]
- Collman J. P., Herrmann P. C., Boitrel B., Zhang X., Eberspacher T. A., Fu L., Wang J., Rousseau D. L., Williams E. R. J. Am. Chem. Soc. 1994;116:9783–9784. [Google Scholar]
- Yamaguchi S., Wada A., Funahashi Y., Nagatomo S., Kitagawa T., Jitsukawa K., Masuda H. Eur. J. Inorg. Chem. 2003;2003:4378–4386. doi: 10.1021/ic035080x. [DOI] [PubMed] [Google Scholar]
- Wada A., Honda Y., Yamaguchi S., Nagatomo S., Kitagawa T., Jitsukawa K., Masuda H. Inorg. Chem. 2004;43:5725–5735. doi: 10.1021/ic0496572. [DOI] [PubMed] [Google Scholar]
- Bhadra M., Lee J. Y. C., Cowley R. E., Kim S., Siegler M. A., Solomon E. I., Karlin K. D. J. Am. Chem. Soc. 2018;140:9042–9045. doi: 10.1021/jacs.8b04671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiladi R. A., Hatwell K. R., Karlin K. D., Huang H.-W., Moënne-Loccoz P., Krebs C., Marzilli L. A., Cotter R. J., Kaderli S., Zuberbühler A. D. J. Am. Chem. Soc. 2001;123:6183–6184. doi: 10.1021/ja010602y. [DOI] [PubMed] [Google Scholar]
- Kim E., Helton M. E., Wasser I. M., Karlin K. D., Lu S., Huang H.-W., Moenne-Loccoz P., Incarvito C. D., Rheingold A. L., Honecker M., Kaderli S., Zuberbuhler A. D. Proc. Natl. Acad. Sci. U. S. A. 2003;100:3623–3628. doi: 10.1073/pnas.0737180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E., Chufán E. E., Kamaraj K., Karlin K. D. Chem. Rev. 2004;104:1077–1134. doi: 10.1021/cr0206162. [DOI] [PubMed] [Google Scholar]
- Chufán E. E., Mondal B., Gandhi T., Kim E., Rubie N. D., Moënne-Loccoz P., Karlin K. D. Inorg. Chem. 2007;46:6382–6394. doi: 10.1021/ic700363k. [DOI] [PubMed] [Google Scholar]
- Halime Z., Kieber-Emmons M. T., Qayyum M. F., Mondal B., Gandhi T., Puiu S. C., Chufán E. E., Sarjeant A. A. N., Hodgson K. O., Hedman B., Solomon E. I., Karlin K. D. Inorg. Chem. 2010;49:3629–3645. doi: 10.1021/ic9020993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Río D., Sarangi R., Chufán E. E., Karlin K. D., Hedman B., Hodgson K. O., Solomon E. I. J. Am. Chem. Soc. 2005;127:11969–11978. doi: 10.1021/ja043374r. [DOI] [PubMed] [Google Scholar]
- Chishiro T., Shimazaki Y., Tani F., Tachi Y., Naruta Y., Karasawa S., Hayami S., Maeda Y. Angew. Chem., Int. Ed. 2003;42:2788–2791. doi: 10.1002/anie.200351415. [DOI] [PubMed] [Google Scholar]
- Ghiladi R. A., Kretzer R. M., Guzei I., Rheingold A. L., Neuhold Y.-M., Hatwell K. R., Zuberbühler A. D., Karlin K. D. Inorg. Chem. 2001;40:5754–5767. doi: 10.1021/ic0105866. [DOI] [PubMed] [Google Scholar]
- Ju T. D., Ghiladi R. A., Lee D.-H., van Strijdonck G. P. F., Woods A. S., Cotter R. J., Young V. G., Karlin K. D. Inorg. Chem. 1999;38:2244–2245. [Google Scholar]
- Braterman P. S., Davies R. C., Williams R. J. P. Adv. Chem. Phys. 1964;7:359–407. [Google Scholar]
- Kim E., Chufán E. E., Kamaraj K., Karlin K. D. Chem. Rev. 2004;104:1077–1133. doi: 10.1021/cr0206162. [DOI] [PubMed] [Google Scholar]
