

Abstract
Delayed hippocampal injury and memory impairments follow neonatal hypoxia-ischemia (HI) despite the use of therapeutic hypothermia (TH). Death of hippocampal pyramidal cells occurs acutely after HI, but characterization of delayed cell death and injury of interneurons (INs) is unknown. We hypothesize that injury of INs after HI is: i) asynchronous to that of pyramidal cells, ii) independent of injury severity, and iii) unresponsive to TH. HI was induced in C57BL6 mice at p10 with unilateral right carotid ligation and 45 min of hypoxia (FiO2=0.08). Mice were randomized to normothermia (36⁰C,NT) or TH (31⁰C) for 4h after HI and anesthesia-exposed shams were use as controls. Brains were studied at 24h (p11) or 8d (p18) after HI. Vglut1, GAD65/67, PSD95, parvalbumin (PV) and calbindin-1 (Calb1) were measured. Cell death was assessed using cresyl violet staining and TUNEL assay. Hippocampal atrophy and astroglyosis at p18 were used to assess injury severity and to correlate with number of PV+INs. VGlut1 level decreased by 30% at 24h after HI, while GAD65/67 level decreased by ~50% in forebrain 8d after HI, a decrease localized in CA1 and CA3. PSD95 levels decreased in forebrain by 65% at 24h after HI and remained low 8 days after HI. PV+INs increased in numbers (per mm2) and branching between p11 and p18 in sham mice but not in NT and TH mice, resulting in 21 to 52% fewer PV+INs in injured mice at p18. Calb1 protein and mRNA were also reduced in HI injured mice. At p18, somatodendritic attrition of INs was evident in all injured mice without evidence of cell death. Neither hippocampal atrophy nor astroglyosis correlated with the number of PV+INs at p18. Thus, HI exposure has long lasting effects in the hippocampus impairing the development of the GABAergic system with only partial protection by TH independent of the degree of hippocampal injury.
Keywords: GABAergic neurons, Parvalbumin, Calbindin, Memory, Developing brain
INTRODUCTION
Neonatal hypoxia-ischemia (HI) is the most common acute insult affecting the brain of full term infants (Johnston, 2005). The standard of care, therapeutic hypothermia (TH), decreases death or severe disability by 33% at 18 months of age in infants suffering HI brain injury (Jacobs et al., 2013; Kurinczuk et al., 2010; Shankaran et al., 2005). However, TH does not decrease the risk of cognitive impairments at school age among survivors of HI (Azzopardi et al., 2014; Shankaran et al., 2012). Since memory disabilities account for a significant proportion of cognitive impairments after neonatal HI (de Vries and Cowan, 2009; Gadian et al., 2000; Marlow et al., 2005) and both, clinical and pre-clinical studies, suggest that memory impairments are not attenuated by TH (Azzopardi et al., 2014; Burnsed et al., 2015; Diaz et al., 2017), the study of the hippocampus, and area essential in memory processing, is important to identify injury mechanisms not fully addressed by TH and to design novel adjuvant therapies.
Within the hippocampus, preserved interaction between pyramidal cells and interneurons (INs) is essential for learning and memory formation and for functional recovery after injury (Coghlan et al., 2012; Heckers and Konradi, 2014; Kalueff, 2007; Reichel et al., 2014). Although neonatal HI brain injury is often deadly to neurons within the hippocampus, (Johnston, 2005) their susceptibility to acute death appears to be subtype-specific. In-culture, pyramidal cells are more susceptible than INs to early cell death following simulated ischemia (Ramamoorthy and Shi, 2014). Similarly, in-vivo neuronal loss within the hippocampal pyramidal cell layer occurs early after HI in both p7 (pre-term) (Aggarwal et al., 2014; Stone et al., 2008) and p10 (near-term) (Burnsed et al., 2015; Northington et al., 2011) mouse models. This early injury of hippocampal pyramidal cells by HI is followed by atrophy that evolves after the insult (Burnsed et al., 2015; Northington et al., 2011) and is linked to memory impairments unresponsive to TH (Burnsed et al., 2015; Diaz et al., 2017). Whether the hippocampal atrophy evolving after neonatal HI is the result of delayed death of specific neuron subtypes or impaired maturation of surviving neurons leading to maladjusted connectivity or a combination of these events remains unclear.
Inhibitory synaptic activity requires fully functional INs to preserve working memory and cognition by limiting “intrusive inputs”. Biochemical, morphological and functional differences among INs within the hippocampus are vast (Somogyi and Klausberger, 2005). Parvalbumin (PV) identifies the greatest proportion of INs whose neuronal bodies are localized within the pyramidal cell layer (axo-axonal, basket, and bi-stratified cells) and calbindin (Calb)1 identifies those INs receiving glutamatergic inputs from the CA3 region (Schaffer collateral associated cell in the CA1 region) and those exclusively innervating INs in the CA1, CA3 and dentate gyrus (DG) (Heckers and Konradi, 2014; Somogyi and Klausberger, 2005). Thus, INs expressing PV and Calb1 provide a significant proportion of the inhibitory synaptic influence, (Hefft and Jonas, 2005; Karadi et al., 2012; Takesian and Hensch, 2013) and decreased numbers or complexity of those INs correlate with memory impairments in mice (Stevens et al., 2012). We hypothesize that injury of hippocampal INs after HI is: i) asynchronous to that of pyramidal cells, ii) independent of severity of injury, and iii) unresponsive to TH.
MATERIALS AND METHODS
Mice.
We used C57BL6 mice of both sexes for the experiments. The research protocol was approved by the Institutional Animal Care and Use Committee at Johns Hopkins University – School of Medicine and followed the Guide for the Care and Use of Laboratory Animals provided by the NIH, US Department of Health and Human Services 85–23, 1985.
Neonatal HI brain injury model and TH.
We used the Rice-Vannucci model adapted for neonatal mice to induce HI in C57BL6 mice at postnatal day (p) 10 as previously published. (Ditelberg et al., 1996; Graham et al., 2004) In brief, mice were exposed to anesthesia using inhaled isoflurane (3% for induction and 1% maintenance) and nitrous oxide to perform permanent unilateral right carotid artery ligation and after 1h recovery, mice were exposed to 45 minutes of hypoxia at FiO2 = 0.08. After HI, mice pups were randomized to normothermia (NT, 36˚C) or exposure to TH (31˚C) for 4h as previously described by our group.(Burnsed et al., 2015) Continuous core body temperature monitoring (Ad Instruments, Inc., Colorado Springs, CO) was performed in one pup in each treatment group using a tissue implantable thermocouple microprobe placed in the rectum prior to hypoxia and continuing throughout the 4h treatments. Sham mice were exposed to inhaled isoflurane and nitrous oxide at p10 for 5 min. Mice pups were returned to the dam after treatment (NT/TH) until they were killed at 24h (p11) and 8d (p18) after HI for mRNA and protein analysis (n = 4 −10 pups/ time/ treatment/ sex) or for immunohistochemistry (IHC) (n = 3 – 9 pups/ time/ treatment/ sex). Mice were killed via exposure to 20% (v/v) mixture of isoflurane in propylene glycol via onedrop exposure method (Markovic and Murasko, 1993) and then decapitated. Right forebrain (containing the hippocampus ipsilateral to carotid ligation) was rapidly microdissected and snap-frozen using isopentane on dry ice for real-time qRT PCR and western blot, while whole brain was perfused with 4% paraformaldehyde (4% PFA) and sectioned for cresyl-violet (CV) staining, TUNEL assay and IHC.
Exclusion criteria.
Hippocampal infarct in this model mostly involves the CA1 and CA3 subfields. If infract is extensive, it leaves little tissue for accurate evaluation. Therefore, forebrain pieces collected from brains showing obliterated hippocampus during microdissection were not used for biochemical analysis (RT-PCR or western blot). With this criterion we excluded, eight (4 NT and 4 TH) and seven (4 NT and 3 TH) at p11 and p18, respectively. Similarly, sections from perfused brains demonstrating hippocampal infarct involving >1/3 of the hippocampus (assessed by CV staining and GFAP IHC vs. contralateral hemisphere) were not included in the analysis (CV staining, TUNEL assay, and IHC). With this criterion, nine (5 NT and 4 TH) and eleven (5 NT and 6 TH) were excluded at p11 and p18, respectively. Mice developing large hippocampal infarcts were not protected by TH. This adds translatablity to the model, since randomized clinical trial of TH in humans also demonstrated lack of protection on severe cases of HI encephalopathy (Shankaran et al., 2005).
Determination of gene expression by real time qRT-PCR.
