

Abstract
Aims
The ability of the right ventricle (RV) to adapt to increased afterload is the major determinant of survival in patients with pulmonary hypertension (PH). In this study, we explored the effect of genetic background on RV adaptation and survival in a rat model of severe pulmonary arterial hypertension (PAH).
Methods and results
PH was induced by a single injection of SU5416 (SU) in age-matched Sprague Dawley (SD) or Fischer rats, followed by a 3-week exposure to chronic hypoxia (SUHx). SD and Fischer rats exhibited similar elevations in RV systolic pressure, number of occlusive pulmonary vascular lesions, and RV hypertrophy (RV/LV+S) in response to SUHx. However, no Fischer rats survived beyond 7 weeks compared with complete survival for SD rats. This high early mortality of Fischer rats was associated with significantly greater RV dilatation and reduced ejection fraction, cardiac output, and exercise capacity at 4 weeks post-SU. Moreover, microarray analysis revealed that over 300 genes were uniquely regulated in the RV in the severe PAH model in the Fischer compared with SD rats, mainly related to angiogenesis and vascular homoeostasis, fatty acid metabolism, and innate immunity. A focused polymerase chain reaction array confirmed down-regulation of angiogenic genes in the Fischer compared with SD RV. Furthermore, Fischer rats demonstrated significantly lower RV capillary density compared with SD rats in response to SUHx.
Conclusion
Fischer rats are prone to develop RV failure in response to increased afterload. Moreover, the high mortality in the SUHx model of severe PAH was caused by a failure of RV adaptation associated with lack of adequate microvascular angiogenesis, together with metabolic and immunological responses in the hypertrophied RV.
Keywords: Pulmonary hypertension , Animal models of human disease, Gene expression and regulation, Right ventricle, Remodelling
1. Introduction
Pulmonary arterial hypertension (PAH) is a devastating disease characterized by increased pulmonary vascular resistance (PVR) resulting from complex arterial remodelling and lung microvascular rarefaction.1,2 These changes lead to elevated pulmonary arterial pressures and increased right ventricle (RV) afterload resulting in RV hypertrophy which is initially compensatory but ultimately progresses to decompensated RV remodelling, RV dysfunction, and right heart failure.3 It is well established that the degree of haemodynamic abnormality does not necessarily predict the level of RV dysfunction, and it is not uncommon for patients with very similar pulmonary haemodynamic abnormalities to display marked differences in the adequacy of RV compensation and function. Importantly, RV function is predictive of clinical status and prognosis in all forms of pulmonary hypertension (PH). For example, 5-year survival in patients with PAH is highly correlated with RV ejection fraction (RVEF), and less so with pulmonary artery pressures or PVR.4 Therefore, the degree to which the RV can adapt to chronic increases in afterload is believed to be a major determinant of functional capacity and long-term prognosis in patients with PH, independent of the severity of haemodynamic abnormalities.5 However, individual patients exhibit remarkable variability in RV adaptation to similar increases in afterload, which has been suggested to be due to differences in genetic make up.6
Previously, we have observed important strain-dependent differences in the severe PAH phenotype in rats exposed to a single injection of the VEGFR2 antagonist, SU5416 (SU), combined with a 3-week exposure to chronic hypoxia (SUHx).7 In particular, Fischer rats exhibited very high mortality in the SUHx model of severe PAH by 7 weeks, whereas Sprague Dawley (SD) rats showed excellent survival for up to 14 weeks in the same model.7 As well, Fischer rats showed greater RV dilatation compared with SD rats, despite comparable haemodynamic severity of PH. Therefore, we hypothesized that the high mortality in the Fischer rat may be due to maladaptation of the RV to increased afterload in response to severe PH, unique to this strain, thereby providing a model for those human PAH patients who appear to be ‘genetically prone’ to develop right heart failure.6
In the present report, we show strain-dependent defective RV adaptation in a Fischer rat model of severe PAH, similar to that observed in many patients with severe PH. RV dysfunction was related to a reduced vascularization of the hypertrophied RV, and associated with reduced expression of genes involved in angiogenesis, as well metabolic and immunological responses. Thus, targeting these pathways may provide novel therapeutic opportunities to improve RV adaption and function, thereby improving functional status and survival in patients with PH.
2. Methods
Please see the Supplementary material online for detailed methods.
2.1 Ethics
All study protocols were approved by the animal ethics and research committee (University of Ottawa, Ontario, Canada) and conducted according to guidelines from the Canadian Council for the Care of on Animal Care (CCAC).
2.2 SUHx model of PAH
Male Sprague Dawley (SD, Harlan laboratories, Indianapolis, IN, USA) and Fischer (CDF, Charles River, Montreal, Quebec, Canada) rats weighing 125–200 g were used for this study. PH was induced by a single subcutaneous injection of SU5416 (SU: 3-(3,5-dimethyl-1H-pyrrol-2-ylmethylene)-1,3-dihydroindol-2-one) (Tocris, Bristol, UK) followed by 3 weeks SUHx, as previously described.7,8
2.3 Non-invasive assessment by echocardiography
Echocardiography was performed using the Vevo2100 ultrasonography system (VisualSonics, Ontario, Canada). Rats were anaesthetized using 2–3% inhaled isoflurane. RV chamber size expressed as the ratio of end-diastolic RV to LV internal diameter (RVID-d/LVID-d), RV free wall (RVFW) thickness, and cardiac output (CO) were measured as described previously.7,9
2.4 Cardiac magnetic resonance imaging
Cardiac magnetic resonance imaging (MRI) was performed using the 7T GE/Agilent MRI system at the Ottawa preclinical imaging core. Rats were anaesthetized using 2% inhaled isoflurane. Please see the Supplementary material online for detailed procedure.