- Kieber-Emmons M. T., Li Y., Halime Z., Karlin K. D., Solomon E. I. Inorg. Chem. 2011;50:11777–11786. doi: 10.1021/ic2018727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogutan D. K., Stoian S. A., McGuire R., Schwalbe M., Teets T. S., Nocera D. G. J. Am. Chem. Soc. 2011;133:131–140. doi: 10.1021/ja108904s. [DOI] [PubMed] [Google Scholar]
- Chatterjee S., Sengupta K., Mondal B., Dey S., Dey A. Acc. Chem. Res. 2017;50:1744–1753. doi: 10.1021/acs.accounts.7b00192. [DOI] [PubMed] [Google Scholar]
- Mittra K., Sengupta K., Singha A., Bandyopadhyay S., Chatterjee S., Rana A., Samanta S., Dey A. J. Inorg. Biochem. 2016;155:82–91. doi: 10.1016/j.jinorgbio.2015.11.013. [DOI] [PubMed] [Google Scholar]
- Mittra K., Chatterjee S., Samanta S., Sengupta K., Bhattacharjee H., Dey A. Chem. Commun. 2012;48:10535–10537. doi: 10.1039/c2cc35162e. [DOI] [PubMed] [Google Scholar]
- Gregory M., Mak P. J., Sligar S. G., Kincaid J. R. Angew. Chem., Int. Ed. 2013;52:5342–5345. doi: 10.1002/anie.201300760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak P. J., Thammawichai W., Wiedenhoeft D., Kincaid J. R. J. Am. Chem. Soc. 2015;137:349–361. doi: 10.1021/ja5107833. [DOI] [PubMed] [Google Scholar]
- Kim E., Kamaraj K., Galliker B., Rubie N. D., Moënne-Loccoz P., Kaderli S., Zuberbühler A. D., Karlin K. D. Inorg. Chem. 2005;44:1238–1247. doi: 10.1021/ic048907b. [DOI] [PubMed] [Google Scholar]
- Kim E., Shearer J., Lu S., Moenne-Loccoz P., Helton M. E., Kaderli S., Zuberbuhler A. D., Karlin K. D. J. Am. Chem. Soc. 2004;126:12716–12717. doi: 10.1021/ja045941g. [DOI] [PubMed] [Google Scholar]
- Liu J.-G., Naruta Y., Tani F. Chem.–Eur. J. 2007;13:6365–6378. doi: 10.1002/chem.200601884. [DOI] [PubMed] [Google Scholar]
- Collman J. P., Sunderland C. J., Berg K. E., Vance M. A., Solomon E. I. J. Am. Chem. Soc. 2003;125:6648–6649. doi: 10.1021/ja034382v. [DOI] [PubMed] [Google Scholar]
- Liu J.-G., Naruta Y., Tani F. Angew. Chem., Int. Ed. 2005;44:1836–1840. doi: 10.1002/anie.200462582. [DOI] [PubMed] [Google Scholar]
- Liu J., Naruta Y., Tani F., Chishiro T., Tachi Y. Chem. Commun. 2004:120–121. doi: 10.1039/b311538k. [DOI] [PubMed] [Google Scholar]
- Sharma V., Wikström M., Kaila V. R. I. Biochim. Biophys. Acta, Bioenerg. 2011;1807:813–818. doi: 10.1016/j.bbabio.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Yoshioka Y., Kawai H., Yamaguchi K. Chem. Phys. Lett. 2003;374:45–52. [Google Scholar]
- Peterson R. L., Himes R. A., Kotani H., Suenobu T., Tian L., Siegler M. A., Solomon E. I., Fukuzumi S., Karlin K. D. J. Am. Chem. Soc. 2011;133:1702–1705. doi: 10.1021/ja110466q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajdor K., Kincaid J. R., Nakamoto K. J. Am. Chem. Soc. 1984;106:7741–7747. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.