Total RNA was extracted from two combined pieces from anterior and posterior right forebrain from all treatment groups. For NT and TH right forebrain was ipsilateral to carotid ligation, as described above. PureLink™ RNA mini kit purification system (Invitrogen, Carlsbad, CA) was used according to manufacturer specifications. One μg of total RNA was used for generation of complementary DNA (cDNA) using iScript cDNA synthesis kit (BioRad, Hercules, CA). Reverse transcription protocol included 5 min at 25°C; 30 min at 42°C and 5 min at 85°C. cDNA was then used to amplify PV, and Calb1 genes by real time qRT-PCR using 300 nM concentration of specific primers (Table 1 (Liu et al., 1996)).The amplification protocol included 40 cycles of 30 sec at 95.0 °C, 1 min at 60.5 °C ending with 30 sec at 72.0 °C. GADPH (forward: 5’-TTGTCAAGCTCATTTCCTGGTATG-3’; reverse: 5’-GCCATGTAGGCCATGAGGTC-3’; 76-bp PCR product) and β-actin (forward: 5’-CCCAACTTGATGTATGAAGG-3’; reverse: 5’-TTGTGTAAGGTAAGGTGTGC-3’; 119-bp PCR product) were evaluated as housekeeping genes using the BestKeeper approach to determine stability of gene expression under experimental conditions(Pfaffl et al., 2004) as previously reported(Chavez-Valdez et al., 2012). Based on their gene stability, β-actin was selected as the housekeeping gene for calculations. Fold difference in gene expression was then corrected to β-actin using the Pfaffl method(Pfaffl, 2001). Melting curves confirmed amplification of single PCR products.
Table 1.
List of primers
| GENE | Primer | Sequences | UniSTS ID [Reference] |
|---|---|---|---|
| Parvalbumin | Sense | 5’- CCTCTTCCTTTCCTTGCAAG-3 | 119724 |
| Antisense | 5’-GGTGTCTGTATCACATTCACCC-3’ | ||
| Calbindin-1 | Sense | 5’ AGTGGTTGTGGTCACCAACTCT 3’ | [29] |
| Antisense | 5’ CCAGGTTACTACCAGTGCAGGA 3’ |
VGlut1, GAD65/67, PSD95, and Calb1 protein levels.
Cytoplasmic and synaptic-enriched fractions were prepared using a piece of forebrain from the same brains used for RNA isolation. A modified Subcellular Protein Fractionation Kit (Thermo Scientific, Rockford, IL, Cat # 87790) protocol was used to obtain these fractions. All buffers were supplied as part of the kit and were supplemented with protease and phosphatase inhibitors prior to use. In brief, ~100 mg of forebrain was washed in ice-cold phosphate-buffered saline (PBS, pH 7.2) prior to homogenization in Cytoplasmic Extraction Buffer at 1:10 (w:v) using a tissue grinder. Homogenized tissue was transferred to a tissue strainer and centrifuged at 500 x g for 5 min. Supernatant (cytoplasmic fraction) was collected and used for Calb1 immunoblotting. Pellet was sequentially re-suspended in ice cold Membrane and Nuclear Extraction Buffers at 1:6.5 and 1:2.25 (w:v), respectively. Supernatants (membrane and nuclear fractions) were stored at −80°C for testing. The final pellet was re-suspended using the Pellet Extraction Buffer at 1:1.25 (w:v) containing protease and phosphatase inhibitors and confirmed to be synaptic-enriched fraction following testing of all fractions with synaptic markers (data not shown). Twenty percent (w/v) glycerol was added to the fractions for cryoprotection and protein concentrations were determined using the Bradford assay (Bradford, 1976). Thirty μg-aliquots of homogenized protein were diluted 3:1 (v:v) in 4X loading buffer under reducing conditions and loaded in 4–20% mini-protean TGX polyacrylamide precast protein gels (Biorad Inc,, Hercules, CA). Protein was transferred to nitrocellulose membrane using TransBlot Turbo Midi-size (Biorad Inc), stained with Ponceau S, blocked with 2.5% bovine serum albumin (BSA) with 0.1% Tween-20 in 50 mM Tris buffered saline (TBST, pH 7.4). Nitrocellulose blots were incubated overnight at 4°C with primary antibodies at 1:200 (Vglut1) or 1:500 (GAD65/67, PSD95 and Calb1). After exposure to each primary antibody, membranes were washed with TBST, exposed to secondary antibodies for 1h and then developed using enhanced chemiluminescence (Clarity Western ECL Substrate, Biorad Inc.). To quantify protein immunoreactivity, optical density (OD) was determined with iVision Software adjusted for background. The reliability of sample loading and protein transfer was verified by staining nitrocellulose membranes with Ponceau S before immunoblotting.
IHC for GAD 65/67, PV, Calb1, and GFAP.
For IHC, pups were killed with an overdose of isoflurane as described above, and exsanguinated with cold 0.1 M PBS (pH7.4) via intra-cardiac perfusion. Brains were perfusion fixed with 4% PFA in 0.1M PBS for 30 min at 4ml/min. Tissues were cryoprotected with graded immersion in 15% and then 30% sucrose in PBS until the tissue sank, frozen and stored in −80° C until cut at 50μm on a freezing microtome (Northington et al., 1996). Floating IHC was performed as previously described (Northington et al., 1996) with whole rabbit antisera anti-PV (ABcam plc, Cambridge, MA; 1:250), anti-Calb1 (Cell Signaling Technology, Inc, Beverly MA; 1:250), anti-GFAP (DAKO / Agilent Technologies, Santa Clara, CA; 1:1000), or anti-GAD65/67 (ABcam PLC; 1:500) followed by goat anti-rabbit antibody (1:200) used as the secondary antibody and DAB as the chromagen (Northington et al., 1996). CV counterstaining was performed in those sections previously immunostained for PV to better assess the nuclear morphology of pyramidal cells surrounding PV+INs.
Single and double labeled IF-IHC for PSD95, α-tubulin, PV, TNF-α and GFAP.
Similar procedures to process the tissue were followed as described for standard IHC. In addition, 50μm sections were washed in TBS (pH 7.2) for 10 min prior to antigen retrieval with sodium citrate buffer (pH 6.0) incubated for 30 min at 80°C. Tissues were permeabilized using 0.2% Triton X in TBS for 15 min at RT, blocked using 4% normal goat serum (NGS) in 0.1% Tween/TBS for 1h at RT, prior to exposure to: i) chicken anti-GFAP (Novus Biologicals USA, Littleton, CO; 1:1500) and rabbit anti-PV (ABcam PLC; 1:1000) or rabbit anti-TNF-α (Cell Signaling Technology, Inc; 1:250) in 4% NGS in TBS overnight at 4°C; or ii) chicken anti-α-tubulin (Abcam PLC; 1:1000) and rabbit anti-PSD95 (Proteintech Group, Inc. Rosemont, IL; 1:200). Following incubation with primary antibodies and washes, tissues were incubated in the dark for 2h at RT exposed to goat anti-chicken IgY Alexa Fluor 488 and goat anti-rabbit Alexa Fluor 568 emitting a green and red fluorescence signal, respectively (Thermo Fisher Scientific, Inc, Waltham, MA). Following exposure to secondary antibodies, tissues were incubated for 5 min in 4’,6-daamidino-2-phenylindole (DAPI; 1μg/mL) in TBS, washed in TBS, and mounted and dried for 30 min prior to coverslip with ProLong Gold Antifade Mountant (Molecular Probes, Life Technologies Corp., Carlsbad, CA).
Antibodies.
For western blot.
Vglut1 (SC377425; RRID: AB_2687960): mouse monoclonal antibody raised against amino acids 1–55 at the N terminus of VGlut1 of human origin detecting a single band at 55 kDa (1μg/ml). GAD65/67 (Ab11070; RRID: AB_297722): rabbit polyclonal antibody raised against a synthetic peptide corresponding to the C terminal amino acids 572–585 of human origin GAD65–67 detecting a doublet band at 67/65 kDa (2μg/ml). PSD95 (Ab18258; RRID:AB_444362): rabbit polyclonal antibody raised against a synthetic peptide derived from residues 50–150 of mouse PSD95 detecting a product a ~85 kDa (2 μg/ml). Calb1 (CS13176; RRID:AB_2687400): Rabbit monoclonal antibody raised against a recombinant protein specific to the N-terminus of whole Calb-1 protein of human origin detecting product at 28 kDa (2μg/ml).
For immunohistochemistry. DAB-based.
GAD65/67 (Ab11070): details as described above for western blotting (2μg/ml). GFAP (DAKO Z0334; RRID:AB_10013382): Rabbit polyclonal antibody raised against GFAP isolated from cow spinal cord with no reported cross reactivity (1μg/ml). PV (Ab11427; RRID:AB_298032): Rabbit polyclonal antibody raised against a full length purified native protein corresponding to rat PV from skeletal muscle and tested using cerebellar Purkinje Cells as positive control in every slide (4μg/ml). Calb1 (CS13176): details as described above for western blotting. Similar to PV IHC, cerebellar Purkinje Cells were used as internal positive controls for IHC (4μg/ml).
Immunofluorescence.