2.5 Exercise capacity testing
On Day 24 post-SU, rats received treadmill training at a speed of 10 m/min (no incline) for 5 min. The endurance test was performed on Day 31 using the following protocol: starting at 10 m/min × 5 min, incremented by 5 m/min every 5 min to a maximum of speed of 25 m/min (no incline). Fatigue was determined by a blinded assessor when the rat accepted the electrical stimulus three times within 10 s.
2.6 Right heart catheterization
At 4 weeks post-SUHx, rats were anaesthetized by an intraperitoneal injection of xylazine (7 mg/kg) and ketamine (35 mg/kg). Right ventricular systolic pressure (RVSP) was measured using high-fidelity pressure catheters (Transonic-Scisense Inc., Ontario, Canada) as describe previously.7
2.7 Lung and heart morphometric and histological measurements
The left lobe of the lung and the whole heart were sectioned, fixed in 4% paraformaldehyde and embedded in paraffin. Tissue blocks were sectioned 5 μm thickness with a microtome (Leica Microsystems, Ontario, Canada). Staining, microscopy, and morphometry were performed as described in the Supplementary material online.
2.8 Immunohistochemistry
Paraffin heart sections were stained with rabbit polyclonal antibody to CD31 (Novus Biologicals, Ontario, Canada) at 1:250 dilution for 1 h at room temperature, followed by DAB staining using the VECTASTAIN ABC Elite kit (Vector Labs, Ontario, Canada) according to manufacturer’s protocol. For natural cytotoxicity receptor-1 (NCR1), paraffin heart sections were stained with rabbit polyconal antibody to NCR1 (Abcam, Ontario, Canada) at 1:400 dilution followed by DAB staining using Rabbit specific HRP/DAB (ABC) Detection IHC Kit (Abcam, Ontario, Canada) according to manufacturer’s protocol. Images were acquired using Nikon Eclipse TE2000 inverted microscopes using 10X and 40X objectives (Nikon C1si, Nikon Instruments Inc., Melville, NY, USA) and NCR1 positive cells were counted using the open-source softwares ImageJ (Fiji package). Total NCR1 positive cells per high magnification (40X) image were calculated and five images per samples were analysed.
2.9 Fluorescence microangiography
Fluorescence microangiography (FMA) was performed as previously described10 with slight modifications. Please see the Supplementary material online for detailed description.
2.10 Microarray gene expression
RNA was isolated from RV tissue using miRCURY RNA isolation kit (Exiqon, Woburn, MA, USA) according to manufacturer’s protocol. Total RNA was assayed using Affymetrix Rat Gene 2.0ST and Affymetrix miRNA 4.0 gene chips. A detailed description of data analysis for microarray can be found in the Supplementary material online.
2.11 PCR array for angiogenic genes
RNA was isolated from lungs as described above and cDNA was prepared using RT2 First Strand Kit (Qiagen, Ontario, Canada) as per manufacturer’s instructions. The cDNA was stored at −80°C until used for the RT2 Profiler PCR array for angiogenic genes (Qiagen, Ontario, Canada). Quantitative RT-PCR was performed using SYBR green qPCR mastermix (Qiagen, Ontario, Canada) as per the manufacturer’s instructions.
2.12 Statistical analysis
Data are represented as mean ± standard error of mean (SEM) unless otherwise stated. Statistical analysis was performed using Student’s t-test or one-way analysis of variance (ANOVA) (>2 groups) followed by Tukey’s multiple comparison test with significance level of P < 0.05, unless otherwise stated. Survival curves were compared using the Log-rank Mantel–Cox analysis.
3. Results
3.1 High mortality in Fischer rats is not due to differences in RV haemodynamic afterload, lung vascular remodelling, or RV hypertrophy
Median survival of Fischer rats was 34 days after induction of severe PH, whereas all of SD rats survived to end-study (56 days) post-SU (P < 0.0002) (Figure 1A). Importantly, the striking discrepancy in survival could not be attributed to differences in RV haemodynamic afterload, as RVSP was similar between SD and Fischer rats (Figure 1B). Histopathological examination of pulmonary arterioles also showed no significant differences in the proportion of pulmonary vessels with Grade 0, Grade 1, or Grade 2 remodelling between SD and Fischer receiving SUHx at 4 weeks post-SU (Figure 1C and D). Moreover, Masson’s Trichrome staining showed no significant differences in total RV cross section area and RV myocyte cross sectional area between Fischer and SD rats in the SUHx model of severe PAH at 4 weeks post-SU (Figure 2A–C).
Figure 1.

(A) Kaplan Meier Survival analysis of Fischer (F) (n = 9) and SD (S) (n = 12) rats after SUHx (Log rank /Mantel-Cox analysis). Pulmonary hemodynamics. (B) Right ventricular systolic pressure (RVSP) was similar in both strains after SUHx. *P < 0.05 vs CTL (matched to strain). N = 4–6 per group. (C) Representative hematoxylin and eosin staining of paraffin-embedded lung sections from animals 4-weeks post-SU depicting Grade 0, Grade 1 or Grade 2 remodelling of pulmonary vessels in SUHx vs control lungs. Black arrows indicate vessels, scale bar = 200 μm. (D) Bar graph shows quantitative analysis of percentage of Grade 0, Grade 1 or Grade 2 vessels. Similar extent of pulmonary vascular remodelling was observed between both strains. Statistical analysis using One-way ANOVA with Tukey’s multiple comparisons test. N = 3–4 per group.
Figure 2.

(A) Macroscopic images of Masson’s Trichrome stained heart sections and microscopic images of RV cross-sections illustrating cardiomyocyte fibre thickness. (B) Quantification of RV area relative to LV+S area. (C) Cardiomyocyte cross sectional area. Statistical analysis using One-way ANOVA with Tukey’s multiple comparisons test. *P < 0.05 vs CTL (matched to strain). N = 3–6 per group.