PSD95 (Proteintech 20665–1-AP; RRID: AB_2687961): Rabbit polyclonal antibody raised against a PSD95 (drosophila) peptide (5μg/ml). α-tubulin (ab89984; AB_10672056): Chicken polyclonal antibody raised against a synthetic peptide conjugated to KLH derived from within residues 1 – 100 of Human α-tubulin (4μg/ml). PV (Ab11427), details as described above for DAB-based IHC (1μg/ml). GFAP (Novus NBP1–05198; RRID:AB_1556315) Chicken polyclonal antibody raised against purified GFAP from bovine spinal cord (0.75μg/ml). TNF-α (CS11948; AB_2687962): Rabbit monoclonal antibody which recognized endogenous levels of total TNF-α protein and produced using recombinant protein (4 μg/ml).
Quantification of PV+INs.
Quantification of PV+INs was performed by two independent investigators (RCV and JGH) blinded to treatment groups. Fifty μm coronal sections of anterior, middle and posterior hippocampus were analyzed as a whole and by regions (CA1, CA3 and DG). For both CA1 and CA3, PV+ INs were quantified within the area extending from the alveus to the lacunosum moleculare layer including the oriens, pyramidal cell and radiatum layers. CA1 region was limited by the fasciola cinereum (anterior sections) or the dorsal subiculum (middle and posterior sections) and the CA2 region (all sections). CA3 region was limited by the CA2 region and the polymorph layer of the DG. The DG region was limited by the microvessels crossing the hippocampus within the lacunosum moleculare layer and the moleculare layer of the dentate and included the polymorph layer limited by the CA3 region. For these experiments hippocampal sections were classified as: i) anterior, if the fasciola cinereum in the vicinity of the dorsal third ventricle was identified; ii) middle, if dorsal subiculum was identified and the CA3 did not cross the horizontal level of the fasciculus retroflexus localized in the midbrain; and iii) posterior, if the CA3 region crossed the level fasciculus retroflexus and the pyramidal cell layer was ventrally continuous. Once the regions were agreed upon, quantification was performed under the light microscope using magnifications of 20X and 40X to include only PV+ INs demonstrating full cell body within the section. Hippocampal regions (CA1, CA3, and DG) and sections (anterior, middle and posterior) were analyzed individually and combined. Image J software was used to determine the area of each of the regions and the whole hippocampus and the number of PV+ INs were adjusted for area (mm2).
TUNEL assay.
Fifty-micron coronal brain sections containing anterior, middle and posterior hippocampus were used in the assay. All buffers and protocols were supplied by the manufacturer as part of the colorimetric TUNEL system kit (Promega Corp, Madison, WI). Tissues were washed in 0.85% NaCl for 5 min followed by immersion in PBS for 5min, additional fixation in 4% PFA for 15min and permeabilization using a 20 μg/ mL solution of proteinase K for 35 min. After further fixation with 4% PFA and wash in PBS, the reaction mix containing a biotinylated nucleotide mix, recombinant terminal deoxynucleotidyl transferase (rTdT) enzyme was applied for 60 min at 37°C. After washing with double strength saline sodium citrate (2X SSC) followed by PBS, sections were blocked with 0.3% hydrogen peroxide, exposed to Streptavidin HRP and stained with DAB. Tissues were counterstained with CV. DNAase I treated sham hippocampal slices were used as positive controls in every experiment. DNAase I (Promega Corp.) was prepared in 4 mM Tri-HCl (pH 7.9), 1 mM NaCl, 0.6 mM MgCl2, and 1mM CaCl2 to produce a 10 unit/mL solution, and was applied for 5 min to tissue sections. Reaction mixture lacking biotinylated nucleotide mix or rTdT, were used as negative controls for the TUNEL reaction.
Assessing hippocampal and CA1 atrophy following HI.
A separate subset of p18 anterior coronal hippocampal sections from sham and HI injured mice were stained using CV following standard methods. These sections were adjacent to those used for GFAP and PV IHC. All sections used to measure the hippocampus were assessed for symmetry between the right and left hemispheres. Assessment of plane symmetry of coronal sections was based on the evaluation of the shape and localization of several structures within the midbrain, including the cerebral peduncle, the fasciculus retroflexus, and the dorsal lateral geniculate nucleus, as well as the evaluation of the striatum laterally. An ocular filar micrometer inserted in the eyepiece of the light microscope was used to measure in the ipsilateral anterior hippocampus the: i) length from the third to the lateral ventricle, ii) height medially from the alveus to the limit with the thalamus, and iii) the height of the CA1 pyramidal cell layer between the oriens and radiatum layers. A hippocampal rhomboid area was calculated using the height (h) and length (l) of the hippocampus (0.5*h*l).
GFAP-derived scoring.
A semi-quantitative GFAP-derived scoring system was developed to grade astrogliosis as a surrogate for hippocampal injury at p18 (Chalak et al., 2014; Cikriklar et al., 2013; Danzer et al., 2011) for the purpose of evaluating correlation between injury and PV+ IN counts (Table 2). The GFAP-derived scoring system quantifies the dispersion IR within the hippocampus and the morphological changes characteristic of astrogliosis (Pekny and Nilsson, 2005; Sofroniew and Vinters, 2010). Evaluation was performed in the CA1, CA3 and DG regions in sections containing anterior, middle and posterior hippocampus. The worse score among the three hippocampal sections determined the grade of injury for the specific hippocampal region. The cumulative score was the sum of the scores for each of the three regions. Scoring was based on the abundance of GFAP staining and glial scars evident at low magnification (4X), astrocyte body size, and number and thickness of their branching, as well as presence of overlap between astrocytic domains at higher magnification (20X) (Sofroniew and Vinters, 2010). GFAP score varied between 1 and 7 and the cumulative score varied from 3 to 21.
Table 2.
GFAP-derived scoring system
| Low magnification (4X) | Higher magnification (20X) | ||||
|---|---|---|---|---|---|
| Staining | Glial scars | Astrocyte body |
Astrocyte branching |
Astrocyte domain overlap |
|
| 1 | Light | 0 | Pinpoint | few and thin | No |
| 2 | Light | 0 | Pinpoint | many but thin | No |
| 3 | Light | 0 | Double | many and thick | Yes |
| 4 | + | 1 | Larger | thick branching | Yes |
| 5 | ++ | 2 | Larger | thick branching | Yes |
| 6 | +++ | ≥3 | Larger | thick branching | Yes |
| 7 | ++++ | diffuse | Larger | thick branching | Yes |
Statistics.
For analysis of two related groups, such as the fold-change in gene expression relative to sham produced by the Pfaffl method, non-parametric Wilcoxon signed-rank test vs. sham stratified by sex was applied. For analysis of multiple groups, non-parametric Kruskal-Wallis H one-way ANOVA stratified by sex was applied with post-hoc pair analysis using Dunn-Bonferroni’s test. All results were presented as box and whisker plot, where the box was limited by the 25th and 75th percentiles and the solid line represented the median. Significance was assigned by p-value ≤ 0.05 in all cases, with adjustment for multiple comparisons if needed. Non-parametric Spearman Rho correlations were applied between the number of PV+ INs and the hippocampal and CA1 measurements as well as the GFAP scoring. Analysis was performed using IBM SPSS Statistics 24v (IBM Corporation, Armonk, NY).
RESULTS
Pyramidal neuron cell death occurs acutely after HI, but there is limited evidence of hippocampal cell death at p18.
We assayed for TUNEL to detect the spatiotemporal distribution of cell death in hippocampus as visualized by DNA fragmentation. Representative positive (Fig 1A) and negative (Supplemental fig 1) controls are shown. A few scattered TUNEL + cells were identified throughout some of the hippocampal sections of sham mice at p11 (Fig 1B) and p18. A large number of cells with DNA fragmentation were observed 24h after the HI insult (p11) throughout the hippocampus from both male (Fig 1C) and female injured mice (Fig 1E). Clusters of TUNEL + cells in the process of dying contained pyknotic and shrunken nucleus, with apparent spillage of nuclear contents to the cytosol (Fig. 1C”). TUNEL + cells were more abundant in the CA1 and CA3 regions than the DG at p11. On the contrary, TUNEL + cells were rarely observed in the hippocampus at eight days after HI (p18) in either male or female mice (Fig 1F-I).
Figure 1. Limited evidence of delayed hippocampal cell death at p18.

Representative photomicrographs of TUNEL stained anterior hippocampus from sham/ DNAase treated (A), sham at p11 (B, D) and at p18 (F, G) and NT at p11 (C, E) and at p18 (H, I) treated male and female mice with details (A’-I’). Additional high magnification detail panels are shown to demonstrate karyorrhexis documented with DNAse treatment (A”) and pyknosis with nuclear shrinkage in a low basal level in the sham (B”) and more prominent in NT injure mice (C”).
Neonatal HI induces early decrease in forebrain Vglut1 followed by delayed decrease in GAD65/67.
Although ~20% of mice developed a large hippocampal ischemic infarct in response to HI as described in “methods” (data not shown), most developed focal or segmental injury within the pyramidal cell layer with various cell death phenotypes typically within the CA1 and CA3 regions at 24h (p11) after neonatal HI (Fig 2A1–2). The timing of pyramidal cell death coincided with the 30% decrease in pre-synaptic glutamatergic marker, Vglut1 in synaptic-enriched protein fractions from NT and TH male mice at 24h after HI (H=7.3, df 2, KW p=0.026; p<0.05 vs. sham; n=5–10 per group, Fig.2B), which persisted by eight days after HI in NT mice (H=7.8, df 2, KW p=0.02; p=0.016 vs. sham) but recovers in TH mice (p=0.18 vs. sham). In contrast, the pre-synaptic GABAergic marker, GAD65/67 was decreased by 37% at eight days, but not at 24h after HI injury ((H=8.5, df 2, KW p=0.014; p=0.03 vs. sham; n=4–8 per group, Fig.2C) in forebrain of male NT mice. IHC revealed that the decrease in GAD65/67 protein was primarily localized to the CA1 and CA3 regions of both, male and female mice (Fig.2 D 1–6 , representative photomicrographs of female hippocampi are shown). While GAD 65/67 + immunoreactivity (IR) in p18 sham mice hippocampi extended from the limit of the oriens layer (Or) towards the radiatum layer (Rd), IR in the HI injured hippocampus was limited to the region adjacent to the Or. These early and delayed changes in glutamatergic and GABAergic markers, respectively, may explain the lack of progressive increase of PSD95 levels between p11 and p18 (Fig. 2E). At eight days after HI, PSD95 remained decreased in synaptic-enriched fractions by 50% and 44% in NT male and female mice, respectively (KW p<0.05 and p<0.05 vs. sham in both; Fig. 2F) and TH attenuated that effect. Double-labeling immunofluorescence for PSD95 and α-tubulin documented a decreased number of puncta on the surface of dendrites and neuronal soma within the CA1 pyramidal cell layer at p18 (Supplemental Fig. 2).
Figure 2. Early decrease in Vglut1, delayed decrease in GAD65/67 and persistent decrease in PSD95 following neonatal HI.

Representative CV staining photomicrographs of hippocampal CA1 (A1) and CA3 (A2) regions from NT mice showing classical columnar pyramidal cell layer (Py) injury with abundant necrotic cell death and apoptotic bodies (arrows) at p11. Box and whiskers plot representing protein expression of VGlut1 (B), GAD 65/67 (C) and PSD95 (F) in forebrain synaptic-enriched fractions from sham (white), NT (black) and TH (grey) treated male and female mice. Boxes are limited by the 75th and 25th percentiles (interquartile range, IQR) and whiskers are limited by the last data point within 1.5 times the IQR from the median (continuous line inside the box). *, p<0.05. Representative immunoblot comparing expression of PSD95 at p11 and p18 in sham and NT male mice (E). Representative photomicrographs of GAD 65/67 showing immunostaining of synaptic buttons (D1 and D2) and differences in IR in the hippocampal CA1 (D1 vs.D5) and CA3 (D4 vs. D6) regions from sham and NT treated female mice at p18. Rd, radiatum layer; PC, pyramidal cell.
Deficit of PV and Calb1 positive INs in the hippocampus at eight days after neonatal HI.
The number of INs expressing PV almost doubled between p11 and p18 in the sham mice whole hippocampus (p=0.006 [male]; p=0.034 [female]), and CA1 (p=0.03 [male]; p=0.034 [female]) and CA3 (p=0.045 [male]; p=0.034 [female]) subfields, but it remained stagnant in HI injured mice treated with NT. HI injury at p10 did not affect the number of PV + INs in either the whole hippocampus or independently in the CA1, CA3 or DG regions 24h after HI (p11) (n=3–5 per group; Fig. 3A-C and 3G). In contrast, the numbers of PV + INs were lower in HI injured mice treated with NT than in sham at eight days after HI (p18) and TH provide inconsistent protection (Fig. 3D-F and 3G). At p18, the number of PV + INs (per mm2) in the whole hippocampus of male (Fig. g1) and female (Fig. g2) HI injured mice treated with NT were 46% and 58% lower than sham, respectively (H=13.2, df 2, KW p=0.001, p=0.001 vs. sham [males]; H=10.1, df 2, KW p=0.006, p=0.004 vs. sham [females]; n=4–9 per group; Fig 3D vs. 3E and Fig 3G). Even with TH the number of PV+ INs were 24% and 39% lower than sham in male and female injured mice, respectively (p<0.05 vs. sham in both, Fig 3F and3G). The number of PV+ INs were similarly deficient in anterior (28%−47%; H=13.1, df 2, KW p=0.001 [male]; H=8.1, df 2, KW p=0.017 [female]; Supplemental fig 3A1–2), middle (51%; H=8.7, df 2, KW p=0.013 [male]; H=10.1, df 2, KW p=0.006 [female]; Supplemental fig 3B1–2) and posterior (48–49%; H=8.9, df 2, KW p=0.01 [male]; H=8.7, df 2, KW p=0.013 [female]; Supplemental fig 3C1–2) sections of the hippocampus of HI injured male and female mice treated with NT, while those treated with TH had variable responses, which were sex-specific. TH appears to attenuate the deficit in PV+ INs in the more posterior hippocampal sections of male injured mice, but not in the anterior sections. TH did not prevent the deficit in PV+ INs in female mice (Supplemental fig 3A-C). Deficits in the number of PV + INs were primarily documented in the CA1 (25–52%) and CA3 regions (42–57%) in NT mice (p<0.05 vs. sham; Fig 3G) and TH only attenuated the deficit of PV+ INs in the CA1 region of the injured male. Deficit in PV + INs in the DG was exclusive of injured female mice (Fig 3g2). In addition to decreased number of PV + INs at eight days after HI, those INs that were positively IR to PV demonstrated somatodendritic attrition with decreased number and complexity of dendrites and shrunken cell bodies but otherwise normal nucleus. (Martin, 1999) TH appeared to partially prevent these changes (Fig 3H-J). The decrease number of PV + INs at eight days after HI and the partial response to TH in male mice matched the decrease in PV mRNA levels (Fig 3K).
Figure 3. Decreased number of PV+INs in hippocampal sections of HI injured mice at p18.

Representative photomicrographs of PV IR of hippocampus from sham (A, D), NT (B, E) and TH (C, F) treated male (A1 to F1) and female (A2 to F2) mice at p11 (A-C) and p18 (D-F) with details (A’-F’, magnification bar = 10μ). Number of PV+INs per mm2 in the hippocampus of male (G1) and female (G2) sham (white boxes; n (males) = 5 at p11 and 6 at p18; n (females) = 3 at p11 and 4 at p18), NT (black boxes; n (males) = 5 at p11 and 9 at p18; n (females) = 3 at p11 and 5 at p18), and TH (grey boxes; n (males) = 5 at p11 and 9 at p18; n (females) = 5 at p11 and 5 at p18) treated mice at p11 and p18. Data represented using box and whisker plots to show the PV+ INs/ mm2 counts in whole hippocampus (HIP), and in CA1, CA3 and DG regions in separate at p11 and p18. Boxes are limited by the 75th and 25th percentiles (interquartile range, IQR) and whiskers are limited by the last data point within 1.5 times the IQR from the median (continuous line inside the box).*, p<0.05. (n=3–9 mice per treatment, time and sex). Representative high magnification photomicrographs showing morphology of PV + INs of sham (H), NT (I), and TH (J) at p18. Fold change in PV mRNA levels in forebrain of male and female mice (K) treated with NT (black) and TH (grey) vs. sham (discontinuous line sitting at 1)*, p<0.05. (n=5 mice per treatment and sex).
In PV IHC sections counterstained with CV, morphologically normal INs in the CA1 (Fig.4A1) and CA3 (Fig.4A2) were observed in close proximity to dying pyramidal neurons showing variable degrees of pyknosis, karyolysis and karyorrhexis. Very few PV+INs manifested pyknoticnuclear changes suggestive of cell death at p11 (Fig.4B). Invariably, these few INs dying at 24h after HI injury were always surrounded by dying pyramidal cells. At p18, most PV+ INs demonstrating somatodendritic attrition did not show nuclear changes suggestive of cell death (Fig.4C).
Figure 4. Morphologically intact INs in close proximity to dying pyramidal cells in the hippocampus at p11.

Twenty-four hours after HI injury, extensive evidence of necrotic cell death of pyramidal cells is documented in the CA1 (A1) and (A2) regions of the hippocampus using CV counterstaining. Large number of nuclei demonstrating variable degrees of karyolysis and karyorrhexis (black arrow heads) and pyknosis (black arrows) are shown just adjacent to a morphologically intact PV+ IN (white asterisk, A1 and A2). A1’ and A2’ photomicrographs were captured at a different depth from same field to better document the morphology of the PV+ IN. In rare instances, nuclear condensation suggesting cell death of some PV+INs (black asterisk, B) is documented in close proximity to morphologically normal INs (white asterisk). Invariably, these few INs dying at 24h after HI injury are always surrounded by injured pyramidal cells with nuclei demonstrating pyknosis (black arrows) or karyorrhexis (black arrow head). Eight days after HI, many PV+ INs demonstrate somatodendritic attrition without nuclear condensation to suggest cell death (white arrow, C). Microglia is still observed 8 days after the HI insult.