3.2 Fischer SUHx model is characterized by RV dysfunction and severe dilatation
Echocardiographic analysis of RV function and structure was performed at 4 weeks post-SU. The RVID-d/LVID-d ratio was similar in healthy control Fischer and SD rats (Figure 3A and B). However, Fischer rats showed more RV dilatation in response to SUHx with a significantly greater increase in RVID-d/LVID-d compared with the SD rats at 4 weeks post-SU (1.06 ± 0.06 vs. 0.74 ± 0.06, P < 0.0001; Figure 3B and Supplementary material online, Table S1). Furthermore, no significant difference in RVFW thickness was observed at 4 weeks between Fischer and SD SUHx rats (Figure 3C). CO was comparable between control Fischer and SD rats (150.9 ± 17.0 mL/min and 184.0 ± 14.9 mL/min, respectively). However, in response to SUHx, Fischer rats exhibited significantly lower CO compared with SD rats (59.9 ± 4.0 mL/min and 119.1 ± 11.3 mL/min, respectively) at 4 weeks post-SU, indicating RV systolic dysfunction in Fischer rats (Figure 3D). No difference in heart rate was observed between SD and Fischer rats (Supplementary material online, Figure S1).
Figure 3.

Assessment of RV structure and function by echo at 4 weeks post SU. (A, B) RVID-d/LVID-d: end diastolic RV/LV internal diameter in M-mode echo. (C) RVFW: RV free wall thickness (M-mode echo). (D) Cardiac output. Data represented as mean ± SEM. Statistical analysis using One-way ANOVA with Bonferroni’s post-hoc test. *P < 0.05 vs CTL (matched to strain), #P < 0.05 vs S-SUHx. N = 5–14 per group.
3.3 Greater RV dilation and reduced RV ejection by fraction cardiac MRI in Fischer rats with severe PAH
Cardiac MRI was used to better define the changes in RV structure and function between the two rat strains in response to severe PH at 4 weeks post-SU. Again, Fischer rats exhibited evidence of defective RV adaptation (Figure 4A and B), with a significantly greater increase in right ventricular/left ventricular end-diastolic volume ratio compared with SD rats (RVEDV/LVEDV: 2.16 ± 0.24 vs. 1.19 ± 0.1, respectively) at 4 weeks of SUHx-induced severe PH (P < 0.001; Figure 4C). As well, RVEF was significantly decreased in SUHx Fischer rats (39 ± 0.04%, P < 0.0001 vs. control; Figure 4D), whereas no reduction in RVEF was seen in the severe PAH model in the SD rats (61.18 ± 0.03%), confirming that RV systolic function was better preserved in the SD rat strain (P < 0.01; Figure 4D).
Figure 4.

Evaluation of RV function by MRI. Representative short-axis views of RV and LV in (A) end-diastole and (B) end-systole using MRI at 4 weeks post SU. (C) End-diastolic RV/LV volumes and (D) RV ejection fraction (%). Statistical analysis using One-way ANOVA with Tukey’s multiple comparisons test. *P < 0.05 vs CTL. #P < 0.05 vs S-SUHx. N = 3–5 per group.
3.4 Exercise capacity is reduced in the Fischer SUHx model
Exercise tolerance was assessed using a standardized treadmill test to better establish the functional importance of abnormalities in cardiac adaptation in PH at 4 weeks (Supplementary material online, Figure S2). Fischer rats demonstrated significantly reduced exercise capacity vs. SD measured by both treadmill distance (59 ± 29 vs. 210 ± 52 m, P < 0.05) and treadmill time (4.9 ± 2.1 vs. 13.8 min ± 2.3, P < 0.05) (Supplementary material online, Figure S2).
3.5 RV global expression profiling reveals dysregulated genes related to immunity, metabolism and circulation processes in Fischer rats
Unbiased, global transcriptional profiling was performed to compare changes in RV gene expression in the SUHx model of severe PAH in Fischer and SD rats. As expected, principle component analysis revealed substantial differences in RV gene expression between control and PH rats, which were greater than the differences observed between the two rat strains under the two conditions (Figure 5A). However, based on unbiased hierarchical clustering, Fischer and SD rats were seen to exhibit distinct transcriptional profiles (Figure 5B and Supplementary material online, Figure S3). Moreover, a plot showing the relative changes in Fischer and SD rats in transcriptional activity induced by severe PH at 4 weeks post-SUHx showed that approximately 400 genes were significantly differentially regulated (Figure 5C). Of the 20 000 genes studied, 318 were found to be uniquely regulated in Fischer rats, whereas only 41 genes were uniquely regulated in SD rats, and 82 genes were similarly regulated in both rat strains (Supplementary material online, Table S2). Gene ontology (GO) analysis revealed that unique changes in expression of genes in the Fischer rats were associated mainly with biological processes such as innate immunity, fatty acid metabolism, immunity and defense, and regulation of vasoconstriction and vasodilation (Table 1, Supplementary material online, Tables S3 and S4). These included genes that corresponded to GO terms related to vasoconstriction and blood circulation included EDNRB (endothelin receptor Type B), PTGIS (prostacyclin synthase), CORIN (Atrial Natriuretic Peptide-Converting Enzyme), EDN1 (endothelin-1), CCR2 (Chemokine (C-C Motif) Receptor 2), and NPPA (Atrial natriuretic peptide, ANP). Down-regulated genes in Fischer rats with severe PH included those related to natural killer (NK)-cells (KLRC3, KLRK1, KLRB1C, KLRD1, and KLRB1A), fatty acid metabolism, and PGC1α (Peroxisome Proliferator-Activated Receptor Gamma, Co-activator 1 Alpha; PPARGC1A), a master regulator of mitochondrial activity.