Calb1 was used to assess changes in a different subset of hippocampal INs after HI injury and TH. This marker for INs is only useful to differentiate INs in the pyramidal cell layer since it is expressed heavily by the DG granular cells as well. Similar to PV+ INs, the number of Calb1+ INs appeared decreased in the CA1 and CA3 regions of the hippocampus eight days after HI (Fig 5A-F). TH did not prevent these deficits after HI. In agreement, western blotting showed that at eight days after HI (p18), Calb1 protein levels in cytosolic fractions decreased by 34% and 28% in HI injured male and female mice, respectively (H=8.5, df2, KW p=0.014 [males]; H=8.0, df 2, KW p=0.018 [females]), and TH attenuated these decreases (Fig 5G1–2). Changes in Calb1 mRNA levels in the forebrain of injured male and female mice at eight days after HI were also found (Fig 5H). As shown for injured PV + INs, somatodendritic attrition was also documented in Calb1 + INs eight days after HI with attenuation of these morphological changes by TH (Fig 5I-K).
Figure 5. Anterior hippocampus showing decreased number of Calb1+INs in injured mice at p18.
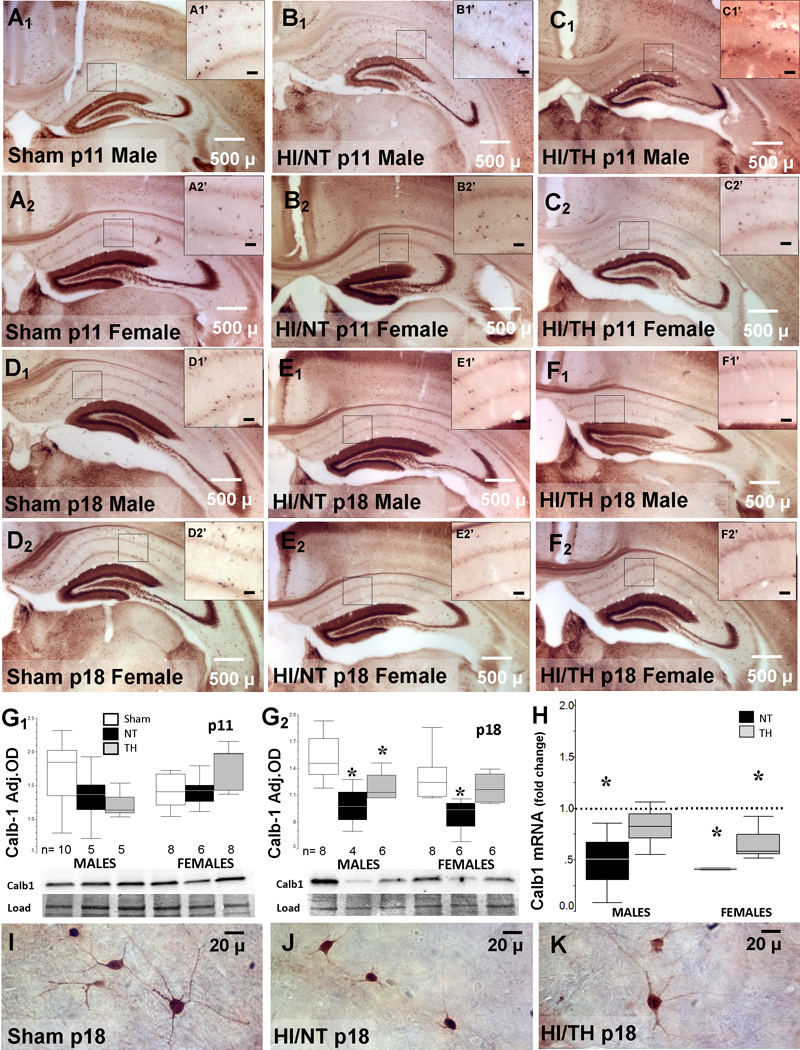
Representative photomicrographs of Calb1 IR of anterior hippocampus from sham (A, D), NT (B, E) and TH (C, F) treated male (A1 to F1) and female (A2 to F2) mice at p11 (A-C) and p18 (D-F) with details (A’-F’, magnification bar represents 20μ). Box and whisker plots representing Calb1 protein levels in forebrain of male and female sham (white), NT (black), and TH (grey) treated mice at p11 (G1) and p18 (G2). Box and whisker plot representing fold change in Calb1 mRNA levels (H) in NT (black) and TH (grey) vs. sham (discontinuous line sitting at 1). Boxes are limited by the 75th and 25th percentiles (interquartile range, IQR) and whiskers are limited by the last data point within 1.5 times the IQR from the median (continuous line inside the box). *, p<0.05. (n=4–10 mice per group). Representative high magnification photomicrographs showing morphology of Calb1 + INs of sham (I), NT (J), and TH (K) at p18.
Number of PV+ INs did not correlate with severity of gliosis or degree of hippocampal atrophy.
GFAP expression increased between p11 and p18 and was more often documented in the lacunosum moleculare layer extending towards the pyramidal cell layer with some sparing of the radiatum and oriens layers (data not shown). Although, increase in hippocampal GFAP + IR and decrease in the number of PV+ INs appeared in concert eight days after HI (Fig 6A vs. 6B), the severity of the deficit of PV+INs in the CA1 pyramidal cell layer did not follow the degree of gliosis estimated by GFAP IR (representative microphotographs, Supplemental fig. 4). Increase in GFAP + IR or decrease in the number of PV+ INs were not caused by hypoxia alone as shown when comparing the contralateral (hypoxia alone) vs. the ipsilateral (carotid ligation side) hippocampi (Fig 6C). The GFAP-derived score (Table 2) and the percent of hippocampal atrophy were directly correlated eight days after HI (R2=0.88, p<0.001; Fig.6D1) and thus, both were used as delayed surrogates of the severity of HI injury. Similar correlations between the GFAP-derived score and the hippocampal height (R2=0.74, p<0.001), length (R2=0.79, p<0.001) and CA1 height (R2=0.80, p<0.001) were documented. Neither the measurements of hippocampal atrophy (Fig. 6D2), nor the GFAP-derived score (Fig.6D3) predicted the number of hippocampal PV+ INs preserved eight days after HI. Of note, no co-localization between TFN-α and GFAP was documented eight days after HI (data not shown).
Figure 6. No correlation between number of PV+ INs and hippocampal injury.
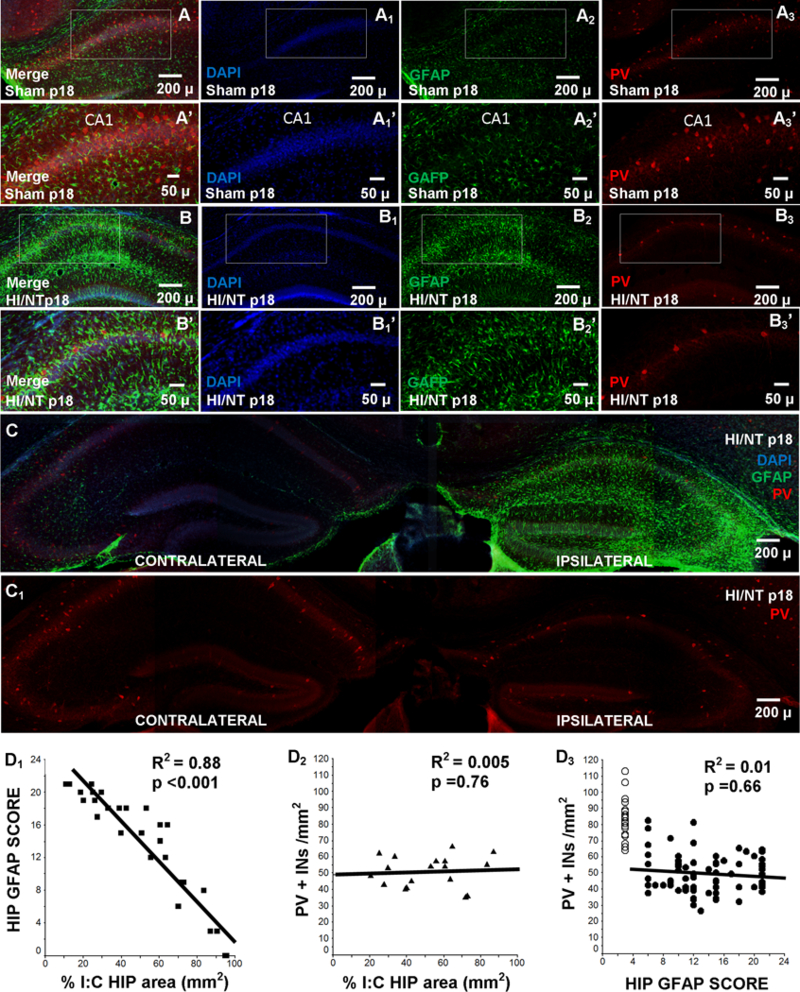
Representative photomicrographs of double immunostained GFAP (green, Alexa Fluor 488) and PV (red, Alexa Fluor 568) IF-IHC counterstained with DAPI. Anterior hippocampus from sham (A) and NT (B) treated mice at p18 are shown with details (A’ and B’). Comparison between anterior hippocampus ipsilateral and contralateral to the injury is shown as a merged GFAP (green) and PV (red) counterstained with DAPI image (C) and as a PV (red) image (C1) at p18. Scatter plots show correlations between: (D1) percent hippocampal atrophy (x-axis) and GFAP severity score (y-axis, table 2), (D2) number of PV+INs/ mm2 (y-axis) and percent hippocampal atrophy (x-axis), and (D3) number of PV+INs/ mm2 (y-axis) and GFAP severity score (x-axis) Best fitted regression line is also shown (continuous line) and R2 and p-values. ○, sham; ■, ▲, ●, injured mice. HIP, hippocampus
DISCUSSION
Here, we show that: i) early decrease in Vglut1 temporally coincides with the loss of hippocampal pyramidal cells 24 h after HI, and ii) delayed decrease in GAD65/67 coincides with reduced numbers of INs expressing PV and Calb1 within CA1 and CA3 and with evidence of somatodendritic attrition in those INs surviving by eight days after HI. Susceptibility to early cell death after HI is greater in pyramidal cells than in INs and even in those INs showing delayed somatodendritic attrition eight days after HI, the evidence of cell death is limited. TH attenuates the decline in the number of PV+ INs, but only in the CA1 region of injured male mice. The severity of injury assessed by the degree of hippocampal atrophy and astroglyosis does not account for the deficit in the number of PV+ INs at eight days after HI. Altogether, these data substantiate that even mild to moderate HI insult has long lasting effects in the hippocampus impairing the development of the GABAergic system with only partial protection by TH in a sex and regional specific manner.