Figure 5.

Microarray analysis of gene expression in the right ventricle at 4 weeks post SU. (A) Principal component analysis of RV gene expression changes in SD and Fischer rats treated with/without SUHx. (B) Heatmap of differentially expressed genes in SD and Fischer RVs in CTL and SUHx-induced PH. Genes with significant fold changes were filtered at a threshold of P < 0.05, >3-fold change. (C) Volcano plot of RV gene expression changes in SD and Fischer rats treated with SUHx relative to CTL. N = 3 per group.
Table 1.
GO term analysis by biological processes unique to PAH in Fischer
| Rank | GO term | P-value | Genes |
|---|---|---|---|
| 1 | Natural killer cell-mediated immunity | 0.002086119 | KLRC3, FCGR1A, KLRK1, KLRB1C, KLRD1, KLRB1A |
| 2 | Fatty acid metabolism | 0.002151395 | SLC27A1, ACADSB, PTGIS, ACSL1, DECR1, CYP2E1, HPGDS, LIPE, HADHA, CYP4B1 |
| 3 | Immunity and defense | 0.002800724 | CTHRC1, IL1R1, CCL2, KLRC3, LITAF, KLRK1, CD1D1, C1QC, HSPA2, CD69, FCGR1A, CD2, GSTZ1, KLRD1, HPGDS, PLAT, C5AR1, LGALS3, SLAMF9, IL1RN, SLAMF8, C1QA, C1QB, CD86, CD55, CCR2, COQ10B, SEMA4D, HSPB3, KLRB1C, GADD45A, F2R, IQUB, KLRB1A |
| 4 | Cell communication | 0.003307644 | RP1, KLRC3, FGF7, CCL2, THRB, GABRB2, EDN1, CD151, FOXS1, SPHKAP, LAMB3, CD2, SEMA3D, KLRD1, PTPRD, IL1RN, PCDH7, ARHGAP26, FOXP2, TNFAIP6, CDH13, MAGIX, CD86, ADAM1A, LAMA5, GRN, CCR2, ADAMTS1, SEMA4D |
| 5 | Regulation of vasoconstriction and dilation | 0.004281473 | EDNRB, PTGIS, EDN1, CCR2, NPPA |
| 6 | Blood circulation and gas exchange | 0.007987292 | EDNRB, PTGIS, CORIN, EDN1, CCR2, NPPA |
| 7 | Complement-mediated immunity | 0.008314347 | C1QA, C1QB, CTHRC1, CD55, C1QC |
| 8 | Lipid, fatty acid, and steroid metabolism | 0.017330542 | SLC27A1, CYP2J4, ACADSB, SPTLC2, CORIN, OSBPL6, CYP2E1, DECR1, CYB5B, CDS1, ANXA5, PPARGC1A, HADHA, CYP4B1, ACSL1, PTGIS, INPP5J, SULT1A1, HPGDS, LIPE |
| 9 | Other homoeostasis activities | 0.030329365 | CCR2, CLIC1, RGD1566189, PPARGC1A, NPPA |
| 10 | Homoeostasis | 0.033749417 | ISCU, EDN1, CCR2, SLC30A3, CLIC1, RGD1566189, PPARGC1A, NPPA |
| 11 | B-cell- and antibody-mediated immunity | 0.038297877 | SLAMF9, CD69, FCGR1A, IL1RN, SLAMF8 |
| 12 | Cell adhesion | 0.040097918 | PTPRD, KLRC3, LGALS3, SLAMF9, KLRK1, PCDH7, MCAM, CD151, HMCN2, ITGA9, CDH13, LAMB3, CD2, LOXL1, KLRD1 |
| 13 | Cell adhesion-mediated signalling | 0.041487794 | CDH13, PTPRD, LAMB3, KLRC3, LAMA5, CD2, ADAMTS1, PCDH7, CD151, KLRD1, ARHGAP26 |
| 14 | Cell proliferation and differentiation | 0.046212612 | FGF7, ERBB4, EDN1, SLAMF8, PCDH7, MEIS1, TGFB1, JUNB, FOXS1, FOXP2, EPHA3, EDNRB, CD86, CCND1, EPHA7, PLK2, NDRG4, RGD1566399, TFDP2, EMP1 |
| 15 | Extracellular matrix protein-mediated signalling | 0.050271633 | TNFAIP6, LAMB3, LAMA5, ADAMTS1 |
| 16 | Muscle contraction | 0.050789496 | EDNRB, CNN3, PHKG1, TAGLN2, HPGDS, TPM3, CKB |
| 17 | Oogenesis | 0.059665763 | LDLR, ERBB4, PIWIL2, LOC691325 |
3.6 SUHx-induced PH is associated with reactivation of RV foetal gene program
SUHx was associated with reduction of α-MHC expression and up-regulation of β-myocin heavy chain, A-type natriuretic peptide, and brain or b-type natriuretic peptide, assessed by quantitative RT-PCR (Supplementary material online, Figure S4). No significant differences in expression of these four genes were observed between SD and Fischer rats in the SUHx model of severe PAH. A greater relative increase in ANP expression in Fischer SUHx was attributable almost entirely to the low basal expression in the control Fischer rats; however, protein levels of ANP and BNP were similar in Fischer and SD RV both at baseline and in response to SUHx (Supplementary material online, Figure S5).
3.7 Down-regulation of angiogenic genes and evidence of reduced RV vascularization in Fischer rats with severe PH at 4 weeks post-SU
A focused PCR array was performed to evaluate alterations in 84 key genes involved in modulating the biological processes of angiogenesis in the RV of Fischer and SD rats. There were significant differences in gene expression at baseline, with higher expression of 11 genes, and lower expression of five genes, in Fischer rats as compared with SD rats (Supplementary material online, Table S5). In response to SUHx, 14 genes were differentially expressed between the two strains relative to their respective baseline controls (Figure 6A and Supplementary material online, Table S6). These included prostaglandin endoperoxidase synthase 1 (Ptgs1) and endothelin 1 (Edn1), which were uniquely increased in the RV of Fischer rats, while tumour necrosis factor (Tnf), chemokine (C-C motif) ligand 2 (Ccl2), vascular endothelial growth factor C (Vegfc), angiopoeitin 1 (Angpt1), and V-akt murine thymoma viral oncogene homologue 1 (Akt1) were decreased in response to SUHx (Figure 6A). Overall, a relative down-regulation of angiogenic genes was observed in the RV of Fischer rats as compared with SD rats.
Figure 6.

Inadequate RV angiogenesis in response to SUHx-induced PH at 4 weeks following SU. (A) Volcano Plot graph of PCR-array of angiogenic genes in right ventricles. The graph shows Log 2 of the ratio of fold change in each gene’s expression in Fischer rats to SD rats. The fold change for each gene’s expression in SUHx group was calculated relative to CTL group. P-values were derived using student t-test. The dashed line indicates P < 0.05. N = 4 per group. (B) Immunohistochemistry staining for CD31 reveals a reduction in capillary density after SUHx in both strains and (D) quantification of capillary density. N = 3 per group. (C) Single slice confocal microscopy images of RV after fluorescence microangiography. (E) 3D capillary volume rendering from FMA stacks was expressed as capillary volume %. N = 3–4 per group. One-way ANOVA with Tukey’s multiple comparisons test where *P < 0.05 vs CTL; #P < 0.05 vs SD-SUHx. (F) Negative control for CD31 IHC. RV section was stained as described in the methods section in absence of primary antibody (CD31) and used as a negative control for CD31 IHC.
We next assessed RV vascularization in Fischer and SD rats with severe PH using endothelial cell specific immunohistochemical staining and FMA. Staining for endothelial cell-specific CD31 revealed a reduction in capillary density in RV cross sections in both strains in the SUHx model at 4 weeks compared with strain-matched controls (Figure 6B, D, and F); however, the reduction in capillary density was significantly greater in Fischer vs. SD (P < 0.05). Functional RV microcirculation was imaged by FMA, to better demonstrate the 3-dimensional architecture of perfused microvessels (Figure 6C). The size of the capillary bed was quantified by measuring capillary volume expressed as a percentage of the total volume imaged by optical sectioning, revealing a significant reduction in the severe PAH model, although, once again, this was more marked in F-SUHx (12 ± 1%) compared with S-SUHx rats (Figure 6E).
3.8 Reduced infiltration of NK cells in the Fischer rat RV compared with the SD rat RV at 4-week post-SU
Immunohistochemistry for NCR-1 showed very few NK cells in the RV of control Fischer and SD rats. In the SUHx model of severe PAH, there was a marked increase in infiltration of NCR-1 positive cells at 4 weeks in the SD but not in the Fischer rats (Figure 7A and B). This is consistent with the relative decrease in NK cell gene expression in the RV of Fischer compared with SD rats in response to SUHx (Table 1 and Supplementary material online, Table S2).
Figure 7.
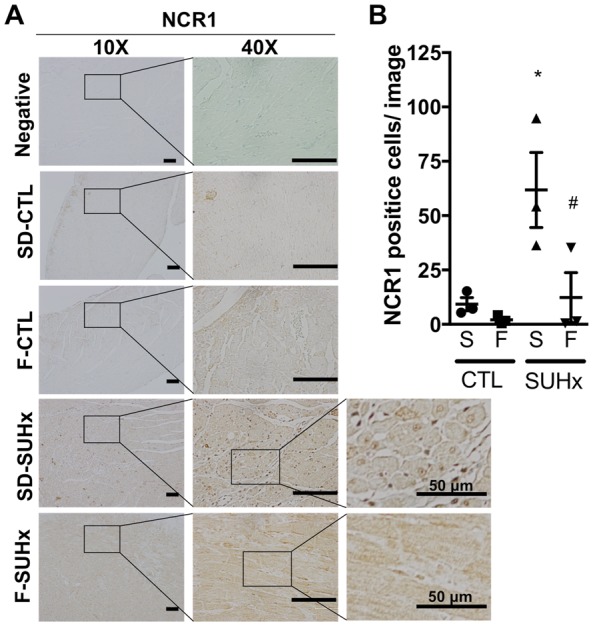
Natural killer (NK) cell infiltration in the RV. (A) Representative images (scale bar = 100 μm for 10X and 40X images) and (B) bar graph demonstrating NCR1 positive natural killer cell infiltration in the RV of Fischer CTL, SD CTL, SD SUHx and Fischer SUHx rats. Statistical analysis using One-way ANOVA with Tukey’s multiple comparisons test. *P < 0.05 vs CTL; #P < 0.05 vs SD-SUHx. N = 3 per group.
4. Discussion
We report that early mortality in Fischer rats in the SUHx model of severe PAH is associated with strain-specific maladaptive RV remodelling and progressive right heart failure. Compared with SD rats, the maladaptive RV changes in the Fischer rat were associated with differential expression of genes involved in angiogenesis, metabolism, and inflammation, all of which have previously been implicated in the development of RV failure. Therefore, Fischer rats provide a RV ‘failure-prone’ model in response to severe pressure overload, which recapitulates the variability in susceptibility to RV failure commonly seen in PAH, patients, and is ideally suited to test novel therapies targeting RV adaptation.