Hippocampal injury progresses for many days after neonatal HI in the mouse (Northington et al., 2011; Northington et al., 2001; Stone et al., 2008). Published work points towards acute injury of pyramidal cells with classical petechial and laminar injury within the pyramidal cell layer evident by 24h after HI, (Aggarwal et al., 2014) which temporally matches the decrease in VGlut1 shown here. These acute events are partially prevented by TH, as we have shown here and elsewhere (Burnsed et al., 2015). In contrast, GAD 65/67 expression does not decrease at 24h after HI in the CA1 and CA3 regions of mice treated with NT, but it does at eight days after HI. Since, GAD 65/67 is an indicator of INs capable to produce and release GABA, (Le Magueresse and Monyer, 2013) our data document that GABAergic INs are more resilient than pyramidal cells to acute death after HI injury, but injury of INs may still occur at delayed points following the insult. Our results are consistent with observations documenting greater susceptibility of pyramidal cells to death than of GAD 65/67 + INs in primary cortical cultures exposed to near anoxia (FiO2 0.01) and in juvenile rats (p21) exposed to ischemia using a middle cerebral artery occlusion model (Ramamoorthy and Shi, 2014). The timing of GABAergic deficits after neonatal HI and the effect of TH have not been previously reported. The persistence of the GABAergic deficits beyond p18 has not been explored, but since the pattern of GABA and GAD expression and the development of GABAergic synapses in rodents at p18 are near to those documented in the adult rodent brain, (Del Rio et al., 1992; Huang et al., 1999; Luhmann and Prince, 1991) the delayed GABAergic deficits observed eight days after HI are likely to carry out to adulthood (Benes and Berretta, 2001).
Interneurons mature biochemically and morphologically during the post-natal period to functionally couple to the changes occurring in pyramidal cells within the hippocampus. In this process expression of biochemical markers such as PV and Calb1 is up-regulated (Le Magueresse and Monyer, 2013). Maturational arrest or delay has been documented in various models of injury in the developing brain (Salmaso et al., 2014). Here, we document that the number of INs expressing PV almost doubles between p11 and p18 in sham mice but it remains stagnant in CA1 and CA3 regions of HI injured mice treated with NT, a finding that may be the product of the down-regulation in PV gene expression eight days after HI injury. As a result, the number of PV+ INs is deficient eight days after neonatal HI when compared to sham mice, matching the delayed decrease in GAD 65/67 + IR. Along with these delayed biochemical deficits, morphological disturbances characterized by somatodendritic attrition are documented in a large number of surviving INs at eight days after neonatal HI. These morphological findings are in agreement with those reported in the CA1 of rats after HI (Mudrick and Baimbridge, 1989). Limited evidence of delayed death of INs is documented by us eight days after neonatal HI and by others in models of chronic hypoxia (Komitova et al., 2013). Subacute death at points not studied before p18 may explain the shortfall in the numbers of PV+INs documented with our experiments. However, our results may point towards a disturbance in the maturation of hippocampal INs following neonatal HI. Whether this potential developmental disturbance consists of maturational arrest or delay as reported in mouse models of prolonged hypoxic brain injury (Salmaso et al., 2014) or of recapitulation of immature biochemistry and phenotype during recovery after injury as documented in response to neurite transection of hippocampal neurons in culture or to kainic acid induced hippocampal seizures (Haas et al., 2004; Szabo et al., 2000), is still unknown.
The protection provided by TH against HI brain injury is sexually dimorphic in the murine model (Burnsed et al., 2015; Diaz et al., 2017). In accord with these observations, TH attenuated the deficit of PV+ INs in the CA1 region of male mice eight days after HI, but it did not protect female mice against loss of PV+ INs. This sexual dimorphism in response to TH may have a behavioral correlate; decreasing the inhibitory influence within the hippocampus of female mice and increasing the risk for hyperactivity/ anxiety behaviors, which are more often documented in injured female mice (Lucas et al., 2014) and are not ameliorated by TH in the same model used in the present experiments (Diaz et al., 2017). However, the protection of the male PV+ INs within the CA1 region by TH eight days after HI was not homogenous throughout the whole hippocampus, with greater protection in the posterior hippocampus than in the anterior hippocampus. Long-axis specialization studies suggest a functional dichotomy between the anterior and posterior hippocampus (Duarte et al., 2016; Duarte et al., 2014; Lepore et al., 2009; Maguire et al., 2000). While anterior hippocampus is involved in encoding and spatial learning; the posterior hippocampus is involved in memory retrieval and spatial navigation. Since spatial memory deficits inversely correlate with the number of hippocampal INs expressing PV (Stevens et al., 2012), our previously reported behavioral findings suggesting that encoding is impaired despite treatment with TH (Diaz et al., 2017), match the lack of protection of the PV+ INs in the anterior hippocampus in both sexes. However, isolation of specific aspects of memory retrieval (Vorhees and Williams, 2014) and thus, the study the sexual dimorphism in the posterior hippocampus requires more specific animal testing.
It is reasonable to assume that the severity of hippocampal injury may predict the degree of deficit of PV+ and Calb1+ INs by eight days after neonatal HI, with these late deficits simply being a late manifestation of the earlier insult to pyramidal cells and the loss of glutamatergic input. However, in humans, memory impairment does not appear to follow the severity of HIE or injury since no differences in memory outcomes at school age between HIE survivors treated or non-treated with TH have been reported despite protection of motor domains (Azzopardi et al., 2014). In the murine model of neonatal HI and TH, we have shown a similar profile of protection (Burnsed et al., 2015; Diaz et al., 2017) and thus, we hypothesized for our experiments that the changes in INs may not be dependent of the severity of hippocampal injury. Hence, two aspects of hippocampal injury were assessed eight days after neonatal HI in this study; first, the severity of hippocampal and CA1 atrophy; and second, the degree of astrogliosis as measured by a GFAP-derived grading system. The validity of GFAP IR as a delayed surrogate of CNS injury is based on a connection between type and severity of CNS injury and a continuum of changes resulting in long-lasting astrocytic disruption (Sofroniew and Vinters, 2010). The strong correlation between the severity of hippocampal and CA1 atrophy and the degree of astrogliosis eight days after HI, further validate the usefulness of the proposed scoring system in the evaluation of severity of hippocampal injury at delayed times. Furthermore, eight days after HI, GFAP and TNF-α did not co-localize and thus degree of astrogliosis at this time point was more a delayed reporter of severity of injury, rather than a marker of chronic neuroinflammation. Here, we found no correlation between the severity of hippocampal atrophy or the grade of astrogliosis and the number of PV+ INs in any hippocampal region eight days after neonatal HI. This lack of association suggests that the cascade of events leading to the decreased number of PV+INs eight days after HI is not solely caused by the lack of input from pyramidal cells or the overall degree of hippocampal disruption, but instead it may be a disruption of specific pathways leading to maturation of INs. Potentially, therapies targeting these unknown pathways enhancing survival or maturation of INs may supplement the effect of TH alone.