It is well established that RV dysfunction is one of the strongest predictors of mortality in PAH patients,4,11,12 and therefore, a better understanding of the molecular mechanisms underlying RV maladaptation to increased afterload is needed. This has been hampered by the lack of animal models that predictably reproduce the salient features of RV remodelling leading to right heart failure in PAH patients. We now show the early development of RV decompensation in severe PH in the Fischer rat strain. Both Fischer and SD rat strains showed increases in RV diameter and volumes and reduction in CO and systolic function at 4 weeks; however, these changes were more pronounced in the Fischer rats. The reproducible and consistent differences in RV adaptation in the SUHx model between Fischer and SD rats are likely due in large part to differences in genetic backgrounds, reflected in differences in patterns of gene expression in response to progressive increases in RV afterload. Microarray analysis revealed a number of differentially regulated pathways in the Fischer rats compared with the SD rats, which offer further insights into the mechanisms underlying adaptive and maladaptive remodelling. These pathways include innate immunity, fatty acid metabolism, and vascular homoeostasis, many of which have all been previously implicated in myocardial remodelling.13,14
Although myocardial inflammation has been strongly implicated in left-sided heart failure,15 less is known about the role inflammation in RV remodelling. Global gene profiling in the RV of Fischer rats with severe PH revealed a marked down-regulation of genes related to NK cell function. In addition to their role in innate immunity and host defense, natural killer cells have been implicated in angiogenesis. It has been long recognized that decidual NK cells promote placental vascularization by inducing growth of spiral arteries during pregnancy, in part by secreting angiogenic factors.16 Moreover, it has been reported that NOD-SCID mice exhibit poor functional recovery and revascularization following myocardial infarction compared with immune-competent mice, which can be rescued by adoptive transfer of IL-2 stimulated NK cells.13 NK cells have also been implicated in the pathobiology of PAH.17,18 Nfil3−/− mice that lack NFIL3 transcription factor and Ncr1-GFP mice that lack the NK cell activating receptor exhibit NK cell deficiency and develop spontaneously develop PH.18 As well, the therapeutic effect of endothelial progenitor cells in the MCT model of PH was shown to be in large part dependent of NK cell activity.17 Interestingly, impaired NK cell function has been reported in patients with hereditable and idiopathic PAH,19 and these cells showed reduced secretion of cytokines. In the present study, we observed a decrease in infiltration of NK cells in the RV of Fischer rats compared with SD rats in response to SUHx. Therefore, altered number of NK cells, and possibly the production of angiogenic cytokines by NK cells, could contribute to the reduced RV vascularisation in Fischer rats in the severe PAH model.
The transcriptomic analysis also revealed down-regulation in genes regulating fatty acid metabolism in Fischer rats in response to severe PH including several enzymes important for the synthesis and metabolism of acyl-CoA such as acyl-CoA dehydrogenase, as well as peroxisome PGC-1α, the master regulator of mitochondrial metabolism.20 It is well established that metabolic changes play an important role in the development of both left and right heart failure.21–23 Increased RV glucose uptake and impaired RV fatty acid metabolism have been observed in patients with PAH using 18F-fludeoxyglucose and 123I-labeled beta-methyl-p-iodophenyl-pentadecanoic acid single photon emission computed tomography.23–25 Since fatty acid oxidation requires 12% more oxygen to generate the same amount of adenosine triphosphate as glucose oxidation, this shift in metabolism may aggravate the state of relative RV ischaemia caused by impaired angiogenesis in maladaptive remodelling.26 Interestingly, both PGC-1α and genes encoding the acyl-CoA dehydrogenases have been previously shown to be reduced in the RV of SD rats exhibiting RV dysfunction in a similar model of severe PAH.27 However, in this report SD rats were kept in hypoxic conditions for 4 weeks, rather than 3 weeks, and gene expression was studied at later time points when RV decompensation might be expected in response to severe PH even in a relatively resistant rat strain. Of note, no reduction in metabolic gene expression or RV decompensation was seen after pulmonary artery banding in this report, despite similar increases in RVSP.27 This suggests that increased RV afterload alone is not sufficient, and additional inflammatory, and other signals associated with PH biology, are required for RV dysfunction to be manifested.
RV decompensation in PAH has been previously attributed to capillary rarefaction resulting in relative myocardial ischaemia within the markedly hypertrophied RV.24,26,28 Consistent with this, decreased coronary flow reserve has been described in RV of patients with PAH,29 and decreased capillary density has been reported to be a distinguishing feature between decompensated and compensated RV remodelling in PAH patients.30 Interestingly, endothelial cells isolated from the RV of patients with right heart failure demonstrated reduced in vitro angiogenic activity,30 suggesting that differences in the neovascularization of the hypertrophied myocardium may be critical for adequate RV adaptation. In our study, we also observed a marked reduction in RV capillary density in the severe PAH model, which was more marked in Fischer compared with SD rats. The decrease in myocardial capillaries seen on thin sections was confirmed by FMA which provides 3-dimensional imaging of the functional microvascular architecture in thick (40 micron) sections of the RV therefore yielding a quantitative assessment of RV capillaries similar to rigorous methods, such as unbiased stereology.31,32 Reduced RV vascularity was associated with a decrease in expression of angiogenic genes in the RV of Fischer rats compared with SD rats, again consistent with the emerging concept that microvascular angiogenesis is critical for adaptive RV remodelling in response to marked increases in afterload associated with severe PH. Indeed, our group has recently demonstrated that cardiotrophin-1, an interleukin cytokine superfamily member which promotes physiological myocardial remodelling and angiogenesis, improved capillary density, reversed dilation, and restored contractile function of the RV in the Fischer rat SUHx model.33
4.1 Limitations
This study has several limitations which are important to note. Although, we provide evidence to support RV dysfunction, including RV dilatation and decreased RVEF and CO using both MRI and echocardiography, we did not assess pressure–volume loops, which is the gold-standard for examining RV-pulmonary arterial coupling and contractility. Also, only male animals were studied and the relative ability of the RV female SD and Fischer rats to adapt to pressure overload needs to be assessed in future studies.