We acknowledge several limitations in this study. Whether PV and Calb1 IHC accurately represent the morphology of INs surviving HI, or instead disturbances in the transport of proteins to remote processes (Martin et al., 1998), is still unclear. We have not determined the mechanism(s) leading to decrease expression of PV and Calb1 or impaired maturation of INs following neonatal HI. However, sex-specific delayed alterations of specific neurotrophic pathways, similar to those reported acutely after HI (Chavez-Valdez et al., 2014), are under investigation. Additionally, the electrophysiological and functional consequences of potential impaired influence from INs in the developing hippocampus after HI and TH are also unknown. Because developmental GABAergic switch from depolarizing to hyperpolarizing is a process preserved across species (Bosman et al., 2005; Pinto et al., 2010), and hyperpolarizing responses to GABA are achieved after the first week of postnatal life in the mouse (Bregestovski and Bernard, 2012), we speculate that GABAergic deficits at p18 would result in long lasting inhibitory impairments. We also acknowledge that our experiments only assess the effects of neonatal HI and TH in a subset of mice not showing large hippocampal infarct, but since the experimental model has a much higher rate of hippocampal infarcts compare to that observed in human newborns after HIE, we believe that our methods strengthen the translatability of our results. Lastly, the translatability of the sexually dimorphic responses to HI and TH from the mouse to the human still requires extensive work. Although there is a growing body of evidence in rodent models (Burnsed et al., 2015; Chavez-Valdez et al., 2014; Diaz et al., 2017; Sanches et al., 2015; Shimizu et al., 2016; Waddell et al., 2016), and pre-cooling data from humans (Cohen and Stonestreet, 2014; Smith et al., 2014) suggesting sexual dimorphism in response to HI, randomized clinical trials for TH has been underpowered to assess sex differences in response.
Susceptibility to acute death is neuronal subtype-specific in the hippocampus following neonatal HI. While pyramidal cells are acutely loss following neonatal HI, INs are mostly preserved. INs develop delayed biochemical and morphological evidence that suggest maturational disruption with low expression of GAD65/67, PV and Calb1 along with somatodendritic attrition without significant evidence of cell death at eight days after HI. The functional implications of these delayed deficits are still unknown, but the known sexually dimorphic memory deficits reported after HIE (Baron et al., 2012; Marlow et al., 2005; Smith et al., 2014) may be explained by our findings. The protection afforded by TH to the hippocampal INs is limited to the posterior CA1 region in male mice. Therefore, modulating the maturational disruption of hippocampal INs after neonatal HI may complement the effects of TH alone in a sex-specific manner.
Supplementary Material
ACKNOWLEDGEMENTS.
The authors thank Mrs. Deborah Flock and Ms. Ariel Mason for their technical support and Mrs. Rosie Silva for her administrative assistance. Experiments and investigators were funded in part by grants from the National Institutes of Health (KO8NS096115 –R.C-V. and RO1HD070996, RO1HD086058 - FJN.), the Johns Hopkins University-School of Medicine Clinician Scientist Award (R.C-V) and by the Sutland-Pakula Endowment for Neonatal Research (R.C-V.).
Footnotes
Ethics statement: We confirm that any aspect of the work covered in this manuscript involving experimental animals has been conducted with the ethical approval of all relevant bodies.
Final approval: All authors have approved the final version of this manuscript.
Conflict of interest: We confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.
REFERENCES
- Aggarwal M, Burnsed J, Martin LJ, Northington FJ, Zhang J. 2014. Imaging neurodegeneration in the mouse hippocampus after neonatal hypoxia-ischemia using oscillating gradient diffusion MRI. Magn Reson Med 72(3):829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzopardi D, Strohm B, Marlow N, Brocklehurst P, Deierl A, Eddama O, Goodwin J, Halliday HL, Juszczak E, Kapellou O and others. 2014. Effects of hypothermia for perinatal asphyxia on childhood outcomes. N Engl J Med 371(2):140–9. [DOI] [PubMed] [Google Scholar]
- Baron IS, Brandt J, Ahronovich MD, Baker R, Erickson K, Litman FR. 2012. Selective deficit in spatial location memory in extremely low birth weight children at age six: the PETIT study. Child Neuropsychol 18(3):299–311. [DOI] [PubMed] [Google Scholar]
- Benes FM, Berretta S. 2001. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology 25(1):1–27. [DOI] [PubMed] [Google Scholar]
- Bosman LW, Heinen K, Spijker S, Brussaard AB. 2005. Mice lacking the major adult GABAA receptor subtype have normal number of synapses, but retain juvenile IPSC kinetics until adulthood. J Neurophysiol 94(1):338–46. [DOI] [PubMed] [Google Scholar]
- Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–54. [DOI] [PubMed] [Google Scholar]
- Bregestovski P, Bernard C. 2012. Excitatory GABA: How a Correct Observation May Turn Out to be an Experimental Artifact. Front Pharmacol 3:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnsed JC, Chavez-Valdez R, Hossain MS, Kesavan K, Martin LJ, Zhang J, Northington FJ. 2015. Hypoxia-ischemia and therapeutic hypothermia in the neonatal mouse brain--a longitudinal study. PLoS One 10(3):e0118889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalak LF, Sanchez PJ, Adams-Huet B, Laptook AR, Heyne RJ, Rosenfeld CR. 2014. Biomarkers for severity of neonatal hypoxic-ischemic encephalopathy and outcomes in newborns receiving hypothermia therapy. J Pediatr 164(3):468–74 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez-Valdez R, Martin LJ, Flock DL, Northington FJ. 2012. Necrostatin-1 attenuates mitochondrial dysfunction in neurons and astrocytes following neonatal hypoxia-ischemia. Neuroscience 219:192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez-Valdez R, Martin LJ, Razdan S, Gauda EB, Northington FJ. 2014. Sexual dimorphism in BDNF signaling after neonatal hypoxia-ischemia and treatment with necrostatin-1. Neuroscience 260:106–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cikriklar HI, Ekici MA, Ozbek Z, Cosan DT, Baydemir C, Yurumez Y. 2013. Investigation of the course of GFAP in blood in the initial 24 hours in rats subjected to minor head trauma. Eur Rev Med Pharmacol Sci 17(24):3391–7. [PubMed] [Google Scholar]
- Coghlan S, Horder J, Inkster B, Mendez MA, Murphy DG, Nutt DJ. 2012. GABA system dysfunction in autism and related disorders: from synapse to symptoms. Neurosci Biobehav Rev 36(9):2044–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SS, Stonestreet BS. 2014. Sex differences in behavioral outcome following neonatal hypoxia ischemia: Insights from a clinical meta-analysis and a rodent model of induced hypoxic ischemic injury. Exp Neurol 256:70–3. [DOI] [PubMed] [Google Scholar]
- Danzer E, Zhang L, Radu A, Bebbington MW, Liechty KW, Adzick NS, Flake AW. 2011. Amniotic fluid levels of glial fibrillary acidic protein in fetal rats with retinoic acid induced myelomeningocele: a potential marker for spinal cord injury. Am J Obstet Gynecol 204(2):178 e1–11. [DOI] [PubMed] [Google Scholar]
- de Vries LS, Cowan FM. 2009. Evolving understanding of hypoxic-ischemic encephalopathy in the term infant. Semin Pediatr Neurol 16(4):216–25. [DOI] [PubMed] [Google Scholar]
- Del Rio JA, Soriano E, Ferrer I. 1992. Development of GABA-immunoreactivity in the neocortex of the mouse. J Comp Neurol 326(4):501–26. [DOI] [PubMed] [Google Scholar]
- Diaz J, Abiola S, Kim N, Avaritt O, Flock D, Yu J, Northington FJ, Chavez-Valdez R. 2017. Therapeutic Hypothermia Provides Variable Protection against Behavioral Deficits after Neonatal Hypoxia-Ischemia: A Potential Role for Brain-Derived Neurotrophic Factor. Dev Neurosci 39(1–4):257–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditelberg JS, Sheldon RA, Epstein CJ, Ferriero DM. 1996. Brain injury after perinatal hypoxia-ischemia is exacerbated in copper/zinc superoxide dismutase transgenic mice. Pediatr Res 39(2):204–8. [DOI] [PubMed] [Google Scholar]
- Duarte IC, Castelhano J, Sales F, Castelo-Branco M. 2016. The anterior versus posterior hippocampal oscillations debate in human spatial navigation: evidence from an electrocorticographic case study. Brain Behav 6(9):e00507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte IC, Ferreira C, Marques J, Castelo-Branco M. 2014. Anterior/posterior competitive deactivation/activation dichotomy in the human hippocampus as revealed by a 3D navigation task. PLoS One 9(1):e86213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadian DG, Aicardi J, Watkins KE, Porter DA, Mishkin M, Vargha-Khadem F. 2000. Developmental amnesia associated with early hypoxic-ischaemic injury. Brain 123 Pt 3:499–507. [DOI] [PubMed] [Google Scholar]
- Graham EM, Sheldon RA, Flock DL, Ferriero DM, Martin LJ, O’Riordan DP, Northington FJ. 2004. Neonatal mice lacking functional Fas death receptors are resistant to hypoxic-ischemic brain injury. Neurobiol Dis 17(1):89–98. [DOI] [PubMed] [Google Scholar]
- Haas MA, Vickers JC, Dickson TC. 2004. Binding partners L1 cell adhesion molecule and the ezrin-radixin-moesin (ERM) proteins are involved in development and the regenerative response to injury of hippocampal and cortical neurons. Eur J Neurosci 20(6):1436–44. [DOI] [PubMed] [Google Scholar]
- Heckers S, Konradi C. 2014. GABAergic mechanisms of hippocampal hyperactivity in schizophrenia. Schizophr Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S, Jonas P. 2005. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci 8(10):1319–28. [DOI] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. 1999. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell 98(6):739–55. [DOI] [PubMed] [Google Scholar]
- Jacobs SE, Berg M, Hunt R, Tarnow-Mordi WO, Inder TE, Davis PG. 2013. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev 1:CD003311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston MV. 2005. Excitotoxicity in perinatal brain injury. Brain Pathol 15(3):234–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalueff AV. 2007. Neurobiology of memory and anxiety: from genes to behavior. Neural Plast 2007:78171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karadi K, Janszky J, Gyimesi C, Horvath Z, Lucza T, Doczi T, Kallai J, Abraham H. 2012. Correlation between calbindin expression in granule cells of the resected hippocampal dentate gyrus and verbal memory in temporal lobe epilepsy. Epilepsy Behav 25(1):110–9. [DOI] [PubMed] [Google Scholar]
- Komitova M, Xenos D, Salmaso N, Tran KM, Brand T, Schwartz ML, Ment L, Vaccarino FM. 2013. Hypoxia-induced developmental delays of inhibitory interneurons are reversed by environmental enrichment in the postnatal mouse forebrain. J Neurosci 33(33):13375–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurinczuk JJ, White-Koning M, Badawi N. 2010. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum Dev 86(6):329–38. [DOI] [PubMed] [Google Scholar]
- Le Magueresse C, Monyer H. 2013. GABAergic interneurons shape the functional maturation of the cortex. Neuron 77(3):388–405. [DOI] [PubMed] [Google Scholar]
- Lepore N, Shi Y, Lepore F, Fortin M, Voss P, Chou YY, Lord C, Lassonde M, Dinov ID, Toga AW and others. 2009. Pattern of hippocampal shape and volume differences in blind subjects. Neuroimage 46(4):949–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Khastgir A, McCauley JM, Dunn ST, Morrissey JH, Christakos S, Hughes MR, Bourdeau JE. 1996. RT-PCR microlocalization of mRNAs for calbindin D28k and vitamin D receptor in the murine nephron. Am J Physiol 270(4 Pt 2):F677–81. [DOI] [PubMed] [Google Scholar]
- Lucas EK, Jegarl A, Clem RL. 2014. Mice lacking TrkB in parvalbumin-positive cells exhibit sexually dimorphic behavioral phenotypes. Behav Brain Res 274:219–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhmann HJ, Prince DA. 1991. Postnatal maturation of the GABAergic system in rat neocortex. J Neurophysiol 65(2):247–63. [DOI] [PubMed] [Google Scholar]
- Maguire EA, Gadian DG, Johnsrude IS, Good CD, Ashburner J, Frackowiak RS, Frith CD. 2000. Navigation-related structural change in the hippocampi of taxi drivers. Proc Natl Acad Sci U S A 97(8):4398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic SN, Murasko DM. 1993. Anesthesia inhibits interferon-induced natural killer cell cytotoxicity via induction of CD8+ suppressor cells. Cell Immunol 151(2):474–80. [DOI] [PubMed] [Google Scholar]
- Marlow N, Rose AS, Rands CE, Draper ES. 2005. Neuropsychological and educational problems at school age associated with neonatal encephalopathy. Arch Dis Child Fetal Neonatal Ed 90(5):F380–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ. 1999. Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol 58(5):459–71. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C. 1998. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: A perspective on the contributions of apoptosis and necrosis. Brain Res Bull 46(4):281–309. [DOI] [PubMed] [Google Scholar]
- Mudrick LA, Baimbridge KG. 1989. Long-term structural changes in the rat hippocampal formation following cerebral ischemia. Brain Res 493(1):179–84. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Chavez-Valdez R, Graham EM, Razdan S, Gauda EB, Martin LJ. 2011. Necrostatin decreases oxidative damage, inflammation, and injury after neonatal HI. J Cereb Blood Flow Metab 31(1):178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northington FJ, Ferriero DM, Graham EM, Traystman RJ, Martin LJ. 2001. Early Neurodegeneration after Hypoxia-Ischemia in Neonatal Rat Is Necrosis while Delayed Neuronal Death Is Apoptosis. Neurobiol Dis 8(2):207–19. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Koehler RC, Traystman RJ, Martin LJ. 1996. Nitric oxide synthase 1 and nitric oxide synthase 3 protein expression is regionally and temporally regulated in fetal brain. Dev Brain Res 95:1–14. [DOI] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. 2005. Astrocyte activation and reactive gliosis. Glia 50(4):427–34. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29(9):e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. 2004. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper--Excel-based tool using pair-wise correlations. Biotechnol Lett 26(6):509–15. [DOI] [PubMed] [Google Scholar]
- Pinto JG, Hornby KR, Jones DG, Murphy KM. 2010. Developmental changes in GABAergic mechanisms in human visual cortex across the lifespan. Front Cell Neurosci 4:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamoorthy P, Shi H. 2014. Ischemia induces different levels of hypoxia inducible factor-1alpha protein expression in interneurons and pyramidal neurons. Acta Neuropathol Commun 2:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichel JM, Nissel S, Rogel-Salazar G, Mederer A, Kafer K, Bedenk BT, Martens H, Anders R, Grosche J, Michalski D and others. 2014. Distinct behavioral consequences of short-term and prolonged GABAergic depletion in prefrontal cortex and dorsal hippocampus. Front Behav Neurosci 8:452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmaso N, Jablonska B, Scafidi J, Vaccarino FM, Gallo V. 2014. Neurobiology of premature brain injury. Nat Neurosci 17(3):341–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanches EF, Arteni N, Nicola F, Aristimunha D, Netto CA. 2015. Sexual dimorphism and brain lateralization impact behavioral and histological outcomes following hypoxia-ischemia in P3 and P7 rats. Neuroscience 290:581–93. [DOI] [PubMed] [Google Scholar]
- Shankaran S, Laptook AR, Ehrenkranz RA, Tyson JE, McDonald SA, Donovan EF, Fanaroff AA, Poole WK, Wright LL, Higgins RD and others. 2005. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med 353(15):1574–84. [DOI] [PubMed] [Google Scholar]
- Shankaran S, Pappas A, McDonald SA, Vohr BR, Hintz SR, Yolton K, Gustafson KE, Leach TM, Green C, Bara R and others. 2012. Childhood outcomes after hypothermia for neonatal encephalopathy. N Engl J Med 366(22):2085–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Dietz RM, Cruz-Torres I, Strnad F, Garske AK, Moreno M, Venna VR, Quillinan N, Herson PS. 2016. Extended therapeutic window of a novel peptide inhibitor of TRPM2 channels following focal cerebral ischemia. Exp Neurol 283(Pt A):151–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AL, Alexander M, Rosenkrantz TS, Sadek ML, Fitch RH. 2014. Sex differences in behavioral outcome following neonatal hypoxia ischemia: insights from a clinical meta-analysis and a rodent model of induced hypoxic ischemic brain injury. Exp Neurol 254:54–67. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. 2010. Astrocytes: biology and pathology. Acta Neuropathol 119(1):7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P, Klausberger T. 2005. Defined types of cortical interneurone structure space and spike timing in the hippocampus. J Physiol 562(Pt 1):9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens HE, Jiang GY, Schwartz ML, Vaccarino FM. 2012. Learning and memory depend on fibroblast growth factor receptor 2 functioning in hippocampus. Biol Psychiatry 71(12):1090–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone BS, Zhang J, Mack DW, Mori S, Martin LJ, Northington FJ. 2008. Delayed neural network degeneration after neonatal hypoxia-ischemia. Ann Neurol 64(5):535–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo G, Kartarova Z, Hoertnagl B, Somogyi R, Sperk G. 2000. Differential regulation of adult and embryonic glutamate decarboxylases in rat dentate granule cells after kainate-induced limbic seizures. Neuroscience 100(2):287–95. [DOI] [PubMed] [Google Scholar]
- Takesian AE, Hensch TK. 2013. Balancing plasticity/stability across brain development. Prog Brain Res 207:3–34. [DOI] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT. 2014. Assessing spatial learning and memory in rodents. ILAR J 55(2):310–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell J, Hanscom M, Shalon Edwards N, McKenna MC, McCarthy MM. 2016. Sex differences in cell genesis, hippocampal volume and behavioral outcomes in a rat model of neonatal HI. Exp Neurol 275 Pt 2:285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.