5. Conclusion
In conclusion, we have demonstrated that Fischer rats develop maladaptive RV remodelling in the SUHx model of severe PAH, which may lead to early mortality. This was associated with impairment in the RV angiogenesis, NK cells and dysregulated expression in a number of gene families associated with vascular homoeostasis, inflammation, and cardiac metabolism. Our findings suggest that the Fischer rat strain may be uniquely suited for the study of RV decompensation in response to severe PH, and provide a RV failure-prone model for the further exploration of molecular mechanisms underlying maladaptive remodelling, as well as for the study of novel RV-targeted therapies.
Supplementary Material
Acknowledgements
The authors would like to thank Anli Yang and Xiaoxue Wen for their technical support in animal procedures. Gareth Pawlidor for assistance in bioinformatics analysis and Gregory Cron and Rebecca Thornhill for assistance with cardiac MRI acquisitions.
Conflict of interest: D.J.S. is a consultant for, and has equity interest in, Northern Therapeutics Inc. All other authors have nothing to disclose.
Funding
This work was funded by a grant from the Canadian Health Research Institute (CIHR; Foundation grant: FDN-143291). Additional funding was provided by an unrestricted grant from Northern Therapeutics Inc. (NT). K.R.C. is a recipient of Research Fellowship from Heart and Stroke Foundation of Canada and Scholar award from Canadian Vascular Network and CIHR.
Footnotes
Time for primary review: 37 days
References
- 1. Jurasz P, Courtman D, Babaie S, Stewart DJ.. Role of apoptosis in pulmonary hypertension: from experimental models to clinical trials. Pharmacol Ther 2010;126:1–8. [DOI] [PubMed] [Google Scholar]
- 2. Chaudhary KR, Taha M, Cadete VJ, Godoy RS, Stewart DJ.. Proliferative versus degenerative paradigms in pulmonary arterial hypertension: have we put the cart before the horse? Circ Res 2017;120:1237–1239. [DOI] [PubMed] [Google Scholar]
- 3. Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF.. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 2009;135:794–804. [DOI] [PubMed] [Google Scholar]
- 4. van de Veerdonk MC, Kind T, Marcus JT, Mauritz GJ, Heymans MW, Bogaard HJ, Boonstra A, Marques KM, Westerhof N, Vonk-Noordegraaf A.. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol 2011;58:2511–2519. [DOI] [PubMed] [Google Scholar]
- 5. Rich S, Pogoriler J, Husain AN, Toth PT, Gomberg-Maitland M, Archer SL.. Long-term effects of epoprostenol on the pulmonary vasculature in idiopathic pulmonary arterial hypertension. Chest 2010;138:1234–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Voelkel NF, Bogaard HJ, Gomez-Arroyo J.. The need to recognize the pulmonary circulation and the right ventricle as an integrated functional unit: facts and hypotheses (2013 Grover Conference series). Pulm Circ 2015;5:81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jiang B, Deng Y, Suen C, Taha M, Chaudhary KR, Courtman DW, Stewart DJ.. Marked strain-specific differences in the SU5416 rat model of severe pulmonary arterial hypertension. Am J Respir Cell Mol Biol 2016;54:461–468. [DOI] [PubMed] [Google Scholar]
- 8. Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM.. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J 2001;15:427–438. [DOI] [PubMed] [Google Scholar]
- 9. Urboniene D, Haber I, Fang YH, Thenappan T, Archer SL.. Validation of high-resolution echocardiography and magnetic resonance imaging vs. high-fidelity catheterization in experimental pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2010;299:L401–L412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dutly AE, Kugathasan L, Trogadis JE, Keshavjee SH, Stewart DJ, Courtman DW.. Fluorescent microangiography (FMA): an improved tool to visualize the pulmonary microvasculature. Lab Invest 2006;86:409–416. [DOI] [PubMed] [Google Scholar]
- 11. D'Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, Goldring RM, Groves BM, Kernis JT, Levy PS, Pietra GG, Reid LM, Reeves JT, Rich S, Vreim CE, Williams GW, Wu M.. Survival in patients with primary pulmonary hypertension. Ann Intern Med 1991;115:343–349. [DOI] [PubMed] [Google Scholar]
- 12. van Wolferen SA, Marcus JT, Boonstra A, Marques KM, Bronzwaer JG, Spreeuwenberg MD, Postmus PE, Vonk-Noordegraaf A.. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J 2007;28:1250–1257. [DOI] [PubMed] [Google Scholar]
- 13. Bouchentouf M, Forner K-A, Cuerquis J, Michaud V, Zheng J, Paradis P, Schiffrin EL, Galipeau J.. Induction of cardiac angiogenesis requires killer cell lectin-like receptor 1 and α4β7 integrin expression by NK cells. J Immunol 2010;185:7014–7025. [DOI] [PubMed] [Google Scholar]
- 14. Sutendra G, Dromparis P, Paulin R, Zervopoulos S, Haromy A, Nagendran J, Michelakis ED.. A metabolic remodeling in right ventricular hypertrophy is associated with decreased angiogenesis and a transition from a compensated to a decompensated state in pulmonary hypertension. J Mol Med 2013;91:1315–1327. [DOI] [PubMed] [Google Scholar]
- 15. Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res 2015;116:1254–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, Prus D, Cohen-Daniel L, Arnon TI, Manaster I, Gazit R, Yutkin V, Benharroch D, Porgador A, Keshet E, Yagel S, Mandelboim O.. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med 2006;12:1065–1074. [DOI] [PubMed] [Google Scholar]
- 17. Ormiston ML, Deng Y, Stewart DJ, Courtman DW.. Innate immunity in the therapeutic actions of endothelial progenitor cells in pulmonary hypertension. Am J Respir Cell Mol Biol 2010;43:546–554. [DOI] [PubMed] [Google Scholar]
- 18. Ratsep MT, Moore SD, Jafri S, Mitchell M, Brady HJ, Mandelboim O, Southwood M, Morrell NW, Colucci F, Ormiston ML.. Spontaneous pulmonary hypertension in genetic mouse models of natural killer cell deficiency. Am J Physiol Lung Cell Mol Physiol 2018;doi:10.1152/ajplung.00477.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ormiston ML, Chang C, Long LL, Soon E, Jones D, Machado R, Treacy C, Toshner MR, Campbell K, Riding A, Southwood M, Pepke-Zaba J, Exley A, Trembath RC, Colucci F, Wills M, Trowsdale J, Morrell NW.. Impaired natural killer cell phenotype and function in idiopathic and heritable pulmonary arterial hypertension. Circulation 2012;126:1099–1109. [DOI] [PubMed] [Google Scholar]
- 20. Handschin C, Spiegelman BM.. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev 2006;27:728–735. [DOI] [PubMed] [Google Scholar]
- 21. Talati M, Hemnes A.. Fatty acid metabolism in pulmonary arterial hypertension: role in right ventricular dysfunction and hypertrophy. Pulm Circ 2015;5:269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sankaralingam S, Lopaschuk GD.. Cardiac energy metabolic alterations in pressure overload-induced left and right heart failure (2013 Grover Conference Series). Pulm Circ 2015;5:15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nagaya N, Goto Y, Satoh T, Uematsu M, Hamada S, Kuribayashi S, Okano Y, Kyotani S, Shimotsu Y, Fukuchi K, Nakanishi N, Takamiya M, Ishida Y.. Impaired regional fatty acid uptake and systolic dysfunction in hypertrophied right ventricle. J Nucl Med 1998;39:1676–1680. [PubMed] [Google Scholar]
- 24. Gomez A, Bialostozky D, Zajarias A, Santos E, Palomar A, Martinez ML, Sandoval J.. Right ventricular ischemia in patients with primary pulmonary hypertension. J Am Coll Cardiol 2001;38:1137–1142. [DOI] [PubMed] [Google Scholar]
- 25. Kim Y, Goto H, Kobayashi K, Sawada Y, Miyake Y, Fujiwara G, Chiba H, Okada T, Nishimura T.. Detection of impaired fatty acid metabolism in right ventricular hypertrophy: assessment by I-123 beta-methyl iodophenyl pentadecanoic acid (BMIPP) myocardial single-photon emission computed tomography. Ann Nucl Med 1997;11:207–212. [DOI] [PubMed] [Google Scholar]
- 26. Ryan JJ, Archer SL.. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res 2014;115:176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gomez-Arroyo J, Mizuno S, Szczepanek K, Van Tassell B, Natarajan R, dos Remedios CG, Drake JI, Farkas L, Kraskauskas D, Wijesinghe DS, Chalfant CE, Bigbee J, Abbate A, Lesnefsky EJ, Bogaard HJ, Voelkel NF.. Metabolic gene remodeling and mitochondrial dysfunction in failing right ventricular hypertrophy secondary to pulmonary arterial hypertension. Circ Heart Fail 2013;6:136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van Wolferen SA, Marcus JT, Westerhof N, Spreeuwenberg MD, Marques KM, Bronzwaer JG, Henkens IR, Gan CT, Boonstra A, Postmus PE, Vonk-Noordegraaf A.. Right coronary artery flow impairment in patients with pulmonary hypertension. Eur Heart J 2007;29:120–127. [DOI] [PubMed] [Google Scholar]
- 29. Vogel-Claussen J, Skrok J, Shehata ML, Singh S, Sibley CT, Boyce DM, Lechtzin N, Girgis RE, Mathai SC, Goldstein TA, Zheng J, Lima JA, Bluemke DA, Hassoun PM.. Right and left ventricular myocardial perfusion reserves correlate with right ventricular function and pulmonary hemodynamics in patients with pulmonary arterial hypertension. Radiology 2011;258:119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Potus F, Ruffenach G, Dahou A, Thebault C, Breuils-Bonnet S, Tremblay E, Nadeau V, Paradis R, Graydon C, Wong R, Johnson I, Paulin R, Lajoie AC, Perron J, Charbonneau E, Joubert P, Pibarot P, Michelakis ED, Provencher S, Bonnet S.. Downregulation of MicroRNA-126 contributes to the failing right ventricle in pulmonary arterial hypertension. Circulation 2015;132:932–943. [DOI] [PubMed] [Google Scholar]
- 31. Kolb TM, Peabody J, Baddoura P, Fallica J, Mock JR, Singer BD, D'Alessio FR, Damarla M, Damico RL, Hassoun PM.. Right ventricular angiogenesis is an early adaptive response to chronic hypoxia-induced pulmonary hypertension. Microcirculation 2015;22:724–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Graham BB, Koyanagi D, Kandasamy B, Tuder RM.. Right ventricle vasculature in human pulmonary hypertension assessed by stereology. Am J Respir Crit Care Med 2017;196:1075–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abdul-Ghani M, Suen C, Jiang B, Deng Y, Weldrick JJ, Putinski C, Brunette S, Fernando P, Lee TT, Flynn P, Leenen FHH, Burgon PG, Stewart DJ, Megeney LA.. Cardiotrophin 1 stimulates beneficial myogenic and vascular remodeling of the heart. Cell Res 2017;27:1195–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.